

Abstract
Background
Deleted in Liver Cancer 1 (DLC1) is a tumor suppressor gene frequently deleted in cancer. However, DLC1 is not known to be deleted in angiosarcoma, an aggressive malignancy of endothelial cell derivation. Additionally, the physiologic functions of DLC1 protein in endothelial cells are poorly defined.
Methods
We investigated the effects of shRNA-induced DLC1 depletion in endothelial cells. Cell growth was measured by 3H thymidine incorporation, IncuCyte imaging, and population doublings; cell death by cell cycle analysis; gene expression by Affimetrix arrays and quantitative polymerase chain reaction; NF-κB activity by reporter assays; and protein levels by immunoblotting and immunofluorescence staining. We tested Tanespimycin/17-AAG and Fasudil treatment in groups of nine to 10 mice bearing ISOS-1 angiosarcoma. All statistical tests were two-sided.
Results
We discovered that DLC1 is a critical regulator of cell contact inhibition of proliferation in endothelial cells, promoting statistically significant (P < .001) cell death when cells are confluent (mean [SD] % viability: control DLC1 = 15.6 [19.3]; shDLC1 = 73.4 [13.1]). This prosurvival phenotype of DLC1-depleted confluent endothelial cells is attributable to a statistically significant and sustained increase of NF-κB activity (day 5, P = .001; day 8, P = .03) associated with increased tumor necrosis factor alpha–induced protein 3 (TNFAIP3/A20) signaling. Consistently, we found that DLC1 is statistically significantly reduced (P < .001 in 5 of 6) and TNFAIP3/A20 is statistically significantly increased (P < .001 in 2 of 3 and P = 0.02 in 1 of 3) in human angiosarcoma compared with normal adjacent endothelium. Treatment with the NF-κB inhibitor Tanespimycin/17-AAG statistically significantly reduced angiosarcoma tumor growth in mice (treatment tumor weight vs control, 0.50 [0.19] g vs 0.91 [0.21] g, P = .001 experiment 1; 0.66 [0.26] g vs 1.10 [0.31] g, P = .01 experiment 2).
Conclusions
These results identify DLC1 as a previously unrecognized regulator of endothelial cell contact inhibition of proliferation that is depleted in angiosarcoma and support NF-κB targeting for the treatment of angiosarcoma where DLC1 is lost.
Deleted in Liver Cancer 1 (DLC1) is a tumor suppressor gene in a variety of cancer types, including liver, lung, colon, and brain (1–3). In cancer cells, DLC1 expression is often lost or reduced due to gene deletion or aberrant DNA methylation, and reintroduction of DLC1 inhibits cancer cell growth (2,4). Most tissues express DLC1, but the functions of DLC1 in normal cells are poorly characterized. DLC1-null embryos die by day 10.5 of gestation with placental blood vessel abnormalities, providing evidence for an essential yet unclear developmental function of DLC1 (1,5–7).
DLC1 is a rho-GTPase-activating protein (GAP), which inactivates rho-A, -B, and -C (3,8), and inhibition of the rho family of small GTPases is critical to DLC1 tumor suppressor function (9). DLC1 possesses additional functional domains that contribute to its full tumor suppressive function (8–13). Cyclin-dependent kinase-5 (CDK5) phosphorylates DLC1, activating its tumor suppressive functions (14).
DLC1 is not currently known to be a tumor suppressor in angiosarcoma, an aggressive vascular tumor arising from blood and lymphatic endothelia (15,16). Previous studies reported that primary endothelial cells express high levels of rho and its effector rho-associated kinase, ROCK (17), suggesting a contribution of ROCK in angiogenesis and angiosarcoma (18). Recently, siRNA depletion of DLC1 promoted endothelial cell motility and reduced tube formation (7).
To gain insight into the potential roles of DLC1 in angiosarcomagenesis, we have explored DLC1-dependent pathways contributing to regulation of endothelial cell growth and survival.
Methods
Cells and Cell Function
Human umbilical vein endothelial cells (HUVEC; passages 4–7 unless otherwise stated; Lifeline Cell Technology, Frederick, MD; FC-0044), human dermal microvascular endothelial cells (HMVEC-d, passages 2–5; Lonza, Allendale, NJ; CC-2543), the human microvascular endothelial cell line (HMEC-1; ATCC, Manassas, VA; CRL-3243), the hybrid endothelial EA.hy926 cell line (ATCC, CRL-2922), human primary fibroblasts (CCD-18Co, passages 10–12; ATCC CCD-18Co), and the mouse angiosarcoma cell line ISOS-1 (19) were propagated as detailed in the Supplementary Methods (available online).
Cell proliferation by 3H-thymidine incorporation, calcein-fluorescence, IncuCyte imaging, population doublings; cell cycle with propidium iodide, and use of Staurosporine (Adipogen, San Diego, CA; AG-CN2-0022), C6-ceramide (Avanti Polar Lipids, Alabaster, AL; 860506P), Tanespimycin/17-AAG (Selleck Chemicals, Houston, TX; S1141), and C3 Transferase from Clostridium botulinum (Cytoskeleton, Denver, CO; CT04) were used as described in Supplementary Methods (available online).
Gene Expression
Lentiviral particles for silencing and overexpression of DLC1 and control shRNAs were prepared using a third-generation system (see the Supplementary Methods, available online) (20). RNA purification, cDNA synthesis, quantitative polymerase chain reaction (qPCR), and qPCR primers are described in the Supplementary Methods and Supplementary Table 1 (available online). Gene expression profiles were analyzed using Affimetrix human microarray Hu Gene ST 2.0 array and analysis of variance (ANOVA). Microarray raw data are deposited in ArrayExpress (E-MTAB-5263). NFκB reporter assays (Supplementary Methods, available online) utilized a lentiviral vector where expression of the firefly luciferase reporter gene is driven by NF-κB activation and dTomato is constitutively expressed for normalization and cell tracking.
Immunoblotting, Protein Array, and Rho Activity
Primary antibodies for immunoblotting are listed in Supplementary Table 2 (available online). Images were acquired using a LAS 4000 imager device (GE). The Supplementary Methods (available online) provide details of immunoblotting, use of the human apoptosis array kit (R&D Systems, Minneapolis, MN; ARY009), and measurement of GTP-bound rho.
Mouse Model
All mouse experiments were conducted in adherence to the National Institutes of Health Guide for Care and Use of Laboratory Animals with protocols approved by the Institutional Animal Care and Use Committee of the Center for Cancer Research, National Cancer Institute. BALB/c female mice (age eight weeks, Charles River Laboratories) were inoculated s.c. with 1×106 ISOS-1 cells isolated from tumors, detailed in the Supplementary Methods (available online). One week after inoculation, mice were randomly assigned to four groups (n = 10/group) for i.p. injections with vehicle, Fasudil (Selleck Chemicals HA-1077; 50 mg/kg), Tanespimycin/17-AAG (Selleck Chemicals S1141; 80 mg/kg), or the combination of Fasudil (50 mg/kg) and 17-AAG (80 mg/kg) three times per week until the specified end point (tumor reaching the estimated volume of 2000 mm3, calculated as V = 1/2 × D × d2, where D and d are the longest and shortest perpendicular tumor diameters) was reached in any mouse. The next day, the mice were killed, and the tumors were evaluated (Supplementary Methods, available online).
Tissue Samples, Immunostaining, and Quantitation
Anonymized tissue samples were obtained from six patients (Supplementary Table 3, available online) under institutionally approved protocols (Kyoto University, Japan) with written informed consent and from mouse tumors. Details of tissue fixation, processing, antigen retrieval, and immunofluorescence staining with primary and secondary antibodies (Supplementary Table 2, available online) and fluorescence quantification are described in Supplementary Methods, available online. Images were acquired using a 710 NLO confocal (Carl Zeiss); 22 fields per slide (63× magnification) were acquired (11 each from normal and angiosarcoma regions). Images were processed using Fiji (21); results of fluorescence quantification are presented as boxplots.
Statistical Analysis
Data are expressed as means (standard deviations). Unpaired/paired two-tailed Student’s t test was used for statistical analysis of the difference between two groups with normal and homoscedastic distribution; otherwise, the Mann-Whitney U test was used. One- or two-way ANOVA with Tukey Honestly Significant Difference (HSD) post hoc was used for statistical analysis of differences comparing three or more groups. A P value of less than .05 was considered statistically significant. All statistical tests were two-sided.
Results
Effects of DLC1 on Endothelial Cell Survival
We found that DLC1 protein levels are statistically significantly (P = .003) higher in primary human umbilical vein endothelial cells propagated at high cell density compared with low cell density (Figure 1A). Consistent with DLC1 being regulated by protein degradation (22), cell density–dependent differences in DLC1 protein levels were not associated with a statistically significant difference (P = .75) in relative DLC1 mRNA levels (Figure 1B). Primary human dermal microvascular endothelial cells (HMVEC-d), HMEC-1 cells, and primary human fibroblasts showed a similar difference in DLC1 protein content when grown at high or low cell density (Figure 1C).
Figure 1.

Deleted in Liver Cancer 1 (DLC1) protein levels in endothelial cells propagated at different densities. Human umbilical vein endothelial cells (HUVEC) grown at low (10%–20% confluent) or high (90%–100% confluent) cell density were tested for DLC1 protein content (representative blot, left; quantitation of results from three independent experiments, right; P = .003 from two-sided paired Student’s t test is shown) (A) and mRNA levels (relative mRNA levels by quantitative polymerase chain reaction normalized to HPRT1, n = 3; P = .75 from two-sided paired Student’s t test is shown) (B). DLC1 protein levels in cell lysates of human dermal microvascular endothelial cells, SV40-immortalized human microvascular endothelial cell line line, and primary human fibroblasts (CCD 18Co) grown at low (1.45 × 103 cells cm-2), medium (mid, 9.10 × 103 cells cm-2), and high (3.64 × 104 cell cm-2) cell densities (C). DLC1 = Deleted in Liver Cancer 1; HMEC-1 = human microvascular endothelial cell line; HMVEC-d = human dermal microvascular endothelial cells; HUVEC = human umbilical vein endothelial cells.
We effectively silenced DLC1 in HUVEC using four shRNAs (Figure 2A). When propagated at subconfluent densities, the loss of DLC1 minimally changed HUVEC population doublings over 120 days, surface CD31 and VE-Cadherin levels, and the ability to form vascular structures on extracellular matrix (Supplementary Figure 1, A–C, available online). However, we found that DLC1-silenced HUVEC incubated as a confluent monolayer for 125 or 150 hours survive better than control cells, as reflected by appreciably greater monolayer density (Figure 2B) and a statistically significantly (P < .001) greater number of residual live cells (Figure 2C). For example, DLC1-silenced HUVEC (shDLC1#3) were 73.4 (13.1)% viable after 125-hour culture compared with control HUVEC that were 15.6 (19.3)% viable (P < .001).
Figure 2.

Effects of Deleted in Liver Cancer 1 (DLC1) on endothelial cell death. A) DLC1 silencing by distinct shRNAs. pLKO identifies the empty vector control for all DLC1 shRNAs; GFP identifies an empty vector where the puromycin marker of pLKO is substituted by the GFP reporter. Immublotting results. B and C) DLC1-silenced and control human umbilical vein endothelial cells (HUVEC) were seeded as confluent monolayers and incubated for 150 hours. B) Representative fluorescent images of calcein-labeled viable cells (10×). C) Percent remaining cells is statistically significantly increased in DLC1-silenced HUVEC compared with control after incubation at confluence (125- and 150-hour time points); means (SD) (one experiment, 32 replicates; results of statistical analysis by two-sided Student’s t test are shown in tabular form below the bar graph; representative of five independent experiments with shDLC1#3; remaining cells ranged between 10% and 40% in control; 40% and 80% in DLC1-silenced cells). D) Cell cycle distribution of DLC1-silenced and control HUVEC seeded as confluent monolayers and evaluated on days 1, 4, 6 and 8; the bar graphs reflect the mean (SD) (n = 4 experiments); the table shows the statistical significance of group differences at each time point. P values by two-sided Student’s t test. E) Cell piling in DLC1-silenced HUVEC after four-week culture at confluency; bright field images (20×); the cytoplasmic membrane of two overlapping cells is contoured (on the right). Scale bars: B = 50 μm; E = 80 μm. DLC1 = Deleted in Liver Cancer 1.
Once confluent, primary endothelial cells maintain viability by an equilibrium between cell division and cell death (23). After HUVEC were maintained as confluent monolayers for one to eight days (Figure 2D), cell cycle analysis showed a statistically significant reduction of cells/cell fragments (sub-G0/G1 peak) in DLC1-silenced HUVEC compared with control at days 4 (P = .002), 6 (P = .002), and 8 (P < .001), consistent with reduced endothelial cell death after DLC1 silencing (Figure 2D). It also showed a statistically significant increase in the proportion of DLC1-silenced HUVEC in G0/G1 compared with control cells at days 6 (P = .04) and 8 (P = .001), consistent with retardation of cell cycle progression from G1 to S (Figure 2D). Furthermore, we observed cell piling in cultures of DLC1-silenced HUVEC after four weeks at high density, consistent with the reduced “cell contact inhibition” of proliferation (Figure 2E). Thus, DLC1 regulates cell survival, cell cycle, and “contact inhibition” of growth in confluent endothelial cells.
DLC1 Target Genes and Signaling in Endothelial Cells
Microarray analysis showed that DLC1 silencing changes the expression of many genes in confluent HUVEC (Supplementary Figure 2A, available online). Among these, we identified reduced expression of genes linked to cell death regulation and increased expression of prosurvival genes (Supplementary Figure 2B, available online). qPCR confirmed that expression of tumor necrosis factor, alpha-induced protein 3 (TNFAIP3/A20, an inhibitor of TNFα-induced apoptosis and product of NF-κB activation) is statistically significantly increased (P = .04) in confluent, DLC1-silenced HUVEC compared with control, whereas expression of BCL2-Like 11 (BCL2L11/BIM, an apoptotic activator in the BCL-2 family) is statistically significantly decreased (P = .03) (Figure 3A;Supplementary Figure 2C, available online). Lysates of DLC1-silenced, confluent HUVEC contained increased levels of TNFAIP3/A20 and reduced levels of the cell death regulators BCL2L11/BIM, clusterin, and cleaved-caspase 3 compared with control (Figure 3B;Supplementary Figure 2D, available online).
Figure 3.
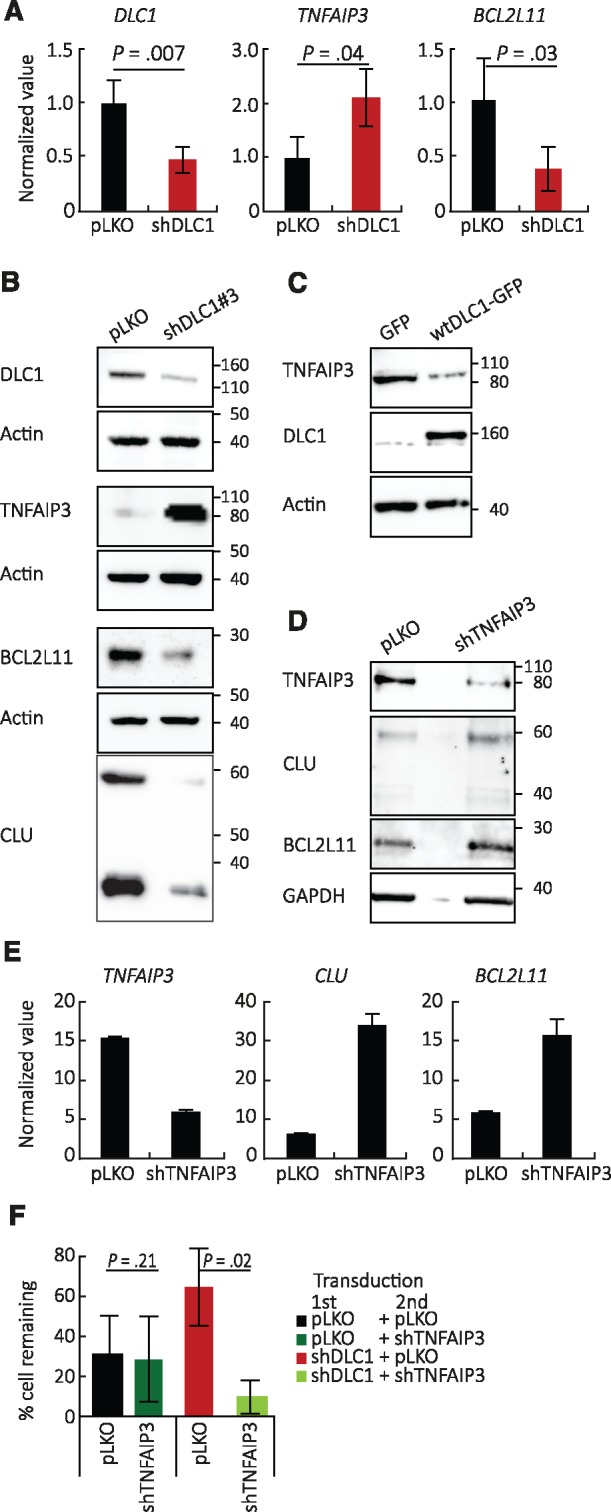
Deleted in Liver Cancer 1 (DLC1) regulation of TNFAIP3/A20, BCL2L11/BIM, and clusterin expression in endothelial cells. A and B) Effects of DLC1 silencing in human umbilical vein endothelial cells (HUVEC); (A) quantitative polymerase chain reaction (mean [SD]; n = 5 different experiments; P values from two-sided Student’s t test are shown) and (B) immunoblotting results; same cell lysates used in the three blots. C) Wild-type DLC1 was overexpressed in EA.hy926 endothelial cells; TNFAIP3/A20 protein levels by immunoblotting. D) Effects of TNFAIP3/A20 silencing on clusterin and BCL2L11/BIM protein levels in human microvascular endothelial cells (HMEC-1 line). E) Effects of TNFAIP3/A20 silencing on clusterin and BCL2L11/BIM mRNAs. F) Effects of TNFAIP3/A20 silencing on the prosurvival advantage conferred by DLC1 silencing in HUVEC (mean [SD], n = 3 independent experiments at day 8, P values from two-sided Student’s t test are shown). CLU = clusterin; DLC1 = Deleted in Liver Cancer 1.
Consistent with the results observed after DLC1 silencing, low-density HUVEC displayed a higher content of TNFAIP3/A20 and a lower content of the pro-apoptotic BCL2L11/BIM and clusterin proteins compared with high-density HUVEC (Supplementary Figure 3, A and B, available online).
We explored biochemical and functional links among DLC1, TNFAIP3/A20, and BCL2L11/BIM. First, we found that wild-type (WT)-DLC1 overexpression reduces TNFAIP3/A20 protein in EA.hy926 cells, which constitutively express TNFAIP3/A20 (Figure 3C), supporting a role for DLC1 as a TNFAIP3/A20 inhibitor. Second, we found that TNFAIP3/A20 silencing increases mRNA and protein levels of BCL2L11/BIM and clusterin in endothelial cells (Figure 3, D and E), supporting a role for TNFAIP3/A20 as a BCL2L11/BIM and clusterin regulator. Third, we found that TNFAIP3/A20 silencing effectively neutralizes the prosurvival effect conferred by DLC1 depletion in endothelial cells (Figure 3F), supporting the role of TNFAIP3/A20 as an essential mediator of endothelial cell survival when DLC1 is silenced.
These experiments outline a pathway whereby DLC1 deficiency in endothelial cells promotes expression of TNFAIP3/A20, which represses the pro-apoptotic proteins BCL2L11/BIM and clusterin.
Rho Contribution to DLC1 Regulation of TNFAIP3/A20 Expression
TNFAIP3/A20 is a product of NF-κB activation (24). We asked whether the NF-κB pathway is active in DLC1-silenced endothelial cells when TNFAIP3/A20 is detected. Reporter assays showed that NF-κB activity is statistically significantly increased in confluent DLC1-silenced endothelial cells compared with control, and this increase is sustained over five (P = .001) and eight (P = .03) days in confluent cultures (Figure 4A).
Figure 4.

NF-κB activity in Deleted in Liver Cancer 1 (DLC1)–silenced endothelial cells. A) NF-κB activity is measured by a luciferase reporter transduced into human umbilical vein endothelial cells (HUVEC); the results reflect means (SD) (n = 3 different experiments; P values shown are from two-sided paired Student’s t test). B) p-IκBα and p-p65 levels in DLC1-silenced HUVEC (vectors #3 and #5). C and D) Effects of Tanespimycin/17-AAG on (C) NF-κB activation (mean [SD] normalized to untreated pLKO; n = 3 different experiments, P values from two-sided paired Student’s t test are shown) and (D) the prosurvival effect induced by DLC1 silencing in HUVEC (means [SD]; n = 3 different experiments, P values from two-sided paired Student’s t test are shown). E) HUVEC were first infected with a control shRNA or with a DLC1 shRNA that targets the 3' untranslated region, and then infected with control GFP vector or wild-type (WT)-DLC1-GFP (WT-DLC1). Results reflect means (SD) (n = 2 different experiments, P values are from two-sided paired Student’s t test). DLC1 = Deleted in Liver Cancer 1 (DLC1); GFP = control GFP vector; HUVEC = human umbilical vein endothelial cells; p-MLC = phosphorylated myosin light chain; shControl = shRNA nonmammalian control; shUTR = shRNA that targets the 3' untranslated region; WT = wild-type.
Consistent with NF-κB activation, phosphorylation of IκBα and p65/RelA (p-p65) are increased in confluent DLC1-silenced endothelial cells compared with control (Figure 4B). The compound Tanespimycin (17-N-allylamino-17-demethoxygeldanamycin, 17-AAG), an inhibitor of the heat shock protein 90 that lowers NF-κB activation in endothelial cells (25), statistically significantly reduced (P = .04) NF-κB activity (Figure 4C) and statistically significantly decreased (P < .001) survival in DLC1-silenced endothelial cells (Figure 4D).
We infected HUVEC with a DLC1 shRNA that targets the untranslated region (shUTR DLC1) and subsequently expressed either GFP vector or WT-DLC1-GFP. Re-expression of WT-DLC1 statistically significantly reduced (P = .02) NF-κB activity (Figure 4E) induced by DLC1 silencing in endothelial cells, providing further evidence that DLC1 regulates NF-κB activity.
The GTPases rho-A, -B, and -C are major targets of DLC1 activity. Consistently, DLC1 silencing in HUVEC increased rho activity and phosphorylated myosin light chain (p-MLC), a target of the rho/ROCK pathway, compared with control (Figure 5, A–C). However, a specific rho inhibitor (the cell-permeable isoenzyme C3 transferase; inhibits all rho forms) minimally altered TNFAIP3 mRNA, TNFAIP3 protein, and clusterin protein levels in DLC1-silenced HUVEC (Figure 5, D and E).
Figure 5.

Role of rho in Deleted in Liver Cancer 1 (DLC1) regulation of endothelial cell survival. A) Active rho (GTP-bound rho) in DLC1–silenced human umbilical vein endothelial cells (HUVEC) compared with control (representative of two experiments). B and C) Phosphorylated (S20) myosin light chain in DLC1-silenced HUVEC compared with control: (B) representative and (C) quantitation (mean [SD]; n = 3 different experiments, P values from two-sided paired Student’s t test). D and E) Effects of the specific rho-A inhibitor (rho-inh, isoenzyme C3 transferase) on mRNA levels of TNFAIP3/A20 mRNA (D, mean [SD]; n = 2 experiments) and protein levels of TNFAIP3 and clusterin (E; representative of two experiments) in DLC1-silenced HUVEC. F and G) HUVEC viability after introduction of wild-type or mutant R718A DLC1 (lacking GAP activity) in DLC1-silenced HUVEC; (F) schematic of the experiments and (G) results from day 8 (mean [SD]; n = 2 different experiments, four replicates each, P values from two-sided paired Student’s t test). DLC1 = Deleted in Liver Cancer 1 (DLC1); GFP = control GFP vector; HUVEC = human umbilical vein endothelial cells; p-MLC = phosphorylated myosin light chain; shControl = shRNA nonmammalian control; shUTR = shRNA that targets the 3' untranslated region; WT = wild-type.
Other experiments (schematic of experiment Figure 5F) tested the mutant DLC1-R718A, which lacks rho-GAP activity (9). We found that the rho-mutant DLC1-R718A was as effective as WT-DLC1 at statistically significantly (control vs WT, P = .02; control vs R718A, P = .01; WT vs R718A, P = .83) reducing the viability of confluent HUVEC where the endogenous DLC1 was silenced (Figure 5G). Thus, a rho-GAP-independent function of DLC1 is likely responsible for regulation of cell death in confluent endothelial cells.
DLC1 in Normal Endothelium and in Angiosarcoma
Increased DLC1 in confluent compared with sparse endothelial cells predicted that the endothelial cells lining an established vasculature as a continuous monolayer would express DLC1. Loss of endothelial cell contact inhibition of growth after DLC1 silencing in vitro predicted that pathological loss of DLC1 in vivo would remove constraints on endothelial cell growth/survival.
We evaluated six biopsies from six patients with angiosarcoma (Supplementary Table 3, available online), a highly proliferative malignancy of endothelial derivation, which included tumor and adjacent normal tissue (Figure 6, A–C). Nuclear DAPI staining showed that cell density is typically greater in angiosarcoma than in the surrounding normal tissue (Figure 6D,E). The tumor cells were CD31+ and proliferative, as evidenced by Ki67 co-immunostaining (Figure 6E). The CD31+ endothelium in the surrounding normal tissue was largely Ki67-, reflecting resting vasculature (Figure 6D). We imaged DLC1 in angiosarcoma specimens: the normal (Figure 6F) and tumor (Figure 6G) tissues displayed DLC1-specific immunostaining (Supplementary Figure 4, A–C, available online) in the CD31+ cells, with differences in intensity. Quantitative staining analysis showed a statistically significant DLC1 reduction in CD31+ tumor cells compared with the CD31+ normal surrounding endothelium (P < .001 in 5 of 6 tumors) (Figure 6H). This statistically significant difference in DLC1 staining intensity was confirmed by a different DLC1-specific antibody (Supplementary Figure 4D, available online). Thus, DLC1 is generally reduced in angiosarcoma compared with normal resting endothelium.
Figure 6.

Deleted in Liver Cancer 1 (DLC1) in human angiosarcoma and normal endothelium. Histology of angiosarcoma section containing normal and tumor cells; hematoxylin and eosin staining (A); boxed areas magnified in ((B) normal) and ((C) tumor). CD31 and Ki67 immunostaining (D and E) shows that the normal CD31+ endothelial cells are mostly nonproliferative (some CD31- cells, likely gland-associated are Ki67+ in (D), whereas CD31+ tumor cells are Ki67+(E)). CD31+ normal endothelium is DLC1+(F); DLC1 levels in angiosarcoma (G). Quantitative analysis of DLC1 fluorescence intensity in CD31+ regions within areas of normal and tumor tissue in individual angiosarcoma tissue biopsy specimens (H). Quantitative analysis of TNFAIP3/A20 (I) and clusterin (J) fluorescence intensity in CD31+ regions of six individual biopsies from six patients, including normal and tumor areas. The box-and-whisker plots in (H and I) reflect the distribution of data points (box bottom: first quartile; box top: third quartile; horizontal line: median; whiskers: lowest and highest data points within 1.5 times the interquartile range). Statistical significance shown in (H–J) is from Mann-Whitney U test. Scale bars: A = 1 mm; B and C = 300 μm; D and E = 200 μm; F and G = 20 μm. CLU = clusterin; DLC1 = Deleted in Liver Cancer 1 (DLC1); N = normal; T = tumor.
Consistent with the results in vitro, specific TNFAIP3/A20 and clusterin immunofluorescence (Supplementary Figure 5, available online) showed that TNFAIP3/A20 fluorescence levels are statistically significantly higher in CD31+ angiosarcoma cells compared with the surrounding normal CD31+ endothelial cells (P < .001 in 2 of 3, P = .02 in the third) (Figure 6I) and that clusterin fluorescence levels are statistically significantly lower in CD31+ angiosarcoma cells compared with the surrounding normal CD31+ endothelial cells (P = .003, P = .001, P = .01) (Figure 6J). Thus, these results support a role for DLC1 depletion as an inducer of prosurvival signaling in angiosarcoma.
Effects of Tanespimycin/17-AAG and Fasudil on Experimental Angiosarcoma Progression
These observations predict that NF-κB inhibition, but not rho inhibition, would inhibit progression of DLC1-deficient angiosarcoma. To test this, we selected an ISOS-1-based model of murine angiosarcoma, which resembles poorly differentiated human angiosarcoma (19). ISOS-1 cells resemble human angiosarcoma cells in having low/undetectable DLC1 and clusterin but detectable TNFAIP3/A20 (Figure 7A), suggestive of NF-κB and TNFAIP3/A20 activity.
Figure 7.

Effects of the NF-κB inhibitor Tanespimycin/17-AAG on angiosarcoma progression in mice. A)Deleted in Liver Cancer 1 (DLC1), clusterin, and TNFAIP3/A20 proteins in the murine angiosarcoma cell line ISOS-1. B and C) Tanespimycin/17-AAG and Fasudil treatment of ISOS-1 tumors established in syngeneic mice. The individual data points reflect tumor weight in individual mice (n = 10 mice/group in experiment #1, B; n = 9 mice/group in experiment #2 (C)); the horizontal lines reflect median tumor weight/group. Statistical significance displayed in (B and C) is calculated by analysis of variance with Tukey HSD post hoc test. D) p-myosin light chain immunostaining in ISOS-1 tumor tissues from control and treated mice. E) Clusterin mRNA levels are statistically significantly higher in ISOS-1 tumor tissues from mice treated with Tanespimycin/17-AAG than in control and Fasudil only–treated mice (mean [SD]; n = 9 mice/group, P values from two-sided paired Student’s t test are shown). Scale bar = 50 μm. DLC1 = Deleted in Liver Cancer 1; CLU = clusterin; pMLC = p-myosin light chain.
We established subcutaneous ISOS-1 tumors in syngeneic mice. When the tumor volume had reached 0.030 to 0.050 cm3, the mice were randomly assigned (10 mice/group) to receive intraperitoneal injections of vehicle, the rho inhibitor Fasudil alone (50 mg/kg, three times/week), the NF-κB inhibitor Tanespimycin/17-AAG alone (80 mg kg, three times/week) or a combination of Fasudil (50 mg/kg, three times/week) plus 17-AAG (80 mg kg, three times/week). After two-week treatment (the largest tumor in the control group was or approached 2000 mm3), the mice were killed. The weight (Figure 7B) of tumors from mice treated with Tanespimycin/17-AAG alone was statistically significantly lower than the weight of tumors in the control group (0.50 [0.19] g vs 0.91 [0.21] g; P = .001). Instead, the weight of tumors from mice treated with Fasudil alone was not statistically significantly different from the weight of tumors in the control group (0.83 [0.29] g vs 0.91 [0.21] g; P = .82). The addition of Fasudil did not reduce statistically significantly the tumor weight of Tanespimycin/17-AAG-only-treated mice (0.43 [0.34] g vs 0.50 [0.19] g; P = .15). These results were confirmed in a separate experiment, showing that the weight of tumors from mice treated with Tanespimycin/17-AAG alone was statistically significantly lower than the weight of tumors in the control group (0.66 [26] g vs 1.10 [0.31] g; P = .01) (Figure 7C).
We looked for evidence of drug activity in the treated mice. p-MLC, which was prominent in control ISOS-1 tumors, was minimally detected in Fasudil-treated tumors, indicating that Fasudil had blocked rho-ROCK signaling (Figure 7D). Clusterin mRNA levels were statistically significantly higher in tumors from Tanespimycin/17-AAG-treated mice compared with control (P = .005) or Fasudil-alone-treated (P = .02) mice (Figure 7E), showing that Tanespimycin/17-AAG inhibited NF-κB/A20 signaling. These results indicate that inhibition of NF-κB signaling is a promising approach for the experimental treatment of angiosarcoma, where DLC1 is low and the NF-κB pathway is active.
Discussion
We report that DLC1 plays a previously unrecognized role as an essential physiologic regulator of cell contact inhibition of proliferation in endothelial cells. In this process, primary cells stop dividing and die when they occupy the entire surface allocated to them (26); depletion of DLC1 impairs this process. We further report that DLC1 is reduced or lost in angiosarcoma, a malignancy of endothelial cell derivation, and provide evidence that the loss of DLC1 drives NF-κB activation, a therapeutically targetable pathway in experimental angiosarcoma.
Cell contact inhibition of proliferation is a complex process initiated by mechanical cues (27,28), key junctional proteins (29), and soluble factors (30) converging on the regulation of the Hippo and YAP/TAZ signaling pathways (31–33). Most normal adult cells are contact inhibited (34). Cancer cells, however, escape cell contact inhibition, which is implicated in cancer cells’ ability to multiply, invade host tissues, and metastasize (35,36).
The tumor suppressor activity of DLC1 has not been previously linked to regulation of cell contact inhibition of growth, but to regulation of the actin cytoskeleton and motility of cells. These functions are dependent on DLC1 rho-GAP activity and binding to several ligands, including tensin, talin, and FAK (12,37). We now established that DLC1 regulation of endothelial cell contact inhibition of growth is rho-independent and relies upon a pathway involving NF-κB/TNFAIP3/A20 signaling and downstream cell death effectors. When DLC1 is silenced in endothelial cells, NF-κB is activated, TNFAIP3/A20 is induced, and expression of the pro-apoptotic clusterin, BIM, and cleaved caspase-3 is reduced, protecting endothelial cells from physiologic death when confluent. Consistent with this, previous studies showed that DLC1 overexpression represses NF-κB activity in prostate cancer cells (38) and nasopharyngeal cancer cells (39,40).
TNFAIP3/A20 is a well-known NF-κB-dependent prosurvival molecule in endothelial cells (41–46), protecting these cells from death induced by tumor necrosis factor α (TNFα) and other signals (41,47). Here we describe a new role of TNFAIP3/A20 as a mediator of prosurvival signals in DLC1-depleted endothelial cells.
The newly identified DLC1 deficiency in human angiosarcoma and new awareness of NF-κB/TNFAIP3/A20 activation in DLC1-deficient cells provided a rational basis for NF-κB targeting in DLC1-negative angiosarcoma. Tanespimycin/17-AAG, an effective NF-κB inhibitor in human microvascular endothelial cells (25), statistically significantly decreased tumor burden in this mouse model. This holds promise for human angiosarcoma, which displays high-level NF-κB activity (48) and has abnormally low levels of DLC1, as shown here.
There are several limitations of this study. First, angiosarcoma is an extremely rare malignancy even among sarcomas (49). Second, Tanespimycin/17AAG, which reduced angiosarcoma growth statistically significantly in mice, is no longer under drug development, despite having shown promising antitumor activity but poor pharmaceutical and toxicity profiles (50). Nonetheless, clinical development of newer HSP90 inhibitors continues (50). Third, the mechanisms that underlie DLC1 activation by cell-to-cell contact and DLC1 regulation of the NFκB pathway need further exploration.
Funding
This work was supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research (DSM, XQ, DRL, GT) and by the Japan Society for the Promotion of Science KAKENHI, Grants-in-Aid for Scientific Research 15H05790 (AO, KK).
Notes
The funders had no role in design of the study; the collection, analysis, or interpretation of the data; the writing of the manuscript; or the decision to submit the manuscript for publication.
The authors declare no competing financial interests.
DSM and GT conceived of the study and designed and evaluated experiments; DSM performed the experiments; XQ provided critical reagents and experimental assistance; AA and KK provided patient samples; DSM, AO, KK, XQ, and GT performed analysis and interpretation of data; DL provided administrative and scientific support; DSM and GT wrote the manuscript; AO, KK, XQ, and DL critically reviewed the manuscript.
We thank Ms. Luowei Li, members of the Laboratory of Cellular Oncology, Mr. Parthav Jailwala, and the Center for Cancer Research Collaborative Bioinformatics Resource of the Advanced Biomedical Computing Center operated by SAIC-Frederick and supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health (YT-10-060), and Dr. Gomez-Robles for help in various aspects of this work.
Supplementary Material
References
- 1. Durkin ME, Yuan BZ, Zhou X et al. , DLC-1: A rho GTPase-activating protein and tumour suppressor. J Cell Mol Med. 2007;115:1185–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Liao YC, Lo SH.. Deleted in liver cancer-1 (DLC-1): A tumor suppressor not just for liver. Int J Biochem Cell Biol. 2008;405:843–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yuan BZ, Miller MJ, Keck CL et al. , Cloning, characterization, and chromosomal localization of a gene frequently deleted in human liver cancer (DLC-1) homologous to rat RhoGAP. Cancer Res. 1998;5810:2196–2199. [PubMed] [Google Scholar]
- 4. Durkin ME, Ullmannova V, Guan M et al. , Deleted in liver cancer 3 (DLC-3), a novel rho GTPase-activating protein, is downregulated in cancer and inhibits tumor cell growth. Oncogene. 2007;2631:4580–4589. [DOI] [PubMed] [Google Scholar]
- 5. Durkin ME, Avner MR, Huh CG et al. , DLC-1, a rho GTPase-activating protein with tumor suppressor function, is essential for embryonic development. FEBS Lett. 2005;5795:1191–1196. [DOI] [PubMed] [Google Scholar]
- 6. Sabbir MG, Wigle N, Loewen S et al. , Identification and characterization of Dlc1 isoforms in the mouse and study of the biological function of a single gene trapped isoform. BMC Biol. 2010;8:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shih YP, Yuan SY, Lo SH.. Down-regulation of DLC1 in endothelial cells compromises the angiogenesis process. Cancer Lett. 2017;398:46–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kim TY, Healy KD, Der CJ et al. , Effects of structure of rho GTPase-activating protein DLC-1 on cell morphology and migration. J Biol Chem. 2008;28347:32762–32770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Qian X, Li G, Asmussen HK et al. , Oncogenic inhibition by a deleted in liver cancer gene requires cooperation between tensin binding and rho-specific GTPase-activating protein activities. Proc Natl Acad Sci U S A. 2007;10421:9012–9017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Du X, Qian X, Papageorge A et al. , Functional interaction of tumor suppressor DLC1 and caveolin-1 in cancer cells. Cancer Res. 2012;7217:4405–4416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Braun AC, Olayioye MA.. Rho regulation: DLC proteins in space and time. Cell Signal. 2015;278:1643–1651. [DOI] [PubMed] [Google Scholar]
- 12. Li G, Du X, Vass WC et al. , Full activity of the deleted in liver cancer 1 (DLC1) tumor suppressor depends on an LD-like motif that binds talin and focal adhesion kinase (FAK). Proc Natl Acad Sci U S A. 2011;10841:17129–17134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shih YP, Sun P, Wang A et al. , Tensin1 positively regulates rhoA activity through its interaction with DLC1. Biochim Biophys Acta. 2015;185312:3258–3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tripathi BK, Qian X, Mertins P et al. , CDK5 is a major regulator of the tumor suppressor DLC1. J Cell Biol. 2014;2075:627–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cioffi A, Reichert S, Antonescu CR et al. , Angiosarcomas and other sarcomas of endothelial origin. Hematol Oncol Clin North Am. 2013;275:975–988. [DOI] [PubMed] [Google Scholar]
- 16. Donnell RM, Rosen PP, Lieberman PH et al. , Angiosarcoma and other vascular tumors of the breast. Am J Surg Pathol. 1981;57:629–642. [DOI] [PubMed] [Google Scholar]
- 17. Ghosh K, Thodeti CK, Dudley AC et al. , Tumor-derived endothelial cells exhibit aberrant Rho-mediated mechanosensing and abnormal angiogenesis in vitro. Proc Natl Acad Sci U S A. 2008;10532:11305–11310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Montalvo J, Spencer C, Hackathorn A et al. , ROCK1 & 2 perform overlapping and unique roles in angiogenesis and angiosarcoma tumor progression. Curr Mol Med. 2013;131:205–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Masuzawa M, Fujimura T, Tsubokawa M et al. , Establishment of a new murine-phenotypic angiosarcoma cell line (ISOS-1). J Dermatol Sci. 1998;162:91–98. [DOI] [PubMed] [Google Scholar]
- 20. Salvucci O, Ohnuki H, Maric D et al. , EphrinB2 controls vessel pruning through STAT1-JNK3 signalling. Nat Commun. 2015;6:6576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schindelin J, Arganda-Carreras I, Frise E et al. , Fiji: An open-source platform for biological-image analysis. Nat Methods. 2012;97:676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kim TY, Jackson S, Xiong Y et al. , CRL4A-FBXW5-mediated degradation of DLC1 Rho GTPase-activating protein tumor suppressor promotes non-small cell lung cancer cell growth. Proc Natl Acad Sci U S A. 2013;11042:16868–16873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zheng L, Dengler TJ, Kluger MS et al. , Cytoprotection of human umbilical vein endothelial cells against apoptosis and CTL-mediated lysis provided by caspase-resistant Bcl-2 without alterations in growth or activation responses. J Immunol. 2000;1649:4665–4671. [DOI] [PubMed] [Google Scholar]
- 24. Yamaguchi N, Oyama M, Kozuka-Hata H et al. , Involvement of A20 in the molecular switch that activates the non-canonical NF-small ka, CyrillicB pathway. Sci Rep. 2013;3:2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Thangjam GS, Dimitropoulou C, Joshi AD et al. , Novel mechanism of attenuation of LPS-induced NF-kappaB activation by the heat shock protein 90 inhibitor, 17-N-allylamino-17-demethoxygeldanamycin, in human lung microvascular endothelial cells. Am J Respir Cell Mol Biol. 2014;505:942–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Eagle H, Levine EM.. Growth regulatory effects of cellular interaction. Nature. 1967;2135081:1102–1106. [DOI] [PubMed] [Google Scholar]
- 27. Dupont S, Morsut L, Aragona M et al. , Role of YAP/TAZ in mechanotransduction. Nature. 2011;4747350:179–183. [DOI] [PubMed] [Google Scholar]
- 28. Kim NG, Gumbiner BM.. Adhesion to fibronectin regulates Hippo signaling via the FAK-Src-PI3K pathway. J Cell Biol. 2015;2103:503–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hirate Y, Hirahara S, Inoue K et al. , Polarity-dependent distribution of angiomotin localizes Hippo signaling in preimplantation embryos. Curr Biol. 2013;2313:1181–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yu FX, Zhao B, Panupinthu N et al. , Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell. 2012;1504:780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Irvine KD. Integration of intercellular signaling through the Hippo pathway. Semin Cell Dev Biol. 2012;237:812–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. McClatchey AI, Yap AS.. Contact inhibition (of proliferation) redux. Curr Opin Cell Biol. 2012;245:685–694. [DOI] [PubMed] [Google Scholar]
- 33. Piccolo S, Dupont S, Cordenonsi M.. The biology of YAP/TAZ: Hippo signaling and beyond. Physiol Rev. 2014;944:1287–12312. [DOI] [PubMed] [Google Scholar]
- 34. Stoker MG. Role of diffusion boundary layer in contact inhibition of growth. Nature. 1973;2465430:200–203. [DOI] [PubMed] [Google Scholar]
- 35. Hanahan D, Weinberg RA.. The hallmarks of cancer. Cell. 2000;1001:57–70. [DOI] [PubMed] [Google Scholar]
- 36. Shinzawa K, Watanabe Y, Akaike T.. Primary cultured murine hepatocytes but not hepatoma cells regulate the cell number through density-dependent cell death. Cell Death Differ. 1995;22:133–140. [PubMed] [Google Scholar]
- 37. Lukasik D, Wilczek E, Wasiutynski A et al. , Deleted in liver cancer protein family in human malignancies. Oncol Lett. 2011;25:763–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tripathi V, Popescu NC, Zimonjic DB.. DLC1 suppresses NF-kappaB activity in prostate cancer cells due to its stabilizing effect on adherens junctions. Springerplus. 2014;3:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hippenstiel S, Schmeck B, Seybold J et al. , Reduction of tumor necrosis factor-alpha (TNF-alpha) related nuclear factor-kappaB (NF-kappaB) translocation but not inhibitor kappa-B (Ikappa-B)-degradation by Rho protein inhibition in human endothelial cells. Biochem Pharmacol. 2002;64(5-6):971–977. [DOI] [PubMed] [Google Scholar]
- 40. Huang W, Liu J, Feng X et al. , DLC-1 induces mitochondrial apoptosis and epithelial mesenchymal transition arrest in nasopharyngeal carcinoma by targeting EGFR/Akt/NF-kappaB pathway. Med Oncol. 2015;324:115. [DOI] [PubMed] [Google Scholar]
- 41. Daniel S, Arvelo MB, Patel VI et al. , A20 protects endothelial cells from TNF-, Fas-, and NK-mediated cell death by inhibiting caspase 8 activation. Blood. 2004;1048:2376–2384. [DOI] [PubMed] [Google Scholar]
- 42. Lee EG, Boone DL, Chai S et al. , Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science. 2000;2895488:2350–2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sakakibara S, Espigol-Frigole G, Gasperini P et al. , A20/TNFAIP3 inhibits NF-kappaB activation induced by the Kaposi's sarcoma-associated herpesvirus vFLIP oncoprotein. Oncogene. 2013;3210:1223–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Won M, Park KA, Byun HS et al. , Novel anti-apoptotic mechanism of A20 through targeting ASK1 to suppress TNF-induced JNK activation. Cell Death Differ. 2010;1712:1830–1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Guan Z, Basi D, Li Q et al. , Loss of redox factor 1 decreases NF-kappaB activity and increases susceptibility of endothelial cells to apoptosis. Arterioscler Thromb Vasc Biol. 2005;251:96–101. [DOI] [PubMed] [Google Scholar]
- 46. Guo S, Messmer-Blust AF, Wu J et al. , Role of A20 in cIAP-2 protection against tumor necrosis factor alpha (TNF-alpha)-mediated apoptosis in endothelial cells. Int J Mol Sci. 2014;153:3816–3833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dixit VM, Green S, Sarma V et al. , Tumor necrosis factor-alpha induction of novel gene products in human endothelial cells including a macrophage-specific chemotaxin. J Biol Chem. 1990;2655:2973–2978. [PubMed] [Google Scholar]
- 48. Yang J, Kantrow S, Sai J et al. , INK4a/ARF [corrected] inactivation with activation of the NF-kappaB/IL-6 pathway is sufficient to drive the development and growth of angiosarcoma. Cancer Res. 2012;7218:4682–4695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Corey RM, Swett K, Ward WG.. Epidemiology and survivorship of soft tissue sarcomas in adults: A national cancer database report. Cancer Med. 2014;35:1404–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jhaveri K, Ochiana SO, Dunphy MP et al. , Heat shock protein 90 inhibitors in the treatment of cancer: Current status and future directions. Expert Opin Investig Drugs. 2014;235:611–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.