

Sporadic cerebral amyloid angiopathy is a common small vessel disease and a largely untreatable cause of ICH and contributor to age-related cognitive decline and Alzheimer’s disease. Charidimou et al. provide an interdisciplinary review of emerging concepts in the field, illustrating mechanisms associated with amyloid cerebrovascular pathology and neurological dysfunction.
Keywords: cerebral amyloid angiopathy, small vessel disease, Alzheimer’s disease, perivascular drainage, intracerebral haemorrhage
Abstract
Sporadic cerebral amyloid angiopathy is a common, well-defined small vessel disease and a largely untreatable cause of intracerebral haemorrhage and contributor to age-related cognitive decline. The term ‘cerebral amyloid angiopathy’ now encompasses not only a specific cerebrovascular pathological finding, but also different clinical syndromes (both acute and progressive), brain parenchymal lesions seen on neuroimaging and a set of diagnostic criteria—the Boston criteria, which have resulted in increasingly detected disease during life. Over the past few years, it has become clear that, at the pathophysiological level, cerebral amyloid angiopathy appears to be in part a protein elimination failure angiopathy and that this dysfunction is a feed-forward process, which potentially leads to worsening vascular amyloid-β accumulation, activation of vascular injury pathways and impaired vascular physiology. From a clinical standpoint, cerebral amyloid angiopathy is characterized by individual focal lesions (microbleeds, cortical superficial siderosis, microinfarcts) and large-scale alterations (white matter hyperintensities, structural connectivity, cortical thickness), both cortical and subcortical. This review provides an interdisciplinary critical outlook on various emerging and changing concepts in the field, illustrating mechanisms associated with amyloid cerebrovascular pathology and neurological dysfunction.
Introduction
Cerebrovascular deposition of amyloid-β (also known as cerebral amyloid angiopathy, CAA) is a strikingly common finding in elderly people and a major cause of spontaneous intracerebral haemorrhage (ICH), as well as an important contributor to age-related cognitive decline (Viswanathan and Greenberg, 2011; Boulouis et al., 2016). However, it is not a long time since CAA was considered a rather obscure pathological curiosity and innocent bystander in the elderly brain, often accompanying Alzheimer's disease. Despite the close molecular relationship between the two entities, CAA remains clinically distinct from Alzheimer disease, but has the potential to link cerebrovascular and neurodegenerative pathways in the ageing brain (Cordonnier and van der Flier, 2011). The CAA story is remarkable in that unlike most neurological disorders, the recognition of the underlying pathology preceded by many years the association with clinical syndromes and disease. In 1954, Stefanos Pantelakis from the University Psychiatric Clinic in Bel-Air, Switzerland, highlighted CAA as a distinct entity (Pantelakis, 1954). In his landmark paper, Pantelakis raised the hypothesis that vascular amyloid may originate in the brain (in line with modern concepts of amyloid pathophysiology) and—among other things—described what are still the pathological hallmarks of CAA: (i) preferential involvement of small arterioles and capillaries of the leptomeninges and cerebral cortex, without necessarily involving adjacent parenchymal; (ii) topographical distribution favouring posterior lobar brain regions (especially the occipital lobes); (iii) lack of involvement of white matter small vessels; (iv) association with increased age and dementia; (v) lack of association with hypertension and arteriosclerosis (the other main small vessel disease, which preferentially affects the basal ganglia and brainstem); and (vi) lack of any link with systemic amyloidosis.
Since these early descriptions, understanding of the pathophysiological mechanisms and clinical imaging manifestations of CAA has increased substantially. CAA denotes not only a specific cerebrovascular pathological trait, but also a clinical syndrome (or syndromes), and a set of clinical imaging diagnostic criteria (the Boston criteria), which have resulted in increasingly detected disease during life (Linn et al., 2010; Charidimou et al., 2012a). Over the past decade we have also come to realise that the integrity of cerebral microvessels is critical for solute efflux from the interstitial fluid of the brain, as these molecules pass from brain into the perivascular drainage system and the systemic lymphatic circulation (Carare et al., 2013). Cerebrovascular accumulation of amyloid can thus be viewed in some ways ‘as a canary in a coal mine’, an indicator of perivascular drainage failure (Carare et al., 2013; Keable et al., 2016). This process may set in motion a damaging disease process that both further exacerbates the clearance defects and triggers clinically relevant haemorrhagic (Zhao et al., 2015) and ischaemic brain injury (Reijmer et al., 2016b).
This review update focuses on selected recent topics that have not been extensively covered in previous reviews and illustrate emerging and changing concepts in sporadic CAA, including the neurological effects and underlying biology of the disease. Given the fast-moving CAA research landscape (Charidimou et al., 2016a) in the current paper we are not covering all the clinical and imaging aspects of CAA in detail, given the more specific published reviews (Greenberg et al., 2009b, 2014; Pantoni, 2010; Smith et al., 2012b; Reijmer et al., 2016b). Instead, emphasis is given to concepts, findings and theoretical frameworks of the disease, with examples from the literature to illustrate these points. Finally, our paper focusses on the more common sporadic CAA—for the rare genetic forms of cerebrovascular amyloid accumulation and CAA-related inflammation, please see other reviews and recent publications for further information (Revesz et al., 2009; Charidimou et al., 2012a; Auriel et al., 2016).
Cerebral amyloid angiopathy clinical aspects
Pathologically-defined CAA is common in the elderly and advancing age is the strongest known risk factor for sporadic CAA (Vinters, 1987). Population-based autopsy studies indicate a CAA prevalence of 20–40% in non-demented, and 50–60% in demented elderly populations (age range for both groups 80–90 years) (Keage et al., 2009). For severe CAA, prevalence in demented and non-demented elderly groups is 30–40% and 7–24%, respectively (Keage et al., 2009). In Alzheimer’s disease brains, CAA is identified in an estimated 85–95% of the cases when sought diligently (Kalaria and Ballard, 1999; Jellinger, 2002). Only a relatively small proportion of these older individuals are diagnosed during life with CAA-related clinical symptomatology (acute or progressive). The sentinel clinical presentations of CAA include spontaneous lobar ICH, cognitive impairment and dementia (either post ICH or in patients without stroke) and transient focal neurological episodes (‘amyloid spells’) often associated with acute convexity subarachnoid haemorrhage or cortical superficial siderosis. The relative frequency of the different CAA clinical presentations is hard to assess, as it is heavily dependent on making an accurate diagnosis of underlying CAA in different settings using appropriate MRI sequences (including blood sensitive sequences). The association of these presentations with objective assessments of clinical frailty, comorbidities or chronological age per se are under investigation. The key neuroimaging signatures of the disease on clinical MRI include multiple strictly lobar cerebral microbleeds, cortical superficial siderosis, white matter hyperintensities, cortical microinfarcts and MRI-visible perivascular spaces in the centrum semiovale (Greenberg et al., 2014). An overview of the key CAA clinical presentations and the significance/role of different MRI markers of small vessel disease in each of these based on the totality of current evidence is provided in Table 1.
Table 1.
Overview of the sentinel clinical presentations of CAA and the role of key MRI small vessel disease biomarkers and imaging signatures of the disease within each of these
| Sentinel clinical presentations of CAA | ||||
|---|---|---|---|---|
| CAA/SVD MRI biomarkers | Spontaneous lobar ICH | Post-ICH dementia | TFNEs (‘amyloid spells’) | MCI/dementia (without ICH) |
| Lobar CMBs |
|
|
|
|
| CSS |
|
|
|
−Significance? |
| Acute cSAH | − | − |
|
− |
| WMH |
|
|
|
|
| CSO-EPVS |
|
|
|
|
| Small DWI lesions |
|
? |
|
|
| Cortical microinfarcts | ? | ? | ? |
|
+/− denotes the relative frequency or absence and overall significance of each of the markers, respectively. The clinical role of MRI biomarkers was considered in the following areas: diagnosis, clinical function, severity/progression of CAA and clinical prognosis.
+ = low; ++ = moderate; +++ = high frequency; − = rare; ? = unknown role; CMB = cerebral microbleed; cSAH = cortical subarachnoid haemorrhage; CSO-EPVS = centrum semioviale-enlarged periventricular space; DWI = diffusion weighted imaging; rICH = recurrent ICH; TFNEs = transient focal neurological episodes; WMH = white matter hyperintensity.
Ratings represent the authors’ overall consensus based on the literature cited in the corresponding sections of the text and clinical practice.
Dementia and cognitive impairment in cerebral amyloid angiopathy
Cognitive impairment in CAA was first reported over two decades ago (Gray et al., 1985; Greenberg et al., 1993). Dissecting the independent impact of CAA on cognition, however, is confounded by co-existing Alzheimer’s disease and other age-related pathologies (e.g. sporadic non-amyloid microangiopathy). Recent studies in older community dwelling persons have suggested an association between moderate-to-severe CAA pathology and lower perceptual speed and episodic memory (Arvanitakis et al., 2011). In these cohorts, CAA is a common finding and appears to independently contribute to the level of cognitive deficit beyond Alzheimer’s disease or other accompanying pathologies (Pfeifer et al., 2002; Boyle et al., 2015). In one community-based cohort, individuals harbouring CAA not only had increased risk of dementia [odds ratio (OR): 1.237; 95% confidence interval (CI): 1.082–1.414] but a faster rate of decline in both global and domain-specific cognition (Boyle et al., 2015).
In a hospital-based setting, patients with CAA without dementia evaluated for stroke or stroke-like symptoms appeared to be at high risk for developing dementia (Moulin et al., 2016). Interestingly, in this study, the history of previous CAA-related ICH was not associated with the degree of cognitive deficits. Indeed previous work has suggested that cognitive decline in CAA may precede ICH (Viswanathan et al., 2008; Cordonnier et al., 2010; Xiong et al., 2016b), thus indicating that CAA-related cognitive decline may occur independently of symptomatic ICH. In another hospital-based study, patients with CAA without dementia evaluated for stroke or stroke-like symptoms appeared to perform significantly worse on nearly all tests in a standard neurocognitive battery compared to a group of healthy controls with similar age and education level (Xiong et al., 2016a). In this study, these deficits were clearly present even though the majority of patients did not have self-reported cognitive concerns or cognitive deficits (Xiong et al., 2016a). In the analyses of the relationship between these cognitive deficits and neuroimaging biomarkers, lower total brain volume was associated with slower processing speed and worse executive function, and white matter hyperintensity volume was related to executive function in CAA patients (Xiong et al., 2016a).
Despite the clinical observation of cognitive impairment in CAA, the frequency, severity, and specific cognitive profile of CAA patients are not well understood (Schrag and Kirshner, 2016). To address this question, a pilot study reported neuropsychological and neuroimaging data from an ongoing prospective cohort (Functional Assessment of Vascular Reactivity in CAA) (Case et al., 2016). This study compared the neuropsychological test results in non-demented CAA participants (n = 34) to participants with dementia caused by Alzheimer’s disease (n = 16), mild cognitive impairment (n = 69), and ischaemic stroke (n = 27) (matched for stroke severity to CAA participants). The mean test scores for CAA patients were significantly lower than norms for memory, executive function, and processing speed. A large proportion of these CAA patients (79%) fulfilled criteria for mild cognitive impairment based on low cognitive performance and cognitive concerns. Compared to Alzheimer’s disease, CAA patients had similarly low executive function scores, but relatively preserved memory. CAA participants’ scores were lower than those of the ischaemic stroke group for executive function and processing speed. Lower processing speed scores in CAA were associated with higher white matter hyperintensity volumes (Case et al., 2016).
Taken together, current evidence suggests that mild cognitive impairment and dementia are very prevalent in clinically diagnosed CAA patients. The overall cognitive profile of CAA is more similar to that seen in classic vascular cognitive impairment than Alzheimer’s disease (i.e. executive dysfunction and impaired processing speed with relatively preserved episodic memory). White matter ischaemic lesions may underlie some of the impaired processing speed seen in CAA. CAA-related dementia and vascular cognitive impairment are likely the result of both small, spatially distributed brain injuries including cerebral microbleeds (Moulin et al., 2016), cortical microinfarcts, and altered structural connectivity. In support of this hypothesis, a recent study examining brain networks in CAA as a surrogate measure of global small-vessel pathology showed that lower global network efficiency was independently related to worse performance on tests of processing speed and executive functioning (Reijmer et al., 2015).
Clinical imaging expression and spectrum of cerebral amyloid angiopathy
CAA is most often recognized in life by symptomatic, spontaneous ICH, preferentially affecting cortical-subcortical (lobar) regions (especially the occipital and posterior temporal lobes) (Rosand et al., 2005). These individuals have predominantly severe CAA (Alonzo et al., 1998), with small burden of neurofibrillary tangles and senile plaques on neuropathology (Charidimou et al., 2015c). Multiple recurrent CAA-related haematomas can occur over months or years (at a rate of recurrence on the order of 10% per year) (Biffi et al., 2010), with progressive neurological decline. CAA may also be an important risk factor or cause for ICH related to oral anticoagulation use and thrombolysis-related haematomas in acute ischaemic stroke (Charidimou et al., 2015d; Mattila et al., 2015). Reliable assessment of an individual’s haemorrhagic burden over time and prevention of future ICH is thus a key component in the clinical management of CAA patients (see below).
In this context, characterization of the expanding clinical imaging spectrum of CAA has been refined substantially in the past years with the broader availability of brain MRI (Boulouis et al., 2016). In addition to lobar ICH, other characteristic MRI biomarkers of CAA now include strictly lobar microbleeds, cortical superficial siderosis, white matter hyperintensities (especially posterior-predominant), MRI-visible perivascular spaces in the cerebral white matter and cortical microinfarcts (Fig. 1). These neuroimaging markers probably reflect related but distinct aspects of CAA pathophysiology (Charidimou and Jager, 2014). An important advance in the field has been the recent publication of international consensus standards for describing and reporting many of these small vessel disease lesion types (Wardlaw et al., 2013). Multiple strictly lobar microbleeds detected on T2*/SWI (susceptibility-weighted imaging) MRI are accepted as one of the hallmark biomarkers for CAA presence and are useful for CAA diagnosis within the Boston criteria (Knudsen et al., 2001; Linn et al., 2010) and to assess disease evolution (Pasquini et al., 2016). Strictly lobar cerebral microbleeds within these criteria have a high positive predictive value for CAA even in individuals without ICH presentations in a hospital-based setting (specificity >90% and positive predictive value >87%) (Martinez-Ramirez et al., 2015). This is of particular clinical relevance, as CAA is increasingly recognized to present without major haemorrhage. Lobar microbleeds also frequently occur among presumably healthy people as a result of previously clinically silent CAA (Mesker et al., 2011), but also in the absence of CAA, yielding low overall specificity in this setting (Martinez-Ramirez et al., 2015). However, the detection of cerebral microbleeds in different clinical settings raises thorny clinical dilemmas regarding their biological significance and how they might affect treatment decisions, especially regarding antithrombotic drug use (Greenberg et al., 2009b; Wang et al., 2014). Clinical radiological criteria for CAA-related inflammation have also been validated (Auriel et al., 2016), allowing non-invasive diagnosis of this potentially treatable meningoencephalitis syndrome (Eng et al., 2004).
Figure 1.

Schematic representation of the spectrum of haemorrhagic and ischaemic manifestations of sporadic CAA, visible on structural MRI, including three key common neuropathological lesions seen in CAA brains. (A) A lobar cerebral microbleed identified on post-mortem 7 T MRI in the brain tissue of an 81-year-old male with dementia and severe CAA on pathology. On haematoxylin and eosin stain, brown and yellow deposits representing haemosiderin, and haematoidin, are seen, indicating that this haemorrhage is not chronic but subacute. (B) Prussian blue stain from an area corresponding to cortical superficial siderosis on in vivo MRI of a patient with advanced CAA. Blue deposits corresponding to haemosiderin are seen in the subarachnoid space (SAS) surrounding a leptomeningeal arteriole (which shows a vessel-within-vessel appearance) and in the superficial layers of the cortex. (C) A cortical microinfarct identified on histopathological examination. Note the severe vascular amyloid deposition (dark brown: immunostained for amyloid-β) in both the overlying leptomeningeal small vessels of different diameters and two cortical arterioles in the vicinity of the microinfarct. Bottom: Imaging CAA in clinical practice. 1.5 T brain MRI sequences. (D) Seventy-five-year-old male; initial evaluation of stereotyped episodes of right sided of tingling in the past year, axial section of SWI demonstrating a focus of cortical superficial siderosis on both banks of the left central sulcus, and multiple strictly lobar microbleeds (arrowheads in magnification). (E) Eighty-eight-year-old female with history of cognitive complaints, 6 months after initial imaging demonstrating multiple lobar microbleeds. Persistent headache and recent episode of left hand numbness and weakness; axial section of FLAIR sequence showing a linear hyperintensity in the right central sulcus, corresponding to acute subarachnoid blood. (F) Seventy-seven-year-old male otherwise meeting Boston criteria for CAA; coronal section (top) of T1-weighted, and axial section of T2-weighted sequences showing multiple dilated perivascular spaces (magnification) in the centrum semiovale, following the path of small calibre arteries. Diffuse widening of cerebral sulci due to marked cortical atrophy can also be appreciated. (G) Sixty-five-year-old male with acute onset of right hemiplegia and impaired consciousness at hospital arrival. Large right lobar (mostly parietal) ICH ruptured into the convexities and subarachnoid space. Contralateral multiple strictly lobar microbleeds are seen. EPVS = enlarged perivascular space; CMBs = cerebral microbleeds; sSAH = spinal subarachnoid haemorrhage; WMH = white matter hyperintensity.
Recent data have implicated acute or chronic haemorrhage within or adjacent to the cortical sulci (often described as cortical superficial siderosis when chronic or as sulcal subarachnoid haemorrhage when acute) as key haemorrhagic neuroimaging signatures of CAA (Charidimou et al., 2015b). They likely reflect repeated episodes of blood leaking into the subarachnoid space from brittle and fragile CAA affected vessels. Siderosis is emerging as perhaps the most clinically relevant manifestation of the disease: (i) it is a trigger of transient focal neurological symptoms (‘amyloid spells’) (Greenberg et al., 1993; Charidimou et al., 2012b), expanding the CAA clinical imaging spectrum (Raposo et al., 2011; Calviere et al., 2016); (ii) it has been shown to carry a very high risk of future symptomatic lobar ICH (Linn et al., 2010; Charidimou et al., 2013c) with important implications for CAA clinical care, such as strict blood pressure control (Gorelick et al., 2011; Biffi et al., 2015; Hemphill et al., 2015; Carcel et al., 2016) and avoiding antithrombotics unless otherwise strongly indicated (Biffi et al., 2010; Charidimou et al., 2012a); and (iii) it may be an independent risk factor for new onset dementia after ICH (Moulin et al., 2016). Of note, siderosis exerts these effects independent of, and above any effect of coincident microbleeds (Greenberg et al., 2004; Biffi et al., 2010). Recently, consensus standards for rating and reporting cortical superficial siderosis have been suggested and are encouraged for use in observational and clinical studies (Charidimou et al., 2015b). The emerging key role of cortical superficial siderosis in neurological dysfunction in CAA might be linked with the neuropathological observation of more severe leptomeningeal CAA compared to parenchymal and between superficial and deep cortical layers in any given brain (Kovari et al., 2013).
Role of molecular imaging in cerebral amyloid angiopathy
While CAA appears detectable by molecular amyloid imaging (Klunk et al., 2004; Bacskai et al., 2007; Greenberg et al., 2008) the clinical diagnostic utility of amyloid PET in suspected CAA remains undefined. Non-demented patients with CAA show higher global Pittsburgh compound B (PiB)-PET retention when compared to healthy control subjects and higher occipital-to-global PiB ratios compared to patients with Alzheimer’s disease (Johnson et al., 2007; Ly et al., 2010). However, due to the frequent occurrence of high PiB-PET uptake in the healthy elderly likely reflecting incipient Alzheimer disease, specificity is limited; one study reported sensitivity of 91%, and specificity of 55% compared to healthy controls (Baron et al., 2014). A negative PiB scan appears to rule out severe CAA and may in the future have clinical research implications for prognostication, treatment decisions, and patient selection for drug trials. A small study recently tested the clinical relevance of florbetapir-PET, another amyloid tracer, in distinguishing CAA-related lobar ICH from hypertensive ICH (Gurol et al., 2016). In a study sample of cognitively normal patients, including 10 lobar haemorrhages with a clinical imaging diagnosis of probable CAA and nine with deep haemorrhages and without evidence for CAA of similar age (66.9 versus 67.1 years), sex, and leukoaraiosis volumes, the mean global cortical florbetapir uptake was higher in CAA versus non-CAA (P = 0.001). Based on visual rating for positive/negative florbetapir, all 10 patients with CAA versus 1/9 non-CAA patients were classified as positive (sensitivity: 100%; 95% CI: 66%–100% and specificity: 89%; 95% CI: 51–99% for determination of probable CAA) (Gurol et al., 2016). The general conclusion is similar to previous studies using PiB-PET: a negative scan almost certainly rules out probable CAA—at least in these specific and well-defined comparison groups used in the study, ‘probable’ CAA with haemorrhage and deep hypertensive-related haemorrhage.
The clinical usefulness of amyloid-PET for CAA diagnosis in everyday practise currently remains limited and under investigation. Further larger prospective studies are needed to provide external validation, and test whether amyloid-PET can be useful for detecting CAA pathology in cases where the diagnosis is uncertain. Examples would include cases with mixed location of ICH (both lobar and deep), single lobar haemorrhage without microbleeds (labelled as ‘possible’ CAA) as well as cases with microbleeds or cortical superficial siderosis without any haematomas. A large prospective study of such uncertain cases with long follow-up is required to determine the sensitivity and specificity of amyloid imaging in these settings and at earlier stages of suspected CAA. Development of a molecular imaging tracer specific for vascular amyloid would be a major step forward for the field and preclinical studies continue to explore possible candidates (Jia et al., 2014, 2015). Other novel methods, such as early-phase PiB-PET (Farid et al., 2015) and tau imaging, might help test similarities and differences between CAA and Alzheimer’s disease in living individuals (Villemagne et al., 2015), although given the strong overlap between these pathologies, it is unlikely that any technique in isolation will fully distinguish these two entities.
Amyloid imaging has recently become an important tool in CAA research to explore mechanisms and test spatial and quantitative correlations between proposed CAA-related brain lesions and the inciting pathology (i.e. vascular amyloid). In a cross-sectional study, higher PiB retention was detected at the sites (i.e. shells concentrically surrounding the bleeds) of lobar microbleeds in CAA when compared to other locations with high-probability to show CAA-related microbleeds (Dierksen et al., 2010). Using a similar approach, in a longitudinal analysis of follow-up MRIs (median of 19 months after baseline), sites of future microbleeds demonstrated increased PiB retention relative to simulated lesions placed according to a probability density map (distribution volume ratio 1.34 versus 1.14, P < 0.0001) with fall-off at increasing distances from sites of future microbleeding (Gurol et al., 2012). Despite the limitations imposed by the relatively poor spatial resolution of amyloid-PET, resulting in some imprecision for co-localizing PiB retention and haemorrhage location, these studies provide some proof of the concept that vascular amyloid deposition might be a key contributor to lobar microbleeds and macrobleeds. However, the exact mechanisms for these associations remain uncertain. Also, another study showed a positive correlation between leukoaraiosis volume and global PiB retention in patients with CAA but not in healthy controls or patients with Alzheimer’s disease, suggesting that predominanlty vascular amyloid deposition rather than parenchymal might drive this association (Gurol et al., 2013). More recently, a study suggested that MRI-visible high degree centrum semiovale perivascular spaces, a novel CAA-related MRI marker, were correlated with a higher median amyloid PiB retention across a wide range of cerebrovascular amyloid deposition, after adjusting for age, microbleeds and white matter hyperintensities (Charidimou et al., 2015a).
Cerebral amyloid angiopathy: the search for clinical trials surrogate markers
Although conventional CAA neuroimaging biomarkers are currently used for diagnosis and clinical monitoring, none has yet emerged as a valid clinical trials surrogate marker (Greenberg et al., 2014). This apparent paradox might be partly due to the fact that these haemorrhagic and non-haemorrhagic structural focal lesions used clinically are only the tip of the iceberg and fail to capture the cumulative large-scale impact of the disease. Also, the MRI-histopathological basis of small vessel disease neuroimaging biomarkers still needs to be defined, representing a remarkable lacune in the field (Gouw et al., 2011). For example, lesion-by-lesion analysis has been reported for fewer than 100 microbleeds (Fazekas et al., 1999; Tatsumi et al., 2008; Schrag et al., 2010) often using relatively insensitive T2*-weighted techniques. Of note, the large numbers of cerebral microbleeds noted by MRI are rarely seen in autopsy brain specimens, a disconnect that likely reflects the ability of T2*-weighted MRI to fully sample the brain to an extent not achievable by histology. Although microbleeds are strongly associated with advanced CAA and it is commonly assumed that amyloid deposition in the walls of cortical vessels directly causes microbleeding, a recent detailed ex vivo 7 T MRI study challenged this notion. In 7/19 identified microbleeds, the presumed involved vessel could be identified on histopathological sectioning. Only one of these vessels was positive for amyloid-β at the site of rupture. Moreover, the density of amyloid-β positive cortical vessels was lower in areas surrounding microbleeds, compared to control areas (van Veluw et al., 2017). Extremely limited data exist for perivascular spaces and siderosis. Brain banks (comprised of large brain donation programmes and collaborative initiatives) will be essential, in combination with new research techniques, in validating biomarkers in the field (Samarasekera et al., 2013; McAleese et al., 2016; Skrobot et al., 2016).
For CAA-related cognitive impairment, a major reason that no single neuroimaging surrogate marker has emerged is the mismatch between the lesion types that are most MRI detectable versus those that are actually numerous enough to substantially damage brain connections and mediate cognitive dysfunction. Cerebral microbleeds or lacunes of presumed vascular origin (Wardlaw et al., 2013) for example, are readily visualized by MRI, but their total burden is typically only one to two per brain (Longstreth et al., 1998; Vernooij et al., 2007, 2008) in community dwelling elderly, and <10–20 even in brains with advanced small vessel disease (Greenberg et al., 2004; Viswanathan et al., 2006a; Herve et al., 2009). Cerebral microinfarcts hypothesized to play a key role in vascular cognitive impairment (Smith et al., 2012b), conversely, are estimated to number in the hundreds or thousands per CAA brain (Westover et al., 2013; Auriel et al., 2015), but at mean diameter of ∼200 µm (Okamoto et al., 2009; Smith et al., 2012a; Westover et al., 2013), are largely invisible to conventional MRI.
Finally, haemorrhagic and non-haemorrhagic structural lesions discussed seem to represent vascular-mediated injury to the brain rather than abnormalities of the vessels themselves. Thus, they probably measure late and irreversible steps in the postulated pathways leading from vascular dysfunction to brain injury and clinical dysfunction.
Neuropathological aspects of cerebral amyloid angiopathy and differences with ‘hypertensive arteriopathy’
CAA results from a chronic degenerative process by which the media of parenchymal arterioles undergoes progressive loss of its smooth muscle cells with simultaneous accumulation of an eosinophilic hyaline material (Attems et al., 2011), mostly composed of the more soluble, amyloid-β40 species. This is in contrast to amyloid plaques found in Alzheimer’s disease, predominantly composed of amyloid-β42 species. The vessels affected by amyloid-β can show secondary vasculopathic changes, including fibrinoid necrosis, loss of smooth muscle cells, wall thickening, microaneurysm formation, and perivascular blood breakdown products deposition (Love et al., 2014). A specific and important subtype of CAA is capillary CAA, which has received lots of attention recently (Attems et al., 2011). This forms the basis for the CAA classification into two pathological types: CAA type 1, characterized by amyloid in cortical capillaries (with or without involvement of other vessels), and CAA type 2, where amyloid deposits are restricted to leptomeningeal and cortical arteries, but not capillaries (Attems et al., 2011). Recently, the neuropathological and clinical characteristics of capillary CAA were investigated for the first time in a subset (n = 300; aged ≥ 85 years) of the Vantaa 85 prospective population-based study (Makela et al., 2015). The authors reported that capillary CAA was most frequent in the occipital lobe, while CAA type1 was associated with the severity of CAA overall (P < 0.001), dementia (P < 0.001), severe Alzheimer’s-type neuropathology (P-value 0.09 for CERAD C and 0.017 for Braak stages V-VI), and APOE ɛ4 (but not ɛ2) allele carrier status (P < 0.001) (Makela et al., 2015). Immunohistochemical analysis in one study revealed that capillary CAA is associated with altered expression of sphingolipids, likely reflecting inflammatory pathways involvement (de Wit et al., 2016).
When CAA is noted in the subarachnoid space, the amyloid deposits are usually adventitial rather than medial in the walls of larger affected arterioles in a chunky, spiral-like fashion, suggesting they may have resulted from aggregates of amyloid-β that came to lodge in arterial adventitia through the perivascular drainage system (Vinters, 2015). The distribution of CAA pathology shows a characteristic patchy pattern (Vinters, 1987), whereby foci of vessels severely affected by CAA may be adjacent to other vessel segments with mild or absent amyloid-β deposition (Vinters, 1987; Attems et al., 2011), contributing to the heterogeneity of the disease. The severity and extent of histological changes provide the basis of several CAA neuropathological scoring systems (Vonsattel et al., 1991b; Olichney et al., 1995; Thal et al., 2003), each with strengths and limitations (Attems, 2005; Attems et al., 2011). Recently, a standardized consensus neuropathological protocol for rating CAA was developed (Love et al., 2014), potentially allowing comparison of pathological data across centres.
In contrast to CAA, which is relatively easy to define, the other common subtype of age-related small vessel disease remains less clearly defined at a diagnostic or molecular level and is broadly described as ‘hypertensive arteriopathy’ (Pantoni, 2010). The umbrella term ‘hypertensive arteriopathy’ groups together a spectrum of sporadic non-amyloid small vessel disease pathologies often associated with advanced age (but not clearly age-driven), hypertension (though the disease is not necessarily, or even often, related specifically to hypertension), diabetes mellitus, and other common vascular risk factors (Pantoni, 2010). Hypertensive arteriopathy is characterized pathologically by collagenous thickening of the vessel wall with narrowing of the lumen and progressive loss of smooth muscle, and sometimes by exudation of fibrin and other serum proteins or by scanty mural deposition of lipid (Pantoni, 2010). Different pathological subtypes include arteriolosclerosis, fibrinoid necrosis, and lipohyalinosis (Pantoni, 2010; Charidimou et al., 2016b). The term ‘sporadic non-amyloid microangiopathy’ has been recently suggested as a more accurate alternative to group together these diseases (Charidimou et al., 2016b).
Despite the limitations in definitions and the lack of widely accepted terms for this small vessel pathology, the rationale for making a clear distinction between CAA and sporadic non-amyloid microangiopathy is 2-fold. First, the two pathological processes are predominantly found in small vessels of different brain areas. In contrast to CAA, which typically affects the small arteries and arterioles in the cerebral cortex and overlying leptomeninges, sporadic non-amyloid microangiopathy predominantly affects the small perforating end arteries of the deep grey nuclei and deep white matter (Pantoni, 2010; Charidimou et al., 2016b). In parallel with this distribution of the underlying pathology, sporadic non-amyloid microangiopathy is commonly associated with cerebral microbleeds and spontaneous ICH in deep brain regions (e.g. basal ganglia, thalamus, and brainstem), whereas CAA is characterized by cerebral microbleeds and spontaneous ICH in a lobar distribution (cortical–subcortical), as well as cortical superficial siderosis. This allows for the clinical distinction of the two disorders based on the Boston diagnostic criteria (Knudsen et al., 2001; Smith and Greenberg, 2003; Linn et al., 2010; Martinez-Ramirez et al., 2015). The key neuropathological, clinical and imaging differences of CAA versus sporadic non-amyloid microangiopathy are summarized in Table 2. Of major clinical relevance, the two small vessel diseases have a different natural history and inherently distinct recurrent haemorrhagic risk in patients presenting with ICH: CAA-related lobar ICH carries a significantly higher risk for recurrence compared to deep ICH related to sporadic non-amyloid microangiopathy (Hill et al., 2000; Bailey et al., 2001; Viswanathan et al., 2006b; Weimar et al., 2011). This is particularly relevant for treatment decisions, especially antithrombotic use in the setting of small vessel disease (see ‘Therapeutic aspects’ section). Subdividing cerebral small vessel disease into CAA and non-CAA components may be an oversimplification as patients often have a mix of both pathologies. However, this distinction serves to distinguish the differences in the putative pathophysiology and different clinical and pathological consequences of the two diseases (Table 2).
Table 2.
Key neuropathological, clinical and neuroimaging characteristics of the two major sporadic cerebral small vessel diseases: CAA and ‘hypertensive arteriopathy’
| Characteristics | CAA | Sporadic non-amyloid microangiopathy (‘hypertensive arteriopathy’) |
|---|---|---|
| Small vessel pathology | Amyloid-β deposition and associated vasculopathy in cortical and leptomeningeal vessels. | A range of different features, e.g. arteriolosclerosis, fibrinoid necrosis, mural damage etc. |
| Risk factors | Age, APOE e4 and e2. | Age, hypertension, diabetes, smoking. |
| Associated clinical syndromes | ||
| ICH | Lobar (cortical-subcortical), cerebellar? | Typically deep: basal ganglia, thalamus, pons cerebellum; sometimes lobar. |
| Associated with high risk of recurrence (7–12% per year). | ||
| Ischaemic stroke | Not typically associated with lacunes. | Lacunar syndromes. |
| Uncertain role other than affecting treatment decisions, e.g. antithrombotic drugs, thrombolysis etc. | ||
| Other clinical syndromes | Transient focal neurological episodes (‘amyloid spells’), cognitive impairment and dementia, inflammatory CAA. | Vascular cognitive impairment and dementia. |
| MRI markers of small vessel disease | ||
| CMBs | Strictly lobar. | Predominantly deep, with or without lobar. |
| cSS | Very common: ∼40% in symptomatic CAA. | Rare: <5% of deep ICH. |
| MRI-visible perivascular spaces | Centrum semiovale (i.e. cerebral white matter). | Basal ganglia. |
| White matter hyperintensities | Posterior predominance, white matter spots. | No predilection for brain region, peribasal ganglia. |
| Lacunes | Not typically present, more superficial in the cerebral white matter. | Common, usually in the basal ganglia or deep white matter. |
| Diagnosis | Boston criteria, based on the presence of multiple strictly lobar CMBs or macrobleeds and cSS. | No established criteria available; diagnosed in patients not fulfilling Boston criteria for CAA, in the appropriate clinical context and based on MRI markers of small vessel damage. |
| Therapeutic implications | Acute treatment according to the clinical syndrome at presentation. Main long term goal is prevention of new lobar ICH (recurrent or incident), by blood pressure management and avoiding antithrombotics unless a completing indication exists. Cognitive rehabilitation in mild cognitive impairment/dementia. | Acute treatment according to the clinical syndrome at presentation. Control of vascular risk factors. Cognitive rehabilitation in vascular cognitive impairment/dementia. Safety of antithrombotic use is of lesser concern compared to CAA patients. |
CMB = cerebral microbleed; cSS = cortical superficial siderosis.
Emerging concepts in cerebral amyloid angiopathy pathophysiology
Perivascular drainage, vascular pathophysiology and cerebral amyloid angiopathy-related brain injury
The pathogenesis of CAA and its downstream effects on the brain are incompletely understood and highly complex. However, there is little evidence that age-associated cerebrovascular accumulation in sporadic CAA is driven by amyloid-β overproduction. Instead, current evidence suggests that amyloid-β accumulation is driven largely by reduced peptide clearance (Deane et al., 2009; Mawuenyega et al., 2010). The small vessels of the brain appear to play a major role in this clearance process, both as sites of efflux across the blood–brain barrier (Zlokovic et al., 2010) and as the site of intramural perivascular drainage of amyloid-β from the brain interstitial fluid (Weller et al., 1998; Marin-Padilla and Knopman, 2011). Hence, the view of CAA as a protein elimination failure angiopathy has been devised (Carare et al., 2013; Carare and Kalaria, 2016) and provides the most conceptually compelling model to explain this failure and the pathophysiological basis of disease progression.
Details on the ‘garbage truck of the brain’, including brain pathways for lymphatic drainage and for the convective influx/glymphatic communication pathways are the focus of intensive scrutiny and not definitively established (Nedergaard, 2013; Tarasoff-Conway et al., 2015). However, the prevailing model of perivascular (peri-arterial) drainage of interstitial fluid relies on vessel pulsations as the motive force and biochemical interactions with basement membranes (Schley et al., 2006; Morris et al., 2016) that drive clearance countercurrent to blood flow (Boche et al., 2008; Carare et al., 2008). As perivascular drainage pathways progressively fail with age (possibly in combination with APOE e4 or e2 alleles or other pathological conditions), amyloid-β is increasingly trapped in the perivascular compartment allowing for increased aggregation and deposition along the basement membranes of small arteries. It is plausible that this failure in the cortex and resultant stagnation of interstitial fluid might lead to enlargement or dilation of perivascular spaces (also termed Virchow-Robin spaces) in the underlying white matter (Weller et al., 2015). In CAA, enlarged perivascular spaces often become visible on standard clinical MRI sequences, preferentially in the centrum semiovale (i.e. cerebral white matter) (Roher et al., 2003; Charidimou et al., 2013b, 2014; Martinez-Ramirez et al., 2013; van Veluw et al., 2016). In line with this, a recent PET-MRI study suggested that high centrum semiovale perivascular spaces are associated with higher median cortical PiB retention across a wide range of cerebrovascular amyloid deposition (Charidimou et al., 2015a).
A model on cause and effect for the perivascular drainage-amyloid accumulation hypothesis is not yet available. Nevertheless, perivascular drainage impairment potentially sets in motion a self-reinforcing pathway by which worsening vascular amyloid-β accumulation leads to activation of vascular injury pathways, impaired vascular physiology, and further increased parenchymal and vascular amyloid-β accumulation (through a ‘feed-forward loop’ of reducing drainage efficiency) (Hawkes et al., 2011; Arbel-Ornath et al., 2013) (Fig. 2). Mouse model studies from multiple research groups have highlighted the potential importance of this pathway. Interruption of vascular pulsation by photocoagulation markedly reduces perivascular clearance (Arbel-Ornath et al., 2013) and increases local amyloid-β accumulation as plaques and CAA (Garcia-Alloza et al., 2011). The kinetics of CAA development are not well established, but vascular amyloid-β progresses mostly by propagation, which results in narrowing of unaffected gaps in small vessels (Robbins et al., 2006; Kimbrough et al., 2015) and expansion of amyloid-β from the basement membranes to progressively replace all tissue elements in the artery wall (Keable et al., 2016).
Figure 2.

Schematic overview of potential mechanisms of CAA pathophysiology as a self-reinforcing process. See text for details. Aβ = amyloid-β; BBB = blood–brain barrier; NVU = neurovascular unit.
Another intriguing possibility is that the evolving pathophysiology of CAA is reflected and partly driven by specific alterations in the array of vascular gene expression (or vasculome) over time (Guo et al., 2012). Recent findings suggest that perturbations in the vasculome may mediate both brain function and injury. For example, under normal conditions, the endothelium can provide signals that support neurogenesis (Shen et al., 2004; Ohab et al., 2006), protect neurons against stress (Guo et al., 2008), and mediate homeostasis in oligodendrocyte precursor pools (Arai and Lo, 2009). When activated by injury, the endothelium can produce pro-inflammatory cytokines that magnify neuroinflammation and secondary injury (Pober and Sessa, 2007), disrupt endothelial tight junctions and basal lamina with blood–brain barrier breakdown (Rosenberg and Yang, 2007; Sandoval and Witt, 2008), and alter vessel physiology. It has also been suggested that amyloid might propagate and be transmitted in a prion-like fashion, but this is an active area of investigation (Jaunmuktane et al., 2015; Frontzek et al., 2016). At what ‘tipping-point’ CAA starts to propagate more CAA, potentially setting in motion this vicious cycle, is currently unknown.
The key molecular and physiological steps involved in damaging small vessels and retarding amyloid clearance, as well as the importance of these pathways in human cerebrovascular amyloid-β accumulation, have yet to be elucidated. At a neuropathological level, advanced CAA is associated with important morphological changes including loss of vascular smooth muscle cells (Dotti and De Strooper, 2009) and replacement with amyloid-β deposits, often markedly thickening the vessel wall (Mandybur, 1986; Vonsattel et al., 1991a; Greenberg et al., 2009a) or even vessel occlusion (Olichney et al., 1995) and loss of compliance leading to brittle, fragile vessels prone to microaneurysm formation and leakage (Attems et al., 2011). Cerebrovascular compliance is an almost entirely uncharacterized aspect of small vessel disease, but a candidate mediator of reduced perivascular clearance (Weller et al., 2008). The loss and distortion of the normal anatomical elements of the vessel wall in CAA presumably trigger the various types of structural focal—mainly cortical—brain lesions such as ICH, cerebral microbleeds, siderosis and microinfarcts.
Capillaries and neurovascular dysfunction in cerebral amyloid angiopathy
Amyloid-β was recently shown to be highly toxic to pericytes, which, in turn, are important to blood–brain barrier function, basement membrane maintenance, and play a key role for neurovascular coupling (Hall et al., 2014). The distribution of capillary flows was also recently demonstrated to determine oxygen extraction efficiency (Jespersen and Ostergaard, 2012), implying that capillary pathology can be a source of tissue hypoxia and neuronal injury—even in the absence of flow-limiting pathology per se or structural alterations (Ostergaard et al., 2016). Whether these findings have specific implications in CAA has not yet been investigated directly, but data from the Alzheimer’s disease field are compelling (Zlokovic, 2011; Nelson et al., 2016). However, they do raise the possibility that amyloid-β deposition at the level of the capillaries might lead to degenerative changes by two mechanisms: (i) interfering with various elimination pathways, including the perivascular drainage pathway, clearance at the blood–brain barrier, or impairing the transendothelial clearance (Fig. 3); and (ii) affecting the metabolism of brain tissue (Kumar-Singh, 2009).
Figure 3.
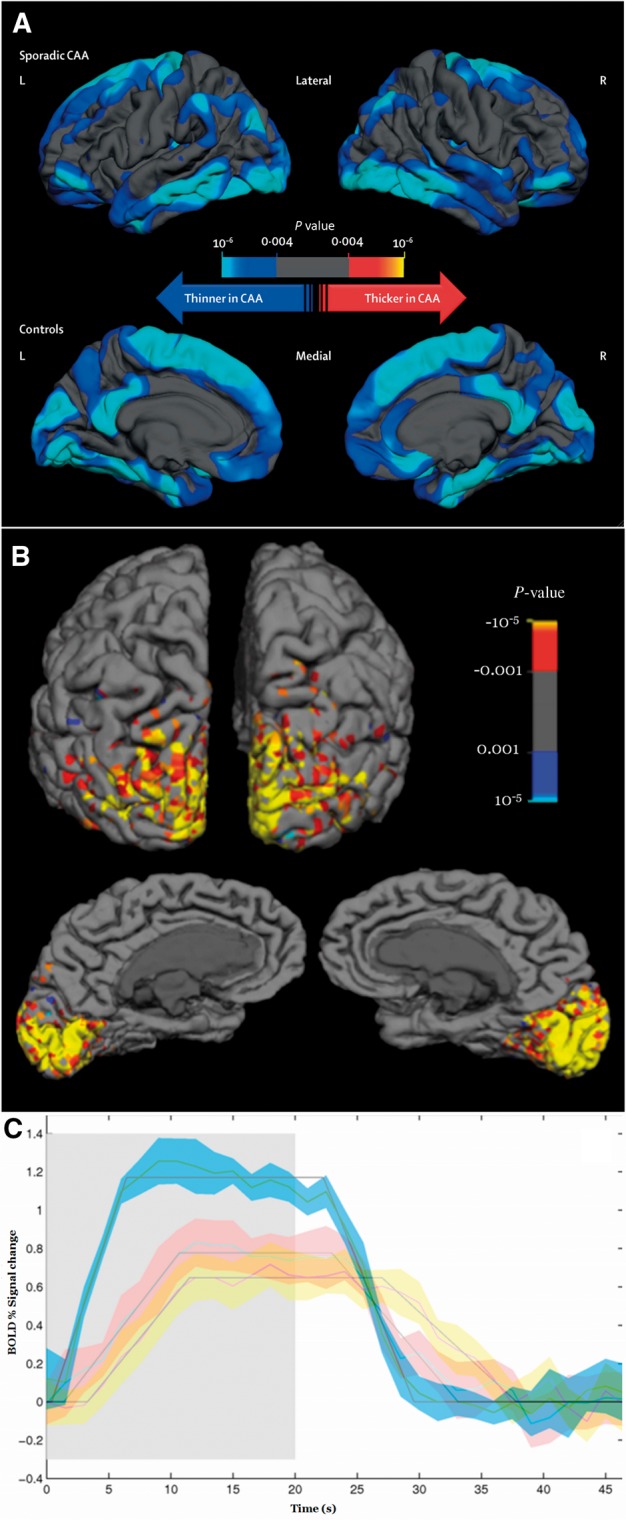
Cortical thickness and functional MRI measures in cerebral amyloid angiopathy. (A) Regional differences in cortical thickness between patients with sporadic CAA and their age-matched controls (Fotiadis et al., 2016, reproduced with permission). Topographic surface maps were generated based on a general linear model (adjusting for age and sex) using a threshold of P < 0.01 (with false discovery rate correction for multiple comparisons) (Fotiadis et al., 2016). The resulting maps show the statistically significant regional differences in cortical thickness between the two groups. (B) Functional MRI visually activated region of interest of a representative probable CAA subject. The colour scale denotes the Z-statistic for activation using the canonical haemodynamic response function. (C) Functional MRI measurements in response to visual stimulation in an elderly probable CAA patient (red error space) versus an age-matched control subject (blue error space). For the CAA patient, functional MRI was done at baseline (red error space) and again with the same scanner and protocol after one clinically asymptomatic year (yellow error space). The solid lines represent the change from baseline BOLD signal averaged over 16 cycles of visual stimulation (on 20 s, shaded region, then off 28 s), with standard deviations of the responses shown in blue, red and yellow spaces and the trapezoidal model fits, as previously described (Dumas et al., 2012a). In the CAA patient, the amplitude of the modelled peak response decreased from 0.80% at baseline to 0.65% at 1 year.
Cortical thinning in cerebral amyloid angiopathy
Of major interest, a recent study provided strong evidence of CAA-related cortical atrophy independent of Alzheimer’s disease (Fotiadis et al., 2016). Though the mechanism for this association is not well defined, the average cortical width of 63 non-demented CAA subjects versus 63 age-matched healthy control subjects was reduced (2.17 ± 0.11 mm in CAA versus 2.31 ± 0.07 mm in controls, P < 0.001) (Fotiadis et al., 2016) (Fig. 3A). A similar finding was observed in subjects with Dutch-type hereditary CAA, a condition characterized by severe CAA but little or no Alzheimer’s disease pathology (Natte et al., 2001). The average cortical thickness of 26 Dutch-type hereditary CAA subjects was significantly lower than age-matched healthy controls (2.31 ± 0.18 mm versus 2.42 ± 0.10 mm, P = 0.006) (Fotiadis et al., 2016). Greatest thinning was noted in medial frontal and posterolateral brain regions, a somewhat different pattern from the primarily limbic and multimodal association regions with greatest thinning in Alzheimer’s disease (Dickerson et al., 2009). The clinical implications of cortical thinning in CAA and its relation to cognitive impairment requires further study.
Altered vascular reactivity
CAA appears to have substantial effects on vascular function/physiology, which might result in more widespread brain injury (Smith and Greenberg, 2009; Pantoni, 2010). Amyloid deposition in the vessel wall may cause gliovascular unit impairment (Kimbrough et al., 2015), endothelial dysfunction (Grinberg et al., 2012), impaired autoregulation and blood–brain barrier disruption (Attems et al., 2011). One key candidate mechanism suggested by transgenic animal studies is impaired cerebrovascular compliance and reactivity to physiological stimuli (Niwa et al., 2000; Shin et al., 2007; Han et al., 2008; Park et al., 2013). The brain microvasculature forms interconnected vascular networks (a 2D network of pial arterioles that connects to a 3D network of subsurface microvessels) (Blinder et al., 2013; Shih et al., 2015); the practical consequence is that apparently unaffected microvessels might still contribute to the overall CAA-related tissue injury by indirect consequences from neighbouring amyloid-laden microvessels (Shih et al., 2013).
The best established physiological change in individuals with advanced CAA is reduced vasodilation to physiological stimuli. This finding has been demonstrated in studies measuring the functional MRI blood oxygen level-dependent (BOLD) response to visual stimulation (Dumas et al., 2012b; Peca et al., 2013) (Fig. 3B), which take advantage of the occipital predominance of CAA (Vinters and Gilbert, 1983). In non-demented CAA patients, the functional MRI response showed 27–28% reduced peak amplitude, 73% longer time to peak, and 42% longer time to return to baseline compared with elderly control subjects matched for age (Dumas et al., 2012b; Peca et al., 2013) (Fig. 3C). Altered BOLD response might be driven by vascular or neuronal dysfunction, but the findings that CAA patients and control individuals had similar visual evoked response amplitude (Peca et al., 2013) and that results were consistent in secondary analyses restricted to responding voxels only (Dumas et al., 2012a), suggest that the differences are largely driven by effects of CAA on the vessels themselves. A longitudinal analysis also showed declining amplitude in BOLD response to visual stimulation among CAA subjects (Switzer et al., 2016) (Fig. 3C). These studies also showed associations between impaired functional MRI response and white matter hyperintensity volume, suggesting mismatched blood supply versus tissue demand as a potential mechanism mediating CAA-related tissue damage. Another finding consistent with this possibility is that cortical thickness is negatively correlated with the time to peak BOLD response to visual stimulation (r = −0.4, P = 0.005 with adjustment for covariates) (Fotiadis et al., 2016).
Microstructural damage and brain network alterations
CAA-related perfusion impairment may be responsible not only for subcortical white matter lesions (Gurol et al., 2013) but also for tissue microstructural changes detected by diffusion tensor imaging (Salat et al., 2006; Reijmer et al., 2015) and microinfarcts (Reijmer et al., 2016b). Analysis (Fig. 4A) of the local network parameter node strength (the sum of fractional anisotropy-weighted connections attached to a given node) found locally altered network structure in CAA subjects compared to controls, particularly in occipital, posterior parietal, and posterior temporal cortex (Fig. 4B), resembling the distribution of CAA pathology (Reijmer et al., 2015). Network disturbances were associated with worse cognitive functioning and amyloid load on PET, providing direct links between vascular amyloid and impairments in white matter connectivity (Reijmer et al., 2015). Punctate diffusion-weighted imaging lesions (presumably corresponding to acute microinfarcts) in subcortical white matter have been demonstrated to produce focal changes in mean diffusivity and fractional anisotropy (Auriel et al., 2014), raising the idea that altered diffusivity may partly reflect the cumulative effect of cerebral microinfarcts or other focal lesions on cortical microstructure. Brain network alterations in patients with CAA (n = 33) appear to worsen measurably over just 1.3-year follow-up, progressing from posterior to frontal regions with increasing disease severity; this decline is associated with worse executive functioning (Beta = 0.41, P = 0.04) (Reijmer et al., 2016a). Of note, CAA patients included in all the aforementioned in vivo studies were symptomatic with high vascular lesion burden. A relevant question is whether advanced imaging techniques can also detect CAA-related abnormalities before the disease becomes symptomatic (Akoudad et al., 2013). This approach is currently limited, however, by our poor understanding of tissue-level pathological basis (Fig. 4C) of the various neuroimaging abnormalities discussed, making it difficult to link these large-scale measures to any specific pathogenic process or parenchymal injuries.
Figure 4.

White matter injury in CAA detected with diffusion tensor imaging. (A) Whole brain fibre tract reconstruction from fractional anisotropy map reflecting the degree of anisotropic diffusion of water molecules. (B) Left hemisphere: visualization of a DTI-based brain network from a patient with severe CAA. Brain regions are depicted by nodes and fibre tracts between regions are depicted by edges. Right hemisphere: local differences in connectivity strength between patients with CAA compared to age-matched controls, as previously described (Reijmer et al., 2015). Results show that tracts projecting to occipital, parietal, and temporal regions are most affected, whereas tracts projecting to frontal and subcortical regions are relatively spared (darker nodes show greater differences from controls). (C) Regions marked in red were selected for histopathological evaluation of an autopsied CAA subject who underwent DTI scanning during life. The white matter near a region of reduced nodal strength showed considerable myelin loss on Luxol® Fast Blue stained sections relative to a region of relatively reserved nodal strength.
Towards mapping different cerebral amyloid angiopathy phenotypes
It is becoming increasingly evident that CAA is not a uniform, but rather a complex and very heterogeneous entity that can follow several different pathways (Figs 5 and 6). While sporadic CAA is commonly found in the elderly (Keage et al., 2009), it is usually mild and clinically silent. In the fraction of patients with CAA that will go on to develop clinical symptoms, the sentinel presentations of the disease vary (Greenberg et al., 1993; Maia et al., 2007), while each presentation is associated with its own cluster of biomarkers (Table 2). It follows that the clinical and imaging heterogeneity and phenotypes of the disease might be reflecting distinct neuropathological subtypes or patterns of cerebrovascular amyloid deposition (Fig. 5) and activation of diverging pathophysiological cascades. Mild CAA, a common finding in elderly and Alzheimer’s disease brains, likely reflects the perivascular drainage impairment accompanying ageing. Severe CAA is generally assumed to result in the symptomatic sentinel presentations of the disease. However, the pattern on amyloid deposition in severe CAA varies greatly with differing anatomical patterns of deposition and degrees of accumulation and progression (Fig. 6) (Keable et al., 2016). The spectrum ranges from amyloid co-localization with basement membranes, to replacement of smooth muscle cells by amyloid with some preservation of basement membrane elements, to complete replacement of the artery wall by amyloid (Fig. 6) (Keable et al., 2016). Yet, in some arterioles, amyloid deposition remains focal, even to the point of complete replacement of smooth muscle layers and basement membrane (Keable et al., 2016). The focal complete replacement of the vessel wall by amyloid might provide a potential site of weakness leading to haemorrhage. In other cases, deposition of amyloid is seen in the tunica adventitia surrounding the perimeter of the vessel (instead of the tunica media), which may represent an additional part of the lymphatic drainage pathway (Keable et al., 2016). Adding another dimension to the spectrum of the disease, these patterns of amyloid deposition may differentially affect leptomeningeal and cortical parenchymal vessels, as well as capillaries (Fig. 6). For example, a systems proteomic analysis demonstrated that clusterin (apolipoprotein J) and tissue inhibitor of metalloproteinases-3, co-localize with amyloid-β and seem to be upregulated in leptomeningeal (but not cortical) arteries from CAA patients compared to young and elderly controls (Manousopoulou et al., 2016).
Figure 5.

Suggested heuristic schematic of the possible different phenotypes of CAA and directions in the expression of the disease. The two main trees (from right to left) depict Alzheimer’s disease (AD) neurodegeneration, in which CAA is commonly found, and sporadic CAA. The third tree, in the background, represents sporadic non-amyloid microangioapathies. All three pathologies often co-exist in different combinations and severity in the ageing brain. Initially, different but overlapping pathophysiological pathways (roots of each tree in the grey box) can result in progressive vascular amyloid accumulation and pathophysiological alterations, which might culminate in structural vascular damage and brain injury. In extreme forms these might result in different neuroimaging and clinical phenotypes of the disease (different branches in each tree). At the one end we have the primarily haemorrhagic types of CAA, predominantly characterized by either multiple lobar cerebral microbleeds (CMB) (i.e. ‘microbleeders’) or cortical superficial siderosis (cSS) (‘superficial bleeders’), and/or symptomatic ICH (i.e. ‘macrobleeders’); at the other end we have the less haemorrhagic phenotypes of CAA, clinically expressed with more cognitive impairment and probably more strongly associated with cortical microinfarcts (CMI), with or without Alzheimer’s disease. Many patients can have intermediate phenotypes of the disease. The APOE ɛ genotype and vascular risk factors likely influence all the critical steps and different directions taken during the course of the disease in the ageing brain. Aβ = amyloid-β; TFNEs = transient focal neurological episodes; WMH = white matter hyperintensity.
Figure 6.

Brain anatomical patterns, evolution and neuropathological phenotypes of cerebrovascular amyloid-β deposition in advanced CAA. (A) Schematic representation of the superficial cortical small vessel disease system in the brain, typically affected by CAA. Leptomeningeal arterioles give off short (S) penetrating arterioles (‘cortical’) reaching three different depths in the cortex (i.e. cortical layer III, V and the grey–white matter junction, S1–3, respectively), while long penetrators (‘medullary’, not usually affected by CAA) continue into the subcortical white matter. In advanced disease, often decreasing severity of CAA is seen between leptomeningeal, superficial and deeper layers in the cortex. (B) Regional severity of CAA with more heavy involvement of occipital and temporal lobes. Severe CAA probably progresses in a posterior-to-anterior fashion in the brain. (C) Neuropathological patterns spectrum of severe amyloid-β deposition. C(a)–C(d). Amyloid co-localization with basement membranes, to replacement of smooth muscle cells with some preservation of basement membrane elements, to complete replacement of the artery wall. C(e) More pronounced CAA-related vessel wall damage resulting in vasculopathic changes. C(f) Very focal amyloid deposition, but with complete replacement of smooth muscle and basement membrane layers at the point of vessel wall cracking, potentially providing a site of weakness and haemorrhage. C(g) Amyloid-β deposition in the tunica adventitia surrounding the perimeter of the vessel (instead of the tunica media). C(h) Capillary CAA, which might range from mild to severe, with dysfunction of pericytes and endothelial cells to complete capillary occlusion. A–C could potentially influence the spectrum of clinical imaging phenotypes of CAA, as neuropathological patterns of amyloid deposition can differentially affect leptomeningeal and cortical parenchymal vessels, with distinct anatomical patterns of deposition, degrees of accumulation, progression and pathophysiological consequences during the disease course. [C was redesigned and modified based on data and figures from (Keable et al., 2016), doi: 10.1007/s00401-016-1555-z, under Creative Commons Attribution License (CC BY)].
The heterogeneity in these pathological patterns and clinical imaging presentations of CAA may be influenced by how different risk factors affect the process of perivascular clearance and brain injury during the disease process (Fig. 2). For example, APOE ɛ4 alters the biochemical composition of basement membranes and is involved in various amyloid degradation systems, whereas APOE ɛ2 promotes structural vasculopathic changes in amyloid-laden vessels, making them more prone to rupture. Mid-life hypertension might alter the biophysical forces acting upon the arterial wall, modifying the motive force for perivascular clearance but also the structural integrity of the vessels (in combination with sporadic non-amyloid microangiopathic alterations). Other potential risk factors and comorbid conditions that result in vessel rupture associated with CAA remain unclear. Of note, the degree of vessel wall replacement by amyloid required before the vessel ruptures is currently unknown.
A number of observations from clinical-MRI CAA cohorts can shed further light on the concept of different disease phenotypes. An interesting study suggested that multiple lobar microbleeds associated with CAA-related pathology might in some respects differ from macrobleeds, since they have a bimodal (rather than continuous) size distribution on MRI (Greenberg et al., 2009a). In a patient subset that underwent autopsy, those with high microbleed counts (‘microbleeders’) had increased wall thickness of amyloid-laden vessels compared to patients with relatively low microbleed counts and lobar ICH (‘macrobleeders’) (Greenberg et al., 2009a). A recent MRI-neuropathological studies recruited 105 patients with CAA pathologic confirmation and in vivo MRI and compared pathologic, imaging, and APOE genotype data between subjects with versus without symptomatic ICH. The two groups had similar CAA burden, almost all pathology samples had neuritic plaques, whereas neurofibrillary tangles were more commonly present in the patients without intracerebral bleeds (87% versus 42%, P < 0.0001). Disseminated cortical superficial siderosis was considerably more common in patients with ICH (33.3% versus 5.9%, P < 0.0001) and was associated with APOE ɛ2 (but not APOE ɛ4) (Charidimou et al., 2015c). Surprisingly, cortical superficial siderosis, which confers a high risk of future lobar ICH, is not associated with lobar microbleeds (Charidimou et al., 2013a; Shoamanesh et al., 2014). A plausible explanation is that siderosis predominantly reflects a severe leptomeningeal pattern of amyloid deposition rather than deeper cortical CAA, which manifests itself more with microbleeds. Although studies of CAA phenotypes often focus on the extreme forms (such as microbleeders, macrobleeders, siderobleeders, etc.), many patients likely fall into a mixed category in which various CAA-related vascular processes co-exist.
Although literature to support firm conclusions remains extremely limited, a ‘phenotype approach’ in CAA may provide a conceptual model allowing greater reliance on different clusters of clinical and pathophysiologically relevant MRI biomarkers in future CAA trials and patient management (Fig. 6) (Greenberg et al., 2014).
Implications for patient management in cerebral amyloid angiopathy
The management of acute CAA-related ICH is the same as any spontaneous ICH, and it is summarized in the American Heart Association/American Stroke Association (Hemphill et al., 2015) and the European Stroke Organization (Steiner et al., 2014) guidelines. To date, there is no disease-specific CAA treatment, and hence the long-term management of CAA patients is centred on the prevention of incident or recurrent haemorrhagic events and dementia. In this context, an important first step in decision-making for certain drug use or avoidance (antithrombotics, statins etc.) and other interventions is identifying the probability that a given patient with spontaneous ICH has CAA. In this setting, an accurate assessment of haemorrhage risk based on the presumed cause of ICH and/or the predominant type/severity of the underlying haemorrhage-prone microangiopathy. A key difference between the two broad types of small vessel disease and aetiologies of spontaneous ICH is the much higher annual recurrence rate in CAA-related lobar ICH compared to sporadic non-amyloid microangiopathy-related ICH (∼10%/year versus 2–3%/year, respectively). Furthermore, the risk of incident dementia after ICH appears to be more than two times higher in patients with lobar (incidence at 1 year: 23.4%; 95% CI: 14.6–33.3) than for patients with non-lobar ICH (incidence at 1 year: 9.2%; 95% CI: 5.1–14.7) (Moulin et al., 2016).
A common approach for CAA patients with a previous history of lobar ICH is to avoid antithrombotic treatment as much as possible, as they might increase the risk of recurrence (Eckman et al., 2003; Hofmeijer et al., 2015). Even for strong indications such as non-valvular atrial fibrillation, oral anticoagulation with warfarin may be generally unsafe following CAA-related ICH (Eckman et al., 2003), although thus far, large observational data are missing. American Heart Association/American Stroke Association guidelines recommend avoiding oral anticoagulation in patients with lobar ICH, but to consider resuming oral anticoagulation in patients with non-lobar ICH (Hemphill et al., 2015). The role of antiplatelet agents and their systematic discontinuation or not in CAA-related ICH patients remains a hotly debated topic (Al-Shahi Salman and Dennis, 2014; Falcone and Rosand, 2014). The RESTART randomized trial (http://www.restarttrial.org/) is underway to provide some answers on whether to avoid antithrombotic use after an ICH. Another option for those CAA-related ICH patients requiring anticoagulation might be newer oral anticoagulants, which have been shown to have lower overall ICH risk. For example, apixaban has a similar intracranial bleeding risk profile as aspirin, but is more effective at reducing ischaemic stroke risk (Connolly et al., 2011). The relative risks are less clear for individuals with asymptomatic lobar microbleeds or cortical superficial siderosis only, fulfilling the Boston criteria for CAA. In this situation, clinicians should carefully weigh risk-benefit for recurrent ischaemic stroke versus haemorrhagic complications. Larger, more definitive studies (Charidimou et al., 2015e) and randomized trials are required to settle these important questions.
Although CAA is not thought to be directly linked to hypertension, strict control of blood pressure within the normal range is recommended, especially after an ICH. A subgroup analysis of the PROGRESS trial (Arima et al., 2010) demonstrated that lowering blood pressure with the antihypertensive drug perindopril (with or without indapamide) reduced the risk of probable CAA-related ICH recurrence by 77% (95% CI, 19%–93%) over a follow-up period of 3.9 years. Recently, inadequate blood pressure control in ICH patients from a large single centre observational study was associated with hazard ratios of 3.53 (95% CI, 1.65–7.54) for recurrent lobar ICH and of 4.23 (95% CI, 1.02–17.52) for non-lobar ICH when compared to those with adequately controlled blood pressure (Biffi et al., 2015). These and other observations strongly support the need for strict and continued blood pressure control in CAA patients, which might impact outcomes such as recurrent ICH, cognitive impairment, or progression of small vessel disease pathology. Finally, supportive care, lifestyle modifications and, in patients with recent CAA-related ICH, cognitive rehabilitation may maximize function and enhance recovery.
Future perspectives
There is currently a growing interest in CAA with an explosion of publications in the last 15 years and dense collaboration networks between research teams in the field (Charidimou et al., 2016a). International efforts aimed at translating mechanistic insights into novel treatment approaches (Al-Shawi et al., 2016), including an ongoing first-of-kind CAA anti-amyloid immunotherapy trial with ponezumab, an anti-amyloid-β40 selective antibody (NCT 01821118) are underway (Greenberg et al., 2014). The rationale for anti-amyloid immunotherapy in CAA is to attenuate amyloid accrual in cerebral vessels and by doing that restore vascular reactivity in the short-term and prevent the occurrence of clinical symptomatology in the long-term (Bales et al., 2016). Rapidly accumulating data on in vivo detection of the pathogenic steps involved in CAA offers substantial promise for future trials aimed at identifying disease-modifying treatments (Saito and Ihara, 2014) for this largely untreatable disease. Nonetheless, there are many remaining ‘known unknowns’ on all the postulated steps in CAA pathogenesis and neurological dysfunction, ranging from vascular amyloid deposition itself to physiological alterations, haemorrhagic and non-haemorrhagic (cortical and subcortical) structural damage, symptomatic ICH, and presentations without major haemorrhage, including cognitive dysfunction. It is anticipated, that shedding light on these aspects may also have implications for other small-vessel disease processes in the ageing brain and vascular cognitive impairment.
Acknowledgements
We thank Dr Yael Reijmer, Panos Fotiadis and Dr Susanne van Veluw for assistance with figures.
Glossary
Abbreviations
- CAA
cerebral amyloid angiopathy
- ICH
intracerebral haemorrhage
Funding
A.C. receives research support from the Bodossaki Foundation. G.B. receives research support from the Fulbright Association and the Monahan Foundation. S.G. receives research support from the National Institutes of Health (R01AG26484, R01NS070834, R01NS096730).
References
- Akoudad S, de Groot M, Koudstaal PJ, van der Lugt A, Niessen WJ, Hofman A. et al. Cerebral microbleeds are related to loss of white matter structural integrity. Neurology 2013; 81: 1930–7. [DOI] [PubMed] [Google Scholar]
- Al-Shahi Salman R, Dennis MS. Antiplatelet therapy may be continued after intracerebral hemorrhage. Stroke 2014; 45: 3149–50. [DOI] [PubMed] [Google Scholar]
- Al-Shawi R, Tennent GA, Millar DJ, Richard-Londt A, Brandner S, Werring DJ. et al. Pharmacological removal of serum amyloid P component from intracerebral plaques and cerebrovascular Abeta amyloid deposits in vivo. Open Biol 2016; 6: 150202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonzo NC, Hyman BT, Rebeck GW, Greenberg SM. Progression of cerebral amyloid angiopathy: accumulation of amyloid-beta40 in affected vessels. J Neuropathol Exp Neurol 1998; 57: 353–9. [DOI] [PubMed] [Google Scholar]
- Arai K, Lo EH. An oligovascular niche: cerebral endothelial cells promote the survival and proliferation of oligodendrocyte precursor cells. J Neurosci 2009; 29: 4351–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbel-Ornath M, Hudry E, Eikermann-Haerter K, Hou S, Gregory JL, Zhao L. et al. Interstitial fluid drainage is impaired in ischemic stroke and Alzheimer’s disease mouse models. Acta Neuropathol 2013; 126: 353–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arima H, Tzourio C, Anderson C, Woodward M, Bousser MG, MacMahon S. et al. Effects of perindopril-based lowering of blood pressure on intracerebral hemorrhage related to amyloid angiopathy: the PROGRESS trial. Stroke 2010; 41: 394–6. [DOI] [PubMed] [Google Scholar]
- Arvanitakis Z, Leurgans SE, Wang Z, Wilson RS, Bennett DA, Schneider JA. Cerebral amyloid angiopathy pathology and cognitive domains in older persons. Ann Neurol 2011; 69: 320–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attems J. Sporadic cerebral amyloid angiopathy: pathology, clinical implications, and possible pathomechanisms. Acta Neuropathol 2005; 110: 345–59. [DOI] [PubMed] [Google Scholar]
- Attems J, Jellinger K, Thal DR, Van Nostrand W. Review: Sporadic cerebral amyloid angiopathy. Neuropathol Appl Neurobiol 2011; 37: 75–93. [DOI] [PubMed] [Google Scholar]
- Auriel E, Charidimou A, Gurol ME, Ni J, Van Etten ES, Martinez-Ramirez S. et al. Validation of clinicoradiological criteria for the diagnosis of cerebral amyloid angiopathy-related inflammation. JAMA Neurol 2016; 73: 197–202. [DOI] [PubMed] [Google Scholar]
- Auriel E, Edlow BL, Reijmer YD, Fotiadis P, Ramirez-Martinez S, Ni J. et al. Microinfarct disruption of white matter structure: a longitudinal diffusion tensor analysis. Neurology 2014; 83: 182–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auriel E, Westover MB, Bianchi MT, Reijmer Y, Martinez-Ramirez S, Ni J. et al. Estimating total cerebral microinfarct burden from diffusion-weighted imaging. Stroke 2015; 46: 2129–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacskai BJ, Frosch MP, Freeman SH, Raymond SB, Augustinack JC, Johnson KA. et al. Molecular imaging with Pittsburgh Compound B confirmed at autopsy: a case report. Arch Neurol 2007; 64: 431–4. [DOI] [PubMed] [Google Scholar]
- Bailey RD, Hart RG, Benavente O, Pearce LA. Recurrent brain hemorrhage is more frequent than ischemic stroke after intracranial hemorrhage. Neurology 2001; 56: 773–7. [DOI] [PubMed] [Google Scholar]
- Bales KR, O’Neill SM, Pozdnyakov N, Pan F, Caouette D, Pi Y. et al. Passive immunotherapy targeting amyloid-beta reduces cerebral amyloid angiopathy and improves vascular reactivity. Brain 2016; 139(Pt 2): 563–77. [DOI] [PubMed] [Google Scholar]
- Baron JC, Farid K, Dolan E, Turc G, Marrapu ST, O’Brien E. et al. Diagnostic utility of amyloid PET in cerebral amyloid angiopathy-related symptomatic intracerebral hemorrhage. J Cereb Blood Flow Metab 2014; 34: 753–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biffi A, Anderson CD, Battey TW, Ayres AM, Greenberg SM, Viswanathan A. et al. Association between blood pressure control and risk of recurrent intracerebral hemorrhage. JAMA 2015; 314: 904–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biffi A, Halpin A, Towfighi A, Gilson A, Busl K, Rost N. et al. Aspirin and recurrent intracerebral hemorrhage in cerebral amyloid angiopathy. Neurology 2010; 75: 693–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blinder P, Tsai PS, Kaufhold JP, Knutsen PM, Suhl H, Kleinfeld D. The cortical angiome: an interconnected vascular network with noncolumnar patterns of blood flow. Nat Neurosci 2013; 16: 889–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boche D, Zotova E, Weller RO, Love S, Neal JW, Pickering RM. et al. Consequence of Abeta immunization on the vasculature of human Alzheimer’s disease brain. Brain 2008; 131(Pt 12): 3299–310. [DOI] [PubMed] [Google Scholar]
- Boulouis G, Charidimou A, Greenberg SM. Sporadic cerebral amyloid angiopathy: pathophysiology, neuroimaging features, and clinical implications. Semin Neurol 2016; 36: 233–43. [DOI] [PubMed] [Google Scholar]
- Boyle PA, Yu L, Nag S, Leurgans S, Wilson RS, Bennett DA. et al. Cerebral amyloid angiopathy and cognitive outcomes in community-based older persons. Neurology 2015; 85: 1930–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calviere L, Cuvinciuc V, Raposo N, Faury A, Cognard C, Larrue V. et al. Acute convexity subarachnoid hemorrhage related to cerebral amyloid angiopathy: clinicoradiological features and outcome. J Stroke Cerebrovasc Dis 2016; 25: 1009–16. [DOI] [PubMed] [Google Scholar]
- Carare RO, Bernardes-Silva M, Newman TA, Page AM, Nicoll JA, Perry VH. et al. Solutes, but not cells, drain from the brain parenchyma along basement membranes of capillaries and arteries: significance for cerebral amyloid angiopathy and neuroimmunology. Neuropathol Appl Neurobiol 2008; 34: 131–44. [DOI] [PubMed] [Google Scholar]
- Carare RO, Hawkes CA, Jeffrey M, Kalaria RN, Weller RO. Review: cerebral amyloid angiopathy, prion angiopathy, CADASIL and the spectrum of protein elimination failure angiopathies (PEFA) in neurodegenerative disease with a focus on therapy. Neuropathol Appl Neurobiol 2013; 39: 593–611. [DOI] [PubMed] [Google Scholar]
- Carare RO, Kalaria R. Cerebrovascular pathology: the dark side of neurodegeneration. Acta Neuropathol 2016; 131: 641–3. [DOI] [PubMed] [Google Scholar]
- Carcel C, Sato S, Anderson CS. Blood pressure management in intracranial hemorrhage: current challenges and opportunities. Curr Treatment Options Cardiovasc Med 2016; 18: 22. [DOI] [PubMed] [Google Scholar]
- Case NF, Charlton A, Zwiers A, Batool S, McCreary CR, Hogan DB. et al. Cerebral amyloid angiopathy is associated with executive dysfunction and mild cognitive impairment. Stroke 2016; 47: 2010–6. [DOI] [PubMed] [Google Scholar]
- Charidimou A, Fox Z, Werring DJ, Song M. Mapping the landscape of cerebral amyloid angiopathy research: an informetric analysis perspective. J Neurol Neurosurg Psychiatry 2016a; 87: 252–9. [DOI] [PubMed] [Google Scholar]
- Charidimou A, Gang Q, Werring DJ. Sporadic cerebral amyloid angiopathy revisited: recent insights into pathophysiology and clinical spectrum. J Neurol Neurosurg Psychiatry 2012a; 83: 124–37. [DOI] [PubMed] [Google Scholar]
- Charidimou A, Hong YT, Jager HR, Fox Z, Aigbirhio FI, Fryer TD. et al. White matter perivascular spaces on magnetic resonance imaging: marker of cerebrovascular amyloid burden? Stroke 2015a; 46: 1707–9. [DOI] [PubMed] [Google Scholar]
- Charidimou A, Jager HR. Developing biomarkers for cerebral amyloid angiopathy trials: do potential disease phenotypes hold promise? Lancet Neurol 2014; 13: 538–40. [DOI] [PubMed] [Google Scholar]
- Charidimou A, Jager RH, Fox Z, Peeters A, Vandermeeren Y, Laloux P. et al. Prevalence and mechanisms of cortical superficial siderosis in cerebral amyloid angiopathy. Neurology 2013a; 817: 626–32. [DOI] [PubMed] [Google Scholar]
- Charidimou A, Jaunmuktane Z, Baron JC, Burnell M, Varlet P, Peeters A. et al. White matter perivascular spaces: an MRI marker in pathology-proven cerebral amyloid angiopathy? Neurology 2014; 82: 57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charidimou A, Linn J, Vernooij MW, Opherk C, Akoudad S, Baron JC. et al. Cortical superficial siderosis: detection and clinical significance in cerebral amyloid angiopathy and related conditions. Brain 2015b; 138(Pt 8): 2126–39. [DOI] [PubMed] [Google Scholar]
- Charidimou A, Martinez-Ramirez S, Shoamanesh A, Oliveira-Filho J, Frosch M, Vashkevich A. et al. Cerebral amyloid angiopathy with and without hemorrhage: evidence for different disease phenotypes. Neurology 2015c; 84: 1206–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charidimou A, Meegahage R, Fox Z, Peeters A, Vandermeeren Y, Laloux P. et al. Enlarged perivascular spaces as a marker of underlying arteriopathy in intracerebral haemorrhage: a multicentre MRI cohort study. J Neurol Neurosurg Psychiatry 2013b; 84: 624–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charidimou A, Nicoll JA, McCarron MO. Thrombolysis-related intracerebral hemorrhage and cerebral amyloid angiopathy: accumulating evidence. Front Neurol 2015d; 6: 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charidimou A, Pantoni L, Love S. The concept of sporadic cerebral small vessel disease: a road map on key definitions and current concepts. Int J Stroke 2016b; 11: 6–18. [DOI] [PubMed] [Google Scholar]
- Charidimou A, Peeters A, Fox Z, Gregoire SM, Vandermeeren Y, Laloux P. et al. Spectrum of transient focal neurological episodes in cerebral amyloid angiopathy: multicentre magnetic resonance imaging cohort study and meta-analysis. Stroke 2012b; 43: 2324–30. [DOI] [PubMed] [Google Scholar]
- Charidimou A, Peeters AP, Jager R, Fox Z, Vandermeeren Y, Laloux P. et al. Cortical superficial siderosis and intracerebral hemorrhage risk in cerebral amyloid angiopathy. Neurology 2013c; 81: 1666–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charidimou A, Wilson D, Shakeshaft C, Ambler G, White M, Cohen H. et al. The Clinical Relevance of Microbleeds in Stroke study (CROMIS-2): rationale, design, and methods. Int J Stroke 2015e; 10: 155–61. [DOI] [PubMed] [Google Scholar]
- Connolly SJ, Eikelboom J, Joyner C, Diener HC, Hart R, Golitsyn S. et al. Apixaban in patients with atrial fibrillation. N Engl J Med 2011; 364: 806–17. [DOI] [PubMed] [Google Scholar]
- Cordonnier C, Leys D, Dumont F, Deramecourt V, Bordet R, Pasquier F. et al. What are the causes of pre-existing dementia in patients with intracerebral haemorrhages? Brain 2010; 133: 3281–9. [DOI] [PubMed] [Google Scholar]
- Cordonnier C, van der Flier WM. Brain microbleeds and Alzheimer’s disease: innocent observation or key player? Brain 2011; 134(Pt 2): 335–44. [DOI] [PubMed] [Google Scholar]
- de Wit NM, Snkhchyan H, den Hoedt S, Wattimena D, de Vos R, Mulder MT. et al. Altered sphingolipid balance in capillary cerebral amyloid angiopathy. J Alzheimers Dis 2016, in press. [DOI] [PubMed] [Google Scholar]
- Deane R, Bell RD, Sagare A, Zlokovic BV. Clearance of amyloid-beta peptide across the blood-brain barrier: implication for therapies in Alzheimer’s disease. CNS Neurol Disord Drug Targets 2009; 8: 16–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickerson BC, Bakkour A, Salat DH, Feczko E, Pacheco J, Greve DN. et al. The cortical signature of Alzheimer’s disease: regionally specific cortical thinning relates to symptom severity in very mild to mild AD dementia and is detectable in asymptomatic amyloid-positive individuals. Cereb Cortex 2009; 19: 497–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dierksen GA, Skehan ME, Khan MA, Jeng J, Nandigam RN, Becker JA. et al. Spatial relation between microbleeds and amyloid deposits in amyloid angiopathy. Ann Neurol 2010; 68: 545–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dotti CG, De Strooper B. Alzheimer’s dementia by circulation disorders: when trees hide the forest. Nat Cell Biol 2009; 11: 114–6. [DOI] [PubMed] [Google Scholar]
- Dumas A, Dierksen GA, Gurol ME, Halpin A, Martinez-Ramirez S, Schwab K. et al. Functional magnetic resonance imaging detection of vascular reactivity in cerebral amyloid angiopathy. Ann Neurol 2012a; 72: 76–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumas A, Dierksen GA, Gurol ME, Halpin A, Martinez-Ramirez S, Schwab K. et al. Functional magnetic resonance imaging detection of vascular reactivity in cerebral amyloid angiopathy. Ann Neurol 2012b; 72: 76–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckman MH, Rosand J, Knudsen KA, Singer DE, Greenberg SM. Can patients be anticoagulated after intracerebral hemorrhage? A decision analysis. Stroke 2003; 34: 1710–6. [DOI] [PubMed] [Google Scholar]
- Eng JA, Frosch MP, Choi K, Rebeck GW, Greenberg SM. Clinical manifestations of cerebral amyloid angiopathy-related inflammation. Ann Neurol 2004; 55: 250–6. [DOI] [PubMed] [Google Scholar]
- Falcone GJ, Rosand J. Aspirin should be discontinued after lobar intracerebral hemorrhage. Stroke 2014; 45: 3151–2. [DOI] [PubMed] [Google Scholar]
- Farid K, Hong YT, Aigbirhio FI, Fryer TD, Menon DK, Warburton EA. et al. Early-Phase 11C-PiB PET in Amyloid Angiopathy-Related Symptomatic Cerebral Hemorrhage: Potential Diagnostic Value? PloS one 2015; 10: e0139926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazekas F, Kleinert R, Roob G, Kleinert G, Kapeller P, Schmidt R. et al. Histopathologic analysis of foci of signal loss on gradient-echo T2*-weighted MR images in patients with spontaneous intracerebral hemorrhage: evidence of microangiopathy-related microbleeds. AJNR Am J Neuroradiol 1999; 20: 637–42. [PMC free article] [PubMed] [Google Scholar]
- Fotiadis P, van Rooden S, van der Grond J, Schultz A, Martinez-Ramirez S, Auriel E. et al. Cortical atrophy in patients with cerebral amyloid angiopathy: a case-control study. Lancet Neurol 2016; 15: 811–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frontzek K, Lutz MI, Aguzzi A, Kovacs GG, Budka H. Amyloid-beta pathology and cerebral amyloid angiopathy are frequent in iatrogenic Creutzfeldt-Jakob disease after dural grafting. Swiss Med Weekly 2016; 146: w14287. [DOI] [PubMed] [Google Scholar]
- Garcia-Alloza M, Gregory J, Kuchibhotla KV, Fine S, Wei Y, Ayata C. et al. Cerebrovascular lesions induce transient beta-amyloid deposition. Brain 2011; 134(Pt 12): 3697–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorelick PB, Scuteri A, Black SE, Decarli C, Greenberg SM, Iadecola C. et al. Vascular contributions to cognitive impairment and dementia: a statement for healthcare professionals from the american heart association/american stroke association. Stroke 2011; 42: 2672–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouw AA, Seewann A, van der Flier WM, Barkhof F, Rozemuller AM, Scheltens P. et al. Heterogeneity of small vessel disease: a systematic review of MRI and histopathology correlations. J Neurol Neurosurg Psychiatry 2011; 82: 126–35. [DOI] [PubMed] [Google Scholar]
- Gray F, Dubas F, Roullet E, Escourolle R. Leukoencephalopathy in diffuse hemorrhagic cerebral amyloid angiopathy. Ann Neurol 1985; 18: 54–9. [DOI] [PubMed] [Google Scholar]
- Greenberg SM, Eng JA, Ning M, Smith EE, Rosand J. Hemorrhage burden predicts recurrent intracerebral hemorrhage after lobar hemorrhage. Stroke 2004; 35: 1415–20. [DOI] [PubMed] [Google Scholar]
- Greenberg SM, Grabowski T, Gurol ME, Skehan ME, Nandigam RN, Becker JA. et al. Detection of isolated cerebrovascular beta-amyloid with Pittsburgh compound B. Ann Neurol 2008; 64: 587–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg SM, Nandigam RN, Delgado P, Betensky RA, Rosand J, Viswanathan A. et al. Microbleeds versus macrobleeds: evidence for distinct entities. Stroke 2009a; 40: 2382–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg SM, Salman RA, Biessels GJ, van Buchem M, Cordonnier C, Lee JM. et al. Outcome markers for clinical trials in cerebral amyloid angiopathy. Lancet Neurol 2014; 13: 419–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg SM, Vernooij MW, Cordonnier C, Viswanathan A, Al-Shahi Salman R, Warach S. et al. Cerebral microbleeds: a guide to detection and interpretation. Lancet Neurol 2009b; 8: 165–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg SM, Vonsattel JP, Stakes JW, Gruber M, Finklestein SP. The clinical spectrum of cerebral amyloid angiopathy: presentations without lobar hemorrhage. Neurology 1993; 43: 2073–9. [DOI] [PubMed] [Google Scholar]
- Grinberg LT, Korczyn AD, Heinsen H. Cerebral amyloid angiopathy impact on endothelium. Exp Gerontol 2012; 47: 838–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo S, Kim WJ, Lok J, Lee SR, Besancon E, Luo BH. et al. Neuroprotection via matrix-trophic coupling between cerebral endothelial cells and neurons. Proc Natl Acad Sci USA 2008; 105: 7582–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo S, Zhou Y, Xing C, Lok J, Som AT, Ning M. et al. The vasculome of the mouse brain. PloS One 2012; 7: e52665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurol ME, Becker JA, Fotiadis P, Riley G, Schwab K, Johnson KA. et al. Florbetapir-PET to diagnose cerebral amyloid angiopathy: a prospective study. Neurology 2016; 87: 2043–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurol ME, Dierksen G, Betensky R, Gidicsin C, Halpin A, Becker A. et al. Predicting sites of new hemorrhage with amyloid imaging in cerebral amyloid angiopathy. Neurology 2012; 79: 320–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurol ME, Viswanathan A, Gidicsin C, Hedden T, Martinez-Ramirez S, Dumas A. et al. Cerebral amyloid angiopathy burden associated with leukoaraiosis: a positron emission tomography/magnetic resonance imaging study. Ann Neurol 2013; 73: 529–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall CN, Reynell C, Gesslein B, Hamilton NB, Mishra A, Sutherland BA. et al. Capillary pericytes regulate cerebral blood flow in health and disease. Nature 2014; 508: 55–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han BH, Zhou ML, Abousaleh F, Brendza RP, Dietrich HH, Koenigsknecht-Talboo J. et al. Cerebrovascular dysfunction in amyloid precursor protein transgenic mice: contribution of soluble and insoluble amyloid-beta peptide, partial restoration via gamma-secretase inhibition. J Neurosci 2008; 28: 13542–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkes CA, Hartig W, Kacza J, Schliebs R, Weller RO, Nicoll JA. et al. Perivascular drainage of solutes is impaired in the ageing mouse brain and in the presence of cerebral amyloid angiopathy. Acta Neuropathol 2011; 121: 431–43. [DOI] [PubMed] [Google Scholar]
- Hemphill JC, 3rd, Greenberg SM, Anderson CS, Becker K, Bendok BR, Cushman M. et al. Guidelines for the management of spontaneous intracerebral hemorrhage: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2015; 46: 2032–60. [DOI] [PubMed] [Google Scholar]
- Herve D, Godin O, Dufouil C, Viswanathan A, Jouvent E, Pachai C. et al. Three-dimensional MRI analysis of individual volume of lacunes in CADASIL. Stroke 2009; 40: 124–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill MD, Silver FL, Austin PC, Tu JV. Rate of stroke recurrence in patients with primary intracerebral hemorrhage. Stroke 2000; 31: 123–7. [DOI] [PubMed] [Google Scholar]
- Hofmeijer J, Kappelle LJ, Klijn CJ. Antithrombotic treatment and intracerebral haemorrhage: between Scylla and Charybdis. Pract Neurol 2015; 15: 250–6. [DOI] [PubMed] [Google Scholar]
- Jaunmuktane Z, Mead S, Ellis M, Wadsworth JD, Nicoll AJ, Kenny J. et al. Evidence for human transmission of amyloid-beta pathology and cerebral amyloid angiopathy. Nature 2015; 525: 247–50. [DOI] [PubMed] [Google Scholar]
- Jellinger KA. Alzheimer disease and cerebrovascular pathology: an update. J Neural Transm 2002; 109: 813–36. [DOI] [PubMed] [Google Scholar]
- Jespersen SN, Ostergaard L. The roles of cerebral blood flow, capillary transit time heterogeneity, and oxygen tension in brain oxygenation and metabolism. J Cereb Blood Flow Metab 2012; 32: 264–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia J, Cui M, Dai J, Liu B. (99 m)Tc(CO)3-Labeled benzothiazole derivatives preferentially bind cerebrovascular amyloid: potential use as imaging agents for cerebral amyloid angiopathy. Mol Pharm 2015; 12: 2937–46. [DOI] [PubMed] [Google Scholar]
- Jia J, Cui M, Dai J, Wang X, Ding Y, Jiaa H. et al. 99mTc-labeled benzothiazole and stilbene derivatives as imaging agents for Aβ plaques in cerebral amyloid angiopathy. Med Chem Commun 2014; 5: 153–8. [Google Scholar]
- Johnson KA, Gregas M, Becker JA, Kinnecom C, Salat DH, Moran EK. et al. Imaging of amyloid burden and distribution in cerebral amyloid angiopathy. Ann Neurol 2007; 62: 229–34. [DOI] [PubMed] [Google Scholar]
- Kalaria RN, Ballard C. Overlap between pathology of Alzheimer disease and vascular dementia. Alzheimer Dis Assoc Disord 1999; 13 (Suppl 3): S115–23. [DOI] [PubMed] [Google Scholar]
- Keable A, Fenna K, Yuen HM, Johnston DA, Smyth NR, Smith C. et al. Deposition of amyloid beta in the walls of human leptomeningeal arteries in relation to perivascular drainage pathways in cerebral amyloid angiopathy. Biochim Biophys Acta 2016; 1862: 1037–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keage HA, Carare RO, Friedland RP, Ince PG, Love S, Nicoll JA. et al. Population studies of sporadic cerebral amyloid angiopathy and dementia: a systematic review. BMC Neurol 2009; 9: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimbrough IF, Robel S, Roberson ED, Sontheimer H. Vascular amyloidosis impairs the gliovascular unit in a mouse model of Alzheimer’s disease. Brain 2015; 138(Pt 12): 3716–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP. et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann Neurol 2004; 55: 306–19. [DOI] [PubMed] [Google Scholar]
- Knudsen KA, Rosand J, Karluk D, Greenberg SM. Clinical diagnosis of cerebral amyloid angiopathy: validation of the Boston criteria. Neurology 2001; 56: 537–9. [DOI] [PubMed] [Google Scholar]
- Kovari E, Herrmann FR, Hof PR, Bouras C. The relationship between cerebral amyloid angiopathy and cortical microinfarcts in brain ageing and Alzheimer’s disease. Neuropathol Appl Neurobiol 2013; 39: 498–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar-Singh S. Hereditary and sporadic forms of abeta-cerebrovascular amyloidosis and relevant transgenic mouse models. Int J Mol Sci 2009; 10: 1872–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linn J, Halpin A, Demaerel P, Ruhland J, Giese AD, Dichgans M. et al. Prevalence of superficial siderosis in patients with cerebral amyloid angiopathy. Neurology 2010; 74: 1346–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longstreth WT Jr., Bernick C, Manolio TA, Bryan N, Jungreis CA, Price TR. Lacunar infarcts defined by magnetic resonance imaging of 3660 elderly people: the Cardiovascular Health Study. Arch Neurol 1998; 55: 1217–25. [DOI] [PubMed] [Google Scholar]
- Love S, Chalmers K, Ince P, Esiri M, Attems J, Jellinger K. et al. Development, appraisal, validation and implementation of a consensus protocol for the assessment of cerebral amyloid angiopathy in post-mortem brain tissue. Am J Neurodegener Dis 2014; 3: 19–32. [PMC free article] [PubMed] [Google Scholar]
- Ly JV, Donnan GA, Villemagne VL, Zavala JA, Ma H, O’Keefe G. et al. 11C-PIB binding is increased in patients with cerebral amyloid angiopathy-related hemorrhage. Neurology 2010; 74: 487–93. [DOI] [PubMed] [Google Scholar]
- Maia LF, Mackenzie IR, Feldman HH. Clinical phenotypes of Cerebral Amyloid Angiopathy. J Neurol Sci 2007; 257: 23–30. [DOI] [PubMed] [Google Scholar]
- Makela M, Paetau A, Polvikoski T, Myllykangas L, Tanskanen M. Capillary amyloid-beta protein deposition in a population-based study (Vantaa 85+). J Alzheimers Dis 2015; 49: 149–57. [DOI] [PubMed] [Google Scholar]
- Mandybur TI. Cerebral amyloid angiopathy: the vascular pathology and complications. J Neuropathol Exp Neurol 1986; 45: 79–90. [PubMed] [Google Scholar]
- Manousopoulou A, Gatherer M, Smith C, Nicoll JA, Woelk CH, Johnson M. et al. Systems proteomic analysis reveals that clusterin and tissue inhibitor of metalloproteinases 3 increase in leptomeningeal arteries affected by cerebral amyloid angiopathy. Neuropathol Appl Neurobiol 2016, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin-Padilla M, Knopman DS. Developmental aspects of the intracerebral microvasculature and perivascular spaces: insights into brain response to late-life diseases. J Neuropathol Exp Neurol 2011; 70: 1060–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Ramirez S, Pontes-Neto OM, Dumas AP, Auriel E, Halpin A, Quimby M. et al. Topography of dilated perivascular spaces in subjects from a memory clinic cohort. Neurology 2013; 80: 1551–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Ramirez S, Romero JR, Shoamanesh A, McKee AC, Van Etten E, Pontes-Neto O. et al. Diagnostic value of lobar microbleeds in individuals without intracerebral hemorrhage. Alzheimers Demen 2015; 11: 1480–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattila OS, Sairanen T, Laakso E, Paetau A, Tanskanen M, Lindsberg PJ. Cerebral amyloid angiopathy related hemorrhage after stroke thrombolysis: case report and literature review. Neuropathology 2015; 35: 70–4. [DOI] [PubMed] [Google Scholar]
- Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC. et al. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science 2010; 330: 1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAleese KE, Alafuzoff I, Charidimou A, De Reuck J, Grinberg LT, Hainsworth AH. et al. Post-mortem assessment in vascular dementia: advances and aspirations. BMC Med 2016; 14: 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesker DJ, Poels MM, Ikram MA, Vernooij MW, Hofman A, Vrooman HA. et al. Lobar distribution of cerebral microbleeds: the rotterdam scan study. Arch Neurol 2011; 68: 656–9. [DOI] [PubMed] [Google Scholar]
- Morris AW, Sharp MM, Albargothy NJ, Fernandes R, Hawkes CA, Verma A. et al. Vascular basement membranes as pathways for the passage of fluid into and out of the brain. Acta Neuropathol 2016; 131: 725–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moulin S, Labreuche J, Bombois S, Rossi C, Boulouis G, Henon H. et al. Dementia risk after spontaneous intracerebral haemorrhage: a prospective cohort study. Lancet Neurol 2016; 15: 820–9. [DOI] [PubMed] [Google Scholar]
- Natte R, Maat-Schieman ML, Haan J, Bornebroek M, Roos RA, van Duinen SG. Dementia in hereditary cerebral hemorrhage with amyloidosis-Dutch type is associated with cerebral amyloid angiopathy but is independent of plaques and neurofibrillary tangles. Ann Neurol 2001; 50: 765–72. [DOI] [PubMed] [Google Scholar]
- Nedergaard M. Neuroscience. Garbage truck of the brain. Science 2013; 340: 1529–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson AR, Sweeney MD, Sagare AP, Zlokovic BV. Neurovascular dysfunction and neurodegeneration in dementia and Alzheimer’s disease. Biochim Biophys Acta 2016; 1862: 887–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa K, Younkin L, Ebeling C, Turner SK, Westaway D, Younkin S. et al. Abeta 1‐40-related reduction in functional hyperemia in mouse neocortex during somatosensory activation. Proc Natl Acad Sci USA 2000; 97: 9735–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohab JJ, Fleming S, Blesch A, Carmichael ST. A neurovascular niche for neurogenesis after stroke. J Neurosci 2006; 26: 13007–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto Y, Ihara M, Fujita Y, Ito H, Takahashi R, Tomimoto H. Cortical microinfarcts in Alzheimer’s disease and subcortical vascular dementia. Neuroreport 2009; 20: 990–6. [DOI] [PubMed] [Google Scholar]
- Olichney JM, Hansen LA, Hofstetter CR, Grundman M, Katzman R, Thal LJ. Cerebral infarction in Alzheimer’s disease is associated with severe amyloid angiopathy and hypertension. Arch Neurol 1995; 52: 702–8. [DOI] [PubMed] [Google Scholar]
- Ostergaard L, Engedal TS, Moreton F, Hansen MB, Wardlaw JM, Dalkara T. et al. Cerebral small vessel disease: aapillary pathways to stroke and cognitive decline. J Cereb Blood Flow Metab 2016; 36: 302–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantelakis S. A particular type of senile angiopathy of the central nervous system: congophilic angiopathy, topography and frequency [in French]. Monatsschr Psychiatr Neurol 1954; 128: 219–56. [PubMed] [Google Scholar]
- Pantoni L. Cerebral small vessel disease: from pathogenesis and clinical characteristics to therapeutic challenges. Lancet Neurol 2010; 9: 689–701. [DOI] [PubMed] [Google Scholar]
- Park L, Zhou P, Koizumi K, El Jamal S, Previti ML, Van Nostrand WE. et al. Brain and circulating levels of Abeta1‐40 differentially contribute to vasomotor dysfunction in the mouse brain. Stroke 2013; 44: 198–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquini M, Benedictus MR, Boulouis G, Rossi C, Dequatre-Ponchelle N, Cordonnier C. Incident cerebral microbleeds in a cohort of intracerebral hemorrhage. Stroke 2016; 47: 689–94. [DOI] [PubMed] [Google Scholar]
- Peca S, McCreary CR, Donaldson E, Kumarpillai G, Shobha N, Sanchez K. et al. Neurovascular decoupling is associated with severity of cerebral amyloid angiopathy. Neurology 2013; 81: 1659–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeifer LA, White LR, Ross GW, Petrovitch H, Launer LJ. Cerebral amyloid angiopathy and cognitive function: the HAAS autopsy study. Neurology 2002; 58: 1629–34. [DOI] [PubMed] [Google Scholar]
- Pober JS, Sessa WC. Evolving functions of endothelial cells in inflammation. Nat Rev Immunol 2007; 7: 803–15. [DOI] [PubMed] [Google Scholar]
- Raposo N, Viguier A, Cuvinciuc V, Calviere L, Cognard C, Bonneville F. et al. Cortical subarachnoid haemorrhage in the elderly: a recurrent event probably related to cerebral amyloid angiopathy. Eur J Neurol 2011; 18: 597–603. [DOI] [PubMed] [Google Scholar]
- Reijmer Y, Fotiadis P, Martinez-Ramirez S, Ayres MA, Vashkevich A, Schwab K. et al. Progression of brain network alterations in patients with cerebral amyloid angiopathy. international stroke conference. Los Angeles: Stroke; 2016a. P. A:124. [Google Scholar]
- Reijmer YD, Fotiadis P, Martinez-Ramirez S, Salat DH, Schultz A, Shoamanesh A. et al. Structural network alterations and neurological dysfunction in cerebral amyloid angiopathy. Brain 2015; 138(Pt 1): 179–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reijmer YD, van Veluw SJ, Greenberg SM. Ischemic brain injury in cerebral amyloid angiopathy. J Cereb Blood Flow Metab 2016b; 36: 40–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revesz T, Holton JL, Lashley T, Plant G, Frangione B, Rostagno A. et al. Genetics and molecular pathogenesis of sporadic and hereditary cerebral amyloid angiopathies. Acta Neuropathol 2009; 118: 115–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins EM, Betensky RA, Domnitz SB, Purcell SM, Garcia-Alloza M, Greenberg C. et al. Kinetics of cerebral amyloid angiopathy progression in a transgenic mouse model of Alzheimer disease. J Neurosci 2006; 26: 365–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roher AE, Kuo YM, Esh C, Knebel C, Weiss N, Kalback W. et al. Cortical and leptomeningeal cerebrovascular amyloid and white matter pathology in Alzheimer’s disease. Mol Med 2003; 9: 112–22. [PMC free article] [PubMed] [Google Scholar]
- Rosand J, Muzikansky A, Kumar A, Wisco JJ, Smith EE, Betensky RA. et al. Spatial clustering of hemorrhages in probable cerebral amyloid angiopathy. Ann Neurol 2005; 58: 459–62. [DOI] [PubMed] [Google Scholar]
- Rosenberg GA, Yang Y. Vasogenic edema due to tight junction disruption by matrix metalloproteinases in cerebral ischemia. Neurosurg Focus 2007; 22: E4. [DOI] [PubMed] [Google Scholar]
- Saito S, Ihara M. New therapeutic approaches for Alzheimer’s disease and cerebral amyloid angiopathy. Front Aging Neurosci 2014; 6: 290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salat DH, Smith EE, Tuch DS, Benner T, Pappu V, Schwab KM. et al. White matter alterations in cerebral amyloid angiopathy measured by diffusion tensor imaging. Stroke 2006; 37: 1759–64. [DOI] [PubMed] [Google Scholar]
- Samarasekera N, Al-Shahi Salman R, Huitinga I, Klioueva N, McLean CA, Kretzschmar H. et al. Brain banking for neurological disorders. Lancet Neurol 2013; 12: 1096–105. [DOI] [PubMed] [Google Scholar]
- Sandoval KE, Witt KA. Blood-brain barrier tight junction permeability and ischemic stroke. Neurobiol Dis 2008; 32: 200–19. [DOI] [PubMed] [Google Scholar]
- Schley D, Carare-Nnadi R, Please CP, Perry VH, Weller RO. Mechanisms to explain the reverse perivascular transport of solutes out of the brain. J Theor Biol 2006; 238: 962–74. [DOI] [PubMed] [Google Scholar]
- Schrag M, Kirshner H. Neuropsychological effects of cerebral amyloid angiopathy. Curr Neurol Neurosci Rep 2016; 16: 76. [DOI] [PubMed] [Google Scholar]
- Schrag M, McAuley G, Pomakian J, Jiffry A, Tung S, Mueller C. et al. Correlation of hypointensities in susceptibility-weighted images to tissue histology in dementia patients with cerebral amyloid angiopathy: a postmortem MRI study. Acta Neuropathol 2010; 119: 291–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Q, Goderie SK, Jin L, Karanth N, Sun Y, Abramova N. et al. Endothelial cells stimulate self-renewal and expand neurogenesis of neural stem cells. Science 2004; 304: 1338–40. [DOI] [PubMed] [Google Scholar]
- Shih AY, Blinder P, Tsai PS, Friedman B, Stanley G, Lyden PD. et al. The smallest stroke: occlusion of one penetrating vessel leads to infarction and a cognitive deficit. Nat Neurosci 2013; 16: 55–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih AY, Ruhlmann C, Blinder P, Devor A, Drew PJ, Friedman B. et al. Robust and fragile aspects of cortical blood flow in relation to the underlying angioarchitecture. Microcirculation 2015; 22: 204–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin HK, Jones PB, Garcia-Alloza M, Borrelli L, Greenberg SM, Bacskai BJ. et al. Age-dependent cerebrovascular dysfunction in a transgenic mouse model of cerebral amyloid angiopathy. Brain 2007; 130(Pt 9): 2310–9. [DOI] [PubMed] [Google Scholar]
- Shoamanesh A, Martinez-Ramirez S, Oliveira-Filho J, Reijmer Y, Falcone GJ, Ayres A. et al. Interrelationship of superficial siderosis and microbleeds in cerebral amyloid angiopathy. Neurology 2014; 83: 1838–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skrobot OA, Attems J, Esiri M, Hortobagyi T, Ironside JW, Kalaria RN. et al. Vascular cognitive impairment neuropathology guidelines (VCING): the contribution of cerebrovascular pathology to cognitive impairment. Brain 2016; 139: 2957–69. [DOI] [PubMed] [Google Scholar]
- Smith EE, Greenberg SM. Clinical diagnosis of cerebral amyloid angiopathy: validation of the Boston criteria. Curr Atheroscler Rep 2003; 5: 260–6. [DOI] [PubMed] [Google Scholar]
- Smith EE, Greenberg SM. Beta-amyloid, blood vessels, and brain function. Stroke 2009; 40: 2601–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith EE, Schneider JA, Wardlaw JM, Greenberg SM. Cerebral microinfarcts: the invisible lesions. Lancet Neurol 2012a; 11: 272–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith EE, Schneider JA, Wardlaw JM, Greenberg SM. Cerebral microinfarcts: the invisible lesions. Lancet Neurol 2012b; 11: 272–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner T, Al-Shahi Salman R, Beer R, Christensen H, Cordonnier C, Csiba L. et al. European Stroke Organisation (ESO) guidelines for the management of spontaneous intracerebral hemorrhage. Int J Stroke 2014; 9: 840–55. [DOI] [PubMed] [Google Scholar]
- Switzer AR, McCreary C, Batool S, Stafford RB, Frayne R, Goodyear BG. et al. Longitudinal decrease in blood oxygenation level dependent response in cerebral amyloid angiopathy. Neuroimage Clin 2016; 11: 461–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarasoff-Conway JM, Carare RO, Osorio RS, Glodzik L, Butler T, Fieremans E. et al. Clearance systems in the brain-implications for Alzheimer disease. Nat Rev Neurol 2015; 11: 457–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatsumi S, Shinohara M, Yamamoto T. Direct comparison of histology of microbleeds with postmortem MR images: a case report. Cerebrovasc Dis 2008; 26: 142–6. [DOI] [PubMed] [Google Scholar]
- Thal DR, Ghebremedhin E, Orantes M, Wiestler OD. Vascular pathology in Alzheimer disease: correlation of cerebral amyloid angiopathy and arteriosclerosis/lipohyalinosis with cognitive decline. J Neuropathol Exp Neurol 2003; 62: 1287–301. [DOI] [PubMed] [Google Scholar]
- van Veluw SJ, Biessels GJ, Bouvy WH, Spliet WG, Zwanenburg JJ, Luijten PR. et al. Cerebral amyloid angiopathy severity is linked to dilation of juxtacortical perivascular spaces. J Cereb Blood Flow Metab 2016; 36: 576–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Veluw SJ, Kuijf HJ, Charidimou A, Viswanathan A, Biessels GJ, Rozemuller AJ. et al. Reduced vascular amyloid burden at microhemorrhage sites in cerebral amyloid angiopathy. Acta Neuropathol 2017; 133: 409–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernooij MW, Ikram MA, Tanghe HL, Vincent AJ, Hofman A, Krestin GP. et al. Incidental findings on brain MRI in the general population. N Engl J Med 2007; 357: 1821–8. [DOI] [PubMed] [Google Scholar]
- Vernooij MW, van der Lugt A, Ikram MA, Wielopolski PA, Niessen WJ, Hofman A. et al. Prevalence and risk factors of cerebral microbleeds: the Rotterdam Scan Study. Neurology 2008; 70: 1208–14. [DOI] [PubMed] [Google Scholar]
- Villemagne VL, Fodero-Tavoletti MT, Masters CL, Rowe CC. Tau imaging: early progress and future directions. Lancet Neurol 2015; 14: 114–24. [DOI] [PubMed] [Google Scholar]
- Vinters HV. Cerebral amyloid angiopathy. A critical review. Stroke 1987; 18: 311–24. [DOI] [PubMed] [Google Scholar]
- Vinters HV. Emerging concepts in Alzheimer’s disease. Annu Rev Pathol 2015; 10: 291–319. [DOI] [PubMed] [Google Scholar]
- Vinters HV, Gilbert JJ. Cerebral amyloid angiopathy: incidence and complications in the aging brain. II. The distribution of amyloid vascular changes. Stroke 1983; 14: 924–8. [DOI] [PubMed] [Google Scholar]
- Viswanathan A, Greenberg SM. Cerebral amyloid angiopathy in the elderly. Ann Neurol 2011; 70: 871–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan A, Guichard JP, Gschwendtner A, Buffon F, Cumurcuic R, Boutron C. et al. Blood pressure and haemoglobin A1c are associated with microhaemorrhage in CADASIL: a two-centre cohort study. Brain 2006a; 129(Pt 9): 2375–83. [DOI] [PubMed] [Google Scholar]
- Viswanathan A, Patel P, Rahman R, Nandigam RN, Kinnecom C, Bracoud L. et al. Tissue microstructural changes are independently associated with cognitive impairment in cerebral amyloid angiopathy. Stroke 2008; 39: 1988–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan A, Rakich SM, Engel C, Snider R, Rosand J, Greenberg SM. et al. Antiplatelet use after intracerebral hemorrhage. Neurology 2006b; 66: 206–9. [DOI] [PubMed] [Google Scholar]
- Vonsattel JP, Myers RH, Hedley-Whyte ET, Ropper AH, Bird ED, Richardson EP. Cerebral amyloid angiopathy without and with cerebral hemorrhages: a comparative histological study. Ann Neurol 1991a; 30: 637–49. [DOI] [PubMed] [Google Scholar]
- Vonsattel JP, Myers RH, Hedley-Whyte ET, Ropper AH, Bird ED, Richardson EP Jr. Cerebral amyloid angiopathy without and with cerebral hemorrhages: a comparative histological study. Ann Neurol 1991b; 30: 637–49. [DOI] [PubMed] [Google Scholar]
- Wang Z, Soo YO, Mok VC. Cerebral microbleeds: is antithrombotic therapy safe to administer? Stroke 2014; 45: 2811–7. [DOI] [PubMed] [Google Scholar]
- Wardlaw JM, Smith EE, Biessels GJ, Cordonnier C, Fazekas F, Frayne R. et al. Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol 2013; 12: 822–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weimar C, Benemann J, Terborg C, Walter U, Weber R, Diener HC. Recurrent stroke after lobar and deep intracerebral hemorrhage: a hospital-based cohort study. Cerebrovasc Dis 2011; 32: 283–8. [DOI] [PubMed] [Google Scholar]
- Weller RO, Hawkes CA, Kalaria RN, Werring DJ, Carare RO. White matter changes in dementia: role of impaired drainage of interstitial fluid. Brain Pathol 2015; 25: 63–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weller RO, Massey A, Newman TA, Hutchings M, Kuo YM, Roher AE. Cerebral amyloid angiopathy: amyloid beta accumulates in putative interstitial fluid drainage pathways in Alzheimer’s disease. Am J Pathol 1998; 153: 725–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weller RO, Subash M, Preston SD, Mazanti I, Carare RO. Perivascular drainage of amyloid-beta peptides from the brain and its failure in cerebral amyloid angiopathy and Alzheimer’s disease. Brain Pathol 2008; 18: 253–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westover MB, Bianchi MT, Yang C, Schneider JA, Greenberg SM. Estimating cerebral microinfarct burden from autopsy samples. Neurology 2013; 80: 1365–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong L, Davidsdottir S, Reijmer YD, Shoamanesh A, Roongpiboonsopit D, Thanprasertsuk S. et al. Cognitive profile and its association with neuroimaging markers of non-demented cerebral amyloid angiopathy patients in a stroke unit. J Alzheimers Dis 2016a; 52: 171–8. [DOI] [PubMed] [Google Scholar]
- Xiong L, Reijmer YD, Charidimou A, Cordonnier C, Viswanathan A. Intracerebral hemorrhage and cognitive impairment. Biochim Biophy Acta 2016b; 1862: 939–44. [DOI] [PubMed] [Google Scholar]
- Zhao L, Arbel-Ornath M, Wang X, Betensky RA, Greenberg SM, Frosch MP. et al. Matrix metalloproteinase 9-mediated intracerebral hemorrhage induced by cerebral amyloid angiopathy. Neurobiol Aging 2015; 36: 2963–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlokovic BV. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat Rev Neurosci 2011; 12: 723–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlokovic BV, Deane R, Sagare AP, Bell RD, Winkler EA. Low-density lipoprotein receptor-related protein-1: a serial clearance homeostatic mechanism controlling Alzheimer’s amyloid beta-peptide elimination from the brain. J Neurochem 2010; 115: 1077–89. [DOI] [PMC free article] [PubMed] [Google Scholar]