

Abstract
Lafora disease (LD) is a fatal form of progressive myoclonus epilepsy characterized by the accumulation of insoluble poorly branched glycogen-like inclusions named Lafora bodies (LBs) in the brain and peripheral tissues. In the brain, since its first discovery in 1911, it was assumed that these glycogen inclusions were only present in affected neurons. Mouse models of LD have been obtained recently, and we and others have been able to report the accumulation of glycogen inclusions in the brain of LD animals, what recapitulates the hallmark of the disease. In this work we present evidence indicating that, although in mouse models of LD glycogen inclusions co-localize with neurons, as originally established, most of them co-localize with astrocytic markers such as glial fibrillary acidic protein (GFAP) and glutamine synthase. In addition, we have observed that primary cultures of astrocytes from LD mouse models accumulate higher levels of glycogen than controls. These results suggest that astrocytes may play a crucial role in the pathophysiology of Lafora disease, as the accumulation of glycogen inclusions in these cells may affect their regular functionality leading them to a possible neuronal dysfunction.
Introduction
Glycogen is a glucose polymer that constitutes the major form of glucose storage in the body. Glycogen deposits offer several advantages: they have low osmotic activity, their synthesis and degradation is very well regulated and they generate Glu-1P without the use of ATP (see 1 for a review). For a long time, brain glycogen has been considered a simple source of glucose in the case of energy demand: glycogen breakdown (glycogenolysis) produces eventually pyruvate which can enter the TCA cycle or be converted into lactate. According to the astrocyte-neuron lactate shuttle (ANLS) hypothesis, astrocyte lactate is transferred to neurons to maintain neuronal metabolism (2). However, recent reports indicate that glycogenolysis plays key additional roles in brain physiology as in long-term potentiation and consolidation of memory (3–5).
Historically, the localization of glycogen in the different brain areas and in cellular neural types has been difficult since acute dissection of brain decreases rapidly glycogen levels, most likely due to the triggering of glycogenolysis as a result of the activation of anaerobic metabolism during the hypoxia state (6,7). In spite of these difficulties, it was proposed that astrocytes were the main neural cellular type that accumulated glycogen (8–13). This assumption has been confirmed recently when a technique that preserves the endogenous levels of glycogen was developed (focused microwave irradiation plus immunodetection with special anti-glycogen antibodies) (14). These authors described that glycogen got accumulated in astrocytes that were mainly distributed in hippocampus, cerebral cortex, striatum and molecular layer of cerebellum (14). Interestingly enough, these areas correspond to those that display the highest metabolic demand due to the highest synaptic activity. It was also indicated that glycogen particles localized in astrocytes that were placed in the vicinity of axonal boutons and suggested that these glycogen stores could be used directly in that area upon energetic demand (11,14).
Lafora progressive myoclonus epilepsy (LD, OMIM#254780) is a fatal neurological disorder characterized by the accumulation of insoluble poorly branched forms of glycogen (polyglucosan inclusions) in brain and other peripheral tissues. No treatment has been established yet for this devastating disease which leads to the death of the patients around 10 years after the onset of the first symptoms (see 15 for a review). LD was described by the Spanish neurologist Gonzalo Rodriguez Lafora in 1911 (16). He depicted the accumulation of what he called ‘amyloid inclusions’ inside the neurons of affected patients (named on his behalf, Lafora bodies, LBs). These inclusions stain positive with the periodic acid Schiff (PAS) reagent, indicating that they are polysaccharides. In fact the staining of axillar samples with this reagent has been used as a diagnostic test of the disease (15). LD is due to mutations in two main genes: EPM2A, which encodes laforin, a glucan phosphatase, and EPM2B, encoding malin, an E3-ubiquitin ligase of the RING type (17–19). The fact that laforin and malin form a functional complex and the fact that patients carrying mutations in either gene are phenotypically indistinguishable indicates that the two proteins participate in similar physiological pathways.
In order to get a deep insight into the pathogenesis of LD, mouse models of the disease have been produced that recapitulate most of the features of this disease (20,21). For example, both Epm2a-/- and Epm2b-/- mice accumulate insoluble polyglucosans in their brain, assessed by the PAS+ staining. In all these studies, it was assumed that the PAS+ inclusions were located into neurons, as no indication about the accumulation of LBs in cells other than neurons in the central nervous system has been described in the literature (15). However, in this work we present strong evidence indicating that most of the glycogen inclusions present in the brain of LD mice co-localize actually with astrocytic markers (glial fibrillary acidic protein, GFAP, and glutamine synthase, GlnS). We then suggest that the accumulation of these glycogen inclusions in astrocytes may affect their functionality, what could lead to neuronal dysfunction.
Results
Most of the glycogen inclusions present in the brain of Epm2b-/- mice co-localize with astrocytic markers
The hallmark of Lafora disease (LD) is the accumulation of insoluble poorly branched glycogen-like inclusions named LBs (see 15 for a review). Mouse models of LD recapitulate this hallmark (20,21). In agreement with these observations, in Figure 1 we show an immunofluorescence analysis of brain sections of control and Epm2b-/- animals of 12 months of age using a specific antibody that recognizes glycogen aggregates (22). Glycogen inclusions were clearly observed in different areas of the brain (i.e. hippocampus, cerebellum) of Epm2b-/- mice, whereas they were absent in similar preparations from control animals of the same age. These results were similar to those traditionally obtained using the PAS staining (20,21), an histochemistry method that detects polysaccharide structures. The immunodetection of glycogen inclusions remained after treating the samples with α-amylase (Fig. 1, bottom panels), indicating that, as already described in the literature (20,21), these structures were resistant to the action of this enzyme. Therefore, Epm2b-/- mice accumulated glycogen inclusions in the brain that were immunoreactive to specific anti-glycogen antibodies and, at the age of 12 months, they were resistant to the action of α-amylase.
Figure 1.

Immunohistofluorescence analyses of glycogen inclusions in the brain of control and Epm2b-/- mice of 12 months of age. Lateral sections of hippocampus and cerebellum of three independent mice of each type were incubated with anti-glycogen antibody and a secondary anti-IgM AlexaFluor (red) 633 conjugated antibody. Sections were then stained with DAPI to visualize nuclei. Images were acquired with a Leica DM RXA2 microscope. Representative images are presented. Scale bars: 100 µm. Bottom panels: sections from Epm2b-/- mice were pretreated with α-amylase as described in Materials and Methods section, and then analyzed by IHF as above. DG, hippocampus dentate gyrus; GL, cerebellar granular layer.
Then, we sought to determine which of the different neural cells accumulated these glycogen inclusions. With this aim we performed immuno co-localization experiments using antibodies that were specific either for neurons (anti-Tuj1, which recognizes neuronal class III β-tubulin, a component of the neuronal cytoskeleton; and anti-NeuN, which recognizes the DNA-binding neuron-specific protein NeuN, which is present in neuronal nuclei, perikarya and some proximal neuronal processes), astrocytes [anti-glutamine synthetase (anti-GlnS), which recognizes a cytosolic enzyme present is astrocytes but not in neurons, and anti-glial fibrillary acidic protein (anti-GFAP), which recognizes a component of the cytoskeleton present in astrocytes but not in neurons], or microglia (anti-Iba1, which recognizes a cytosolic ionized calcium-binding adapter molecule 1, not present in astrocytes or neurons). As shown in Figure 2, the anti-Tuj1 antibody stained neuronal cells in the different tested areas (hippocampus, cerebellum and pontine), some of which presented a glycogen inclusion in the soma (pointed with a white arrow), as it was expected according to the initial findings of LD (16). However, we observed that most of the glycogen inclusions were present in areas that were not stained with the anti-Tuj1 antibody, suggesting an alternative localization. In order to confirm these results we used an alternative specific neuronal marker, anti-NeuN antibody (see Supplementary Material, Fig. S1). Again, we observed that most of the glycogen inclusions were absent from cell structures stained with this marker. These results suggested that most of the glycogen inclusions present in the brain of Epm2b-/- mice were located in structures different from neurons.
Figure 2.

Immuno co-localization experiments using anti-Tuj1 and anti-glycogen antibodies. Lateral sections of hippocampus, cerebellum and pontine from three independent Epm2b-/- mice of 12 months of age were immunodetected using a combination of anti-Tuj1 (neuronal marker) and anti-glycogen antibodies, and secondary anti-rabbit AlexaFluor (green) 488 and anti-IgM AlexaFluor (red) 633 conjugated antibodies, respectively. Sections were then stained with DAPI to visualize nuclei. Images were acquired with a Leica DM RXA2 microscope. Representative images are presented. Scale bars: 100 µm and 10 μm. CA1, hippocampus cornus ammonis 1; GL: cerebellar granular layer; PC, Purkinje cells. White arrows indicate neuronal cells containing glycogen inclusions.
Next, we performed immuno co-localization experiments using specific astrocytic markers (anti-GlnS and anti-GFAP). As observed in Figure 3A, most of the glycogen inclusions co-localized with the anti-GlnS marker, both in the hippocampus and the cerebellum (Supplementary Material, Fig. S2A). A magnification of these cells using confocal microscopy (Fig. 3B and Supplementary Material, Fig. S2B) indicated that glycogen inclusions were surrounded by GlnS+ structures, which suggests that they were inside the astrocytes. We also noticed that glycogen inclusions were more abundant in astrocytic processes, being less abundant in the soma of the astrocyte (Fig. 3A and B). In the same way, glycogen inclusions co-localized with GFAP+ cell structures (Fig. 3C and D) in the hippocampus. In this case, the number of glycogen + GFAP + cells was less abundant than in the case of glycogen + GlnS + cells (see above), perhaps because anti-GFAP antibody stains mainly reactive astrocytes, whereas anti-GlnS antibody stains all types of astrocytes. Confocal analysis confirmed the presence of glycogen inclusions inside the astrocytes, being more abundant in astrocytic processes than in the soma (Fig. 3D). Similar results were obtained when we analyzed the cerebellum (Supplementary Material, Fig. S2C and D).
Figure 3.

Immuno co-localization experiments using anti-GlnS or anti-GFAP and anti-glycogen antibodies. Lateral sections of hippocampus from three independent Epm2b-/- mice of 12 months of age were immunodetected using a combination of anti-GlnS (astrocytic marker) (A) or anti-GFAP (astrocytic marker) (C), anti-glycogen antibodies, and secondary anti-rabbit AlexaFluor (green) 488 and anti-IgM AlexaFluor (red) 633 conjugated antibodies, respectively. Sections were then stained with DAPI to visualize nuclei. Images were acquired with a Leica DM RXA2 microscope. Representative images are presented. Scale bars: 10 μm. CA1, hippocampus cornus ammonis 1; DG, hippocampus dentate gyrus. White arrows indicate examples of astrocytes containing glycogen inclusions. Images were also acquired from similar preparations using a Leica TCS SP8 confocal microscope (B, anti-GlnS; D, anti-GFAP). Four consecutive images from the Z-stacks are shown.
Finally, we performed immuno co-localization experiments using a specific microglia marker (anti-Iba1). In the hippocampus, although Iba1+ cells contained glycogen inclusions (Fig. 4A and B), most of these polysaccharide structures did not co-localize with the microglia marker (Fig. 4A). Similar results were observed in the cerebellum (Supplementary Material, Fig. S3A and B).
Figure 4.
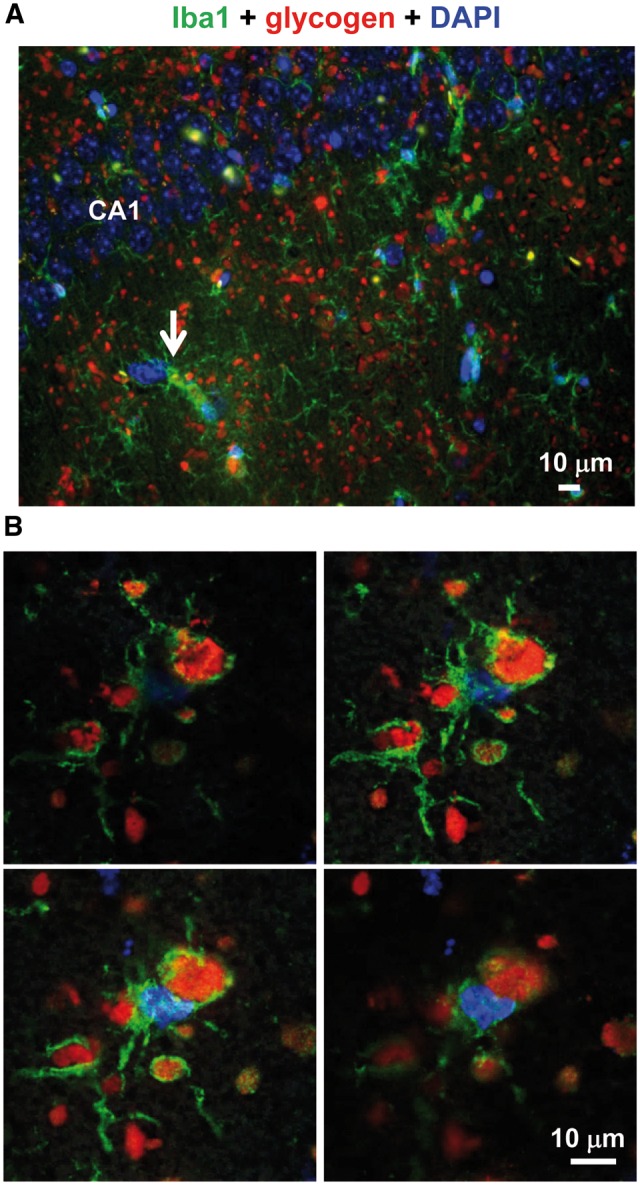
Immuno co-localization experiments using anti-Iba1 and anti-glycogen antibodies. (A) Lateral sections of hippocampus from three independent Epm2b-/- mice of 12 months of age were immunodetected using a combination of anti-Iba1 (microglia marker) and anti-glycogen antibodies, and secondary AlexaFluor (green) 488 and anti-IgM AlexaFluor (red) 633 conjugated antibodies, respectively. Sections were then stained with DAPI to visualize nuclei. Images were acquired with a Leica DM RXA2 microscope. Representative images are presented. Scale bars: 10 μm. CA1, hippocampus cornus ammonis 1. White arrows indicate examples of microglia containing glycogen inclusions. (B) Images were also acquired from similar preparations using a Leica TCS SP8 confocal microscope. Four consecutive images from the Z-stacks are shown.
We repeated the analyses in Epm2a-/- mice lacking laforin (Supplementary Material, Fig. S4). Again, most of the glycogen inclusions co-localized with astrocytic markers but not with the neuronal markers. All these results indicate that the majority of the glycogen inclusions present in the brain of Epm2a-/- and Epm2b-/- mice were located in astrocytes, and only a minor number of glycogen inclusions were present in neuronal cells and microglia. We quantified the presence of glycogen inclusions in neurons and astrocytes and observed that, in both mouse models of LD, only 4% of neurons contained these inclusions, whereas 99% of astrocytes contained this material.
In order to know whether the accumulation of glycogen in astrocytes was a late event in the evolution of the disease (all the experiments presented so far were performed in animals of 12 months of age), we repeated the same experiments in animals that were only 3 months of age. As shown in Supplementary Material, Figure S5, brain sections from Epm2b-/- mice at this age already contained glycogen inclusions although they were less numerous than in the case of older animals (compare Supplementary Material, Fig. S5 and Fig. 1). Most importantly, the majority of the glycogen inclusions present at 3 months of age were sensitive to α-amylase treatment (Supplementary Material, Fig. S5, bottom panel), indicating probably that their structure was not as complex as in the inclusions present in older animals. Then, we performed similar immuno co-localization experiments as above and observed similar results: most of the glycogen inclusions did not co-localize with neuronal markers [anti-Tuj1 (Supplementary Material, Fig. S6A) and anti-NeuN (Supplementary Material, Fig. S6B)], whereas they co-localized with astrocytic markers [anti-GlnS (Supplementary Material, Fig. S7A) and anti-GFAP (Supplementary Material, Fig. S7C)]. Confocal analyses of the samples confirmed that glycogen inclusions were located inside the astrocytes (Supplementary Material, Fig. S7B and D). All these results indicated that the accumulation of glycogen in astrocytes occurs at early stages of the disease, at least when the animals are 3 months old.
Primary astrocytes from mouse models of LD accumulate higher levels of glycogen, but they are able to degrade the polysaccharide upon a nutritional stress via glycogen phosphorylase
Since the results presented above suggested a role of astrocytes in accumulating glycogen inclusions, we obtained astrocytes from cortex and cerebellum from control, Epm2a-/- and Epm2b-/- mice and cultured them in a medium containing high levels of glucose (25 mm) for 24 h. Then, we collected the cells and measured the levels of accumulated glycogen. Primary cortical astrocytes from LD mice of 7 days of age accumulated significantly more glycogen than astrocytes from control mice (Fig. 5A) (similar results were obtained from cerebellar astrocytes; Supplementary Material, Fig. S8A). As glycogen levels are in a dynamic equilibrium between synthesis and degradation (1,23), this result could indicate that astrocytes from LD mice have an increased synthesis of the polysaccharide or, alternatively, that its degradation is partially impaired. To solve this question, cortical astrocytes growing in high glucose (25 mm) were shifted to a medium without glucose for different periods of time (6 and 24 h) and then, we measured the levels of glycogen in each case. In Figure 5B we show that glucose starvation induced the degradation of glycogen in all the cases and that the rate of degradation was similar in astrocytes from LD and control mice (similar results were obtained from cerebellar astrocytes; Supplementary Material, Fig. S8B). These results suggest that astrocytes from LD mice have functional machinery for glycogen degradation upon a nutritional stress condition.
Figure 5.

Primary cortical astrocytes from mouse models of LD accumulate higher levels of glycogen. (A) Primary cultures of cortical astrocytes from 7 days old control, Epm2a-/- and Epm2b-/- mice were grown in high glucose containing medium (25 mm glucose) for 24 h. Then, glycogen content was measured as described in Materials and Methods section. (B) Primary cultures of astrocytes as above were grown in high glucose conditions for 24 h. Then, they were shifted to a medium without added glucose for 6 and 24 h and glycogen content was measured as described in Materials and Methods section. (C) Primary cultures of astrocytes as above were grown in high glucose conditions for 24 h. Then, they were pretreated or not with 300 μm DAB or a combination of 20 mm NH4Cl and 100 μm leupeptine for 1 extra hour. Then, cells were shifted to a medium without added glucose containing or not the corresponding inhibitor for 6 h. Next, glycogen content was measured as described in Materials and Methods section. Results expressed are means of at least three independent batches of astrocytes in each genotype. In (B) and (C), the results are normalized to the levels of glycogen present in the respective basal conditions (high glucose; dotted line). Bars indicate standard deviation. In (A), significant differences between the LD astrocytes and control are indicated (**P< 0.01, ***P< 0.001). In (B) and (C), significant differences between the levels of glycogen respect to basal conditions (high glucose) are indicated (**P< 0.01, ***P< 0.001).
It has been described that two main pathways are involved in the degradation of glycogen: glycogen phosphorylase and lysosomal degradation (glycophagy) (24,25). In order to know which pathway was responsible for the degradation of glycogen upon nutritional stress, cells growing in high glucose (25 mm) were pretreated for 1 h with an inhibitor of glycogen phosphorylase (300 μm DAB; 1,4-dideoxy 1,4 iminoarabinitol) (26) or with a combination of ammonium chloride and leupeptine (20 mm NH4Cl plus 100 μm leupeptine) to inhibit lysosomal function (27) and then shifted to a media without glucose in the presence of the corresponding inhibitor. After 6 h, cells were recovered and the levels of glycogen measured. As shown in Figure 5C, the presence of 300 μm DAB prevented the degradation of glycogen in astrocytes from control and Epm2a-/- mice. However, this inhibitor had a poor effect on the degradation of glycogen in astrocytes from Epm2b-/- mice. On the contrary, when the cells were grown in the presence of the lysosomal inhibitors, the degradation of glycogen was enhanced in comparison to the levels obtained in cells subjected to nutritional stress but without lysosomal inhibitors. These results indicate that the blockage of lysosomal function does not prevent glycogen degradation upon nutritional stress. The enhanced degradation of glycogen under these conditions could be a compensatory mechanism to produce energy since it has been described that ammonium is able to induce mitochondrial dysfunction (28). Thus, the only way to obtain energy is the up-regulation of glycogenolysis and glycolysis.
Taking all these results together, we suggest that primary astrocytes from 7 days old LD mice accumulate higher levels of glycogen than control cells when they are growing in high glucose conditions. However, this glycogen is degradable in all cases upon nutritional stress, glycogen phosphorylase playing a major role in this induced degradation.
In order to know why astrocytes from LD mice accumulate higher levels of glycogen than control cells we analyzed by western blotting key enzymes in glycogen homeostasis (Fig. 6). The total levels of glycogen synthase (GS) were similar in all the cells (control and LD mice astrocytes) in all the conditions, and the phosphorylation status of this enzyme was regulated similarly in control and LD astrocytes (the presence of faster mobility bands, related to un-phosphorylated forms of the protein, were present in samples obtained from astrocytes growing without glucose and without glucose + lysosome inhibitors, what was in agreement with a tendency to lower levels of pSer641 forms of GS). No major differences were observed in the total levels of glycogen phosphorylase, and similar significant levels of phosphorylated forms (related to active forms of the enzyme) were present in growth conditions without glucose + DAB and without glucose + lysosome inhibitors, in the three different types of astrocytes. No changes in the levels of GS kinase 3 or in its phosphorylation status were also observed in any case. The levels of phosphorylated AMPK showed a tendency to increase upon glucose starvation, but no differences were observed among the different types of astrocytes. Finally, the levels of p62, an autophagy marker, showed a tendency to increase upon glucose starvation, being this increase statistically significant upon addition of lysosome inhibitors, but no differences were observed among the different type of astrocytes. Therefore, we observed no major changes in the levels of key proteins involved in glycogen biosynthesis that could explain the higher levels of glycogen present in astrocytes from LD mice.
Figure 6.

Western blot analyses of primary cortical astrocytes grown under the conditions described in the legend of Figure 5. Crude extracts (30 μg) from primary cultures of cortical astrocytes treated as described in Figure 5 were analyzed by western blotting using appropriated antibodies (see Materials and Methods section). C, control; L,: Epm2a-/-; M, Epm2b-/- astrocytes. Molecular weights are indicated in the left. A representative blot of the analysis of at least three independent batches of astrocytes is presented. The intensity of the corresponding bands was measured using the ImageJ software (version 2.0.0). Diagrams of the intensities of the different proteins are presented. Fold change values are means of the different measurements, bars indicate standard deviation and significant differences between the levels of the proteins with respect to basal conditions (high glucose) in each genotype are indicated (#P< 0.05, $P< 0.05, &P< 0.05, for control, Epm2a-/- and Epm2b-/-, respectively). HG, high glucose; NG, no glucose added; NG DAB, no glucose added plus 300 μm DAB; NG N/L, no glucose added plus 20 mm NH4Cl and 100 μm leupeptine.
As glycogen synthesis is enhanced if the levels of glycolytic intermediates such as Glu-6P increase, any process that could affect the levels of these intermediates such as impairment in the mitochondrial respiratory capacity could lead to glycogen production. For this reason we checked this parameter in astrocytes growing in high glucose (25 mm) and then shifted to a media without glucose for 12 h, using the Oxygraph-2K equipment. However, we did not find major differences in any analyzed respiratory parameters [basal, ATP-linked, proton leak, maximal OCR (oxygen consumption rate, OCR), reserve respiratory capacity and non-mitochondrial respiration (now named as residual oxygen consumption)], among the different tested astrocytes growing in either high or without glucose conditions (Fig. 7). Therefore, the higher glycogen levels present in LD astrocytes were not a consequence of altered respiration profile of these cells.
Figure 7.

Analysis of the respiratory capacity of primary cultures of cortical astrocytes grown in high glucose (25 mm glucose) or shifted to a medium without glucose for 12 h. The recorded parameters are: initial respiration (basal), coupling efficiency (ATP linked), proton leak, maximal respiratory rate, reserve capacity and non-mitochondrial respiration (now named as residual oxygen consumption). They were calculated as described in Materials and Methods section. Results, normalized to mtDNA content, are expressed as means of at least three independent batches of astrocytes in each genotype. Bars indicate standard deviation.
Discussion
Glycogen is an excellent substrate to provide a rapid way to obtain energy under the conditions of metabolic demand. In the brain, glycogen is mainly located in astrocytes (8–13), although a recent report indicates that neurons have also an active glycogen turnover (29). One of the difficulties in studying glycogen homeostasis in the brain is the fact that it is rapidly removed under the conditions of stress, so the access to the brain has to be very fast to avoid glycogen degradation (6,7). However, a recent report describes the use of the focused microwave irradiation technique to stop glycogen degradation, allowing in this way a more reliable and quantitative measurement of the levels of glycogen in the brain (14). These authors confirmed that astrocytes are the main cells that accumulate glycogen in the brain since they found that only cells expressing GFAP (an astrocyte marker) contained glycogen particles (14). In addition, they reported that the distribution of glycogen inside astrocytes is not uniform. In fact, glycogen deposits are more abundant in fine astrocytic processes that are close to synapses, being less abundant in the soma of these cells (11,12,14). Perhaps this uneven distribution of glycogen allows a rapid access to energy in areas of high synaptic activity.
Lafora disease is a fatal form of progressive myoclonus epilepsy characterized by the accumulation of insoluble poorly branched glycogen-like inclusions named LBs (see 15 for a review). Using a regular technique to access to the brain from control and LD mice, we and others have been able to report an accumulation of glycogen inclusions in the brain of LD animals, in comparison to controls (see Fig. 1 as an example). Traditionally, the presence of these inclusions has been demonstrated by using the PAS technique. However, in our hands the immunohistofluorescence (IHF) technique using appropriated anti-glycogen antibodies is more sensitive that the PAS technique and perhaps for this reason, we detected more small glycogen inclusions. Nevertheless, even using the IHF technique we were not able to detect glycogen deposits in the brain from control mice, what may suggest that during the processing of the samples it gets degraded as indicated above. Therefore, the glycogen inclusions we detect in brains from LD mice could be either due to an initially higher amount of glycogen than in controls (so after a partial degradation of the polysaccharide, there is still some glycogen remaining) and/or to the fact that the glycogen inclusions present in the brain of LD mice are less competent to be degraded. In any case, the most important evidence we present in this work is that astrocytes are the main neural cells that accumulate glycogen inclusions in mouse models of LD. Most of these inclusions co-localize with astrocytic markers such as GFAP and GlnS and they are mainly located in processes rather than in the astrocytic soma. We would like to point out the strong similarities between our results and those presented by the Hirase laboratory, both in the definition of astrocytes as the main cell type that accumulate glycogen in the brain and also in the subcellular localization of glycogen deposits (see Fig. 4 in 14). In addition to the main astrocytic location, we also found that some neurons accumulated glycogen inclusions, a fact recognized as a hallmark of LD. However, the number of neurons containing these glycogen inclusions was minor in comparison to the huge number of glycogen-containing astrocytes.
A confirmation of the higher accumulation of glycogen in LD astrocytes came from the observation that in primary cultures of astrocytes coming from animals of only 7 days of age, we detected higher levels of glycogen in LD astrocytes than in control cells. Interestingly, the accumulated glycogen under these conditions was fully degradable under the conditions of nutritional stress, mainly by glycogen phosphorylase. This may indicate that LD astrocytes accumulate initially a form of degradable glycogen which, with age, becomes less competent to be degraded. In fact, in this work we present data indicating that glycogen inclusions present in LD mice of 3 months of age is partially degradable by α-amylase. On the contrary, glycogen inclusions present in LD animals of 12 months of age become resistant to the action of α-amylase, probably because of the poorer branching of the glycogen species that make them insoluble (30).
In our opinion, the results presented in this work open a new way to consider the pathogenicity of LD. The huge difference in the amount of glycogen inclusions present in astrocytes versus neurons suggests a prominent role of astrocytes in LD. In agreement with this suggestion, we have recently reported that brain from Epm2b-/- mice shows a progressive accumulation of reactive astrocytes and microglia, and higher levels of pro-inflammatory markers (TNFα, IL-6, IL-1β, CXC10, etc.) (31). Here, we also present evidence of the presence of glycogen inclusions in microglia. Perhaps this presence is due to microglial phagocytosis of astrocytes or neurons. We suggest that the accumulation of glycogen inclusions in astrocytes and microglia may alter the regular functionality of these two cell types, which may contribute to the pathogenesis of the disease.
Our results provide the experimental evidence to an early hypothesis which suggested that in LD, as in other disorders that accumulate glycogen inclusions (i.e. adult polyglucosan body disease), polyglucosans should appear first in astrocytes and later on in neurons (32,33). These authors also suggested that, as polyglucosan inclusions are not metabolizable, this could produce alterations in K+ and glutamate uptake, since the energy that maintains these processes comes mainly from glycogenolysis (32–34). Recently, we have also provided evidence to sustain this hypothesis. We demonstrated that the homeostasis of the astrocytic glutamate transporter GLT-1 is affected in primary cultures of astrocytes from LD mice (35). Perhaps, the accumulation of glycogen inclusions in LD astrocytes may affect its general metabolic properties and this could result in changes in the function of GLT-1 transporter. This is particularly interesting since alterations in the functionality of the glutamate transporter are associated with excitotoxicity, which may induce epilepsy, one of the traits of LD (36). Thus, our results indicate that astrocytes may play a major role in the pathophysiology of LD.
Materials and Methods
Ethic statement, animal care, mice and husbandry
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the Consejo Superior de Investigaciones Cientificas (CSIC, Spain) and approved by the Consellería de Agricultura, Medio Ambiente, Cambio Climático y Desarrollo Rural from the Generalitat Valenciana. All mouse procedures were approved by the animal committee of the Instituto de Biomedicina de Valencia-CSIC [Permit Number: INTRA12 (IBV-14), 2015/VSC/PEA/00029]. All efforts were made to minimize animal suffering. To eliminate the effect of differences in the genetic background of the animals, we backcrossed Epm2a-/- and Epm2b-/- mice (with a mixed background 129sv: C57BL/6) as described previously (21,37) with control C57BL/6JRccHsd mice obtained from Harlan laboratories (Barcelona, Spain) for 10 generations to obtain homozygous Epm2a-/- and Epm2b-/- in a pure background. Mice were maintained in the IBV-CSIC facility on a 12/12 light/dark cycle under constant temperature (23°C) with food and water provided ad libitum.
Male mice of 3 and 12 months of age were sacrificed by cervical dislocation. Brain was recovered and fixed in 4% paraformaldehyde in phosphate buffered saline (PBS) for immunohistochemical analyses. To obtain primary cultures of astrocytes, mice of 7 days of age were decapitated and either both cortical hemispheres or the cerebellum were isolated under aseptic conditions, pressed through a 90 μm pore nylon mesh and immersed in modified DMEM medium.
Obtaining primary cultures of astrocytes and treatments
Primary cultures of cortical and cerebellar astrocytes from control, Epm2a-/- and Epm2b-/- mice of 7 days of age were obtained as in (38,39). Cells were grown in Dulbecco’s modified eagle medium (Lonza, Barcelona, Spain) containing 20% of inactivated fetal bovine serum (FBS), supplemented with 1% l-glutamine, 6 mm glucose, 100 units/ml penicillin and 100 μg/ml streptomycin, in a humidified atmosphere at 37°C with 5% CO2. After the first week, FBS was reduced to 15%. In the third week, FBS was reduced to 10% and 0.25 mm dibutyryl-cAMP (dbcAMP) was added to the cultures (38). Cell cultures were used for experiments after 3 weeks in vitro. For the glycogen analysis, astrocytes were grown in 25 mm glucose containing medium for 24 h in the absence of dbcAMP since it influences glycogen turnover (40). The purity of astrocytes was confirmed by immunofluorescence using anti-GFAP (1/500, Abcam ab7260), anti-NeuN (1/200, Millipore ABN78) and anti-Iba1 (1/200; Waco 019–19741).
Immunohistofluorescence (IHF) analyses
Dehydrated tissues were embedded in paraffin and sectioned at 4 µm. Sections were deparaffined, rehydrated, incubated for 40 min in sodium borohydrate (1 mg/ml, in PBS) to lower background and warmed at 95°C for 20 min in 10 mm citrate buffer (pH 6.0) for antigen retrieval. When indicated, samples were treated with 1 mg/ml α-amylase (Sigma A3176) in PBS during 1 h at 37°C; then they were washed three times (10 min) with PBS. Sections were immersed in blocking buffer (1% BSA, 10% FBS, 0.2% Triton X100, in PBS) and incubated O/N at 4°C with primary antibody diluted in blocking buffer: anti-glycogen (1/500; a generous gift from Dr Otto Baba; Tokyo Medical and Dental University, Tokyo, Japan), anti-Tuj1 (1/500, Sigma T2200), anti-NeuN (1/200, Millipore ABN78), anti-GlnS (1/200, Abcam ab73593), anti-GFAP (1/500, Abcam ab7260) and anti-Iba1 (1/200, Waco 019-19741). After three washes of 10 min in PBS, sections were incubated for 1 h at room temperature with the appropriate secondary antibody diluted at 1/500 in blocking buffer without Triton X-100, washed once with PBS, incubated with DAPI (Sigma, Madrid, Spain), washed twice with PBS and mounted in AquaPolymount (Polysciences Inc., USA). Images were acquired either with a vertical Leica DM RXA2 microscope with a Leica DFC350FX high sensitivity monochrome camera, or with a Confocal Spectral Leica TCS SP8 microscope (Leica, Wetzlar, Germany). Images were treated with the ImageJ 1.43c software (Wayne Rasband, National Institutes of Health, Bethesda, MD). In order to quantify the number of cells containing glycogen inclusions, we counted a total of 100 cells that were stained with the appropriated cellular marker (Tuj1 for neurons; GFAP and Gln synthase for astrocytes) from several images, from three independent mice of each genotype.
Determination of glycogen levels
Cells were scraped on ice into 30% KOH and then heated at 100°C for 15 min. The quantitative amounts of glycogen were assessed in a coupled enzymatic assay by measuring the production of NADPH in the conversion of glucose-6-phosphate to 6-phosphogluconolactone as described in (41,42). The reaction was initiated by the addition of hexokinase and glucose-6-phosphate dehydrogenase to final concentrations of 1.52 U/ml and 0.54 U/ml, respectively. The fluorescence was measured after 30 min of incubation at 37°C using 340 and 460 nm as excitation and emission wavelengths, respectively, and expressed as nmol of glucose per mg of protein.
Western blotting
Crude extracts from primary culture of astrocytes were obtained in lysis buffer [25 mm Tris–HCl, pH 7.5; 150 mm NaCl; 15 mm EDTA, pH 8; 0.6 m sucrose; 50 mm NaF; 15 mm Na4P2O7; 1 mm PMSF; 15 mm 2-mercaptoethanol and complete protease inhibitor cocktail (Roche, Spain)]. A total of 30 µg protein was subjected to SDS-PAGE, transferred into PVDF membrane and revealed with the appropriated antibodies: anti-pS15-glycogen phosphorylase (1/1000, University of Dundee), anti-glycogen phosphorylase (1/1000, Abcam ab88078), anti-pS641-GS (1/1000, Cell Signaling #3891), anti-GS (1/1000, Abcam ab40810), anti-pS9-GS kinase (1/1000, Cell Signaling #9336), anti-GS kinase (1/2000, Cell Signaling #9315), anti-pT172-AMPKα (1/1000, Cell Signaling #2535), anti-AMPKb1/b2 (1/1000, Cell Signaling #4150), anti-p62 (1/3000, Abcam ab56416) and anti-GAPDH (1/5000, Santa Cruz Biotechnologies sc32233). Primary antibodies were incubated O/N at 4°C. Then membranes were incubated with the corresponding secondary antibody [anti-sheep-HRP for the pGP-S15; goat anti-mouse IRDye 680LT (LI-COR Biosciences, Germany) or goat anti-rabbit IRDye 800CW (LI-COR Biosciences, Germany)] for 1 h at room temperature. Images obtained either with a FujiLAS4000 using ECL or ECL-Prime (Amersham Biosciences, GR-Healthcare) or with the Odyssey Infrared Imaging System (LI-COR Biosciences, Germany). The results were analyzed using the software Image Studio version 3.1 (LI-COR Biosciences, Germany).
High-resolution respirometry in intact cells using Oxygraph-2K (Oroboros)
OCR in astrocytes was measured using a high-resolution respirometer (Oxygraph-2k, Oroboros Instruments, Innsbruck, Austria), as described in (43). In brief, 80% confluent primary astrocytes obtained from control, Epm2a-/- and Epm2b-/- mice of 7 days of age, were detached at 37°C with trypsin-EDTA, resuspended in fresh growth media without antibiotics (DMEM with or without glucose) and analyzed in 2 ml Oxygraph chambers. A real-time OCR measurement was performed at 37°C in each chamber at basal conditions and after sequential addition of inhibitors for different mitochondrial respiratory complexes: oligomycin (2.5 µg/ml) to inhibit complex V (to assess the non-phosphorylating resting state or leak respiration), carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (CCCP) uncoupler, with stepwise titration from 5 to 0.5 μm (to assess maximal electron transport system respiratory capacity rate), rotenone (0.5 µm) to inhibit complex I and antimycin A (2.5 µm) to inhibit complex III. Data was analyzed using DatLab6 software (Oroboros, Austria). The recorded parameters were: initial respiration rate (basal), non-mitochondrial respiration [now named as residual oxygen consumption (Rox); it refers to the respiration rate obtained under rotenone and antimycin A treatment], proton leak (corresponding to respiration under oligomycin inhibition minus Rox) and maximal respiratory rate (respiration under CCCP treatment minus Rox). Coupling efficiency (ATP linked) was evaluated as the difference between initial respiration rate and respiration under oligomycin inhibition (both after Rox subtraction). Reserve capacity was defined as the difference between maximal respiration rate and coupling efficiency. The relative copy number of mitochondrial DNA (mtDNA) per diploid nuclear genome was measured by real-time quantitative PCR analysis. For each sample analyzed, two independent PCR reactions were set up to amplify the cytochrome b mtDNA and the ApoB nuclear DNA targets. The primer sets used were: Cytb-forward: 5′-GCTTTCCACTTCATCTTACCATTTA-3′; Cytb-reverse: 5′-TGTTGGGTTGTTTGATCCTG-3′; ApoB-forward: 5′-CGTGGGCTCCAGCATTCTA-3′; ApoB-reverse: 5′-TCACCAGTCATTTCTGCCTTTG-3′. The copy number ratio of mtDNA to ApoB gene (mtDNA/ApoB) was calculated using delta Ct method, and referred as mtDNA content. Fold changes in relative mtDNA copy number refer to relative mtDNA copy number of control animals, which was given a value of 1.0, as in (44,45).
Statistical analysis
The data were expressed as mean ± standard deviation. Statistical significance of differences between the groups was evaluated by a paired Student’s t-test with two-tailed distribution. The significance has been considered at *P < 0.05, **P < 0.01, ***P < 0.001, as indicated in each case.
Supplementary Material
Supplementary Material is available at HMG online.
Supplementary Material
Acknowledgements
We want to thank Dr Otto Baba for the generous gift of the anti-glycogen antibody.
Conflict of Interest statement. None declared.
Funding
This work was supported by grants from the Spanish Ministry of Economy and Competitiveness SAF2014–54604-C3–1-R, a grant from Generalitat Valenciana (PrometeoII/2014/029), a grant form Fundación Ramón Areces (XVIII Concurso Nacional Ayudas Investigacion Ciencias Vida y Materia) and a grant from the National Institute of Health (NIH-NINDS) P01NS097197, which established the Lafora Epilepsy Cure Initiative (LECI), to PS and grants SAF2016–75004-R and Contribution to COST Action CA15203 MITOEAGLE to MC.
References
- 1. Dienel G.A., Cruz N.F. (2015) Contributions of glycogen to astrocytic energetics during brain activation. Metab. Brain Dis., 30, 281–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pellerin L., Bouzier-Sore A.K., Aubert A., Serres S., Merle M., Costalat R., Magistretti P.J. (2007) Activity-dependent regulation of energy metabolism by astrocytes: an update. Glia, 55, 1251–1262. [DOI] [PubMed] [Google Scholar]
- 3. Gibbs M.E., Anderson D.G., Hertz L. (2006) Inhibition of glycogenolysis in astrocytes interrupts memory consolidation in young chickens. Glia, 54, 214–222. [DOI] [PubMed] [Google Scholar]
- 4. Suzuki A., Stern S.A., Bozdagi O., Huntley G.W., Walker R.H., Magistretti P.J., Alberini C.M. (2011) Astrocyte-neuron lactate transport is required for long-term memory formation. Cell, 144, 810–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gibbs M.E. (2016) Role of glycogenolysis in memory and learning: regulation by noradrenaline, serotonin and ATP. Front. Integr. Neurosci., 9, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kong J., Shepel P.N., Holden C.P., Mackiewicz M., Pack A.I., Geiger J.D. (2002) Brain glycogen decreases with increased periods of wakefulness: implications for homeostatic drive to sleep. J. Neurosci., 22, 5581–5587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fiala J.C., Kirov S.A., Feinberg M.D., Petrak L.J., George P., Goddard C.A., Harris K.M. (2003) Timing of neuronal and glial ultrastructure disruption during brain slice preparation and recovery in vitro. J. Comp. Neurol., 465, 90–103. [DOI] [PubMed] [Google Scholar]
- 8. Brown A.M. (2004) Brain glycogen re-awakened. J. Neurochem., 89, 537–552. [DOI] [PubMed] [Google Scholar]
- 9. Brown A.M., Ransom B.R. (2007) Astrocyte glycogen and brain energy metabolism. Glia, 55, 1263–1271. [DOI] [PubMed] [Google Scholar]
- 10. Brown A.M., Ransom B.R. (2015) Astrocyte glycogen as an emergency fuel under conditions of glucose deprivation or intense neural activity. Metab. Brain Dis., 30, 233–239. [DOI] [PubMed] [Google Scholar]
- 11. Takado Y., Knott G., Humbel B.M., Escrig S., Masoodi M., Meibom A., Comment A. (2015) Imaging liver and brain glycogen metabolism at the nanometer scale. Nanomedicine, 11, 239–245. [DOI] [PubMed] [Google Scholar]
- 12. Cali C., Baghabra J., Boges D.J., Holst G.R., Kreshuk A., Hamprecht F.A., Srinivasan M., Lehvaslaiho H., Magistretti P.J. (2016) Three-dimensional immersive virtual reality for studying cellular compartments in 3D models from EM preparations of neural tissues. J. Comp. Neurol., 524, 23–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Waitt A.E., Reed L., Ransom B.R., Brown A.M. (2017) Emerging roles for glycogen in the CNS. Front. Mol. Neurosci., 10, 73.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Oe Y., Baba O., Ashida H., Nakamura K.C., Hirase H. (2016) Glycogen distribution in the microwave-fixed mouse brain reveals heterogeneous astrocytic patterns. Glia, 64, 1532–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Turnbull J., Tiberia E., Striano P., Genton P., Carpenter S., Ackerley C.A., Minassian B.A. (2016) Lafora disease. Epileptic Disord., 18, 38–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lafora G.R., Glueck B. (1911) Beitrag zur histogpathologie der myoklonischen epilepsie. Gesamte Neurol. Psychiatr., 6, 1–14. [Google Scholar]
- 17. Minassian B.A., Lee J.R., Herbrick J.A., Huizenga J., Soder S., Mungall A.J., Dunham I., Gardner R., Fong C.Y., Carpenter S.. et al. (1998) Mutations in a gene encoding a novel protein tyrosine phosphatase cause progressive myoclonus epilepsy. Nat. Genet., 20, 171–174. [DOI] [PubMed] [Google Scholar]
- 18. Serratosa J.M., Gomez-Garre P., Gallardo M.E., Anta B., de Bernabe D.B., Lindhout D., Augustijn P.B., Tassinari C.A., Malafosse R.M., Topcu M.. et al. (1999) A novel protein tyrosine phosphatase gene is mutated in progressive myoclonus epilepsy of the Lafora type (EPM2). Hum. Mol. Genet., 8, 345–352. [DOI] [PubMed] [Google Scholar]
- 19. Chan E.M., Young E.J., Ianzano L., Munteanu I., Zhao X., Christopoulos C.C., Avanzini G., Elia M., Ackerley C.A., Jovic N.J.. et al. (2003) Mutations in NHLRC1 cause progressive myoclonus epilepsy. Nat. Genet., 35, 125–127. [DOI] [PubMed] [Google Scholar]
- 20. Ganesh S., Delgado-Escueta A.V., Sakamoto T., Avila M.R., Machado-Salas J., Hoshii Y., Akagi T., Gomi H., Suzuki T., Amano K.. et al. (2002) Targeted disruption of the Epm2a gene causes formation of Lafora inclusion bodies, neurodegeneration, ataxia, myoclonus epilepsy and impaired behavioral response in mice. Hum. Mol. Genet., 11, 1251–1262. [DOI] [PubMed] [Google Scholar]
- 21. Criado O., Aguado C., Gayarre J., Duran-Trio L., Garcia-Cabrero A.M., Vernia S., San Millan B., Heredia M., Roma-Mateo C., Mouron S.. et al. (2012) Lafora bodies and neurological defects in malin-deficient mice correlate with impaired autophagy. Hum. Mol. Genet., 21, 1521–1533. [DOI] [PubMed] [Google Scholar]
- 22. Baba O. (1993) Production of monoclonal antibody that recognizes glycogen and its application for immunohistochemistry. Kokubyo Gakkai Zasshi, 60, 264–287. [DOI] [PubMed] [Google Scholar]
- 23. Falkowska A., Gutowska I., Goschorska M., Nowacki P., Chlubek D., Baranowska-Bosiacka I. (2015) Energy metabolism of the brain, including the cooperation between astrocytes and neurons, especially in the context of glycogen metabolism. Int. J. Mol. Sci., 16, 25959–25981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dringen R., Gebhardt R., Hamprecht B. (1993) Glycogen in astrocytes: possible function as lactate supply for neighboring cells. Brain Res., 623, 208–214. [DOI] [PubMed] [Google Scholar]
- 25. Pfeifer U. (1972) Lysosomal glycogen breakdown and hepatocellular glycogen metabolism. Cellular autophagy induced by intraperitoneal injection of hypertonic solution of glucose. Virchows Arch. B. Cell. Pathol., 10, 108–117. [PubMed] [Google Scholar]
- 26. Walls A.B., Sickmann H.M., Brown A., Bouman S.D., Ransom B., Schousboe A., Waagepetersen H.S. (2008) Characterization of 1,4-dideoxy-1,4-imino-d-arabinitol (DAB) as an inhibitor of brain glycogen shunt activity. J. Neurochem., 105, 1462–1470. [DOI] [PubMed] [Google Scholar]
- 27. Yang Y.P., Hu L.F., Zheng H.F., Mao C.J., Hu W.D., Xiong K.P., Wang F., Liu C.F. (2013) Application and interpretation of current autophagy inhibitors and activators. Acta Pharmacol. Sin., 34, 625–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lerchundi R., Fernandez-Moncada I., Contreras-Baeza Y., Sotelo-Hitschfeld T., Machler P., Wyss M.T., Stobart J., Baeza-Lehnert F., Alegria K., Weber B.. et al. (2015) NH4(+) triggers the release of astrocytic lactate via mitochondrial pyruvate shunting. Proc. Natl. Acad. Sci. U.S.A., 112, 11090–11095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Saez I., Duran J., Sinadinos C., Beltran A., Yanes O., Tevy M.F., Martinez-Pons C., Milan M., Guinovart J.J. (2014) Neurons have an active glycogen metabolism that contributes to tolerance to hypoxia. J. Cereb. Blood Flow. Metab., 34, 945–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nitschke F., Sullivan M.A., Wang P., Zhao X., Chown E.E., Perri A.M., Israelian L., Juana-Lopez L., Bovolenta P., Rodriguez de Cordoba S.. et al. (2017) Abnormal glycogen chain length pattern, not hyperphosphorylation, is critical in Lafora disease. EMBO Mol. Med., 9, 906–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lopez-Gonzalez I., Viana R., Sanz P., Ferrer I. (2017) Inflammation in Lafora disease: evolution with disease progression in laforin and malin knock-out mouse models. Mol. Neurobiol., 54, 3119–3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. DiNuzzo M., Mangia S., Maraviglia B., Giove F. (2014) Physiological bases of the K+ and the glutamate/GABA hypotheses of epilepsy. Epilepsy Res., 108, 995–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. DiNuzzo M., Mangia S., Maraviglia B., Giove F. (2015) Does abnormal glycogen structure contribute to increased susceptibility to seizures in epilepsy? Metab. Brain Dis., 30, 307–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xu J., Song D., Xue Z., Gu L., Hertz L., Peng L. (2013) Requirement of glycogenolysis for uptake of increased extracellular K+ in astrocytes: potential implications for K+ homeostasis and glycogen usage in brain. Neurochem. Res., 38, 472–485. [DOI] [PubMed] [Google Scholar]
- 35. Munoz-Ballester C., Berthier A., Viana R., Sanz P. (2016) Homeostasis of the astrocytic glutamate transporter GLT-1 is altered in mouse models of Lafora disease. Biochim. Biophys. Acta, 1862, 1074–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nagai T., Takata N., Shinohara Y., Hirase H. (2015) Adaptive changes of extracellular amino acid concentrations in mouse dorsal striatum by 4-AP-induced cortical seizures. Neuroscience, 295, 229–236. [DOI] [PubMed] [Google Scholar]
- 37. Aguado C., Sarkar S., Korolchuk V.I., Criado O., Vernia S., Boya P., Sanz P., de Cordoba S.R., Knecht E., Rubinsztein D.C. (2010) Laforin, the most common protein mutated in Lafora disease, regulates autophagy. Hum. Mol. Genet., 19, 2867–2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hertz L., Peng L., Lai J.C. (1998) Functional studies in cultured astrocytes. Methods, 16, 293–310. [DOI] [PubMed] [Google Scholar]
- 39. Muller M.S., Fox R., Schousboe A., Waagepetersen H.S., Bak L.K. (2014) Astrocyte glycogenolysis is triggered by store-operated calcium entry and provides metabolic energy for cellular calcium homeostasis. Glia, 62, 526–534. [DOI] [PubMed] [Google Scholar]
- 40. Magistretti P.J., Manthorpe M., Bloom F.E., Varon S. (1983) Functional receptors for vasoactive intestinal polypeptide in cultured astroglia from neonatal rat brain. Regul. Pept., 6, 71–80. [DOI] [PubMed] [Google Scholar]
- 41. Brown A.M., Tekkok S.B., Ransom B.R. (2003) Glycogen regulation and functional role in mouse white matter. J. Physiol., 549, 501–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhu A., Romero R., Petty H.R. (2009) An enzymatic fluorimetric assay for glucose-6-phosphate: application in an in vitro Warburg-like effect. Analytical Biochem., 388, 97–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pesta D., Gnaiger E. (2012) High-resolution respirometry: OXPHOS protocols for human cells and permeabilized fibers from small biopsies of human muscle. Methods Mol. Biol., 810, 25–58. [DOI] [PubMed] [Google Scholar]
- 44. Ylikallio E., Page J.L., Xu X., Lampinen M., Bepler G., Ide T., Tyynismaa H., Weiss R.S., Suomalainen A. (2010) Ribonucleotide reductase is not limiting for mitochondrial DNA copy number in mice. Nucleic Acids Res., 38, 8208–8218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fuke S., Kubota-Sakashita M., Kasahara T., Shigeyoshi Y., Kato T. (2011) Regional variation in mitochondrial DNA copy number in mouse brain. Biochim. Biophys. Acta, 1807, 270–274. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.