

Abstract
Objective
Neurodegeneration with brain iron accumulation (NBIA) represents a distinctive phenotype of neurodegenerative disease for which several causative genes have been identified. The spectrum of neurologic disease associated with mutations in NBIA genes is broad, with phenotypes that range from infantile neurodegeneration and death in childhood to adult-onset parkinsonism-dystonia. Here we report the discovery of a novel gene that leads to a distinct form of NBIA.
Methods
Using autozygosity mapping and candidate gene sequencing, we identified mutations in the fatty acid hydroxylase gene FA2H, newly implicating abnormalities of ceramide metabolism in the pathogenesis of NBIA.
Results
Neuroimaging demonstrated T2 hypointensity in the globus pallidus, confluent T2 white matter hyperintensities, and profound pontocerebellar atrophy in affected members of two families. Phenotypically, affected family members exhibited spastic quadriparesis, ataxia, and dystonia with onset in childhood and episodic neurological decline. Analogous to what has been reported previously for PLA2G6, the phenotypic spectrum of FA2H mutations is diverse based on our findings and those of prior investigators, because FA2H mutations have been identified in both a form of hereditary spastic paraplegia (SPG35) and a progressive familial leukodystrophy.
Interpretation
These findings link white matter degeneration and NBIA for the first time and implicate new signaling pathways in the genesis of NBIA.
Neurodegeneration with brain iron accumulation (NBIA) characterizes a group of neurodegenerative disorders that feature progressive extrapyramidal deterioration and excessive iron deposition in several brain regions, most consistently the globus pallidus.1 Subtypes of NBIA have been associated with mutations in the genes encoding ferritin light chain2 and ceruloplasmin.3 Our group has previously identified the causative genes for pantothenate kinase-associated neurodegeneration (PKAN)4 and neuroaxonal dystrophy (NAD),5 NBIA subtypes associated with inherited defects of lipid metabolism, as is a newly described NBIA disorder.6
Neuropathologically, NBIA features significant deposits of extracellular and perivascular iron, typically in the globus pallidus, but variably including the substantia nigra.7 These regions of iron deposition correspond directly to areas of paramagnetic signal hypointensity on T2-weighted magnetic resonance imaging (MRI) images. Neuroaxonal spheroids are also a prominent feature of both PKAN and INAD.8 Other neuropathologic findings observed in NBIA, such as the presence of synuclein-positive Lewy bodies and dystrophic neurites9,10 and tau-reactive neurofibrillary tangles,8,11 indicate that the mechanisms of neurodegeneration in NBIA overlap with those at work in more common sporadic neurodegenerative disorders, suggesting that NBIA may serve as a valuable monogenic model of neurodegeneration.
Onset is often in childhood but can vary widely with forms of NBIA reported in the elderly.12 Extrapyramidal findings typically include dystonia and/or parkinsonism. A progressive dementia may be present. Ophthalmologic features, such as pigmentary retinopathy in PKAN or optic atrophy in INAD, are characteristic.
Many patients with clinical and/or neuroimaging features of NBIA do not have demonstrable mutations in any of the genes known to cause NBIA, even when duplications/deletions are considered,8 indicating that further genes await discovery in this subpopulation with “idiopathic NBIA.” Phenotypic overlap is common and it is likely that several disorders with distinct genetic etiologies are currently classified as idiopathic NBIA. In addition, mutations in the NBIA gene PLA2G6 have recently been shown to lead to a complex movement disorder phenotype without iron accumulation,13 highlighting the heterogeneity within this class of disorders. Although the variability associated with the condition challenges diagnosis and classification at the current time, with further delineation of the genetic causes of subtypes of NBIA previously unappreciated links between various forms of the disorder have begun to emerge and will likely lead to novel insights into shared pathomechanisms underlying neurodegeneration.
In the present studies we used autozygosity mapping to identify a novel gene in a multiplex consanguineous family with idiopathic NBIA. A homozygous mutation in FA2H was then identified in a second family with clinical NBIA. The identification of this gene further expands the NBIA phenotype while also suggesting previously unrecognized links between forms of NBIA.
Patients and Methods
Clinical Data
FAMILY 1
The probands were three affected brothers from a multiplex consanguineous Italian family referred by local physicians with a clinical diagnosis of idiopathic NBIA and a remarkably similar clinical course (Fig 1). All had normal gestation and birth, and normal early attainment of language and motor milestones. However, in all three brothers gait impairment and disequilibrium leading to frequent falls was noted between age four and five. Neurological examination at that time disclosed mild spastic paraparesis and dysmetria. An acquired alternating divergent strabismus was noted shortly thereafter. Dysarthric speech became evident, associated with abnormal prosody leading to a “singsong” voice. Marked, persistent xeroderma evolved, worst in the lower extremities. Despite this neuromotor decline, cognitive function was preserved and the children performed quite well in school with adaptive measures until age 10.
FIGURE 1.
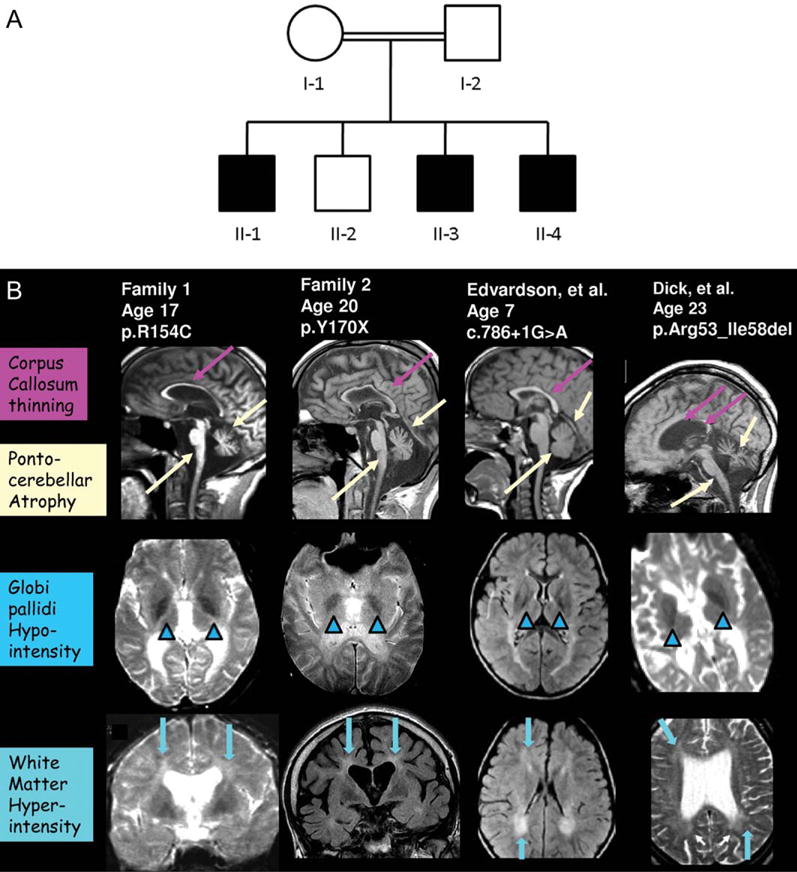
Features of FAHN. (A) Partial pedigree of family 1. (B) MRI features of FAHN. Column 1 depicts MRI images from the index family; Column 2 illustrates findings in the second family identified; Column 3 depicts findings from the case previously published by Edvardson et al;19 Column 4 features MRI findings from Dick et al.20 Hypointensity of the globus pallidus, consistent with iron deposition, is seen on coronal and sagittal T2-weighted images (arrowheads). White matter hyperintensity is demonstrable as well (arrows). Profound pontocerebellar atrophy, mild cerebral atrophy, and thinning of the corpus callosum is also evident (long arrows).
Over the next several years a progressive decline ensued, with worsening ataxia, dysmetria, and spastic quadriparesis. Long periods of relative stability were punctuated by paroxysmal episodes of deterioration associated with unexplained hypopyrexia. There was no clear association of these declines with intercurrent illness. Ophthalmologic examination disclosed asymmetric optic atrophy, with more prominent temporal pallor. Repeat examination during the teenage years demonstrated lateral-beating nystagmus, ocular apraxia, dysphonia, dysmetria, pyramidal tract signs (prolonged clonus, bilateral Babinski signs) and spastic quadriplegia that prohibited independent ambulation. The condition led to progressive scoliosis, dysphagia, and recurrent episodes of aspiration pneumonitis that ultimately necessitated gastrostomy tube placement. At the time of last follow-up, two of the affected siblings had died from respiratory complications of their neurologic disease in their late 20s. No postmortem examination was performed. One of the brothers survives; neurologically, he has severe spastic quadriplegia, anarthria, and acquired epilepsy. However, cognition is relatively spared, although formal testing has not been performed.
MRI demonstrated T2 hypointensity in the globus pallidus bilaterally, consistent with iron deposition. A profound pontocerebellar atrophy was evident, in addition to mild generalized cortical atrophy. Subsequent MRIs demonstrated atrophy of the medulla as well. Confluent periventricular T2 white matter hyperintensities were also observed along with thinning of the corpus callosum (Fig 1).
Electroencephalogram (EEG) was unremarkable in childhood, although two brothers developed seizures in their 20s during the later stages of their disease. Electromyography (EMG) and nerve conduction velocities were normal. Bone marrow biopsy, performed in the oldest brother, demonstrated a coarse, PAS-positive granular cytoplasm in clumped macrophages (Supplementary Figure 1), clinically thought to indicate a lysosomal storage disorder, although follow-up testing of lysosomal enzymes was normal. “Sea-blue histiocytes” have previously been reported in NBIA14 and may be a feature of PKAN.15 Serum lipids and glucose were normal, as were lactate and pyruvate. Ferritin, ceruloplasmin, and copper levels were normal. Immunoglobulin subclasses and α-fetoprotein were unremarkable. Acanthocytes were not observed on routine blood smear. Karyotype was 46, XY.
FAMILY 2
The second family was an Albanian kindred with two affected brothers from the same small village but without known consanguinity. The first brother was born at term without complications, with normal early developmental milestones. He presented with seizures at age 2 years and began having falls at age 4. He developed progressive ataxia, spastic quadriparesis, and dystonia that lead to loss of independent ambulation at age 9. His speech began to deteriorate at 8 years old. At age 20 he had spastic quadriparesis with wheelchair dependence and profound ataxia and generalized dystonia. Mild cognitive impairment was present. Examination disclosed pyramidal tract signs with hyperreflexia, scoliosis, and bowel and bladder incontinence. Bradylalia and significant dysarthria were evident. Ophthalmologic examination demonstrated divergent strabismus, abnormal ocular motility, and optic nerve pallor with normal retina. EEG demonstrated diffuse slowing with superimposed excessive fast activity without epileptiform features.
The clinical presentation and course of the second brother was remarkably similar. He was born without complications, and also developed normally until age 3, when increasing falls were noted. He lost the ability to walk independently at age 4. Seizures never occurred. Examination at age 15 demonstrated spastic quadriparesis with pyramidal tract signs and scoliosis, and significant ataxia and dystonia. Speech was bradylalic and dysarthric. Mild cognitive impairment was observed, as was bowel and bladder incontinence. Strabismus and abnormal eye movements were noted, but optic nerve appearance was felt to be normal.
Human Subjects
Subjects were enrolled after approval was granted by the Institutional Review Board of the Oregon Health & Science University. Clinical information was provided by referring physicians.
Genotyping and Autozygosity Mapping
DNA was isolated from whole blood using standard methods. Affected and unaffected family members were genotyped using the Illumina HumanCNV370-Quad BeadChip per the manufacturer’s instructions. Hybridized arrays were scanned using the iScan system. Autozygosity mapping was performed as previously described using the Homozygosity Detector plug-in software within the BeadStudio suite.16 Regions of shared homozygosity that segregated with disease were visually identified using the Illumina Genome Viewer tool within the BeadStudio suite. Candidate genes were then ranked according to biological function and homology to known NBIA genes.
DNA Sequencing
Primers spanning all seven intron–exon boundaries of FA2H were designed and used to amplify the regions of interest (sequences available upon request). Amplicons were produced from genomic DNA, and sequencing was performed using an ABI 3730 DNA Sequencer (Applied Biosystems, Foster City, CA) as previously described.13 Sequence comparison to reference sequence was performed using Sequencher, v.4.9 (GeneCodes, Ann Arbor, MI).
Protein Function Prediction
We used several in silico applications to predict the effects of the amino acid substitutions identified on protein function, including SIFT (Sorting Intolerant From Tolerant, http://blocks.fhcrc.org/sift/SIFT), PolyPhen (Polymorphism Phenotyping, http://coot.embl.de/PolyPhen/), and SNAP (http://cubic.bioc.columbia.edu/services/snap/).
Histology
Six-μm-thick sections were cut from the available bone marrow tissue block and stained with hematoxylin and eosin.
FA2H Enzyme Activity Assay
Enzyme activity was measured using established methods18. In brief, site-directed mutagenesis was performed using the Quik-Change kit (Agilent, Santa Clara, CA) per the manufacturer’s instructions and subcloned into pcDNA3 as previously described.19 Sequence identity was verified by DNA sequencing. Liquid chromatography-mass spectrometry (LC/MS) was performed as previously described.19
Western Blotting
Epstein–Barr virus (EBV)-transformed lymphoblasts were established from peripheral blood and maintained using standard methods. D6P2T and HeLa cells were transfected with an R154C mutant construct (pcDNA3-hFA2H R154C) or wildtype FA2H (pcDNA3-hFA2H) using FuGENE 6 per the manufacturer’s instructions. Western blotting was performed using anti-FA2H primary antibody as previously described.17
Results
Call rates were >99%. Forty-five autozygous segments were identified in genotyped samples using a cutoff value of at least 100 consecutive single nucleotide polymorphisms (SNPs). Seventeen of these regions of identity by descent were common to at least two affected family members. This number was further decreased by including only those segments with a length ≥1 Mb. Of these blocks of homozygosity, only a single autozygous segment (1.84 Mb in size) on chromosome 16q22-23 was common to all three affected siblings. This block was not present in the unaffected brother. This region of interest, flanked by markers rs28759365 and rs981231, contained 96 known genes and predicted transcripts (Fig 2).
FIGURE 2.
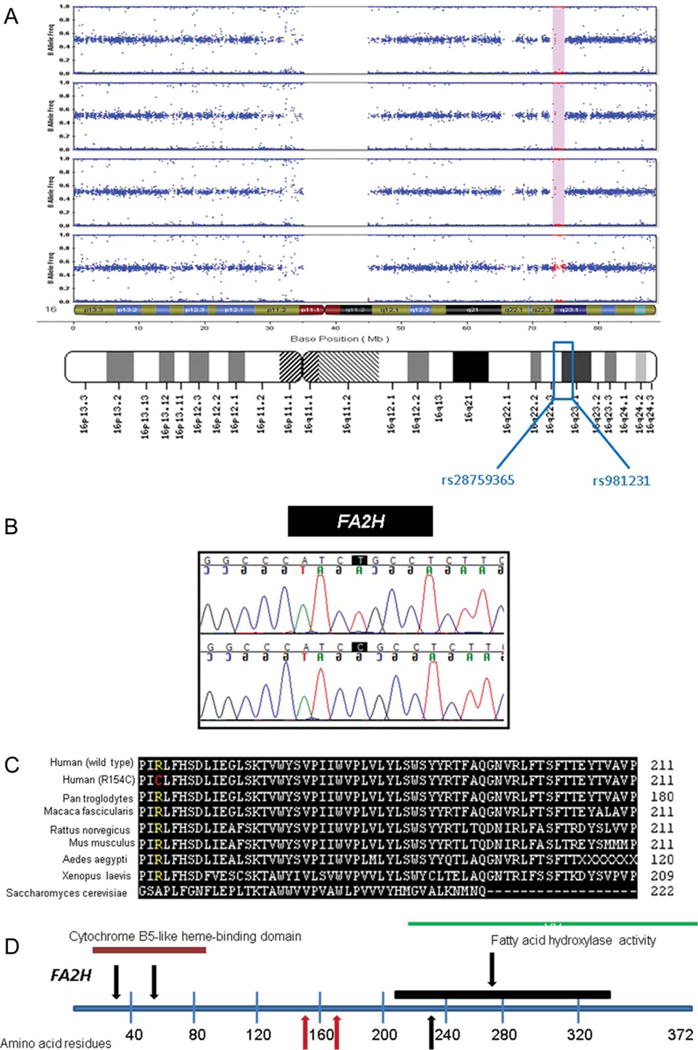
Identification and analysis of a novel FA2H mutation. (A) A shared block of homozygosity nearly 2 Mb in length, common to all three affected members of the index family but absent in the unaffected brother genotyped, was identified. (B) FA2H was selected as a prominent candidate gene given its critical role in lipid metabolism; sequencing identified a c.460C>T homozygous transition in all three affected family members. (C) By aligning homologs across species using ClustalW (http://www.ebi.ac.uk/Tools/clustalw2/index.html), amino acid sequence conservation across species was compared; the mutation identified (R154C) was predicted to alter a highly conserved arginine residue. (D) Comparison of this novel mutation with other identified mutations19,20 (red arrows = Family 1 and 2; black arrows = previously reported mutations; black line indicates extent of splice site mutation predicted to result in skipping of exons 5 and 6).
FA2H was chosen as a likely candidate gene based on its biologic function in lipid homeostasis. DNA sequencing revealed a homozygous c.460C>T missense mutation in the three affected brothers of the index family, whereas the healthy brother was a heterozygote. This mutation resulted in a homozygous p.R154C substitution in exon 3 of FA2H, implicating this gene for the first time in the pathogenesis of NBIA (Fig 2).
The resultant amino acid substitution places a cysteine at the site of a normally highly conserved arginine residue. This transition was predicted to be deleterious by all three modeling algorithms employed (PolyPhen, SIFT, and SNAP), presumably via an effect on protein folding. This nucleotide change was absent in 250 individuals of Northern or Southern European descent, consistent with a pathogenic variant.
Subsequently, sequencing of FA2H was performed in the second family and demonstrated a homozygous c.509_510delAC mutation in both affected patients. This mutation causes a frameshift and resultant premature truncation of the protein (Y170X).
LC/MS analysis of COS7 cells transfected with pcDNA-hFA2H R154C demonstrated no significant difference in enzyme activity (data not shown). However, western blot performed in D6P2T cells transfected with pcDNA-hFA2H R154C demonstrated decreased protein abundance, suggesting the mutation reduces stability of the mRNA or translated protein (Supplementary Figure 2).
In order to determine the relative frequency of FA2H mutations in idiopathic NBIA, sequencing was performed in a cohort of 43 patients with PANK2, PLA2G6-negative idiopathic NBIA and a second cohort of seven patients with PLA2G6-negative INAD. The majority of this cohort had neither white matter hyperintensities nor cerebellar atrophy, and none of these patients had apparent pathogenic sequence alterations in FA2H. Two additional consanguineous pedigrees with PLA2G6-negative INAD were also genotyped; neither showed linkage to 16q22-23. These results indicate that FA2H mutations are likely to represent a rare cause of NBIA.
Discussion
The present studies identify mutations in FA2H as a cause of NBIA. We propose that this disorder represents a distinct subtype of NBIA, which we refer to as fatty acid hydroxylase-associated neurodegeneration (FAHN). Phenotypically, affected patients demonstrated features similar to those observed in NAD (Table). However, the age of onset is typically later and progression slower than seen in NAD. Intellect is relatively spared as well. The peripheral neuropathy typical of NAD was not observed in FAHN, and atypical features (confluent white matter lesions, brainstem atrophy) were observed in FAHN, but are not seen in INAD. The lack of peripheral neuropathy may be related to the presence of a second fatty acid hydroxylase activity in peripheral tissue, but not in the central nervous system (CNS).18 The stepwise deterioration observed in our patients is often seen in PKAN, and is a common feature in many neurometabolic disorders, although the precise trigger remains unknown.
TABLE.
Clinical and Neuroimaging Comparison of NAD and FAHN
| NAD | FAHN | |
|---|---|---|
| Neuromotor features | ||
| Ataxia, dysmetria | Profound | Profound |
| Dystonia | Mild | Mild-severe |
| Spastic quadriplegia | Prominent | Prominent |
| Pyramidal tract signs | Prominent | Prominent |
| Axial hypotonia | Prominent | Not noted |
| Neuro-ophthalmologic features | ||
| Optic atrophy | Prominent | Mild-severe |
| Nystagmus | Present | Present |
| Acquired strabismus | Present | Present |
| Neuroimaging findings | ||
| Cerebellar atrophy | Prominent | Prominent |
| Brainstem atrophy | Absent | Prominent |
| Globus pallidus iron deposition | Variable | Variable |
| Other features | ||
| Seizures | Occasional | Occasional |
| Peripheral neuropathy | Prominent | Absent |
| Neuroaxonal spheroids | Prominent | ??? |
| Excessive beta activity (EEG) | Prominent | Present |
FA2H produces 2-hydroxylated fatty acids for incorporation into 2-hydroxydihydroceramide and 2-hydroxyceramide.18 These ceramide species in turn serve as precursors for the synthesis of galactosylceramides and sulfatides, essential lipid components of normal myelin. Mutations in FA2H have previously been associated with both a familial leukodystrophy19 and, more recently, with a hereditary spastic paraplegia (HSP) phenotype (SPG35).20 It is noteworthy that a leukodystrophy, hereditary spastic paraplegia, and/or NBIA phenotype may all result from mutations in FA2H, as clinically these disorders appear distinct and likely would not have been suspected to be allelic diseases.
A critical role of FA2H in normal myelin maintenance is supported by these findings in human neurological disease, as affected patients developed normally initially then gradually accumulated disability in conjunction with white matter hyperintensities. Ferritin has been shown to associate with myelin in normal cortex21 and to be altered in other conditions associated with brain iron deposition, such as Huntington’s disease22 and multiple sclerosis.23 It is intriguing to speculate that the anomalous myelin produced in FA2H mutations might also alter CNS iron homeostasis by disrupting myelin integrity. However, a heme-binding role and a nonheme di-iron active site are also predicted for FA2H based on sequence homology (amino acids 39–46, including a conserved H-P-G-G motif, and a conserved histidine motif in the C-terminal membrane-bound domain, respectively), and a more direct interaction with iron-containing moieties may also play a role in the iron accumulation observed in FAHN.
In addition to its role in the structural maintenance of myelin, a role for FA2H in lipid signal transduction has more recently been highlighted.18 In particular, FA2H-mediated signaling has been shown to regulate cell cycle exit in rat D6P2T schwannoma cells via effects on cyclin-dependent kinase (cdk) inhibitor expression.24 By altering cdk inhibitor expression in terminally differentiated cells such as neurons, mutations in FA2H may lead to premature apoptosis.25 In addition, through effects on 2-hydroxyceramide production, mutations in FA2H could affect intracellular ceramide pool composition,18 with pleiotropic downstream consequences on fundamental cellular processes including protein and lipid turnover (Fig 3), which may be altered in NBIA. Furthermore, given the emerging importance of ceramide signaling in Lewy body disease pathophysiology,26 an important role for ceramide-mediated modulation of synuclein metabolism is becoming evident, with clear relevance to neurodegenerative disease.27,28 Because no postmortem tissue was available for analysis in our patients, the neuropathologic features of FAHN remain to be defined.
FIGURE 3.
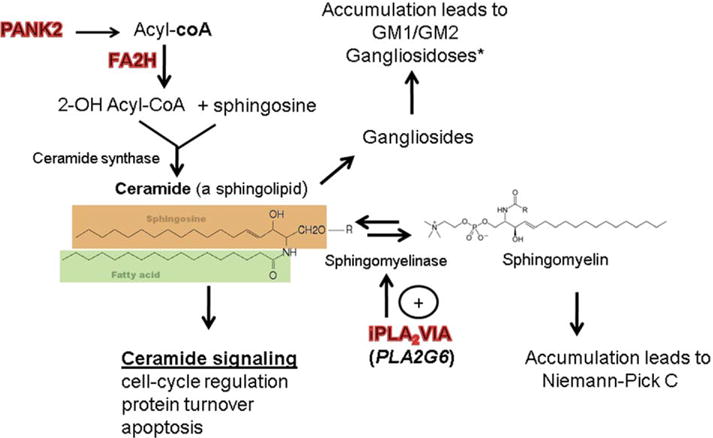
Ceramides connect several neurodegenerative disorders. *Some lysosomal storage disorders, such as the gangliosidoses, features significant alterations in brain iron composition.34,35
The recent identification of Kufor Rakeb (KR) syndrome as a form of NBIA29 has a number of interesting implications. The ATP13A2 gene, mutated in KR disease, has been implicated as a lysosomal divalent cation transporter.30 Impaired ganglioside signaling may impair normal lysosomal function,31 resulting in impaired membrane lipid recycling, and impaired autophagy and accentuated α-synuclein toxicity.32 These potential connections provide tantalizing links between various aspects of NBIA biology, but await further elucidation and characterization.
Iron deposition in patients affected by FA2H mutations appears to be variable,19,20 but may become more evident over time. It was observed in both patients with the R154C and the Y170X mutations. In addition, upon review of the MRIs from previously published cases of FA2H mutation, there was definite evidence for iron deposition noted in one of the cases from the family reported by Edvardson et al19 (Fig 1). There was more equivocal evidence for iron deposition in the cases reported by Dick et al20 (Fig 1); notably, these cases also featured a milder phenotype. This scenario is analogous to what is observed with mutations in PLA2G6, where iron accumulation may be less striking than in PKAN, may occur later in the course after symptoms have already become evident, and may not occur in all patients with demonstrable mutations.10,13
Future work will seek to better determine the frequency of FA2H mutations within the population of patients with idiopathic NBIA, and will include duplication/deletion analysis in order to detect any patients that may have been missed by conventional sequencing techniques. The availability of a mouse model33 (Hama, unpubl.) will facilitate ongoing efforts to better define the pathophysiologic consequences of mutations in FA2H.
In conclusion, the present study identifies mutations in FA2H as a cause of NBIA for the first time, and extends the phenotypic spectrum associated with mutations in this gene while defining a new subtype of NBIA. Our findings, when combined with earlier studies implicating FA2H mutations in both hereditary spastic paraplegia and leukodystrophy, challenge clinical diagnostic algorithms while suggesting a provocative new pathophysiologic link between degenerative white matter disorders and NBIA.
Supplementary Material
Acknowledgments
Portions of this work were funded by pilot grant awards from the NBIA Disorders Association and Associazione Italiana Sindromi Neurodegenerative da Accumulo di Ferro (M.C.K., S.J.H.), the Oregon Medical Research Foundation (M.C.K.), an American Philosophical Society Daland Award (M.C.K.), an American Academy of Neurology Clinical Research Training Fellowship (M.C.K.), and a Parkinson’s Disease Society (UK) award to C.P.R. Supported also by the Association Internationale de Dystrophie Neuro Axonale Infantile (S.J.H.), the National Organization for Rare Disorders (NORD) (S.J.H.), NIH grants R01EY12353 and R01HD050832 (S.J.H.), and the Joint Research Fund of the Hebrew University and Hadassah Medical Organization (S.E.). H.H. and J.H. are supported by MRC Returning Scientist awards. H.H. is supported by MRC grant G0802760. M.C.K. is supported by NIH Pediatric LRP award UWXY3099. This publication was made possible with support from the Oregon Clinical and Translational Research Institute (OCTRI), grant number UL1 RR024140 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH), and NIH Roadmap for Medical Research.
We thank the patients and their families; without their participation this work would not have been possible. We thank the referring physicians and D. Weeks and K. Larkin for assistance with histologic analysis of the bone marrow specimens.
Footnotes
Additional Supporting Information can be found in the online version of this article.
Potential Conflicts of Interest
Nothing to report.
References
- 1.Thomas M, Hayflick SJ, Jankovic J. Clinical heterogeneity of neurodegeneration with brain iron accumulation (Hallervorden-Spatz syndrome) and pantothenate kinase-associated neurodegeneration. Mov Disord. 2004;19:36–42. doi: 10.1002/mds.10650. [DOI] [PubMed] [Google Scholar]
- 2.Curtis ARJ, Fey C, Morris CM, et al. Mutation in the gene encoding ferritin light polypeptide causes dominant adult-onset basal ganglia disease. Nat Genet. 2001;28:350–354. doi: 10.1038/ng571. [DOI] [PubMed] [Google Scholar]
- 3.Harris ZL, Takahashi Y, Miyajima H, et al. Aceruloplasminemia: molecular characterization of this disorder of iron metabolism. Proc Natl Acad Sci USA. 1995;92:2539–2543. doi: 10.1073/pnas.92.7.2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhou B, Westaway SK, Levinson B, et al. A novel pantothenate kinase gene (PANK2) is defective in Hallervorden-Spatz syndrome. Nat Genet. 2001;28:345–349. doi: 10.1038/ng572. [DOI] [PubMed] [Google Scholar]
- 5.Morgan NV, Westaway SK, Morton JE, et al. PLA2G6, encoding a phospholipase A2, is mutated in neurodegenerative disorders with high brain iron. Nat Genet. 2006;38:752–754. doi: 10.1038/ng1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hartig MB, Iuso A, Hempel M, et al. Identification of a second major locus for neurodegeneration with brain iron accumulation. Presented at the 59th Annual Meeting of the American Society of Human Genetics; Honolulu, HI. [Google Scholar]
- 7.Hayflick SJ, Hartman M, Coryell J, et al. Brain MRI in neurodegeneration with brain iron accumulation with and without PANK2 mutations. AJNR Am J Neuroradiol. 2006;27:1230–1233. [PMC free article] [PubMed] [Google Scholar]
- 8.Gregory A, Polster BJ, Hayflick SJ. Clinical and genetic delineation of neurodegeneration with brain iron accumulation. J Med Genet. 2009;46:73–80. doi: 10.1136/jmg.2008.061929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Galvin JE, Giasson B, Hurtig HI, et al. Neurodegeneration with brain iron accumulation, type 1 is characterized by alpha-, beta-, and gamma-synuclein neuropathology. Am J Pathol. 2000;157:361–368. doi: 10.1016/s0002-9440(10)64548-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gregory A, Westaway SK, Holm IE, et al. Neurodegeneration associated with genetic defects in phospholipase A(2) Neurology. 2008;71:1402–1409. doi: 10.1212/01.wnl.0000327094.67726.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wakabayashi K, Fukushima T, Koide R, et al. Juvenile-onset generalized neuroaxonal dystrophy (Hallervorden-Spatz disease) with diffuse neurofibrillary and lewy body pathology. Acta Neuropathol. 2000;99:331–336. doi: 10.1007/s004010050049. [DOI] [PubMed] [Google Scholar]
- 12.Santillo AF, Skoglund L, Lindau M, et al. Frontotemporal dementia-amyotrophic lateral sclerosis complex is simulated by neurodegeneration with brain iron accumulation. Alzheimer Dis Assoc Disord. 2009;23:298–300. doi: 10.1097/WAD.0b013e3181a2b76b. [DOI] [PubMed] [Google Scholar]
- 13.Paisan-Ruiz C, Bhatia KP, Li A, et al. Characterization of PLA2G6 as a locus for dystonia-parkinsonism. Ann Neurol. 2009;65:19–23. doi: 10.1002/ana.21415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Swaiman KF, Smith SA, Trock GL, Siddiqui AR. Sea-blue histiocytes, lymphocytic cytosomes, movement disorder and 59Fe-uptake in basal ganglia: Hallervorden-Spatz disease or ceroid storage disease with abnormal isotope scan? Neurology. 1983;33:301–305. doi: 10.1212/wnl.33.3.301. [DOI] [PubMed] [Google Scholar]
- 15.Cangül H, Ozdemir O, Yakut T, et al. Pantothenate kinase-associated neurodegeneration: molecular confirmation of a Turkish patient with a rare frameshift mutation in the coding region of the PANK2 gene. TurkJ Pediatr. 2009;51:161–165. [PubMed] [Google Scholar]
- 16.Gibbs JR, Singleton A. Application of genome-wide single nucleotide polymorphism typing: simple association and beyond. PLoS Genet. 2006;2:e150. doi: 10.1371/journal.pgen.0020150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alderson NL, Rembiesa BM, Walla MD, et al. The human FA2H gene encodes a fatty acid 2-hydroxylase. J Biol Chem. 2004;279:48562–48568. doi: 10.1074/jbc.M406649200. [DOI] [PubMed] [Google Scholar]
- 18.Hama H. Fatty acid 2-hydroxylation in mammalian sphingolipid biology. Biochim Biophys Acta. 2010;1801:405–414. doi: 10.1016/j.bbalip.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Edvardson S, Hama H, Shaag A, et al. Mutations in the fatty acid 2-hydroxylase gene are associated with leukodystrophy with spastic paraparesis and dystonia. Am J Hum Genet. 2008;83:643–648. doi: 10.1016/j.ajhg.2008.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dick KJ, Eckhardt M, Paisán-Ruiz C, et al. Mutation of FA2H underlies a complicated form of hereditary spastic paraplegia (SPG35) Hum Mutat. 2010;31:E1251–1260. doi: 10.1002/humu.21205. [DOI] [PubMed] [Google Scholar]
- 21.Fukunaga M, Li TQ, van Gelderen P, et al. Layer-specific variation of iron content in cerebral cortex as a source of MRI contrast. Proc Natl Acad Sci USA. 2010;107:3834–3839. doi: 10.1073/pnas.0911177107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bartzokis G, Lu PH, Tishler TA, et al. Myelin breakdown and iron changes in Huntington’s disease: pathogenesis and treatment implications. Neurochem Res. 2007;32:1655–1664. doi: 10.1007/s11064-007-9352-7. [DOI] [PubMed] [Google Scholar]
- 23.Levine SM, Chakrabarty A. The role of iron in the pathogenesis of experimental allergic encephalomyelitis and multiple sclerosis. Ann N YAcad Sci. 2004;1012:252–266. doi: 10.1196/annals.1306.021. [DOI] [PubMed] [Google Scholar]
- 24.Alderson NL, Hama H. Fatty acid 2-hydroxylase regulates cAMP-induced cell cycle exit in D6P2T schwannoma cells. J Lipid Res. 2009;50:1203–1208. doi: 10.1194/jlr.M800666-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sumrejkanchanakij P, Tamamori-Adachi M, Matsunaga Y, et al. Role of cyclin D1 cytoplasmic sequestration in the survival of postmitotic neurons. Oncogene. 2003;22:8723–8730. doi: 10.1038/sj.onc.1206870. [DOI] [PubMed] [Google Scholar]
- 26.Bras J, Singleton A, Cookson MR, Hardy J. Emerging pathways in genetic Parkinson’s disease: potential role of ceramide metabolism in Lewy body disease. FEBS J. 2008;275:5767–5773. doi: 10.1111/j.1742-4658.2008.06709.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stoica BA, Movsesyan VA, Knoblach SM, Faden AI. Ceramide induces neuronal apoptosis through mitogen-activated protein kinases and causes release of multiple mitochondrial proteins. Mol Cell Neurosci. 2005;29:355–371. doi: 10.1016/j.mcn.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 28.Jana A, Hogan EL, Pahan K. Ceramide and neurodegeneration: susceptibility of neurons and oligodendrocytes to cell damage and death. J Neurol Sci. 2009;278:5–15. doi: 10.1016/j.jns.2008.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schneider SA, Paisan-Ruiz C, Quinn NP, et al. ATP13A2 mutations (PARK9) cause neurodegeneration with brain iron accumulation. Mov Disord. 2010;25:979–984. doi: 10.1002/mds.22947. [DOI] [PubMed] [Google Scholar]
- 30.Schmidt K, Wolfe DM, Stiller B, Pearce DA. Cd2+, Mn2+, Ni2+ and Se2+ toxicity to Saccharomyces cerevisiae lacking YPK9p the orthologue of human ATP13A2. Biochem Biophys Res Commun. 2009;383:198–202. doi: 10.1016/j.bbrc.2009.03.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wei J, Fujita M, Nakai M, et al. Protective role of endogenous gangliosides for lysosomal pathology in a cellular model of synucleinopathies. Am J Pathol. 2009;174:1891–1909. doi: 10.2353/ajpath.2009.080680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gitler AD, Chesi A, Geddie ML, et al. Alpha-synuclein is part of a diverse and highly conserved interaction network that includes PARK9 and manganese toxicity. Nat Genet. 2009;41:308–315. doi: 10.1038/ng.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zoller I, Meixner M, Hartmann D, et al. Absence of 2-hydroxylated sphingolipids is compatible with normal neural development but causes late-onset axon and myelin sheath degeneration. J Neurosci. 2008;28:9741–9754. doi: 10.1523/JNEUROSCI.0458-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Autti T, Joensuu R, Aberg L. Decreased T2 signal in the thalami may be a sign of lysosomal storage disease. Neuroradiology. 2007;49:571–578. doi: 10.1007/s00234-007-0220-6. [DOI] [PubMed] [Google Scholar]
- 35.Jeyakumar M, Williams I, Smith D, et al. Critical role of iron in the pathogenesis of the murine gangliosidoses. Neurobiol Dis. 2009;34:406–16. doi: 10.1016/j.nbd.2009.01.015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.