

Patients with PTSD exhibit hypersensitivity to a broad range of threat-neutral stimuli. Clancy et al. demonstrate intrinsic sensory hyperactivity and sensory-driven inhibition deficits, which could overwhelm frontal processing and disrupt executive control. This sensory pathology, implicating dysregulated triangular sensory-prefrontal cortex-amygdala circuitry, suggests potential utility of sensory-based interventions in PTSD.
Keywords: post-traumatic stress disorder (PTSD), sensory hyperactivity, resting state, alpha/gamma oscillations, Granger causality
Abstract
Post-traumatic stress disorder is characterized by exaggerated threat response, and theoretical accounts to date have focused on impaired threat processing and dysregulated prefrontal-cortex-amygdala circuitry. Nevertheless, evidence is accruing for broad, threat-neutral sensory hyperactivity in post-traumatic stress disorder. As low-level, sensory processing impacts higher-order operations, such sensory anomalies can contribute to widespread dysfunctions, presenting an additional aetiological mechanism for post-traumatic stress disorder. To elucidate a sensory pathology of post-traumatic stress disorder, we examined intrinsic visual cortical activity (based on posterior alpha oscillations) and bottom-up sensory-driven causal connectivity (Granger causality in the alpha band) during a resting state (eyes open) and a passive, serial picture viewing state. Compared to patients with generalized anxiety disorder (n = 24) and healthy control subjects (n = 20), patients with post-traumatic stress disorder (n = 25) demonstrated intrinsic sensory hyperactivity (suppressed posterior alpha power, source-localized to the visual cortex—cuneus and precuneus) and bottom-up inhibition deficits (reduced posterior→frontal Granger causality). As sensory input increased from resting to passive picture viewing, patients with post-traumatic stress disorder failed to demonstrate alpha adaptation, highlighting a rigid, set mode of sensory hyperactivity. Interestingly, patients with post-traumatic stress disorder also showed heightened frontal processing (augmented frontal gamma power, source-localized to the superior frontal gyrus and dorsal cingulate cortex), accompanied by attenuated top–down inhibition (reduced frontal→posterior causality). Importantly, not only did suppressed alpha power and bottom-up causality correlate with heightened frontal gamma power, they also correlated with increased severity of sensory and executive dysfunctions (i.e. hypervigilance and impulse control deficits, respectively). Therefore, sensory aberrations help construct a vicious cycle in post-traumatic stress disorder that is in action even at rest, implicating dysregulated triangular sensory-prefrontal-cortex-amygdala circuitry: intrinsic sensory hyperactivity and disinhibition give rise to frontal overload and disrupt executive control, fuelling and perpetuating post-traumatic stress disorder symptoms. Absent in generalized anxiety disorder, these aberrations highlight a unique sensory pathology of post-traumatic stress disorder (ruling out effects merely reflecting anxious hyperarousal), motivating new interventions targeting sensory processing and the sensory brain in these patients.
Introduction
Post-traumatic stress disorder (PTSD) is a highly debilitating mental illness characterized by hypersensitivity to threat (Dalgleish et al., 2001). Nevertheless, unlike most anxiety disorders that are also characterized by hypersensitivity to threat, such as generalized anxiety disorder (GAD) and social anxiety disorder (Mennin et al., 2005; Vassilopoulos, 2005; Garner et al., 2006; Cisler and Koster, 2010; Weinberg and Hajcak, 2011), PTSD exhibits hypersensitivity to a broad range of sensory stimuli beyond trauma-related or threatening cues (Ehlers and Clark, 2000; Hayes et al., 2012). Patients with PTSD often complain about being bothered or feeling ‘flooded’ by everyday stimuli, including background noises that others would not notice (Stewart and White, 2008). Systematic profiling of basic sensory processes demonstrates sensory filtering/gating deficits and sensory hypersensitivity in PTSD (Stewart and White, 2008; Engel-Yeger et al., 2013), while prepulse inhibition of baseline startle response further specifies sensorimotor gating deficiency in PTSD (Grillon et al., 1996). Furthermore, using non-startle, trauma-neutral cues, multiple electrophysiological studies show attenuated repetition suppression in P50 (reflecting impaired sensory gating) and exaggerated sensory evoked brain potentials and mismatch negativity (reflecting sensory hyper-reactivity) in PTSD (Gillette et al., 1997; Morgan and Grillon, 1999; Neylan et al., 1999; Skinner et al., 1999; Shalev et al., 2000; Lewine et al., 2002; Ghisolfi et al., 2004; Holstein et al., 2010; Ge et al., 2011; Javanbakht et al., 2011; Gjini et al., 2013).
This sensory hyperactivity and deficient sensory gating closely aligns with a defining symptom of PTSD—hypervigilance, namely, excessive and constant ‘scanning’ of the environment for potential threat (American Psychiatric Association, 2013). In fact, sensory anomalies and hypervigilance may stem from the same biological root: chronic, tonic increases of dopaminergic and noradrenergic levels in PTSD are known to suppress sensory gating and heighten postsynaptic activity in the sensory cortex (i.e. increase sensory cortical excitability) (Adler et al., 1988; Aston-Jones et al., 1994; Southwick et al., 1997; Berridge and Waterhouse, 2003; Sherin and Nemeroff, 2011; Baisley et al., 2012), which would in turn sustain hypervigilance in these patients. With heightened sensory cortical excitability combined with deficient sensory gating, excessive sensory feedforward input would propagate to higher-order brain regions, inundating higher-order processing and potentially causing global hyperactivity/disinhibition and widespread dysfunctions. In fact, sensory hyperactivity and gating failure due to dopamine and noradrenaline dysregulation have been well documented in schizophrenia and mania; moreover, such sensory anomalies have been linked to various executive deficits (e.g. attention and working memory), constituting a pathological mechanism for these disorders (Freedman et al., 1987; Adler et al., 1990; Geyer et al., 2001; Thoma et al., 2003; Park et al., 2015). Therefore, it stands to reason that sensory hyperactivity and gating failure may play a similar role in PTSD pathology and thus warrant further research.
Our study aimed to elucidate this sensory pathology of PTSD and its neural pathophysiology using EEG. To isolate the broad, threat-neutral sensory hyperactivity in PTSD from anxiety disorders related to threat-specific hyperactivity (and to rule out effects due to mere anxious hyperarousal), we contrasted patients with PTSD versus patients with GAD, in addition to a healthy control group. Akin to the constant, ongoing sensory scanning and vigilance in PTSD, even in the absence of clear threat, we specifically interrogated intrinsic, ‘resting state’ sensory activity with no task demand. Towards that end, we measured alpha (8–12 Hz) oscillations, a well-established inverse marker of sensory activity (including sensory cortical excitability and sensory gating) such that reduced resting state alpha oscillatory power (at occipitoparietal sites) would index intrinsic sensory hyperactivity (Worden et al., 2000; Shaw, 2003; Palva and Palva, 2007; Bollimunta et al., 2008; Foxe and Snyder, 2011; Klimesch, 2012). In addition, alpha oscillatory activities are particularly critical for long-range rhythmic synchronization mediating inhibitory inter-regional interactions (Engel et al., 2001; Tang et al., 2007; Sadaghiani et al., 2012; Hillebrand et al., 2016; Wang et al., 2016). We thus assayed alpha causal interaction between occipitoparietal and frontal sites using Granger causality (GC) analysis (Geweke, 1982; Ding et al., 2006), thereby elucidating deficient inhibition of sensory propagation (i.e. excessive sensory projection) to the frontal cortex in PTSD. Relatedly, we measured frontal gamma oscillations (30–50 Hz), which increase with local neuronal excitation, to examine frontal, higher-order processing (Fell et al., 2003; Jia and Kohn, 2011) and test the hypothesis of frontal overload due to excessive sensory projection in PTSD. Finally, highlighting the clinical implications of this neural pathophysiology, we interrogated the association between aberrations in these neural activities and dysfunctions in low-order, sensory processing and higher-order, executive control.
Materials and methods
Participants
Outpatients defined by a current primary diagnosis of PTSD (n = 28) or GAD (n = 24), and healthy control subjects (n = 23) with no current or past diagnoses (in the past year) participated in the study after providing written, informed consent approved by the Florida State University Institutional Review Board and the Department of Defense Human Research Protection Official’s Review. Diagnoses were made by trained clinicians using the Structured Clinical Interview for DSM-V (American Psychiatric Association, 2013). Participants were 19–60 years old with no history of severe neurological disorders or traumatic brain injury, no current or past psychotic spectrum or bipolar disorders, and no current substance dependence or abuse of opioids, stimulants, or cocaine.
Three participants with PTSD and three healthy control subjects were excluded due to excessive EEG artefact or failure to follow instructions, resulting in a final sample of 25 PTSD, 24 GAD and 20 healthy control participants. Among patients with PTSD, one patient did not have standard resting state (S-RS) data, and four patients did not have modified resting state (M-RS) data, and were thus excluded from analyses considering both states simultaneously. The three groups were equivalent in age and gender ratio (P’s > 0.1), but the PTSD group had higher substance use than the other groups (P’s < 0.005). Substance use was thus entered as a covariate in all relevant analyses. Table 1 provides additional demographic information and questionnaire scores for the three groups. Further details are provided in the Supplementary material.
Table 1.
Participant demographics
| PTSD | GAD | HC | |
|---|---|---|---|
| n | 25 | 24 | 20 |
| Age (years) | 34.6 ± 10.4 | 30.0 ± 12.5 | 32.5 ± 13.8 |
| Gender (female/male) | 16/9 | 17/7 | 8/12 |
| Substance use, %a | 64*** | 0 | 15 |
| Medication use, % | 40 | 50 | 20 |
| DERS-impulse control | 15.1 ± 1.1** | 13.5 ± 1.1 | 10.5 ± 1.2 |
| PCL total | 61.0 ± 16.1** | 43.2 ± 14.0** | 30.7 ± 9.0 |
| PCL arousal cluster | 18.5 ± 5.2** | 14.7 ± 4.6** | 9.2 ± 2.7 |
| PCL-hypervigilance | 4.1 ± 1.0** | 2.4 ± 1.3* | 1.7 ± 0.8 |
| BAI | 26.2 ± 15.7* | 15.8 ± 9.1* | 7.0 ± 7.5 |
| STAI-T | 54.6 ± 9.5** | 54.9 ± 6.7** | 40.3 ± 8.2 |
| BDI | 26.8 ± 12.5** | 22.3 ± 7.5** | 11.6 ± 8.8 |
| Axis I comorbidity | n (%) | n (%) | |
| No comorbid diagnoses | 4 (16) | 10 (42) | |
| Comorbid diagnoses | 21 (84) | 14 (58) | |
| PDD | 12 (48) | 3 (13) | |
| SAD | 9 (36) | 8 (33) | |
| MDD | 5 (20) | 3 (13) | |
| Panic disorder | 3 (12) | 1 (4) | |
| Specific phobia | 2 (8) | 3 (13) | |
| OCD | 2 (8%) | 0 | |
| Alcohol use | 3 (12) | 0 | |
| Substance use | 3 (12) | 0 | |
| Otherb | 10 (40) | 5 (21) | |
| Mean diagnoses, n (SD) | 2.22 (1.47) | 1.00 (1.25) |
BAI = Beck Anxiety Inventory; STAI-T = State and Trait Anxiety Inventory – Trait; BDI = Beck Depression Inventory; DERS = Difficulties in Emotion Regulation Scale; PCL = PTSD Checklist; PDD = Persistent Depressive Disorder; SAD = Social Anxiety Disorder; OCD = Obsessive Compulsive Disorder.
Both PTSD and GAD groups showed higher scores on all questionnaires compared to the healthy control group (P’s < 0.05), except for DERS-Impulse Control, which was specifically elevated in PTSD. The PTSD group further showed higher scores on the PCL total, PCL-arousal cluster and PCL-hypervigilance, and the DERS-Impulse control and BAI questionnaires, relative to the GAD group (P’s < 0.05).
aSubjects with opioid, stimulant and cocaine use were excluded. *P < 0.05; **P < 0.01, ***P < 0.005.
bOther diagnoses refer to diagnoses endorsed by no more than one subject per group, including: binge eating disorder, agoraphobia, hoarding, depressive-not otherwise specified, bulimia, trichotillomania, excoriation, anxiety-not otherwise specified, anorexia nervosa, and other trauma-related disorders.
Questionnaires
PTSD checklist for DSM-IV: civilian version
The PTSD checklist (PCL) (Blanchard et al., 1996) is a widely used measure of PTSD symptom severity across four clusters: intrusion; avoidance; negative alterations in mood and cognition; and hyperarousal. Item 16 assessing symptoms of hypervigilance or ‘being watchful or on guard’ was used to index hypervigilance. In the present study, internal consistency was high for the total questionnaire (α = 0.95), as well as the individual hyperarousal cluster (α = 0.84).
Difficulties in Emotion Regulation: impulse control subscale
The Difficulties in Emotion Regulation (DERS) (Gratz and Roemer, 2004) is a 36-item measure of emotion dysregulation across six domains. The impulse control subscale is particularly pertinent to the current investigation by tapping into executive inhibition when distressed. The impulse control subscale consists of six items on a 5-point Likert scale (e.g. ‘When I am upset, I become out of control’). The DERS and its subscales have strong internal consistency and construct validity within both clinical and non-clinical samples (Gratz and Roemer, 2004).
Other questionnaires included Beck Anxiety Inventory, State-Trait Anxiety Inventory—Trait and Beck Depression Inventory, details of which are provided in the Supplementary material. As indicated in Table 1, there was a main effect of Group for all the questionnaires (F’s > 4.50, P’s < 0.05). PTSD and GAD groups showed higher scores compared to the healthy control group (P’s < 0.05) in all questionnaires, except for DERS-Impulse Control, which was specifically elevated in PTSD (GAD versus healthy controls, P = 0.06). The PTSD group showed further elevations on the Beck Anxiety Inventory, PCL total, PCL-hyperarousal cluster, and PCL-hypervigilance symptom, compared to the GAD group (P’s < 0.005).
Experimental paradigm
Participants were seated in a comfortable recliner in a dimly lit, sound attenuated and electrically shielded room. EEG data were recorded during eyes-open resting state (2 min, eyes fixating on a crosshair on the screen), followed by a session of passive viewing of scenes (5 min; 921 pictures, neutral, positive and negative randomly intermixed; 333 ms/image) (details in the Supplementary material). While the first EEG session assessed intrinsic sensory cortical activity and inter-regional connectivity, the latter session measured (mal)adaptive responses in the context of rich, unceasing sensory input from the environment. For simplicity, we named the first session the standard resting state (S-RS) and the second session the modified resting state (M-RS) as it simulated a real-life resting state in a sensory-rich world.
EEG acquisition and preprocessing
EEG data were recorded from a 96 channel BrainProducts actiCap system with Neuroscan SynAmps RT amplifiers (1000 Hz sampling rate, 0.05–200 Hz online bandpass filter, referenced to the FCz channel). Electro-oculogram (EOG) was recorded using four electrodes with vertical and horizontal bipolar derivations. EEG/EOG data were downsampled to 250 Hz, high-pass (1 Hz) and notch (60 Hz) filtered. We applied Fully Automated Statistical Thresholding for EEG artefact Rejection (FASTER) algorithm for artefact detection and rejection (Nolan et al., 2010). EEG oscillation powers were computed for individual channels for each epoch (1-s) using the multitaper spectral estimation technique (Thomson, 1982; Mitra and Pesaran, 1999). Alpha (8–12 Hz) and gamma (30–50 Hz) powers (higher gamma frequencies were avoided to prevent mircosaccade-related signal contamination) were normalized by the mean power for the global spectrum (1–50 Hz) within each epoch. Alpha powers were extracted from occipitoparietal (right, middle, and left) electrodes, where they were maximally distributed (Palva and Palva, 2007; Foxe and Snyder, 2011; Klimesch, 2012); gamma powers were extracted from frontal sites (of the left and right hemispheres) to reflect frontal, higher-order processing (Fell et al., 2003; Nyhus and Curran, 2010; Jia and Kohn, 2011) (Fig. 1). Lastly, we ascertained the neural sources of alpha and gamma oscillations using Exact Low Resolution Electromagnetic Tomography (eLORETA) based on oscillation powers across the entire scalp electrodes (Pascual-Marqui et al., 2011). Specific technical details are provided in the Supplementary material.
Figure 1.

Alpha and gamma power. (A) Power spectra averaged over occipitoparietal electrodes for each group at each state. Shaded ribbons = standard error of the mean (SEM). (B) Alpha power magnitudes at left, middle and right posterior sites. Error bars = SEM. (C) Scalp topographical maps of alpha powers, with electrodes included in posterior (occipitoparietal) sites bolded and circled. (D) eLORETA localized the source of alpha activity to the bilateral cuneus/precuneus. (E) Power spectra averaged over frontal electrodes for each group at each state. Shaded ribbons = SEM. (F) Gamma power averages at left and right frontal sites. Error bars = SEM. (G) Scalp topographical maps of gamma powers, with frontal electrodes bolded and circled. (H) eLORETA localized the source of gamma activity to the bilateral prefrontal cortex, including superior frontal gyrus and dorsal anterior cingulate cortex. *P < 0.05; **P < 0.01. HC = healthy controls.
Causal connectivity analyses
Inter-regional interactions are thought to be mediated by long-range synchronized neural oscillations, particularly alpha oscillations that underpin both higher-level, cognitive inhibition via top-down projections (Engel et al., 2001; Sadaghiani et al., 2012; Wang et al., 2016) and low-level, automatic inhibition via bottom-up projections (Tang et al., 2007; Hillebrand et al., 2016). Therefore, we applied Granger causality analysis (Geweke, 1982; Ding et al., 2006) to assess frontal→posterior alpha Granger causality to index top-down executive inhibition and posterior→frontal Granger causality to index bottom-up sensory inhibition. Following transformation into reference-free, current source density (CSD) data, scalp powers of ipsilateral frontal-posterior pairs were submitted to bivariate autoregressive (AR) modelling, from which Granger causality spectra were derived (Ding et al., 2000, 2006). Further technical details are provided in the Supplementary material.
Statistical analyses
We performed omnibus repeated-measures analyses of variance (rANOVAs; with Greenhouse-Geisser corrections) on alpha and gamma powers and alpha Granger causality parameters. Pearson correlations were examined among these variables to explore their inherent associations. Clinical associations were assessed by correlating these neural activities with low- and high-order dysfunctions (hypervigilance and difficulty in impulse control). Substance use was included as a covariate for all relevant tests. All P-values (unless otherwise noted) were two-tailed. Multiple-comparison correction was performed on all follow-up contrasts using the false discovery rate (P < 0.05 FDR). Lastly, Supplementary Table 1 summarizes the results pertinent to the hypotheses.
Results
Oscillation powers
An omnibus rANOVA (State × Site × Group) for posterior alpha power confirmed a main effect of state—alpha was reduced in M-RS relative to S-RS [F(1,61) = 11.88, P = 0.001, ηp2 = 0.17], akin to alpha blocking by visual stimulation (Foxe and Snyder, 2011). Importantly, a main effect of group [F(2,61) = 3.96, P = 0.024, ηp2 = 0.12] demonstrated alpha suppression (i.e. sensory hyperactivity) in PTSD (versus GAD/healthy controls, P’s = 0.021/0.012, FDR corrected P’s < 0.05, d’s = 0.75/0.82) (Fig. 1A–C), which was further qualified by a three-way (State × Site × Group) interaction [F(3.46, 103.89) = 2.81, P = 0.036, ηp2 = 0.09]. Follow-up two-way ANOVAs (State × Group) at left and middle sites confirmed the state and group effects above (P’s < 0.05). While confirming the group effect above [F(2,60) = 4.04, P = 0.023, ηp2 = 0.12], the right posterior site also showed a marginal effect of State × Group interaction [F(2,61) = 2.65, P = 0.079, ηp2 = 0.08], represented by the absence of state effect in PTSD (P = 0.288) in contrast to GAD and healthy controls (P’s < 0.05), suggesting a lack of alpha adaptation switching from S-RS to M-RS. Akin to visual cortical processing, neural sources of alpha power (collapsed across groups and states) were identified in the bilateral dorsal occipital visual cortex (cuneus and precuneus; peak voxel: −5, −60, 30; t = 11.8, P < 0.0001) (Fig. 1D).
An omnibus three-way rANOVA (State × Site × Group) for frontal gamma power revealed a marginal effect of group [F(2,61) = 2.48, P = 0.093, ηp2 = 0.08], reflecting greater gamma power in PTSD [versus GAD: t(42) = 2.16, P = 0.037, FDR corrected P < 0.05 one-tailed, d = 0.67; versus healthy control: t(38) = 1.98, P = 0.055, FDR corrected P < 0.05 one-tailed, d = 0.64] (Fig. 1E–G). There was a Site × Group interaction effect [F(2,60) = 3.07, P = 0.054, ηp2 = 0.09], explained by a main effect of site in healthy controls (right > left; P = 0.034, ηp2 = 0.23) but not the patient groups (PTSD, GAD P’s > 0.21). No other significant effects emerged (P’s > 0.13). Gamma power (collapsed across groups and states) was source-localized to the bilateral superior frontal gyrus and dorsal cingulate cortex (peak voxel:−25, 45, 30; t = 17.1, P < 0.00001), linking heightened gamma activity in PTSD to excessive higher-order processing, even at rest (Fig. 1H).
Causal interactions
An omnibus rANOVA (State, Direction, Hemisphere and Group) for alpha Granger causality (Fig. 2) revealed a main effect of direction [bottom-up/posterior→frontal > top-down/frontal→posterior alpha Granger causality, F(1,60) = 68.16, P < 0.001, ηp2 = 0.53], confirming the dominance of posterior→frontal alpha influence in large-scale resting state network interaction (Tang et al., 2007; Hillebrand et al., 2016). A main effect of state also emerged [S-RS > M-RS, F(1,60) = 5.62, P = 0.021, ηp2 = 0.09], suggesting weakened inhibitory influences with increased visual input. Importantly, a main effect of group [F(2,60) = 5.21, P = 0.008, ηp2 = 0.15] indicated reduced inhibitory influences in PTSD (versus GAD/healthy controls, P’s < 0.01, FDR corrected P’s < 0.05, d’s > 0.85). These main effects were qualified by a marginal four-way interaction [F(2,60) = 2.69, P = 0.076, ηp2 = 0.08], which was explicated in follow-up three-way (State × Direction × Group) ANOVAs in each hemisphere.
Figure 2.

Granger causality. (A) S-RS/M-RS: Reduction in right hemisphere posterior→frontal and frontal→posterior alpha causality in PTSD (versus GAD/healthy controls). (B) Average Granger causality values for both directions in both hemispheres. BU = bottom-up; TD = top-down. Error bars = SEM. *P < 0.05; **P < 0.01; ***P < 0.005; †P < 0.1.
The left hemisphere showed only a significant effect of direction as discussed above [F(1,60) = 58.55, P < 0.001, ηp2 = 0.49]. The right hemisphere exhibited all main effects (Group, State and Direction; P’s < 0.01, ηp2 > 0.12), which were qualified by a marginal State × Direction × Group interaction [F(2,60) = 2.64, P = 0.08, ηp2 = 0.08]. A Direction × Group ANOVA for the S-RS indicated a Direction × Group interaction [F(2, 64) = 5.24, P = 0.008, ηp2 = 0.14]: Granger causality reduction in PTSD (versus GAD/healthy controls) was more salient in the bottom-up/posterior→frontal [F(2,64) = 6.47, P = 0.003, ηp2 = 0.17] than top-down/frontal→posterior direction [F(2,64) = 2.62, P = 0.080, ηp2 = 0.08]. A similar ANOVA for the M-RS confirmed a main group effect as above [F(2,61) = 4.73, P = 0.012, ηp2 = 0.13] without Group × Direction interaction (P = 0.198). In sum, patients with PTSD exhibited deficits in bidirectional intrinsic inhibition in the right hemisphere.
Power-connectivity correlations
Correlations among alpha/gamma power and alpha Granger causality were then analysed for the entire sample (collapsed across the groups) to elucidate associations among the corresponding processes these indices each reflected, thereby contextualizing the group effects in the interwoven relations of these key processes (Fig. 3A). We focused on the right hemisphere where Granger causality effects were salient. Posterior alpha power positively correlated with bottom-up/posterior→frontal Granger causality (S-RS/M-RS: r’s = 0.69/0.80, P’s < 0.001): greater sensory activity was associated with greater bottom-up, sensory projection. Importantly, both posterior alpha power and bottom-up Granger causality negatively correlated with frontal gamma power (S-RS/M-RS: r’s = −0.63/−0.54, P’s < 0.001 and S-RS/M-RS: r’s = −0.31/−0.38, P’s < 0.01, respectively), linking sensory hyperactivity and deficient bottom-up inhibition to excessive frontal activity. Nevertheless, frontal gamma power did not correlate with top-down/frontal→posterior Granger causality (S-RS/M-RS: r’s = 0.08/−0.09, P’s > 0.47), suggesting the disconnection of frontal activity from top-down inhibition. Lastly, top-down/frontal→posterior Granger causality positively correlated with posterior alpha power in M-RS (r = 0.35, P = 0.004) but not S-RS (r = 0.09, P = 0.473), linking top-down inhibition to sensory activity reduction in the presence of busy visual input.
Figure 3.

Correlation analyses. (A) Correlations across alpha/gamma powers and Granger causality. Pearson r’s for pairwise correlations in both states (S-RS/M-RS). Solid black lines indicate significant correlations in both states; the solid grey line indicates correlation in M-RS only (r = 0.35). Dashed grey lines indicate non-significant correlations in either state. (B) Correlations between bottom-up Granger causality and severity in hypervigilance and difficulty in impulse control (correlations remained significant after co-varying out substance use). BU = bottom-up; TD = top-down. *P < 0.05; **P < 0.01; ***P < 0.005.
Clinical association
In support of our prediction, intrinsic posterior alpha power and bottom-up Granger causality (at the S-RS state) were negatively correlated with severity of ‘hypervigilance’ (PCL-hypervigilance score); namely, sensory hyperactivity and deficient bottom-up inhibition were associated with greater hypervigilance (r = −0.21, P = 0.094 and r = −0.28, P = 0.019, respectively) (Fig. 3B). Bottom-up Granger causality also negatively correlated with difficulty in impulse control (DERS-Impulse Control Subscale score), r = −0.26, P = 0.036, linking deficient bottom-up inhibition to executive dysfunction.
Discussion
Compared with patients with GAD and healthy control subjects, patients with PTSD exhibited broad suppression of posterior alpha activity and bottom-up alpha Granger causality, indicating excessive sensory activity and deficient inhibition of sensory feedforward projections in PTSD. As these neural activities closely coupled with frontal gamma oscillations, patients with PTSD also showed exaggerated frontal gamma activity. While these anomalies appeared at rest with minimal sensory input, reflecting intrinsic neural pathophysiology, they also manifested in a passive image viewing state with a continuous image stream (M-RS), highlighting their impact in a real-life, sensory-rich environment. Moreover, in spite of enhanced frontal gamma activity, patients with PTSD demonstrated diminished top-down alpha Granger causality (i.e. deficient top-down inhibition), especially in the presence of busy visual input at M-RS. Taken together, these findings suggest that sensory hyperactivity combined with deficient inhibition of sensory projection in PTSD could contribute to excessive frontal activation. Indeed, these sensory anomalies (especially bottom-up alpha Granger causality) correlated not only with severity in hypervigilance (reflecting low-order sensory surveillance of the environment) but also impulse control deficits (reflecting higher-order executive regulation). Importantly, that patients with GAD and healthy controls were equivalent in these indices rules out the possibility that these effects were simply due to general anxious hyperarousal. Finally, comparisons between the PTSD group and a traumatized non-PTSD group confirmed these PTSD-related anomalies, ruling out trauma-related confounds (see details in the Supplementary material).
Research (including simultaneous functional MRI-EEG studies) has isolated a negative correlation between human posterior alpha power and visual cortical activity (Laufs et al., 2003, 2006; Bollimunta et al., 2008). In keeping with that, our eLORETA source analysis localized the alpha activity to the dorsal visual cortex (primarily, cuneus and precuneus), which being critical for visual spatial perception, could represent the direct neural underpinning of excessive visual scanning of the environment (i.e. hypervigilance) in PTSD. Notably, intrinsic (S-RS) alpha power in PTSD approximates the alpha power at M-RS (with rich visual input) in GAD and healthy control groups, exemplifying an exaggerated intrinsic (resting state) sensory activity in patients with PTSD as if they were constantly bombarded by busy sensory input. Furthermore, unlike the other two groups, the PTSD group failed to show alpha adaptation (in the right hemisphere that is critical for visuospatial attention) (Corbetta and Shulman, 2011) as visual input increased, highlighting a rigid, set mode of sensory hypervigilance in PTSD. This lack of sensory adaptation can cause not only the frequent complaints of sensory hypersensitivity and distress (Stewart and White, 2008) but also, in the long run, blunt sensory registration, resulting in paradoxical problems of sensory numbing in PTSD (Engel et al., 2001; Stewart and White, 2008). Along this line, somatosensory analogue of posterior alpha oscillations, the mu oscillations, also showed power suppression in patients with PTSD (versus GAD/healthy control groups; see details in the Supplementary material). While accentuating multisensory hyperactivity, this finding raises an intriguing possibility that chronic somatosensory hyperactivity could lead to somatosensory blunting, ironically contributing to analgesia in patients with PTSD.
These sensory anomalies can have impacts beyond the sensory system. Posterior alpha power and bottom-up inhibition were closely correlated with frontal gamma activity, which, source-localized to the prefrontal cortex, was elevated in PTSD. Our previous finding suggests that stress-induced sensory hyperactivity propagates across the sensory cortical hierarchy to trigger responses in the prefrontal cortex (Krusemark and Li, 2013). Similarly, the excessive sensory projection to the frontal cortex in PTSD could instigate exaggerated frontal neural activity in these patients, even during a resting state. The absence of a significant association between frontal gamma activity and top-down inhibition further emphasized the notion that this frontal activity pertains to passive reaction to sensory overflow as opposed to effective deployment of executive control. In fact, that top-down inhibition was impaired in PTSD relative to GAD and healthy control (based on DERS-Impulse Control Score; Table 1), in spite of enhanced frontal gamma activity, argues that as sensory overflow inundates the frontal cortex and depletes frontal cognitive resources, higher-order executive function is compromised. In keeping with that, the level of suppressed bottom-up sensory inhibition closely coupled with the severity of impulse control deficits.
This impaired executive function would complete a vicious cycle of PTSD pathology by failing to downregulate the overly active sensory brain and prevent it from overloading frontal processing, resulting in widespread symptoms in PTSD. Therefore, current findings of regional brain activity combined with long-range network interactions implicate a vicious cycle of sensory hyperactivity and executive disinhibition in PTSD, pointing to a sensory hypothesis of PTSD: constant, spontaneous sensory hyperactivity leads to frontal overload and cognitive depletion, which breaks down executive control, fuelling and perpetuating PTSD symptoms (Fig. 4). This sensory hypothesis of PTSD expands the current affective conceptualization of PTSD (Dalgleish et al., 2001; Hofmann et al., 2012), which focuses on prefrontal-cortex-amygdala circuit pathology in response to threat; namely, PTSD is characterized by hypoactive ventromedial prefrontal and anterior cingulate cortices and hyper-reactive amygdala in response to threat (Rauch et al., 2006; Etkin and Wager, 2007; Patel et al., 2012). The inclusion of this sensory mechanism would thus form a triangular sensory-prefrontal-cortex-amygdala circuit that is aberrant in PTSD. Moreover, this intrinsic sensory pathology not only poses impacts on a plethora of operations, being low-level and early in the information processing stream, but also represents a tonic, endogenous dysfunction even when the patient is at ‘rest’. Such anomalies, spontaneously occurring, could be responsible for the widespread and severe symptomatology in patients with PTSD.
Figure 4.
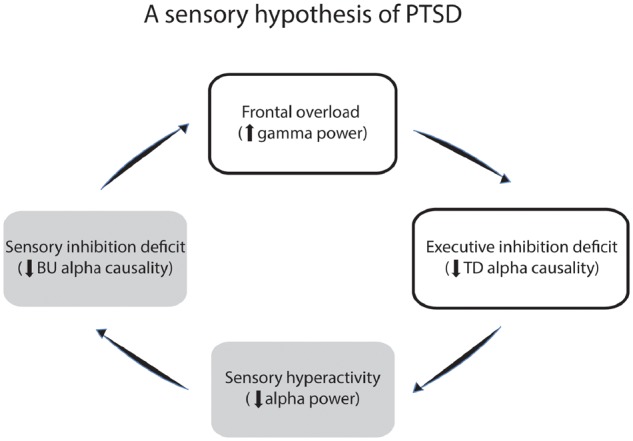
Schematic presentation of a sensory hypothesis of PTSD. A vicious cycle in action during resting state, entailing (i) sensory hyperactivity; (ii) deficient bottom-up (BU), sensory inhibition; (iii) frontal overload (due to sensory overflow); and (iv) deficient top-down (TD), executive inhibition and regulation.
Although both PTSD and GAD groups showed more severe hypervigilance and hyperarousal symptoms than healthy control subjects (P’s < 0.05), the marked neural differences between PTSD and GAD in the current study emphasize a sensory-based pathology that is unique to PTSD, contrasting threat-specific hyperactivity (or general anxious hyperarousal) and potentially limbic-centric pathology in GAD. While echoing the increasingly recognized distinction between PTSD and anxiety disorders (American Psychiatric Association, 2013), this sensory pathology in PTSD draws an interesting parallel to more severe mental illnesses such as psychosis. Sensory anomalies have gained substantial support as an important pathology in schizophrenia (Geyer et al., 2001; Thoma et al., 2003; Park et al., 2015). In fact, a bottom-up, sensory model of schizophrenia has been proposed, implicating low-level sensory anomalies in the proliferation of brain-wide dysfunctions, including downstream cognitive and executive dysfunctions (Javitt, 2009). Similar to schizophrenia, PTSD is associated with dysregulation of the hypothalamic-pituitary-adrenal and noradrenergic systems (Yehuda, 1998; Seedat et al., 2003). As mentioned earlier, these neurochemical imbalances, especially dysregulated dopamine-adrenergic systems, can directly influence sensory gating and processing (Aston-Jones et al., 1994; Berridge and Waterhouse, 2003; Valentino and Van Bockstaele, 2008), contributing to the sensory anomalies manifested in these patients. Importantly, the discovery of direct neural underpinnings of sensory anomalies and hypervigilance in PTSD—attenuated alpha oscillations and alpha causal connectivity—would inspire novel, mechanism-based PTSD treatment. For instance, by incorporating recent advances in neuromodulation via transcranial magnetic or electrical stimulation, new treatments can directly modulate neural oscillations in the sensory cortex and thus eradicate this sensory root in PTSD pathology.
Funding
This research was supported by the National Institute of Mental Health R01MH093413 (W.L.). The work was also supported in part by the subaward of the United States Army award W81XWH-10-2-018 (N.S. and E.B.), and does not necessarily represent the views of the Department of Defense, Department of Veterans Affairs, or the United States Government, nor does it constitute or imply endorsement, sponsorship, or favouring of the study design, analysis, or recommendations.
Supplementary material
Supplementary material is available at Brain online.
Supplementary Material
Glossary
Abbreviations
- GAD
generalized anxiety disorder
- M-RS
modified resting state
- PTSD
post-traumatic stress disorder
- S-RS
standard resting state
References
- Adler LE, Gerhardt GA, Franks R, Baker N, Nagamoto H, Drebing C. et al. Sensory physiology and catecholamines in schizophrenia and mania. Psychiatry Res 1990; 31: 297–309. [DOI] [PubMed] [Google Scholar]
- Adler LE, Pang K, Gerhardt G, Rose GM. Modulation of the gating of auditory evoked potentials by norepinephrine: pharmacological evidence obtained using a selective neurotoxin. Biol Psychiatry 1988; 24: 179–90. [DOI] [PubMed] [Google Scholar]
- American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 5th ednWashington, DC: American Psychiatric Association; 2013. [Google Scholar]
- Aston-Jones G, Rajkowski J, Kubiak P, Alexinsky T. Locus coeruleus neurons in monkey are selectively activated by attended cues in a vigilance task. J Neurosci 1994; 14: 4467–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baisley SK, Fallace KL, Rajbhandari AK, Bakshi VP. Mutual independence of 5-HT(2) and alpha1 noradrenergic receptors in mediating deficits in sensorimotor gating. Psychopharmacology 2012; 220: 465–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge CW, Waterhouse BD. The locus coeruleus-noradrenergic system: modulation of behavioral state and state-dependent cognitive processes. Brain Res Brain Res Rev 2003; 42: 33–84. [DOI] [PubMed] [Google Scholar]
- Blanchard EB, Jones-Alexander J, Buckley TC, Forneris CA. Psychometric properties of the PTSD Checklist (PCL). Behav Res Ther 1996; 34: 669–73. [DOI] [PubMed] [Google Scholar]
- Bollimunta A, Chen Y, Schroeder CE, Ding M. Neuronal mechanisms of cortical alpha oscillations in awake-behaving macaques. J Neurosci 2008; 28: 9976–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cisler JM, Koster EH. Mechanisms of attentional biases towards threat in anxiety disorders: an integrative review. Clin Psychol Rev 2010; 30: 203–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbetta M, Shulman GL. Spatial neglect and attention networks. Annu Rev Neurosci 2011; 34: 569–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalgleish T, Moradi AR, Taghavi MR, Neshat-Doost HT, Yule W. An experimental investigation of hypervigilance for threat in children and adolescents with post-traumatic stress disorder. Psychol Med 2001; 31: 541–7. [DOI] [PubMed] [Google Scholar]
- Ding M, Bressler SL, Yang W, Liang H. Short-window spectral analysis of cortical event-related potentials by adaptive multivariate autoregressive modeling: data preprocessing, model validation, and variability assessment. Biol Cybern 2000; 83: 35–45. [DOI] [PubMed] [Google Scholar]
- Ding M, Chen Y, Bressler SL. Granger causality: basic theory and application to neuroscience. In: Schelter M, Winderhalder M, Timmer J, editors. Handbook of Time Series Analysis. Weinheim: Wiley-VCH Verlag; 2006. p. 437–60. [Google Scholar]
- Ehlers A, Clark DM. A cognitive model of posttraumatic stress disorder. Behav Res Ther 2000; 38: 319–45. [DOI] [PubMed] [Google Scholar]
- Engel AK, Fries P, Singer W. Dynamic predictions: oscillations and synchrony in top-down processing. Nat Rev Neurosci 2001; 2: 704–16. [DOI] [PubMed] [Google Scholar]
- Engel-Yeger B, Palgy-Levin D, Lev-Wiesel R. The sensory profile of people with post-traumatic stress symptoms. Occup Ther Ment Health 2013; 29: 266–78. [Google Scholar]
- Etkin A, Wager TD. Functional neuroimaging of anxiety: a meta-analysis of emotional processing in PTSD, social anxiety disorder, and specific phobia. Am J Psychiatry 2007; 164: 1476–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fell J, Fernandez G, Klaver P, Elger CE, Fries P. Is synchronized neuronal gamma activity relevant for selective attention? Brain Res Brain Res Rev 2003; 42: 265–72. [DOI] [PubMed] [Google Scholar]
- Foxe JJ, Snyder AC. The role of alpha-band brain oscillations as a sensory suppression mechanism during selective attention. Front Psychol 2011; 2: 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freedman R, Adler LE, Gerhardt GA, Waldo M, Baker N, Rose GM. et al. Neurobiological studies of sensory gating in schizophrenia. Schizophr Bull 1987; 13: 669–78. [DOI] [PubMed] [Google Scholar]
- Garner M, Mogg K, Bradley BP. Orienting and maintenance of gaze to facial expressions in social anxiety. J Abnorm Psychol 2006; 115: 760–70. [DOI] [PubMed] [Google Scholar]
- Ge Y, Wu J, Sun X, Zhang K. Enhanced mismatch negativity in adolescents with posttraumatic stress disorder (PTSD). Int J Psychophysiol 2011; 79: 231–5. [DOI] [PubMed] [Google Scholar]
- Geweke J. Measurement of linear dependence and feedback between multiple time series. J Am Stat Assoc 1982; 77: 304–13. [Google Scholar]
- Geyer MA, Krebs-Thomson K, Braff DL, Swerdlow NR. Pharmacological studies of prepulse inhibition models of sensorimotor gating deficits in schizophrenia: a decade in review. Psychopharmacology 2001; 156: 117–54. [DOI] [PubMed] [Google Scholar]
- Ghisolfi ES, Margis R, Becker J, Zanardo AP, Strimitzer IM, Lara DR. Impaired P50 sensory gating in post-traumatic stress disorder secondary to urban violence. Int J Psychophysiol 2004; 51: 209–14. [DOI] [PubMed] [Google Scholar]
- Gillette GM, Skinner RD, Rasco LM, Fielstein EM, Davis DH, Pawelak JE. et al. Combat veterans with posttraumatic stress disorder exhibit decreased habituation of the P1 midlatency auditory evoked potential. Life Sci 1997; 61: 1421–34. [DOI] [PubMed] [Google Scholar]
- Gjini K, Boutros NN, Haddad L, Aikins D, Javanbakht A, Amirsadri A. et al. Evoked potential correlates of post-traumatic stress disorder in refugees with history of exposure to torture. J Psychiatr Res 2013; 47: 1492–8. [DOI] [PubMed] [Google Scholar]
- Gratz KL, Roemer L. Multidimensional assessment of emotion regulation and dysregulation: development, factor structure, and initial validation of the difficulties in emotion regulation scale. J Psychopathol Behav 2004; 26: 41–54. [Google Scholar]
- Grillon C, Morgan CA, Southwick SM, Davis M, Charney DS. Baseline startle amplitude and prepulse inhibition in Vietnam veterans with posttraumatic stress disorder. Psychiatry Res 1996; 64: 169–78. [DOI] [PubMed] [Google Scholar]
- Hayes JP, Vanelzakker MB, Shin LM. Emotion and cognition interactions in PTSD: a review of neurocognitive and neuroimaging studies. Front Integr Neurosci 2012; 6: 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillebrand A, Tewarie P, van Dellen E, Yu M, Carbo EW, Douw L. et al. Direction of information flow in large-scale resting-state networks is frequency-dependent. Proc Natl Acad Sci USA 2016; 113: 3867–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann SG, Ellard KK, Siegle GJ. Neurobiological correlates of cognitions in fear and anxiety: a cognitive-neurobiological information-processing model. Cogn Emot 2012; 26: 282–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holstein DH, Vollenweider FX, Jancke L, Schopper C, Csomor PA. P50 suppression, prepulse inhibition, and startle reactivity in the same patient cohort suffering from posttraumatic stress disorder. J Affect Disord 2010; 126: 188–97. [DOI] [PubMed] [Google Scholar]
- Javanbakht A, Liberzon I, Amirsadri A, Gjini K, Boutros NN. Event-related potential studies of post-traumatic stress disorder: a critical review and synthesis. Biol Mood Anxiety Disord 2011; 1: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javitt DC. When doors of perception close: bottom-up models of disrupted cognition in schizophrenia. Annu Rev Clin Psychol 2009; 5: 249–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia X, Kohn A. Gamma rhythms in the brain. PLoS Biol 2011; 9: e1001045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klimesch W. Alpha-band oscillations, attention, and controlled access to stored information. Trends Cogn Sci 2012; 16: 606–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krusemark EA, Li W. From early sensory specialization to later perceptual generalization: dynamic temporal progression in perceiving individual threats. J Neurosci 2013; 33: 587–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laufs H, Holt JL, Elfont R, Krams M, Paul JS, Krakow K. et al. Where the BOLD signal goes when alpha EEG leaves. Neuroimage 2006; 31: 1408–18. [DOI] [PubMed] [Google Scholar]
- Laufs H, Krakow K, Sterzer P, Eger E, Beyerle A, Salek-Haddadi A. et al. Electroencephalographic signatures of attentional and cognitive default modes in spontaneous brain activity fluctuations at rest. Proc Natl Acad Sci USA 2003; 100: 11053–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewine JD, Thoma RJ, Provencal SL, Edgar C, Miller GA, Canive JM. Abnormal stimulus-response intensity functions in posttraumatic stress disorder: an electrophysiological investigation. Am J Psychiatry 2002; 159: 1689–95. [DOI] [PubMed] [Google Scholar]
- Mennin DS, Heimberg RG, Turk CL, Fresco DM. Preliminary evidence for an emotion dysregulation model of generalized anxiety disorder. Behav Res Ther 2005; 43: 1281–310. [DOI] [PubMed] [Google Scholar]
- Mitra PP, Pesaran B. Analysis of dynamic brain imaging data. Biophys J 1999; 76: 691–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan CA III, Grillon C. Abnormal mismatch negativity in women with sexual assault-related posttraumatic stress disorder. Biol Psychiatry 1999; 45: 827–32. [DOI] [PubMed] [Google Scholar]
- Neylan TC, Fletcher DJ, Lenoci M, McCallin K, Weiss DS, Schoenfeld FB. et al. Sensory gating in chronic posttraumatic stress disorder: reduced auditory P50 suppression in combat veterans. Biol Psychiatry 1999; 46: 1656–64. [DOI] [PubMed] [Google Scholar]
- Nolan H, Whelan R, Reilly RB. FASTER: Fully Automated Statistical Thresholding for EEG artifact Rejection. J Neurosci Methods 2010; 192: 152–62. [DOI] [PubMed] [Google Scholar]
- Palva S, Palva JM. New vistas for alpha-frequency band oscillations. Trends Neurosci 2007; 30: 150–8. [DOI] [PubMed] [Google Scholar]
- Park HRP, Lim VK, Kirk IJ, Waldie KE. P50 sensory gating deficits in schizotypy. Pers Individ Dif 2015; 82: 142–7. [Google Scholar]
- Pascual-Marqui RD, Lehmann D, Koukkou M, Kochi K, Anderer P, Saletu B. et al. Assessing interactions in the brain with exact low-resolution electromagnetic tomography. Philos Trans A Math Phys Eng Sci 2011; 369: 3768–84. [DOI] [PubMed] [Google Scholar]
- Patel R, Spreng RN, Shin LM, Girard TA. Neurocircuitry models of posttraumatic stress disorder and beyond: a meta-analysis of functional neuroimaging studies. Neurosci Biobehav Rev 2012; 36: 2130–42. [DOI] [PubMed] [Google Scholar]
- Rauch SL, Shin LM, Phelps EA. Neurocircuitry models of posttraumatic stress disorder and extinction: human neuroimaging research–past, present, and future. Biol Psychiatry 2006; 60: 376–82. [DOI] [PubMed] [Google Scholar]
- Sadaghiani S, Scheeringa R, Lehongre K, Morillon B, Giraud AL, D’Esposito M. et al. Alpha-band phase synchrony is related to activity in the fronto-parietal adaptive control network. J Neurosci 2012; 32: 14305–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seedat S, Stein MB, Oosthuizen PP, Emsley RA, Stein DJ. Linking posttraumatic stress disorder and psychosis: a look at epidemiology, phenomenology, and treatment. J Nerv Ment Dis 2003; 191: 675–81. [DOI] [PubMed] [Google Scholar]
- Shalev AY, Peri T, Brandes D, Freedman S, Orr SP, Pitman RK. Auditory startle response in trauma survivors with posttraumatic stress disorder: a prospective study. Am J Psychiatry 2000; 157: 255–61. [DOI] [PubMed] [Google Scholar]
- Shaw JC. The brain’s alpha rhythms and the mind. Amsterdam: Elsevier; 2003. [Google Scholar]
- Sherin JE, Nemeroff CB. Post-traumatic stress disorder: the neurobiological impact of psychological trauma. Dialogues Clin Neurosci 2011; 13: 263–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner RD, Rasco LM, Fitzgerald J, Karson CN, Matthew M, Williams DK. et al. Reduced sensory gating of the P1 potential in rape victims and combat veterans with posttraumatic stress disorder. Depress Anxiety 1999; 9: 122–30. [DOI] [PubMed] [Google Scholar]
- Southwick SM, Krystal JH, Bremner JD, Morgan CA III, Nicolaou AL, Nagy LM. et al. Noradrenergic and serotonergic function in posttraumatic stress disorder. Arch Gen Psychiatry 1997; 54: 749–58. [DOI] [PubMed] [Google Scholar]
- Stewart LP, White PM. Sensory filtering phenomenology in PTSD. Depress Anxiety 2008; 25: 38–45. [DOI] [PubMed] [Google Scholar]
- Tang AC, Sutherland MT, Sun P, Zhang Y, Nakazawa M, Korzekwa A. et al. top-down versus bottom-up processing in the human brain: distinct directional influences revealed by integrating SOBI and Granger causality. In: Davies ME, James CJ, Abdallah SA, Plumbley MD, editors. 7th International Conference on Independent Component Analysis and Signal Separation, ICA 2007, London, UK, September 9–12, 2007 Proceedings. Berlin, Heidelberg: Springer Berlin Heidelberg; 2007. p.802–9. [Google Scholar]
- Thoma RJ, Hanlon FM, Moses SN, Edgar JC, Huang M, Weisend MP. et al. Lateralization of auditory sensory gating and neuropsychological dysfunction in schizophrenia. Am J Psychiatry 2003; 160: 1595–605. [DOI] [PubMed] [Google Scholar]
- Thomson DJ. Spectrum estimation and harmonic analysis. Proc IEEE 1982; 70: 1055–96. [Google Scholar]
- Valentino RJ, Van Bockstaele E. Convergent regulation of locus coeruleus activity as an adaptive response to stress. Eur J Pharmacol 2008; 583: 194–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilopoulos SP. Social anxiety and the vigilance-avoidance pattern of attentional processing. Behav Cogn Psychother 2005; 33: 13–24. [Google Scholar]
- Wang C, Rajagovindan R, Han SM, Ding M. Top-down control of visual alpha oscillations: sources of control signals and their mechanisms of action. Front Hum Neurosci 2016; 10: 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg A, Hajcak G. Electrocortical evidence for vigilance-avoidance in generalized anxiety disorder. Psychophysiology 2011; 48: 842–51. [DOI] [PubMed] [Google Scholar]
- Worden MS, Foxe JJ, Wang N, Simpson GV. Anticipatory biasing of visuospatial attention indexed by retinotopically specific alpha-band electroencephalography increases over occipital cortex. J Neurosci 2000; 20: RC63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yehuda R. Psychoneuroendocrinology of post-traumatic stress disorder. Psychiatr Clin North Am 1998; 21: 359–79. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.