

Abstract
Recurrent, de novo, meiotic non-allelic homologous recombination events between low copy repeats, termed LCR22s, leads to the 22q11.2 deletion syndrome (22q11.2DS; velo-cardio-facial syndrome/DiGeorge syndrome). Although most 22q11.2DS patients have a similar sized 3 million base pair (Mb), LCR22A-D deletion, some have nested LCR22A-B or LCR22A-C deletions. Our goal is to identify additional recurrent 22q11.2 deletions associated with 22q11.2DS, serving as recombination hotspots for meiotic chromosomal rearrangements. Here, using data from Affymetrix 6.0 microarrays on 1680 22q11.2DS subjects, we identified what appeared to be a nested proximal 22q11.2 deletion in 38 (2.3%) of them. Using molecular and haplotype analyses from 14 subjects and their parent(s) with available DNA, we found essentially three types of scenarios to explain this observation. In eight subjects, the proximal breakpoints occurred in a small sized 12 kb LCR distal to LCR22A, referred to LCR22A+, resulting in LCR22A+-B or LCR22A+-D deletions. Six of these eight subjects had a nested 22q11.2 deletion that occurred during meiosis in a parent carrying a benign 0.2 Mb duplication of the LCR22A-LCR22A+ region with a breakpoint in LCR22A+. Another six had a typical de novo LCR22A-D deletion on one allele and inherited the LCR22A-A+ duplication from the other parent thus appearing on microarrays to have a nested deletion. LCR22A+ maps to an evolutionary breakpoint between mice and humans and appears to serve as a local hotspot for chromosome rearrangements on 22q11.2.
Introduction
The 22q11.2 deletion syndrome (22q11.2DS), also named DiGeorge syndrome (MIM# 188400) and velo-cardio-facial syndrome (VCFS, MIM# 192430), is the most common chromosomal microdeletion disorder in humans with an estimated incidence of 1 in 4000 live births (1–6). The major clinical characteristics of the syndrome include learning disabilities and psychiatric disorders, characteristic facial appearance, hypernasal speech due to velo-pharyngeal insufficiency, neonatal hypocalcemia, immune deficiency and congenital heart malformations (7–10).
Approximately 90% of individuals affected with the syndrome have a similarly sized de novo, ∼3 million base pair (Mb) hemizygous deletion on chromosome 22q11.2 (11,12). The recurrent deletion is caused by meiotic non-allelic homologous recombination (NAHR) events, when chromosomes misalign and undergo unequal crossing over, between flanking low copy repeats (LCRs; or segmental duplications) termed LCR22. There are several large sized LCR22s that map to the 3 Mb region on 22q11.2 (13–16), termed, LCR22A, -B, -C and -D (15). The 3 Mb deletion is caused by NAHR events between LCR22A and LCR22D and is frequently referred to as the LCR22A-D deletion (13). A subset has nested deletions between LCR22A-B and LCR22A-C, resulting in smaller 1.5 Mb LCR22A-B or 2 Mb LCR22A-C deletions, respectively, although the LCR22A-C deletion is relatively rare (13–15). Each LCR22 is comprised of subunits or modules forming a complex mosaic pattern of >99% sequence identity within modules of different orientations and copy number (16). These modules formed during primate evolution and are not present in mice (17). Recently, an inversion polymorphism was discovered between LCR22B-D or LCR22C-D (18). This inversion is required for the LCR22A-D deletion to take place. It is present in the healthy parent in which the de novo deletion occurs (18). The presence of an inversion occurs commonly in normal individuals and it reflects the complexity of the 22q11.2 region. It also provides new insights into the mechanism that leads to the LCR22A-D deletion (18).
Besides the four main LCR22s associated with the characteristic 22q11.2DS phenotype, there are additional dispersed modules of LCRs (segmental duplications) that are smaller, which map within this 3 Mb interval (19). Genomic architecture is a key mutational mechanism for causing human congenital anomaly disorders and also for promoting genetic variation (20). The role of these LCRs or other possible sequence elements leading towards recurrent rearrangements and resulting in 22q11.2DS, has not been determined. Such investigation requires a sufficiently large sized cohort where DNA or genetic data are available.
In this report, we processed and examined Affymetrix 6.0 array data from 1680 unrelated probands with 22q11.2DS to better delineate the prevalence of novel recurrent nested 22q11.2 deletions. We chose to investigate recurrent deletions as a priority because the region of chromosome breakage might shed light on molecular mechanisms responsible for abnormal meiotic chromosome rearrangements. With available material from patient-parent trios, we performed quantitative PCR, microsatellite marker analysis and FISH mapping studies to define a novel deletion type. We also compared the local genomic architecture where breakpoints occurred between humans and mice, to better understand the potential role in how the 22q11.2 region evolved.
Results
Nested 22q11.2 deletions
The first goal of the current study is to identify novel recurrent nested deletions within the LCR22A-D region by generating and analyzing Affymetrix 6.0 microarray data from 1680, 22q11.2DS subjects. We first identified deletion sizes in the cohort and found 1519 (90.4%) had a 3 Mb LCR22A-D deletion, 88 (5.2%) had an LCR22A-B deletion and 31 (1.9%) had an LCR22A-C deletion (Table 1). This is similar to what has been found before with smaller sample sizes. The LCR22A-B, A-C and A-D deletions were concordant in 539 samples that were also assayed using MLPA (data not shown). We found one new type of recurrent 22q11.2 deletion from analysis of the microarrays. In 38 (2.3%) subjects, we identified possible recurrent nested deletions of 1.3 Mb (n = 2) and 2.8 Mb (n = 36) (Table 1) with a similar appearing proximal (centromeric) breakpoint distal to PRODH (Proline dehydrogenase 1) and proximal to DGCR2 (DiGeorge critical region gene 2). The distal breakpoints mapped to LCR22B or LCR22D, respectively. Representative log2 ratio plot data are shown in Supplementary Material, Figure S2A and illustrated in Supplementary Material, Figure S2B. Upon investigating the literature, a few reports described individual subjects with a similar type of nested deletion (15,26,29,30). Based upon Affymetrix 6.0 data for all 38 samples (data not shown), the proximal breakpoint interval appeared to be in a similar location among all subjects. We next wanted to narrow the proximal deletion endpoints to confirm this possibility. We had DNA available from 19 of the 38 subjects with nested deletions. In addition, we had DNA from three different 22q11.2DS individuals that were not subjected to Affymetrix 6.0 analysis but had evidence from microsatellite markers that they had the 1.3 or 2.8 Mb nested deletion(s) (27), making a total of 22.
Table 1.
Number of samples in each deletion category from 1680 Affymetrix 6.0 arrays. The deletion types are indicated in the left most column obtained from analysis of 1680 Affymetrix 6.0 arrays on subjects with 22q11.2DS. The LCR22A-B deletion is indicated as A-B, the LCR22A-C deletion is indicated as A-C and LCR22A-D is indicated as A-D. The % of the total cohort with the particular class of deletion is shown, as is the breakdown by sex. We identified two subjects with a presumed nested 1.3 Mb LCR22A+-B deletion and 36 with the 2.8 Mb LCR22A+-D deletion. We found 2.3% had a LCR22+-B or LCR22A+-D deletion combined. A total of four subjects had unique deletions within the LCR22A-D region. The % of total is indicated in the bottom row
The nested proximal deletion is in a 12 kb LCR, termed LCR22A+
To map the position of the proximal chromosome breakpoints in the 22 DNA samples, we performed qPCR assays with primers spanning the interval found by Affymetrix 6.0 arrays. Results from a representative trio are shown in Figure 1. In this trio, the female proband, KD23, has a de novo 22q11.2 deletion as illustrated in Figure 1A (left). A cartoon of the different possible alleles is also shown in Figure 1A (right). Primers pairs for qPCR (Supplementary Material, Table S2) to unique sequences in the DGCR5-DGCR2 region are shown with respect to LCRs and genes in the UCSC Genome Browser snapshot in Figure 1A (hg19 assembly; bottom). We then performed qPCR assays on 22 samples with available DNA along with control samples that did not have a 22q11.2 deletion or had a typical 3 Mb 22q11.2 deletion (Supplementary Material, Fig. S3). Results from qPCR analysis of the KD23 trio is shown in Figure 1B. The breakpoints in all 22 subjects mapped between DGCR10–1 and DGCR2–1 qPCR products (Supplementary Material, Fig. S3). The two qPCR products flank a 12 kb LCR that we termed LCR22A+ (Fig. 1A). We could not further narrow the interval within LCR22A+ because the distal breakpoints are embedded within complex LCR22 modules. MLPA was performed using the SALSA P356 kit on eight subjects with insufficient DNA amount for qPCR, and the deletion in these occurred distal to PRODH, however, the precise breakpoint interval could not be defined using this method (Supplementary Material, Fig. S4).
Figure 1.

Identification of LCR22A+-D deletion by qPCR and haplotype analyses. (A, Top left) Trio of the KD23 pedigree in which a female proband is affected with 22q11.2DS (black circle), while the parents are normal (open shapes). (Top right) Illustration of the position of the LCR22A+-D deletion and the LCR22A-A+ interval with respect to the 22q11.2 region, shown in bp coordinates (hg19 assembly; the position of the chromosome breakpoints is estimated). Illustration of haplotypes of the 22q11.2 region in the trio in which individual alleles are depicted in different colors. The two coding genes, DGCR6 and PRODH as well as the non-coding gene, DGCR5 is indicated as gray filled ovals, distal to LCR22A and proximal to the LCR22A+-D deleted region, illustrated as an open box. The position of the DGCR5 gene in the illustration of the haplotypes in the trio is shown as a gray filled oval. The mother carries an LCR22A-A+ duplication CNV, indicated as a dark gray box. Below is a zoomed in snapshot from the UCSC Genome browser (hg19 assembly) showing the segmental duplication (LCR) track in dense format with LCR22A+ as indicated. Neighboring genes are shown along with a subset of splice variants. The DNA from the fosmid clone used as a probe for FISH mapping (Fig. 2) is indicated by a red star. The position of primer pairs used for qPCR assays is shown. The two primer sets flanking LCR22A+, where the breakpoints occur, are indicated by red color arrows (see primers in Supplementary Material, Table S2). The position of the microsatellite marker, D22S1638 used to identify haplotypes is indicated in blue font. (B) Bar graph of qPCR results for the proband (KD23), mother and father is shown. The y-axis indicates the relative quantity with respect to the control DNA, HapMap sample, GM12878. The x-axis indicates the primer pairs used for qPCR assay using primers illustrated above. Error bars are shown. More details are shown in Supplementary Material, Figure S2 and Table S3. (C) Table listing the sample name, family relation, qPCR results and size in bp of each allele of each microsatellite marker for the proband (Pro), mother (M) and father (F). The colors indicate from which parent (pink from mother and blue from father) had the particular sized PCR product in bp for a given microsatellite marker. Unfilled cells indicate that the alleles for a particular microsatellite marker could not be assigned to be derived from one or the other parent, and was therefore uninformative. (D) Chromatogram profiles of D22S1638 are shown as a representative example. The peaks identify the size, in bp of the PCR products from 1C. Adjacent stutter peak artifacts for each PCR product are typical for microsatellite markers. Details of all results for all samples are shown in Supplementary Material, Figure S4 and summarized in Supplementary Material, Table S4.
Haplotype and FISH analyses to examine 22q11.2 alleles
Microsatellite marker based genotyping has been previously used to determine the presence and extent of the 22q11.2 deletion (12,27) or parent of origin of the deletion (31). Fortunately, one microsatellite marker in the 22q11.2 region, termed D22S1638, maps within the breakpoint interval (Fig. 1A;Supplementary Material, Table S2). The sequence of the D22S1638 PCR product is not present elsewhere in the genome, as determined by BLAST (Basic Local Alignment Search Tool) and BLAT (BLAST-like Alignment Tool; in the UCSC Genome Browser) analyses. We performed haplotype analysis using microsatellite genetic markers spanning the 22q11.2 region on DNA samples from 14 probands and their parents (Supplementary Material, Fig. S5). A representative example of this analysis for the KD23 trio is illustrated in Figure 1C. The parent of origin of a particular allele can be identified based upon the presence of particular sized alleles. Failure to inherit either allele from one parent indicates that the deletion occurred in that particular parent as illustrated in Figure 1A (right). For D22S1638, KD23 is informative for inheriting one allele from either parent (Fig. 1C and D). When included with results from qPCR assays, in eight of 14 families, haplotype analysis confirmed that a de novo LCR22A+-B or LCR22A+-D deletion had occurred (Supplementary Material, Table S4).
We next performed metaphase and interphase FISH mapping analyses on Epstein-barr virus transformed cell lines available for BM1428 and BM355, which are normal, BM293, BM465 and BM1045.001, which had the 3 Mb LCR22A-D deletion, and BM1332.001 as well as BM1024.001, which are two probands with a nested LCR22A+-D deletion. FISH mapping was performed using large insert fosmid genomic clones. We used two sets of control fosmids to detect the pericentromeric region on the p-arm (green fluorescence color) and the TBX1 region, mapping between LCR22A and LCR22B (aqua color; Supplementary Material, Fig. S1). One clone (red fluorescence; Supplementary Material, Fig. S1), mapped to the breakpoint interval between LCR22A-A+ (Fig. 1A-red star; Supplementary Material, Fig. S1). We observed a green-red-aqua pattern on the normal allele of chromosome 22, a green-only pattern in the chromosome with the LCR22A-D deletion and a green-red pattern on the chromosome with the LCR22A+-D deletion (Fig. 2A;Supplementary Material, Fig. S1) as illustrated in Figure 2B. The counts of hybridization signals in the metaphase spreads and interphase nuclei are shown in Supplementary Material, Table S5. FISH mapping visually confirmed the occurrence of different types of 22q11.2 deletions found by qPCR assays and haplotype analysis using microsatellite markers.
Figure 2.
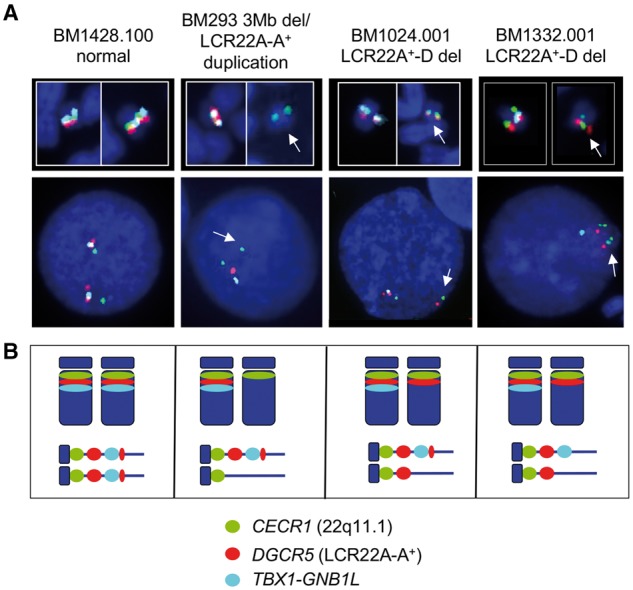
FISH mapping of the 22q11.2 region. (A) FISH mapping was performed on Epstein-barr virus transformed lymphoblastoid cell lines from subjects BM1428.100 (normal), BM293 (3 Mb deletion/0.2 Mb LCR22A-A+ duplication), BM1024.001 (LCR22A+-D deletion) and BM1332.001 (LCR22A+-D deletion). Representative metaphase images of chromosome 22 and interphase nuclei are shown for each. The unique region on 22q11.1 near the pericentromeric region, containing CECR1 is shown by the green fluorescence signals (FITC), DGCR5 (within the LCR22A-A+ CNV) by the red fluorescence signals (S.O. dye) and TBX1-GNB1L by the aqua fluorescence signals. The appearance of yellow or white fluorescence in metaphase spreads indicates overlap of probe signals. White arrows in the metaphase spreads and interphase nuclei indicate the chromosome with the deletion. The fosmids and details of their map position are shown in Supplementary Material, Figure S1. (B) Illustration of the probes, metaphase (above) and interphase (below) images from the four different cell lines. There are two sets of red signals for the normal cell line and one very strong red signal from BM293 that has a 3 Mb deletion on one allele and a genomic duplication on one allele. There are red signals on both alleles for BM1024.001 and BM1332.001. For both samples, the aqua signals are absent from the deleted allele and present on the normal allele. Complete metaphase spreads and interphase nuclei are shown for seven cell lines used for FISH mapping in Supplementary Material, Figure S1.
Occasionally, a smaller sized, red signal was observed mapping distal (telomeric) to the TBX1 probe (aqua) on the non-deleted chromosome, giving a green-red-aqua-red pattern on interphase chromosomes (Fig. 2; Supplementary Material, Fig. S1). The second signal is most likely due to hybridization to a second locus that maps just proximal (centromeric) to LCR22B (Supplementary Material, Fig. S5) (32). This second set of signals was not present in the allele with the LCR22A-D and LCR22A+-D deletions.
Parental samples with a 0.2 Mb duplication of LCR22A-a+
We investigated the genome structure in available parents of subjects with the nested LCR22A+-B or LCR22A+-D deletions. Surprisingly, among the 14 trios, 12 probands with the nested deletion had one parent that carried a 0.2 Mb duplication, based upon qPCR and microsatellite marker analyses, where the proximal deletion endpoint was in LCR22A and the distal was within LCR22A+ (Fig. 1A;Supplementary Material, Figs S1 and S2). We refer to this as the LCR22A-A+ duplication copy number variation (CNV; Fig. 1A, top right). We examined the haplotypes from microsatellite marker analysis in all families in which one parent carried the LCR22A-A+ duplication. For six of the eight families, the de novo deletion occurred in the parent that carried the LCR22A-A+ duplication (Supplementary Material, Fig. S4; Table S4). We do not have grandparental DNA samples, and thus, we cannot unambiguously assign haplotypes of D22S1638 in the parent with the duplication CNV. In those six cases, the most parsimonious conclusion is that the deletion occurred on the allele other than the one harboring the duplication (Fig. 1C;Supplementary Material, Fig. S4; Table S4). For two cases, the nested deletion occurred in the meiosis of one of the normal parents, indicating that the presence of the LCR22A-A+ duplication CNV is not required to cause the nested deletion.
Based upon the presence of a duplication of LCR22A-A+ in one parent, an alternative possibility for the remaining six families with a presumed nested deletion, was that the proband inherited the LCR22A-A+ duplication on one allele and had a de novo typical 3 Mb deletion on the other allele. Indeed, we found by qPCR assays and haplotype analysis using microsatellite markers, that the probands in these six families inherited one allele with the LCR22A-A+ duplication and had a de novo 3 Mb deletion on the other allele derived from the other parent (Supplementary Material, Fig. S4; Table S4). Subject, BM293, had a presumed LCR22A+-D deletion based upon qPCR assays (Supplementary Material, Fig. S3). Only one parent was available for haplotype analysis using microsatellite markers, which was uninformative (Supplementary Material, Fig. S2). However, we performed FISH mapping and found that BM293 had a 3 Mb LCR22A-D deletion on one allele because there was only the centromeric signal present on one allele of 22q11.2 (Supplementary Material, Fig. S1). However, subject BM293 had two copies of the LCR22A-A+ allele based upon qPCR (Supplementary Material, Fig. S3) and had two different alleles for marker, D22S1638 (Supplementary Material, Fig. S5). Upon examination of the other allele by FISH mapping, it appeared that the red signal (DGCR5; LCR22A-A+ region) was larger in diameter, which would be consistent with the presence of a duplication of the LCR22A-A+ region on one allele (Supplementary Material, Fig. S1).
Overall, there are three types of deletions based upon the data we presented, and these are illustrated in the cartoon in Figure 3. Each allele is color-coded to visualize the haplotypes in the proband and where they are derived. For the type A deletion, the proband had a typical de novo 3 Mb deletion and inherited a LCR22A-A+ duplication on the other allele, from the other parent. For the type B deletion, the proband had a de novo LCR22A+-B or LCR22A+-D deletion that occurred in the parent that had a LCR22A-A+ duplication. The most parsimonious explanation is that the deletion occurred on the other allele from the allele with the LCR22A-A+ duplication. For the type C deletion, the proband had a de novo LCR22A+-B or LCR22A+-D deletion from parents that did not have a LCR22A-A+ duplication, but instead had two normal copies of chromosome 22. In all these examples, all subjects had two copies of three genes, coding genes DGCR6 and PRODH as well as the non-coding gene, DGCR5 (Fig. 1A).
Figure 3.
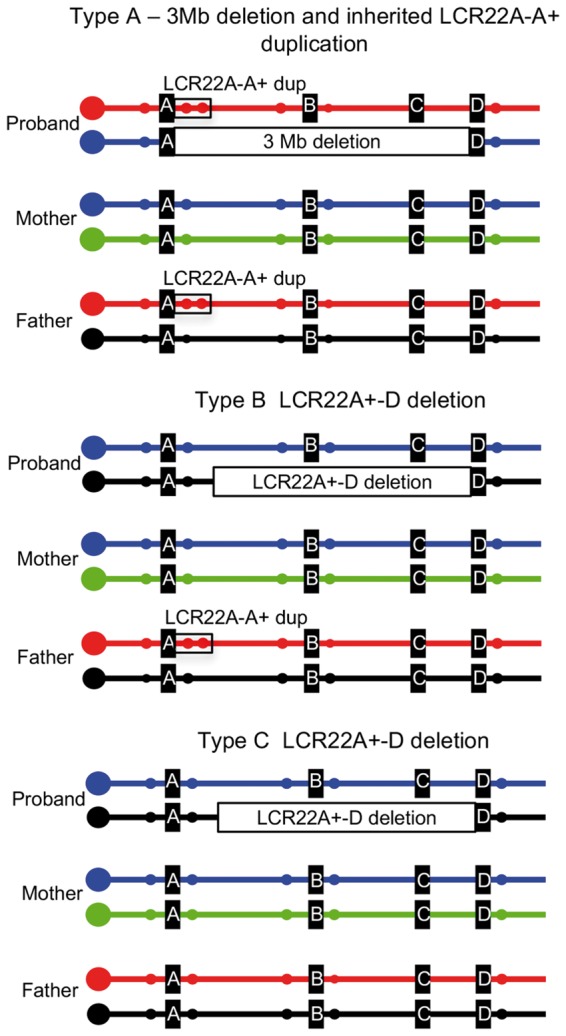
Summary of three types of recurrent 22q11.2 deletions. Illustration of three representative trios highlighting recurrent rearrangements based upon qPCR, microsatellite marker analysis and FISH mapping. The different alleles of chromosome 22q11.2 in each individual are shown in individual colors. Representative microsatellite markers are shown as small filled circles. For the type A deletion, the proband inherited the LCR22A-LCR22A+ duplication (two red filled circles marking D22S1638, surrounded by a black open box) from one parent and had a de novo 3 Mb LCR22A-D (or 1.5 Mb, LCR22A-B) deletion from the other parent. For the type B deletion, the proband had a nested LCR22A+-B or LCR22A+-D deletion, possibly on the opposite allele in the parent carrying the LCR22A-A+ duplication as shown. For the type C deletion, the proband had a nested LCR22A+-B or LCR22A+-D deletion from parents with normal alleles for 22q11.2.
Population frequency of the LCR22A-A+ deletion and duplication
We next were interested in determining the relative frequency of the seemingly benign LCR22A-A+ duplication CNV in the general population. Representative examples of log2 ratio plots from Affymetrix 6.0 arrays of two normal individuals with a deletion or duplication of the LCR22A-A+ CNV are shown in Supplementary Material, Figure S7. A recurrent LCR22A-A+ deletion of this interval has been previously described, in rare cases of hyperprolinemia when accompanied with an inactivating point mutation of PRODH on the other allele (33,34). To determine the frequency of the LCR22A-A+ deletion or duplication CNV, we examined existing published and unpublished microarray data as summarized in Table 2 and shown separated by cohort in Supplementary Material, Table S6. The LCR22A-A+ deletion CNV occurred in 0.3% of 15 579 normal individuals from the general population and 0.3% of 20 987 individuals with developmental delay, autism or other developmental disorders. Since the frequencies for the occurrence of the deletion were the same in normal individuals versus individuals with phenotypic abnormalities, it is possible that the deletion is benign. The LCR22A-A+ duplication CNV on the other hand occurred in 1.3% of normal or affected individuals (Table 2; Supplementary Material, Table S6). As for the LCR22A-A+ deletion, the common duplication CNV seems benign. The duplication occurs 4.3-fold more frequently than the LCR22A-A+ deletion CNV. Based upon this, it is possible that the deletion CNV has some biological function and some effect on phenotype, but it was not identified using this population data.
Table 2.
Summary of 0.2 Mb LCR22A-A+ deletion and duplication CNVs in human subjects from multiple cohorts tested on microarrays. Cases have been diagnosed with medical or behavioral conditions and controls are unaffected individuals (see Supplementary Material, Table S6 for more details on phenotypes from individual cohorts). Cases and controls for each cohort, combined for this table, are provided in Supplementary Material, Table S6. The number of subjects with the LCR22A-A+ deletion (# Dels) LCR22A-A+ duplication (# Dups) is indicated as is their frequency (Del f; Dup f)
| Number of samples | # Dels | # Dups | Del f | Dup f | Phenotype |
|---|---|---|---|---|---|
| 15 579 | 49 | 216 | 0.0034 | 0.0132 | control |
| 20 987 | 98 | 329 | 0.0028 | 0.0121 | casesa |
cases = developmental delay, autism spectrum, attention deficit, other; see Supplementary Material, Table S6.
Genomic architecture of LCR22A+
Next, we wanted to investigate the architectural features of LCR22A+ (Fig. 4A). There is one CpG island within LCR22A+, but no obvious active or repressive chromatin marks that are apparent in ENCODE tracks from the UCSC Genome Browser (data not shown). We examined possible genes that might map within LCR22A+. A splice variant of the long interspersed non-coding RNA (lincRNA) encoded by DGCR5 (35), extends within LCR22A+ as illustrated in Figure 4A. Two other non-coding RNA genes, DGCR9 and DGCR10 map just proximal to LCR22A+, and appear to be alternative splice forms of DGCR5 (Fig. 4A, UCSC Gene and lincRNA tracks in the UCSC Genome Browser). Upon examination of GTEx (Genotype-Tissue Expression project; https://www.gtexportal.org/home/; data not shown) and lincRNA RNA-seq data from adult human tissues (36), DGCR5, DGCR9 and DGCR10 (AC000095) are highly specifically expressed in the brain (Fig. 4A). LCR22A+ contains a non-functional partial copy of the Car15 gene, which encodes a carbonic anhydrase protein in the mouse and rat (37) (Fig. 4A). It does not appear to be complete or functional in humans.
Figure 4.

Genomic locus containing LCR22A+ and evolutionary breakpoint. (A) The position of the LCR22A+-D deletion and LCR22A-A+ duplication with respect to the 22q11.2 region with individual LCR22s are shown. The local region around LCR22A+ is illustrated as is the position of the ADU/VDU balanced translocation breakpoint within (coordinates are provided; hg19 assembly). Alternative splice variants of the non-coding gene, DGCR5 is shown in the snapshot from the UCSC Genome Browser. The lincRNA track for RNA-seq reads is shown for representative tissues. Dark blue indicates high expression in adult human tissues relative to other tissues with lower expression (light blue). The position of the Car15 gene in rodents is shown with respect to the 22q11.2 region, but this gene is not present in humans. The DGCR5 gene is not present in the mouse genome. (B) UCSC Genome Browser snapshot of the region of synteny in the mouse genome is shown (GRCm38/mm10, 2011). The right-hand side of the image maps to the LCR22A+ region, while the orange side maps 1.7 Mb away in the human region of synteny and contains genes Klhl22 and Scarf2. The evolutionary breakpoint between mouse and human maps between Scarf2 and Car15.
The PRDM9 protein is essential for homologous recombination in humans (38,39). There are 13 potential PRDM9 sites in LCR22A+ (Supplementary Material, Table S7), suggesting that at least some of them might be involved in the recombination process that leads to recurrent meiotic rearrangements involving LCR22A+.
LCR22A+ maps in two other locations on 22q11.2 as determined by BLAT analysis using the UCSC Genome Browser hg19 assembly (19,40). One of the two copies maps within LCR22B and the second maps within LCR22D (Supplementary Material, Fig. S8). We can conclude that this LCR has been quite active in recombination processes during primate evolution.
Balanced translocation maps to LCR22A+
It is unknown whether there is a hotspot for chromosome rearrangements within LCR22A+. There could be a clue from other rearrangements involving this LCR. We examined the literature for other types of rearrangements in LCR22A+ and found previous reports of a family whose breakpoint was cloned and sequenced (41) that carried a balanced t(2; 22)(q14; q11.21) translocation in which the proband and mother, ADU and VDU, respectively, had features of 22q11.2DS (42). This family historically was the subject of much interest in the field because the affected proband and mother did not have a deletion but had a balanced translocation, which disrupted DGCR5 (35). Of interest, the translocation breakpoint is within the center of LCR22A+ as illustrated in Figure 4A. We may consider this region in the future when we are able to narrow LCR22A+-B or A+-D deletion endpoints that it might serve as a hotspot for chromosome rearrangements.
LCR22A+ maps to an evolutionary breakpoint
The largest region of synteny to human 22q11.2 maps to mouse chromosome 16. There are inversions and some shuffling of genes between the two species (39,43,44), and some of the evolutionary breakpoints between sets of genes appear to be where the LCR22s map in humans as shown in Figure 5 (39,43,44). The mouse genome does not contain LCR22s as can be visualized in Figure 5. In the human genome, the order of the adjacent genes is: DGCR6, PRODH, DGCR5 and DGCR2 (DGCR5 is not shown in Figure 5, since it is a non-coding gene). In the mouse genome, the order of genes on chromosome 16 is Klhl22 (Kelch like family member 22), Scarf2 (scavenger receptor class F member 2), Car15 and Dgcr2 (DiGeorge critical region gene 2) (Fig. 4B). In humans, the KLHL22 and SCARF2 genes map 1.7 Mb away from LCR22A+, distal (telomeric) to LCR22B. The DGCR5 non-coding gene does not appear to be present in the mouse genome. Thus, LCR22A+ maps to a region of an evolutionary breakpoint between the two species.
Figure 5.
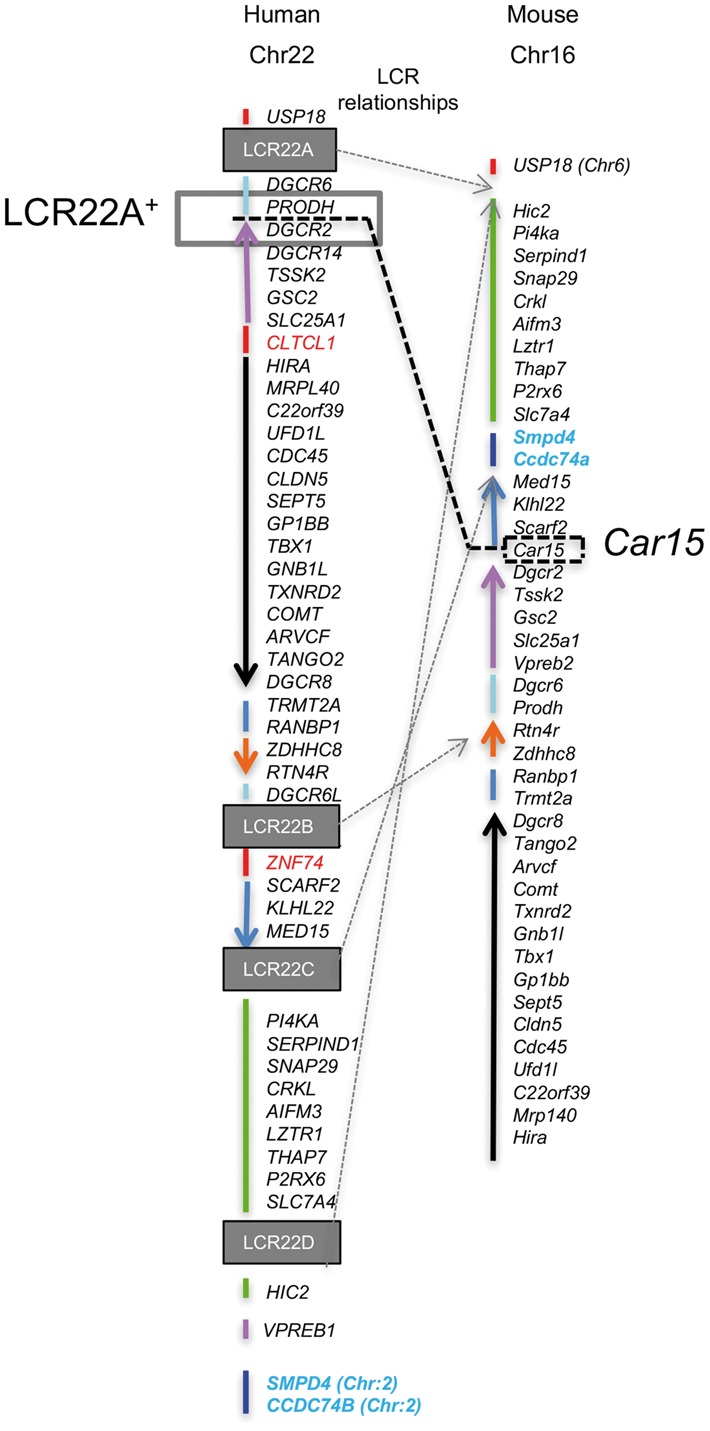
Evolutionary breakpoints between human and mouse regions of synteny on 22q11.2. The known coding genes in the 22q11.2 region are aligned in order from the most centromeric end at the top to the most telomeric end. The region of synteny on mouse chromosome 16 is shown, with genes aligned in accordance to their map position. Note, that non-coding genes are not all predicted between humans and mice and are not shown (e.g. DGCR5 cannot be found in the mouse genome). Two genes in the region of synteny on mouse chromosome 16 map to human chromosome 2 (blue font). The USP18 gene maps to mouse chromosome 6 but not chromosome 16. Changes in the relative order of individual genes or sets of genes between mice and humans are indicated by different color-coded lines and arrows. The LCR22s are indicated as boxes in the human genome but they are not present in the mouse genome. The expected position in the mouse genome for the LCR22s, if they existed, are shown by the light gray dotted arrows. The thick black dotted line indicates the position of the Car15 gene in the mouse, which is within the LCR22A-LCR22A+ region, whose position is shown in the 22q11.2 region.
Discussion
In this report, we identified a recurrent chromosome 22q11.2 rearrangement hotspot in a small, 12 kb segmental duplication or low copy repeat, referred to as LCR22A+. Breakpoints within LCR22A+ are associated with rare nested de novo deletions in 22q11.2DS patients. Breakpoints in LCR22A+ also occur within an inherited common seemingly benign duplication CNV of 0.2 Mb termed LCR22A-A+, harboring two coding genes, DGCR6 and PRODH and one non-coding gene, DGCR5. The LCR22A+-B or LCR22A+-D deletions occur twice as often from a parent with the LCR22A+ duplication CNV, implicating increased risk for this type of deletion. Of interest, a balanced translocation reported previously in one family, ADU/VDU (41), maps within LCR22A+. It is possible that the position of the breakpoint might be similar to the recurrent one in the subjects with LCR22A+-B or LCR22A+-D deletions. We suggest that LCR22A+ may be vulnerable to meiotic chromosomal rearrangements. The results underscore the importance of structural genomic features in mediating genomic variation and disease (20). The breakpoint is of interest evolutionarily because LCR22A+ maps to an evolutionary breakpoint between mouse and human genomes, in which the non-coding DGCR5 gene seems to be human specific and the Car15 gene seems to be mouse specific. Thus, this region has apparent disease-related importance as well as importance in the evolution of complex regions of the human genome.
Understanding mechanisms for 22q11.2 rearrangements
One of the important goals in the field is to identify regions of chromosome breakage within LCR22s in individuals with the typical 3 Mb LCR22A-D deletion. This might help identify potential recombination hotspots and molecular mechanisms. The typical 22q11.2DS deletion occurs largely by de novo NAHR events between the two LCR22s. It has not been possible to identify the chromosome breakpoints within LCR22A and LCR22D, despite many efforts, due to their complex structure and high sequence homology (13,16,31,45). To add to the complexity, a predisposing inversion polymorphism was discovered between LCR22B-D or LCR22C-D, in which 94% of transmitting parents of individuals with de novo 22q11.2 deletions possess the inversion (18).
Since the proximal breakpoint in the 22q11.2DS subjects with nested LCR22A+-B or LCR22A+-D deletions occurred in LCR22A+, it would be of interest to determine whether the inversion polymorphism occurs in parents or whether the nested deletion occurs as a result of a different mechanism. The data we have support a different mechanism. There are two additional copies of LCR22A+, one in LCR22B and the other in LCR22D, with 97.7% sequence identity. The inversion promotes a specific direct orientation of modules or subunits within these duplications, for other NAHR mediated disorders (46–50). The orientation of LCR22A+ in LCR22B is the same as for the one between PRODH-DGCR2, but is in the opposite orientation in LCR22D. If the inversion existed in the parent in which the deletion occurs, the presence of an inversion between LCR22C-D or B-D would cause LCR22A+ in LCR22B or –D to be in the opposite orientation needed for a NAHR event between LCR22A+-D. Thus, it is difficult to envision a simple NAHR mechanism between copies of LCR22A+ that is responsible for these atypical recurrent nested deletions.
We were not able to narrow the breakpoints within LCR22A+ in our cohort. This is because we found that the copies of LCR22A+ in LCR22B and LCR22D are embedded within other LCR22 sequences. This situation makes it very challenging to clone and sequence the 12 kb, LCR22A+ fragment at this time since flanking primers are needed on both sides. Further, the genome architecture of the LCR22s are still not completely defined. Currently, the copy of LCR22A+ within LCR22B in the hg19 genome assembly from 2009, maps within LCR22A in the hg38 genome assembly from 2013 (chr22: 18627728–18639943). In addition, there are several non-sequenced gaps, both within LCR22A in hg38 that are not present in hg19, while there is a similar sized gap in LCR22B in both assemblies. When taken together, it appears that the genome architecture of the LCR22s is still not defined. Another possibility is that there is extensive variation of the structure or sequence of LCR22s within individuals that can add to their complexity. One clue comes from the cloned and sequenced breakpoint of the balanced translocation patients ADU/VDU that maps in the center of LCR22A+. However, it would only be possible to draw a conclusion once the chromosome breakpoints for individuals with nested LCR22A+-B or LCR22A+-D deletions are cloned and sequenced.
We previously performed whole genome sequencing of DNA from two trio families with the LCR22A-D deletion and was able to narrow, but not able to precisely define the position of the chromosome breakpoints (31). Currently, Bionano optical next-generation structural mapping is being used to define the architecture of the genome (18). It is likely that this sequencing method, or others that will likely emerge, will be required to determine the genome architecture of the region, chromosome breakpoints and the precise mechanisms of chromosome rearrangements within the 22q11.2 region.
Homologous recombination in humans requires function of the PRDM9 gene (38,39). We scanned the DNA sequence in this region and found the hotspot motif, CCNCCNTNNCCNC (38,39), present in three locations and the hotspot motif, CCTCCCT (38,39) present in ten locations in LCR22A+. The hotspot motifs can potentially mediate mammalian homologous recombination (Supplementary Material, Table S7). However, it is not known whether these sites are active in this process. We did not find a PRDM9 site at the position of the ADU/VDU breakpoint. Nonetheless, it is possible that the relevant breakpoint events may occur due to other mechanisms.
Of interest, six of the eight subjects with LCR22A+ to LCR22B or LCR22D deletions occurred as a de novo meiotic event from a parent with a 0.2 Mb LCR22A-LCR22A+ duplication. The qPCR, microsatellite and FISH results suggest that the nested 22q11.2 deletion may occur on the opposite allele that carried the 0.2 Mb LCR22A-LCR22A+ duplication CNV, although the mechanism is unknown.
The Williams-Beuren syndrome (WBS) is a rare neurodevelopmental disorder occurring in 1/7500 individuals, caused by NAHR events between complex, region specific LCRs on 7q11.23. A chromosome inversion polymorphism flanked by LCRs occurs in 25% of transmitting parents compared with 5% in the population (49,50). There are further structural polymorphisms within the LCRs on chromosome 7q11.23 (51). It was found in a cohort of 180 WBS subjects that 4.4% of the parents have a deletion of one of the LCR blocks, containing pseudogenes, versus 1% that do not, resulting in a significant increase in risk for the deletion [odds ratio of 4.6; P = 0.002; or risk of 1/1500 individuals (52)]. The positions of the chromosome breakpoints could be mapped to a particular region using paralogous sequence variants but they did not map to a particular site within the LCR block (52). It was suggested that this LCR block might contain higher sequence identity therefore increasing the frequency of NAHR events (52). Structural polymorphisms in the duplications might predispose individuals to further meiotic NAHR events (53,54). This would be consistent with the possibility that a duplication of LCR22A+ might predispose individuals to further NAHR events or other mechanisms. This will have to be investigated in the future by large population studies.
Differences in phenotypes between LCR22A-D versus LCR22A+-D deletions
It is possible that there are phenotypic consequences as to whether or not there is one versus two copies of PRODH, DGCR6 or DGCR5 genes. This is for all subjects with a presumed LCR22A+-D deletion. The heterozygous LCR22A-LCR22A+ deletion occurring in 0.3% in the general population is present in approximately 20% of individuals with hyperprolinemia when accompanied by a sequence mutation in the other allele of PRODH (33,34,55). This is one example how there could be phenotypic consequences whether PRODH is deleted on one allele or both alleles. Elevated levels of proline in the blood plasma are sometimes associated with clinical manifestations including seizures and neurological defects (55–58). This is another example of a possible phenotypic consequence, but here it would be in individuals that carry the LCR22A-LCR22A+ duplication CNV. The function of DGCR6 is not very well known as compared with PRODH. DGCR6 maps just distal to LCR22A. However, there is a paralog, termed DGCR6L, which is 97% identical in sequence and it maps just proximal to LCR22B (32). The DGCR6/DGCR6L genes are widely expressed in most tissues (32). In chicken, Dgcr6 may have a function in embryonic development, based upon antisense RNA studies (59), but there are no reports in which these genes have been inactivated. There is very little known about the function of the long intergenic non-coding RNA gene, DGCR5. This gene appears to be absent in non-human primate species.
Approximately, 25% of 22q11.2DS patients develop schizophrenia in adulthood (60,61). Although there are many genes hemizygously deleted on 22q11.2, PRODH has been of interest in its association with schizophrenia in patients with 22q11.2DS (57,62). Glutamine metabolism downstream of hyperprolinemia in affected subjects may be disrupted in 22q11.2DS (63). Studies using mouse models to understand the function of PRODH have shown that hyperprolinemia might affect glutamatergic neurotransmission (64). More recent work in mouse models has shown that loss of PRODH may affect GABAergic transmission leading to synaptic dysfunction and possibly to behavioral disorders (65). In genetic studies of schizophrenia in the general population, there has been a reported association between variants in PRODH, a role in hyperprolinemia and the occurrence of schizophrenia (55,66–70). This association remains controversial since other studies do not support a connection between PRODH and schizophrenia in the general, non-22q11.2DS population (52,66). In the future, it would be of interest to determine whether 22q11.2DS individuals with both two copies of PRODH have reduced risk to schizophrenia. Data for psychiatric phenotypes for this cohort are still being collected. Such an evaluation would need to be accompanied by measurement of proline levels to confirm that they are normal.
Conclusions
Using high-resolution Affymetrix 6.0 microarray analysis in a large cohort of 1680 unrelated 22q11.2DS subjects, we identified LCR22A+ serving as a hotspot region for 22q11.2 meiotic and evolutionary rearrangements. The interval where the nested proximal breakpoint occurs in individuals with a LCR22A+-B or A+-D deletion is in the same interval as for the distal deletion breakpoint for a common LCR22A-A+ duplication CNV that contains DGCR6-PRODH-DGCR5 genes. Further a balanced translocation in a family with some clinical signs of 22q11.2DS maps to specific sequences within LCR22A+. It is possible that these sequences might be important for chromosome rearrangements. Overall, identification of chromosome rearrangement breakpoints in LCR22A+ underscores the complexity of the genomic architecture of the 22q11.2 region. It also highlights the importance of examining the genome architecture of parental chromosomes because this might confer risk to such rearrangements.
Materials and Methods
Human subjects
A total of >1800 genomic DNA samples from unrelated, de-identified probands with 22q11.2DS were ascertained retrospectively from >25 international clinical and research sites as part of the International 22q11.2 Consortium and the International 22q11.2 Brain and Behavior Consortium (Supplementary Material, Table S1), with their informed consent (Internal Review Board at Albert Einstein College of Medicine, 1999–201). All study subjects were unrelated and diagnosed with 22q11.2DS by clinical evaluation and by the presence of a 22q11.2 deletion using FISH mapping, clinical microarray analysis or Multiplex Ligation-dependent Probe Amplification (MLPA) assays (SALSA MLPA kit P250 DiGeorge; MRC Holland, The Netherlands).
Affymetrix SNP 6.0 array processing
The DNA samples were processed on Affymetrix GeneChip Genome Wide SNP 6.0 arrays in the Genomics Core of Albert Einstein College of Medicine with the following exceptions. A subset had unprocessed Affymetrix 6.0 array data that were already available [37 samples from the Advanced Genomics (AGEN) laboratory core at the Children's Research Institute CRI/MCW, Milwaukee, WI, (21) and 225 Chilean samples from the Center for Human Genetics, Clinica Alemana- University Desarrollo, Santiago, Chile].
Quality control (QC) measures were performed on the raw data obtained from Affymetrix 6.0 arrays on all samples to ensure good data quality, as specified by the microarray manufacturer as previously described (22). The genotype calling methods and further QC measures has been previously described (22). In total, 1680 arrays from unrelated subjects that passed QC measures were used in this study.
Affymetrix SNP 6.0 array 22q11.2 CNV analysis
Raw intensity data (Affymetrix SNPs 6.0 array CEL file) from each genotyping site (Genomics Core; Albert Einstein College of Medicine, CRI/MCW, Milwaukee, WI, and Santiago, Chile) were independently processed and analysed to account for potential batch effects. Probe intensities were extracted from CEL files and analysed using the Copy Number Analysis Module (CNAM) that is part of the Golden Helix Powerseat Package (http://www.goldenhelix.com/index.html). In brief, the normalized intensity data for each probe (both SNP-single nucleotide polymorphism probes and CNV-copy number variant probes) from both 22q11.2DS samples, and the reference control (‘baseline’) CEL files were used to calculate the log2 intensity ratio (LR). A total of 90 different reference healthy control samples were processed at the same time as two batches of 22q11.2DS samples at Albert Einstein College of Medicine. For the 22q11.2DS samples processed at CRI/MCW, Milwaukee, WI, the 270 Affymetrix HapMap pre-computed intensity data were used to calculate the log2 ratio. Similar to samples at Albert Einstein College of Medicine, 30 and 47 reference control samples, respectively, were used for the two batches of GR samples from Santiago, Chile. All the data were analyzed as separate batches. Although the raw signal intensities were normalized prior to the log ratio (LR) calculation to identify deletion size, some variation usually remained. To further remove batch effects and other technical artifacts, principal component analysis (PCA) was used to detect and correct the data from batch effects. A univariate analysis was implemented using the Copy Number Analysis Method (CNAM) of Golden Helix to determine the optimal segmentation of the PCA-corrected LR for each measured subject. Two measurements including Derivative log ratio spread (DLRS) (23) and Waviness factor (24), which derived from Log2 ratio were used as a quality control. The samples with greater than 1.5*Interquartile range (IQR) above the upper quartile, or less than 1.5*IQR below the lower quartile were removed for further univariate analysis to determine the optimal segmentation. Removed samples were analyzed by visualizing the log2 ratio plot manually.
MLPA analysis
A total of 539 samples received since 2015, among the 1680, were tested by MLPA using the SALSA P250-B2 kit (MRC-Holland, Amsterdam, The Netherlands) containing 15 probes from the LCR22A through –D region. This kit does not contain a probe in the LCR22A-A+ region. The SALSA P356 kit for the q arm of chromosome 22 was used on the subset of DNA samples from Santiago, Chile. In this kit, there are two probes for PRODH, mapping to the LCR22A-A+ region and five other probes in the LCR22A-D region. A total of 100–250 ng of DNA in a final volume of 5 µl was heated at 98°C and mixed with 1.5 µl probe mix and 1.5 µl SALSA hybridization buffer. Probe hybridization, ligation and amplification reactions were carried out according to the protocols supplied by MRC Holland in a standard thermal cycler (Tpersonal, Biometra®, Göttingen, Germany). The amplification products were analyzed by capillary electrophoresis in an ABI 3730XL genetic analyzer, using the GeneMapper software v3.5 (Applied Biosystems), following the recommended protocol supplied with the MLPA kit. Raw data were exported and normalization was performed using internal control and reference probes as previously described (25,26). This set values for diploid gene dosage to 1, with deletion and duplication thresholds established at below 0.75 and above 1.25, respectively.
Quantitative PCR analysis
Confirmation of deletion endpoints was performed by quantitative PCR (qPCR) using the SYBR Green-based detection method. The qPCR was carried out on an ABI StepOnePlusTM System. Primer 3.0 software (http://simgene.com/Primer3) was used to design primers to amplify the regions of interest listed in Supplementary Material, Table S2. Each qPCR assay included amplification of three endogenous control samples with known copy number (control reference assay: telomerase reverse transcriptase, TERT; Ribonuclease P RNA component H1, RPPH1; RnaseP). One DNA sample with normal copy number (CEPH HapMap sample GM12878) was used as a control in each experiment. A total of 10 μL reactions were performed using 10 ng of DNA following the manufacturer’s recommended protocol. Results were analyzed using StepOnePlusTM Software based on the ΔΔCT method.
Microsatellite marker assays and haplotype analysis
Microsatellite marker polymorphism data were collected using a panel of eight simple tandem repeat markers (D22S427, D22S1638, D22S941, D22S944, D22S264, D22S311, D22S1709A, D22S308) (27,28). The forward primer for each marker was labeled with FAM for D22S427, NED for D22S1638, FAM for D22S941, TET for D22S944, VIC for D22S264, HEX for D22S1709A, PET for D22S311, PET for D22S308 (Supplementary Material, Table S2). We generated a new microsatellite marker that we named BCRP2 and it was labeled with NED (Supplementary Material, Table S2). PCR was performed using standard conditions optimized for each marker. The amplified products were diluted with water, to obtain a fluorescence signal strength between 1000 and 18 000 relative fluorescence units and the samples were run on an ABI PRISM 3730 sequencer. The .fsa files from the ABI 3730 sequencer were analyzed with GeneMarker® V1.75 software (SoftGenetics, State College; PA http://www.softgenetics.com/GeneMarker.php) to size and call alleles in base pairs (bp). All allele calls were manually reviewed.
FISH mapping
Fosmid clones were identified using the UCSC Genome Browser for the human genome, assembly GRCh37/hg19. The bacterial colonies containing the fosmid clones were purchased from BAC-PAC Resources (https://bacpacresources.org/WIBR-2: Human Fosmid Library). The sequence of the purchased fosmid clones was confirmed using PCR with primers designed from unique sequence within each clone (Supplementary Material, Table S2). Two fosmids were chosen to mark unique sequences just telomeric to the pericentromeric region on 22q11.1 containing the CECR1 gene (DY-495-dUTP; Spectrum green; Fluorescein isothiocyanate-FITC), one in the LCR22A-A+ region, containing DGCR5 (DiGeorge syndrome Critical Region 5) (DY-590-dUTP; Spectrum orange; S.O.) and two in the TBX1-GNB1L region (DY-415-dUTP; spectrum aqua). Dyes were purchased from Dyomics (Jena, GE). Detailed information about the map position of the fosmids are shown in Supplementary Material, Figure S1. FISH mapping was performed on actively growing Epstein Barr virus (EBV) transformed lymphoblastoid cell lines from blood samples using the probes. Briefly, the slides for FISH mapping were denatured with 50% Formamide/2xSSC at 72°C for 1.5 min and then dehydrated with serial ethanol washing steps (ice cold 70, 90 and 100% for 3 min each). Probes were denatured in the hybridization solution (50% dextran sulfate/2xSSC) at 80°C for 7 min, applied to the slides and incubated overnight at 37°C in a humidified chamber. The slides were then washed three times for 5 min with 50% formamide/2X SSC, 1X SSC and 4XSSC/0.1%Tween. Slides were dehydrated with serial ethanol washing steps (see above) and mounted with ProLong Gold antifade reagent with DAPI (4, 6-Diamidino-2-phenylindole; Invitrogen, Carlsbad, CA) for imaging. FISH images were acquired with a manual inverted fluorescence microscope (Axiovert 200, Zeiss) with a fine focusing oil immersion lens (x40, NA 1.3 oil and “60, NA 1.35 oil). Multiple focal planes were acquired for each channel to ensure that signals on different focal planes were included. The resulting fluorescence emissions were collected using 350-to-470 nm (for DAPI), 436-to-480 nm (for DY-415-dUTP), 470-to-540 nm (for DY-495-dUTP and Alexa Fluor 488) and 546-to-600 nm (for DY-590-dUTP an Alexa Fluor 555) filters. The microscope was equipped with a Camera Hall 100 and we used Applied Spectral Imaging software.
Supplementary Material
Supplementary Material is available at HMG online.
Supplementary Material
Acknowledgements
We would like to thank the families with 22q11.2DS who provided DNA and clinical information. We acknowledge the Genomics and Molecular Cytogenetics Cores at Einstein. We also acknowledge Mark Zeffren, Nousin Haque and Antoneta Preldakaj for project management and John Bruppacher, Dan Arroyo, Michael Gleeson, Dominique Calandrillo and Frédérique Bena for technical support at Einstein. We also greatly appreciate the laboratory effort of Dr. Frédérique Bena who works with SE and SEA (Institute of Genetics and Genomics of Geneva, Switzerland).
Conflict of Interest statement. None declared.
Funding
NIH R01 HL084410 (BSE, BEM, DMM, TG, TW), P01 HD070454 (BSE, BEM, DMM, TG, TW, HN, LZ, DZ, YZ, TBC), U01 MH101720 (TG, AD, DMM, MH, WD, YZ, FU, LK-W, CEB, WRK, DG, MS, SE, JB, AS, JV, FBG, GMR, BSE, ASB, JRV, CRM, BEM), R21HL118637 (TW, BEM, TG). GMR was also supported by the FONDECYT-Chile (grants 1100131 and 1130392). ASB was supported by the Dalglish Chair in 22q11.2 Deletion Syndrome, the Canada Research Chair in Schizophrenia Genetics and Genomic Disorders, Canadian Institutes of Health Research funding (MOP-97800 and MOP-89066), and the University of Toronto McLaughlin Centre. CEB was supported by the NIH (R01 MH085903). SE was supported by the Swiss National Science Foundation (FNS 324730_121996; FNS 324730_144260). JV was supported by the Flemish Science Foundation (FWO G.0E1117N).
Availability of Data and Materials
The .cel and genotype files from Affymetrix 6.0 arrays have been deposited to NCBI dbGAP phs001339.v1.p1.
References
- 1. Tezenas Du Montcel S., Mendizabai H., Ayme S., Levy A., Philip N. (1996) Prevalence of 22q11 microdeletion. J. Med. Genet., 33, 719.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Devriendt K., Fryns J.P., Mortier G., van Thienen M.N., Keymolen K. (1998) The annual incidence of DiGeorge/velocardiofacial syndrome. J. Med. Genet., 35, 789–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Botto L.D., May K., Fernhoff P.M., Correa A., Coleman K., Rasmussen S.A., Merritt R.K., O'Leary L.A., Wong L.Y., Elixson E.M.. et al. (2003) A population-based study of the 22q11.2 deletion: phenotype, incidence, and contribution to major birth defects in the population. Pediatrics, 112, 101–107. [DOI] [PubMed] [Google Scholar]
- 4. Goodship J., Cross I., LiLing J., Wren C. (1998) A population study of chromosome 22q11 deletions in infancy. Arch. Dis. Child., 79, 348–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Scambler P.J. (2000) The 22q11 deletion syndromes. Hum. Mol. Genet., 9, 2421–2426. [DOI] [PubMed] [Google Scholar]
- 6. Oskarsdottir S., Vujic M., Fasth A. (2004) Incidence and prevalence of the 22q11 deletion syndrome: a population-based study in Western Sweden. Arch. Dis. Child., 89, 148–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schneider M., Debbane M., Bassett A.S., Chow E.W., Fung W.L., van den Bree M., Owen M., Murphy K.C., Niarchou M., Kates W.R.. et al. (2014) Psychiatric disorders from childhood to adulthood in 22q11.2 deletion syndrome: results from the International Consortium on Brain and Behavior in 22q11.2 Deletion Syndrome. Am. J. Psychiatry, 171, 627–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yu S., Graf W.D., Shprintzen R.J. (2012) Genomic disorders on chromosome 22. Curr. Opin. Pediatr., 24, 665–671. [DOI] [PubMed] [Google Scholar]
- 9. Swillen A., McDonald-McGinn D. (2015) Developmental trajectories in 22q11.2 deletion. Am. J. Med. Genet. C, Semin. Med. Genet., 169, 172–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. McDonald-McGinn D.M., Sullivan K.E., Marino B., Philip N., Swillen A., Vorstman J.A., Zackai E.H., Emanuel B.S., Vermeesch J.R., Morrow B.E.. et al. (2015) 22q11.2 deletion syndrome. Nat. Rev. Dis. Primers, 1, 15071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Morrow B., Goldberg R., Carlson C., Das Gupta R., Sirotkin H., Collins J., Dunham I., O'Donnell H., Scambler P., Shprintzen R.. et al. (1995) Molecular definition of the 22q11 deletions in velo-cardio-facial syndrome. Am. J. Hum. Genet., 56, 1391–1403. [PMC free article] [PubMed] [Google Scholar]
- 12. Lindsay E.A., Goldberg R., Jurecic V., Morrow B., Carlson C., Kucherlapati R.S., Shprintzen R.J., Baldini A. (1995) Velo-cardio-facial syndrome: frequency and extent of 22q11 deletions. Am. J. Med. Genet., 57, 514–522. [DOI] [PubMed] [Google Scholar]
- 13. Edelmann L., Pandita R.K., Morrow B.E. (1999) Low-copy repeats mediate the common 3-Mb deletion in patients with velo-cardio-facial syndrome. Am. J. Hum. Genet., 64, 1076–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Edelmann L., Pandita R.K., Spiteri E., Funke B., Goldberg R., Palanisamy N., Chaganti R.S., Magenis E., Shprintzen R.J., Morrow B.E. (1999) A common molecular basis for rearrangement disorders on chromosome 22q11. Hum. Mol. Genet., 8, 1157–1167. [DOI] [PubMed] [Google Scholar]
- 15. Shaikh T.H., Kurahashi H., Saitta S.C., O'Hare A.M., Hu P., Roe B.A., Driscoll D.A., McDonald-McGinn D.M., Zackai E.H., Budarf M.L.. et al. (2000) Chromosome 22-specific low copy repeats and the 22q11.2 deletion syndrome: genomic organization and deletion endpoint analysis. Hum. Mol. Genet., 9, 489–501. [DOI] [PubMed] [Google Scholar]
- 16. Bailey J.A., Yavor A.M., Viggiano L., Misceo D., Horvath J.E., Archidiacono N., Schwartz S., Rocchi M., Eichler E.E. (2002) Human-specific duplication and mosaic transcripts: the recent paralogous structure of chromosome 22. Am. J. Hum. Genet., 70, 83–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Babcock M., Yatsenko S., Hopkins J., Brenton M., Cao Q., de Jong P., Stankiewicz P., Lupski J.R., Sikela J.M., Morrow B.E. (2007) Hominoid lineage specific amplification of low-copy repeats on 22q11.2 (LCR22s) associated with velo-cardio-facial/digeorge syndrome. Hum. Mol. Genet., 16, 2560–2571. [DOI] [PubMed] [Google Scholar]
- 18. Demaerel W., Hestand M.S., Vergaelen E., Swillen A., Lopez-Sanchez M., Perez-Jurado L.A., McDonald-McGinn D.M., Zackai E., Emanuel B.S., Morrow B.E.. et al. (2017) Nested inversion polymorphisms predispose chromosome 22q11.2 to meiotic rearrangements. Am. J. Hum. Genet., 101, 616–622. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 19. Bailey J.A., Yavor A.M., Massa H.F., Trask B.J., Eichler E.E. (2001) Segmental duplications: organization and impact within the current human genome project assembly. Genome Res., 11, 1005–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Carvalho C.M., Lupski J.R. (2016) Mechanisms underlying structural variant formation in genomic disorders. Nat. Rev. Genet., 17, 224–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tomita-Mitchell A., Mahnke D.K., Struble C.A., Tuffnell M.E., Stamm K.D., Hidestrand M., Harris S.E., Goetsch M.A., Simpson P.M., Bick D.P.. et al. (2012) Human gene copy number spectra analysis in congenital heart malformations. Physiol. Genomics, 44, 518–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Guo T., Repetto G.M., McDonald McGinn D.M., Chung J.H., Nomaru H., Campbell C.L., Blonska A., Bassett A.S., Chow E.W.C., Mlynarski E.E.. et al. (2017) Genome-wide association study to find modifiers for tetralogy of fallot in the 22q11.2 deletion syndrome identifies variants in the GPR98 locus on 5q14.3. Circ. Cardiovasc. Genet., 10, pii: e001690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pinto D., Darvishi K., Shi X., Rajan D., Rigler D., Fitzgerald T., Lionel A.C., Thiruvahindrapuram B., Macdonald J.R., Mills R.. et al. (2011) Comprehensive assessment of array-based platforms and calling algorithms for detection of copy number variants. Nat. Biotechnol., 29, 512–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Diskin S.J., Li M., Hou C., Yang S., Glessner J., Hakonarson H., Bucan M., Maris J.M., Wang K. (2008) Adjustment of genomic waves in signal intensities from whole-genome SNP genotyping platforms. Nucleic Acids Res., 36, e126.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yobb T.M., Somerville M.J., Willatt L., Firth H.V., Harrison K., MacKenzie J., Gallo N., Morrow B.E., Shaffer L.G., Babcock M.. et al. (2005) Microduplication and triplication of 22q11.2: a highly variable syndrome. Am. J. Hum. Genet., 76, 865–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Evers L.J., Engelen J.J., Houben L.M., Curfs L.M., van Amelsvoort T.A. (2016) The use of two different MLPA kits in 22q11.2 deletion syndrome. Eur. J. Med. Genet., 59, 183–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Carlson C., Sirotkin H., Pandita R., Goldberg R., McKie J., Wadey R., Patanjali S.R., Weissman S.M., Anyane-Yeboa K., Warburton D.. et al. (1997) Molecular definition of 22q11 deletions in 151 velo-cardio-facial syndrome patients. Am. J. Hum. Genet., 61, 620–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Delio M., Guo T., McDonald-McGinn D.M., Zackai E., Herman S., Kaminetzky M., Higgins A.M., Coleman K., Chow C., Jalbrzikowski M.. et al. (2013) Enhanced maternal origin of the 22q11.2 deletion in velocardiofacial and DiGeorge syndromes. Am. J. Hum. Genet., 92, 439–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Weksberg R., Stachon A.C., Squire J.A., Moldovan L., Bayani J., Meyn S., Chow E., Bassett A.S. (2007) Molecular characterization of deletion breakpoints in adults with 22q11 deletion syndrome. Hum. Genet., 120, 837–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hestand M.S., Nowakowska B.A., Vergaelen E., Van Houdt J., Dehaspe L., Suhl J.A., Del-Favero J., Mortier G., Zackai E., Swillen A.. et al. (2016) A catalog of hemizygous variation in 127 22q11 deletion patients. Hum. Genome Var, 3, 15065.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Guo X., Delio M., Haque N., Castellanos R., Hestand M.S., Vermeesch J.R., Morrow B.E., Zheng D. (2016) Variant discovery and breakpoint region prediction for studying the human 22q11.2 deletion using BAC clone and whole genome sequencing analysis. Hum. Mol. Genet., 25, 3754–3767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Edelmann L., Stankiewicz P., Spiteri E., Pandita R.K., Shaffer L., Lupski J.R., Morrow B.E. (2001) Two functional copies of the DGCR6 gene are present on human chromosome 22q11 due to a duplication of an ancestral locus. Genome Res., 11, 208–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Guilmatre A., Legallic S., Steel G., Willis A., Di Rosa G., Goldenberg A., Drouin-Garraud V., Guet A., Mignot C., Des Portes V.. et al. (2010) Type I hyperprolinemia: genotype/phenotype correlations. Hum. Mutat., 31, 961–965. [DOI] [PubMed] [Google Scholar]
- 34. Richard A.C., Rovelet-Lecrux A., Delaby E., Charbonnier C., Thiruvahindrapuram B., Hatchwell E., Eis P.S., Afenjar A., Dussardier B.G., Scherer S.W.. et al. (2016) The 22q11 PRODH/DGCR6 deletion is frequent in hyperprolinemic subjects but is not a strong risk factor for ASD. Am. J. Med. Genet. Part B, Neuropsychiatr. Genet., 171, 377–382. [DOI] [PubMed] [Google Scholar]
- 35. Sutherland H.F., Wadey R., McKie J.M., Taylor C., Atif U., Johnstone K.A., Halford S., Kim U.J., Goodship J., Baldini A.. et al. (1996) Identification of a novel transcript disrupted by a balanced translocation associated with DiGeorge syndrome. Am. J. Hum. Genet., 59, 23–31. [PMC free article] [PubMed] [Google Scholar]
- 36. Lonsdale J., Thomas J., Salvatore M., Phillips R., Lo E., Shad S., Hasz R., Walters G., Garcia F., Young N.. et al. (2013) The Genotype-Tissue Expression (GTEx) project. Nat. Genet., 45, 580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pan P.W., Rodriguez A., Parkkila S. (2007) A systematic quantification of carbonic anhydrase transcripts in the mouse digestive system. BMC Mol. Biol., 8, 22.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Myers S., Bowden R., Tumian A., Bontrop R.E., Freeman C., MacFie T.S., McVean G., Donnelly P. (2010) Drive against hotspot motifs in primates implicates the PRDM9 gene in meiotic recombination. Science, 327, 876–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Berg I.L., Neumann R., Sarbajna S., Odenthal-Hesse L., Butler N.J., Jeffreys A.J. (2011) Variants of the protein PRDM9 differentially regulate a set of human meiotic recombination hotspots highly active in African populations. Proc. Natl Acad. Sci. U.S.A., 108, 12378–12383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bailey J.A., Gu Z., Clark R.A., Reinert K., Samonte R.V., Schwartz S., Adams M.D., Myers E.W., Li P.W., Eichler E.E. (2002) Recent segmental duplications in the human genome. Science, 297, 1003–1007. [DOI] [PubMed] [Google Scholar]
- 41. Budarf M.L., Collins J., Gong W., Roe B., Wang Z., Bailey L.C., Sellinger B., Michaud D., Driscoll D.A., Emanuel B.S. (1995) Cloning a balanced translocation associated with DiGeorge syndrome and identification of a disrupted candidate gene. Nat. Genet., 10, 269–278. [DOI] [PubMed] [Google Scholar]
- 42. Augusseau S., Jouk S., Jalbert P., Prieur M. (1986) DiGeorge syndrome and 22q11 rearrangements. Hum. Genet., 74, 206.. [DOI] [PubMed] [Google Scholar]
- 43. Puech A., Saint-Jore B., Funke B., Gilbert D.J., Sirotkin H., Copeland N.G., Jenkins N.A., Kucherlapati R., Morrow B., Skoultchi A.I. (1997) Comparative mapping of the human 22q11 chromosomal region and the orthologous region in mice reveals complex changes in gene organization. Proc. Natl Acad. Sci. U.S.A., 94, 14608–14613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Babcock M., Pavlicek A., Spiteri E., Kashork C.D., Ioshikhes I., Shaffer L.G., Jurka J., Morrow B.E. (2003) Shuffling of genes within low-copy repeats on 22q11 (LCR22) by Alu-mediated recombination events during evolution. Genome Res., 13, 2519–2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shaikh T.H., Kurahashi H., Emanuel B.S. (2001) Evolutionarily conserved low copy repeats (LCRs) in 22q11 mediate deletions, duplications, translocations, and genomic instability: an update and literature review. Genet. Med., 3, 6–13. [DOI] [PubMed] [Google Scholar]
- 46. Koolen D.A., Vissers L.E., Pfundt R., de Leeuw N., Knight S.J., Regan R., Kooy R.F., Reyniers E., Romano C., Fichera M.. et al. (2006) A new chromosome 17q21.31 microdeletion syndrome associated with a common inversion polymorphism. Nat. Genet., 38, 999–1001. [DOI] [PubMed] [Google Scholar]
- 47. Visser R., Shimokawa O., Harada N., Kinoshita A., Ohta T., Niikawa N., Matsumoto N. (2005) Identification of a 3.0-kb major recombination hotspot in patients with Sotos syndrome who carry a common 1.9-Mb microdeletion. Am. J. Hum. Genet., 76, 52–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Giglio S., Calvari V., Gregato G., Gimelli G., Camanini S., Giorda R., Ragusa A., Guerneri S., Selicorni A., Stumm M.. et al. (2002) Heterozygous submicroscopic inversions involving olfactory receptor-gene clusters mediate the recurrent t(4; 8)(p16; p23) translocation. Am. J. Hum. Genet., 71, 276–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Osborne L.R., Li M., Pober B., Chitayat D., Bodurtha J., Mandel A., Costa T., Grebe T., Cox S., Tsui L.C.. et al. (2001) A 1.5 million-base pair inversion polymorphism in families with Williams-Beuren syndrome. Nat. Genet., 29, 321–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bayes M., Magano L.F., Rivera N., Flores R., Perez Jurado L.A. (2003) Mutational mechanisms of Williams-Beuren syndrome deletions. Am. J. Hum. Genet., 73, 131–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Perez Jurado L.A., Peoples R., Kaplan P., Hamel B.C., Francke U. (1996) Molecular definition of the chromosome 7 deletion in Williams syndrome and parent-of-origin effects on growth. Am. J. Hum. Genet., 59, 781–792. [PMC free article] [PubMed] [Google Scholar]
- 52. Cusco I., Corominas R., Bayes M., Flores R., Rivera-Brugues N., Campuzano V., Perez-Jurado L.A. (2008) Copy number variation at the 7q11.23 segmental duplications is a susceptibility factor for the Williams-Beuren syndrome deletion. Genome Res., 18, 683–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Human Genome Structural Variation Working G., Eichler E.E., Nickerson D.A., Altshuler D., Bowcock A.M., Brooks L.D., Carter N.P., Church D.M., Felsenfeld A., Guyer M.. et al. (2007) Completing the map of human genetic variation. Nature, 447, 161–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lupski J.R. (2007) Structural variation in the human genome. N. Engl J. Med., 356, 1169–1171. [DOI] [PubMed] [Google Scholar]
- 55. Jacquet H., Raux G., Thibaut F., Hecketsweiler B., Houy E., Demilly C., Haouzir S., Allio G., Fouldrin G., Drouin V.. et al. (2002) PRODH mutations and hyperprolinemia in a subset of schizophrenic patients. Hum. Mol. Genet., 11, 2243–2249. [DOI] [PubMed] [Google Scholar]
- 56. Afenjar A., Moutard M.L., Doummar D., Guet A., Rabier D., Vermersch A.I., Mignot C., Burglen L., Heron D., Thioulouse E.. et al. (2007) Early neurological phenotype in 4 children with biallelic PRODH mutations. Brain Dev., 29, 547–552. [DOI] [PubMed] [Google Scholar]
- 57. Raux G., Bumsel E., Hecketsweiler B., van Amelsvoort T., Zinkstok J., Manouvrier-Hanu S., Fantini C., Breviere G.M., Di Rosa G., Pustorino G.. et al. (2007) Involvement of hyperprolinemia in cognitive and psychiatric features of the 22q11 deletion syndrome. Hum. Mol. Genet., 16, 83–91. [DOI] [PubMed] [Google Scholar]
- 58. Vorstman J.A., Turetsky B.I., Sijmens-Morcus M.E., de Sain M.G., Dorland B., Sprong M., Rappaport E.F., Beemer F.A., Emanuel B.S., Kahn R.S.. et al. (2009) Proline affects brain function in 22q11DS children with the low activity COMT 158 allele. Neuropsychopharmacology, 34, 739–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hierck B.P., Molin D.G., Boot M.J., Poelmann R.E., Gittenberger-de Groot A.C. (2004) A chicken model for DGCR6 as a modifier gene in the DiGeorge critical region. Pediatr. Res., 56, 440–448. [DOI] [PubMed] [Google Scholar]
- 60. Shprintzen R.J., Goldberg R., Golding-Kushner K.J., Marion R.W. (1992) Late-onset psychosis in the velo-cardio-facial syndrome. Am. J. Med. Genet., 42, 141–142. [DOI] [PubMed] [Google Scholar]
- 61. Bassett A.S., Marshall C.R., Lionel A.C., Chow E.W., Scherer S.W. (2008) Copy number variations and risk for schizophrenia in 22q11.2 deletion syndrome. Hum. Mol. Genet., 17, 4045–4053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zarchi O., Carmel M., Avni C., Attias J., Frisch A., Michaelovsky E., Patya M., Green T., Weinberger R., Weizman A.. et al. (2013) Schizophrenia-like neurophysiological abnormalities in 22q11.2 deletion syndrome and their association to COMT and PRODH genotypes. J. Psychiatr. Res., 47, 1623–1629. [DOI] [PubMed] [Google Scholar]
- 63. Evers L.J., van Amelsvoort T.A., Bakker J.A., de Koning M., Drukker M., Curfs L.M. (2015) Glutamatergic markers, age, intellectual functioning and psychosis in 22q11 deletion syndrome. Psychopharmacology (Berl), 232, 3319–3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Paterlini M., Zakharenko S.S., Lai W.S., Qin J., Zhang H., Mukai J., Westphal K.G., Olivier B., Sulzer D., Pavlidis P.. et al. (2005) Transcriptional and behavioral interaction between 22q11.2 orthologs modulates schizophrenia-related phenotypes in mice. Nat. Neurosci., 8, 1586–1594. [DOI] [PubMed] [Google Scholar]
- 65. Crabtree G.W., Park A.J., Gordon J.A., Gogos J.A. (2016) Cytosolic accumulation of L-proline disrupts GABA-ergic transmission through GAD blockade. Cell Rep., 17, 570–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Willis A., Bender H.U., Steel G., Valle D. (2008) PRODH variants and risk for schizophrenia. Amino Acids, 35, 673–679. [DOI] [PubMed] [Google Scholar]
- 67. Clelland C.L., Read L.L., Baraldi A.N., Bart C.P., Pappas C.A., Panek L.J., Nadrich R.H., Clelland J.D. (2011) Evidence for association of hyperprolinemia with schizophrenia and a measure of clinical outcome. Schizophr. Res., 131, 139–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Jacquet H., Demily C., Houy E., Hecketsweiler B., Bou J., Raux G., Lerond J., Allio G., Haouzir S., Tillaux A.. et al. (2005) Hyperprolinemia is a risk factor for schizoaffective disorder. Mol. Psychiatry, 10, 479–485. [DOI] [PubMed] [Google Scholar]
- 69. Clelland C.L., Drouet V., Rilett K.C., Smeed J.A., Nadrich R.H., Rajparia A., Read L.L., Clelland J.D. (2016) Evidence that COMT genotype and proline interact on negative-symptom outcomes in schizophrenia and bipolar disorder. Transl. Psychiatry, 6, e891.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ghasemvand F., Omidinia E., Salehi Z., Rahmanzadeh S. (2015) Relationship between polymorphisms in the proline dehydrogenase gene and schizophrenia risk. Genet. Mol. Res., 14, 11681–11691. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The .cel and genotype files from Affymetrix 6.0 arrays have been deposited to NCBI dbGAP phs001339.v1.p1.