

Abstract
Building on structural and mechanistic studies of lithiated enolates derived from acylated oxazolidinones (Evans enolates) and chiral lithiated amino alkoxides, we found that amino alkoxides amplify the enantioselectivity of aldol additions. The pairing of enantiomeric series affords matched and mismatched stereoselectivities. The structures of mixed tetramers showing 2:2 and 3:1 (alkoxide-rich) stoichiometries are determined spectroscopically. Rate and computational studies provide a viable mechanistic and stereochemical model based on the direct reaction of the 3:1 mixed tetramers, but they raise unanswered questions for the 2:2 mixed aggregates.
Graphical abstract

Introduction
This paper describes the influence of chiral lithiated amino alkoxides on the aldol addition of oxazolidinone-derived lithium enolates (eq 1). The story begins with the seminal 1981 paper by Evans and co-workers describing remarkably selective aldol additions by the boron enolate of a chiral oxazolidinone.1 The impact of Evans’ paper would be difficult to overstate. Applications and subsequent refinements of the so-called Evans aldol and related stereoselective functionalizations of “Evans enolates” are legion.2 One might surmise that lithiated Evans enolates would be central to the plotline,3 but they reside between the more reactive sodium enolates used in alkylations4 and the more selective transition metal and boron enolates used for aldol additions.5 Nonetheless, there are a handful of lithium-based Evans aldol additions, and surprisingly, many are additions to ketones6 rather than aldehydes.7
 |
(1) |
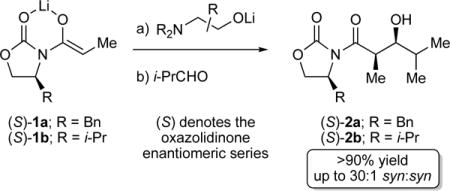
During recent studies, we found that aldol additions to isobutyraldehyde (i-PrCHO) and cyclohexanone afford credible selectivities and yields (Scheme 1).8 Pronounced aging effects9—the influence of time and warming on enolate reactivity before the addition of the electrophile—may have bedeviled synthetic chemists, including a Pfizer process group working on plant scales.6 Independent of the enolate aging effects and in stark contrast to conventional wisdom based on simpler aldol additions,10 the aldolate derived from i-PrCHO is susceptible to a retroaldol reaction that causes a loss of yield and selectivity. However, the cyclohexanone-derived aldolate is robust.8
Scheme 1.
Aldol additions of lithiated Evans enolates.
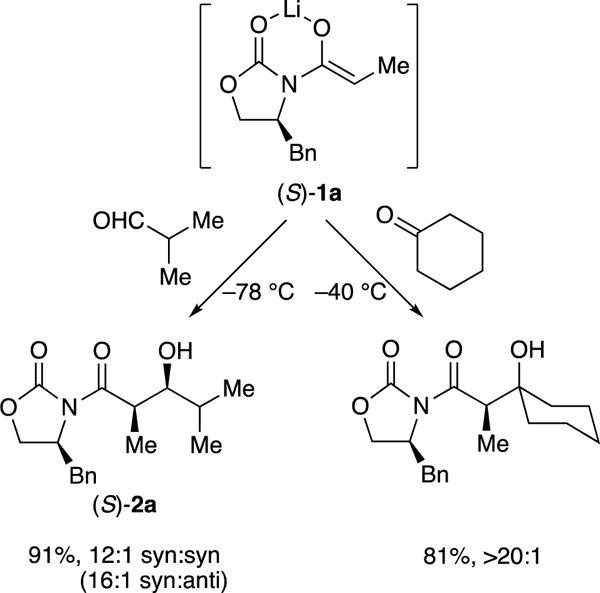
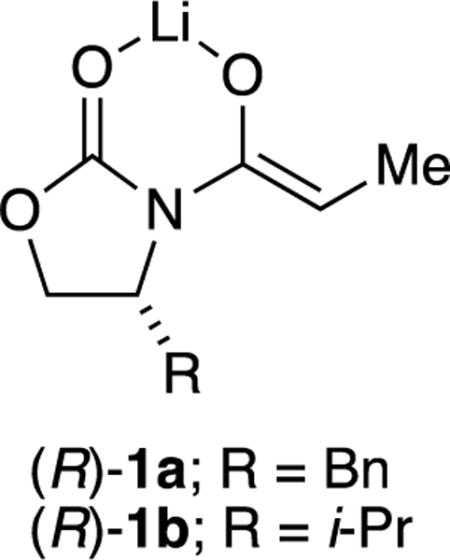
The aging effects of enolate (S)-1a were traced to kinetically formed and highly reactive trisolvated dimers 3 and 4, which formed unreactive unsolvated tetramer 5 on standing at –78 °C or with gentle warming (Scheme 2).8 Rate and computational studies implicated monomer-based transition structure 6. We would be remiss if we did not also mention that computational studies suggested that the H–H interaction highlighted in 6 causes facial selectivity rather than an explicit interaction of the aldehyde with the benzyl moiety. This outcome, too, appears to be incongruent with conventional thinking.
Scheme 2.
Structures of Evans enolates and mechanism of aldol additions.
An increasingly popular and altogether different means of controlling enantioselectivities involves non-covalent auxiliaries—chiral salts that form mixed aggregates in situ.11,12 Such a strategy relies on controlling the complex coordination chemistry of mixed aggregates, which usually requires a combination of determination and luck. An iconic example was reported by the process group at Merck in 1995: an equimolar (2:2) mixture of lithium acetylide 7 and chiral amino alkoxide 8 were added to a ketone with an astonishing 50:1 enantioselectivity en route to 50,000 kg of the reverse transcriptase inhibitor efavirenz (eq 2).13 The Merck group also noted aging effects in which warming before the addition of the ketone was required to optimize the selectivity. In a collaborative effort, we traced the selectivities and aging effects to the slow formation of C2-symmetric 2:2 mixed aggregate 9 and direct 1,2-addition via transition structure 10.14
 |
(2) |
Second-generation reverse transcriptase inhibitors developed at DuPont Pharmaceuticals (now Bristol-Myers Squibb) were prepared by adding lithium acetylides to quinazolinones (eq 3).15 Despite its apparent similarity to the Merck chemistry, the DuPont process relied on caranolate-derived amino alkoxide 11 using 3:1 rather than 2:2 stoichiometry. Structural studies showed the intermediacy of 3:1 (alkoxide-rich) mixed aggregate 12 with a proposed extra-aggregate addition of lithium acetylide.16 Optimizations afforded 2,000 kg of the adduct in >200:1 enantioselectivity before the program was terminated.17
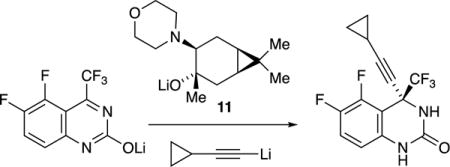 |
(3) |
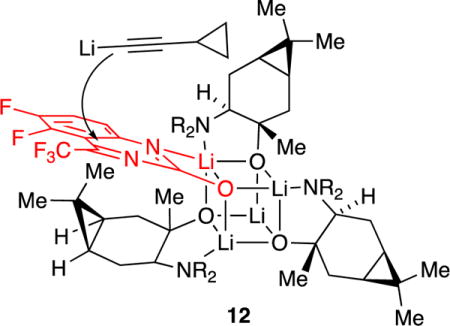
We neither had the skills contemporaneous to the DuPont and Merck studies to characterize amino alkoxides 8 and 11 nor were they needed.13–17 A dozen years later, however, we characterized them as fully chelated tetramers 13 and 14 with S4-symmetric cores (Scheme 3).18 The structures prompted us to ask whether the inherent selectivity of Evans enolates can be amplified by their incorporation into cubic mixed aggregates with chiral alkoxides. It was unclear at the outset whether mixed aggregation would afford discrete structures given the difference in the core symmetries of the enolates and alkoxides, D2d versus S4. Moreover, favorable stereochemical influence was in doubt given that homotetrameric enolate 5 reacts sluggishly via a fleeting monomer rather than via a tetramer.19 In short, we got lucky: tetrameric mixed aggregates form with excellent structural control and react rapidly without dissociation (Scheme 4). This success is by no means simple when viewed in detail. The non-specialist will find summaries of the results in the Discussion.
Scheme 3.

Structures of amino alkoxide homotetramers.
Scheme 4.

Evans aldol addition of lithium enolate mixed aggregates.
Results
Structure Determination: General
The characterization of the enolate–alkoxide mixed tetramers required a multi-pronged approach. Mixed aggregates often display characteristic symmetries (or lack thereof). Symmetry can be broken further using the method of continuous variations,20 also known as the method of Job,21 which has been central to our studies of homoaggregates.22 Although heteroaggregates and mixed aggregates23 often distribute statistically,22 several unusual nonstatistical examples are described below. Pairings of (S)-1a,b and alkoxides 8 or 11 and the subsequent formation of aldol adducts (S)-2a,b are complemented by pairings with antipodal enolates (R)-1a,b.
Stereoisomers of aggregates can be distinguished using two-dimensional NMR spectroscopies (COSY, TOCSY, HSQC, HMBC, and ROESY).24 Detailed discussions of the cross-correlations without an interactive computer interface are beyond the scope of this (or essentially any) paper; the analyses are archived in Supporting Information. We have, however, provided a series of videos rendering the process more tractable. Li–N contacts are shown with 6Li–15N coupling using [ 6Li,15N]11. Density functional theory (DFT) calculations to probe both reactant and transition structures were carried out at the B3LYP/6–31G(d) level with energies from single-point calculations at the MP2 level of theory.25
Most of our efforts focused on carane-derived alkoxide 11 and antipodes of enolates 1a,b, but we also undertook more limited studies of ephedrate-derived alkoxide 8. We begin with a brief description of the results for a simple enolate to establish baseline behaviors. All of the samples described herein required aging at 25 °C for 10 min to ensure the full equilibration of the aggregates.26
Lithium cyclohexenolate (CyOLi)–caranolate derived mixed tetramers
Mixtures of CyOLi and lithium alkoxide (ROLi) 11 in tetrahydrofuran (THF) at varying alkoxide:enolate proportions produced 6Li NMR spectra (Figure 1) consistent with 1:3 (enolate-rich) and 2:2 mixed tetramers (17 and 18) in analogy with the ROLi-acetylide described above.13–17 Tetramer 17 was uniquely consistent with the 1:3 stoichiometry and symmetry. There were, however, two computationally less viable isomers of 18 (not shown). No 3:1 (alkoxide-rich) aggregates were detectable. A Job plot shows some preference for 17 (Figure 2). A distribution of homotetramers and two of three possible mixed tetramers that is otherwise statistical is shown in Figure 3 for comparison.
Figure 1.

6Li NMR spectra of 0.10 M solutions of lithium cyclohexenolate (CyOLi) and 11 in toluene and 0.11 M tetrahydrofuran (THF) at −80 °C with 0.11 M [ 6Li] lithium diisopropylamide ([ 6Li]LDA). The measured mole fractions27 of CyOLi (χCyOLi) are 0.00, 0.13, 0.32, 0.78, and 1.00 for A−E, respectively.
Figure 2.

Job plot showing relative integration of the 6Li resonances (CyOLi)422 (gray), 14 (black), 17 (green), and 18 (red) versus the measured mole fraction of CyOLi (χCyOLi) monitored with 6Li NMR spectroscopy in 0.11 M THF/toluene at −80 °C. The total lithium alkoxide titer (enolate and alkoxide) is constant (0.10 M).
Figure 3.

Simulated Job plot showing a statistical distribution of aggregates A4 (black), A2B2 (red), AB3 (green), and B4 (gray) versus the measured mole fraction of B (χB) for an ensemble.

Structures of caranolate-derived mixed tetramers
Combining an Evans enolate—(S)-1a, (R)-1a, (S)-1b, or (R)-1b—with amino alkoxide 11 produced mixed tetramers (S)-15a,b (R)-15a,b, (S)-16a,b, and (R)-16a,b (Chart 1) in proportions that depended intimately on the choice of toluene versus THF. The phenylalanine-derived “a” series and valine-derived “b” series displayed similar spectroscopic behaviors. Mixed aggregates in the a series are described emblematically as follows.
Chart 1.

Mixed tetramers with 2:2 and 3:1 (alkoxide-rich) stoichiometry.
An equimolar (1:1) mixture of (S)-1a and alkoxide 11 in toluene (Figure 4A,E) afforded a complex distribution of species that, on warming to 25 °C for 10 min and cooling to –80 °C, converged to 2:2 mixed tetramer (S)-15a with high fidelity (Figure 4C). Surprisingly, neither 3:1 nor 1:3 mixed tetramers were observed at any stoichiometry (Figure 4B,D). When [15N]11 was used, the downfield resonance of (S)-15a appeared as a doublet (1.39 ppm, JLi–N = 2.8 Hz) with an accompanying 1:1:1 triplet in the 15N spectrum. This result is consistent with fully chelating amino alkoxide subunits in (S)-15a.
Figure 4.

6Li NMR spectra of 0.10 M solutions of (S)-1a and 11 in toluene and 0.11 M THF/toluene at −80 °C with 0.11 M [ 6Li]LDA. The measured mole fractions of (S)-1a (χ(S)-1a) are 0.00, 0.24, 0.50, 0.74, and 1.00 for A−E, respectively.
The 1:1 pair of 6Li resonances did not distinguish symmetric mixed tetramer (S)-15a from a simpler mixed dimer. To this end, the 6Li spectra of a 1:1 mixture of alkoxide 11 and lithium enolate in which the enolate was equal parts (S)-1a and (R)-1a showed four new resonances (1:1:1:1; Figure 5) consistent with desymmetrized mixed tetramer 19 (eq 4). We completed a Job plot by holding the alkoxide/enolate ratio constant (1:1) and monitoring the 6Li spectra versus mole fractions of enolates (S)-1a and (R)-1a (Figure 6).
 |
(4) |
Figure 5.
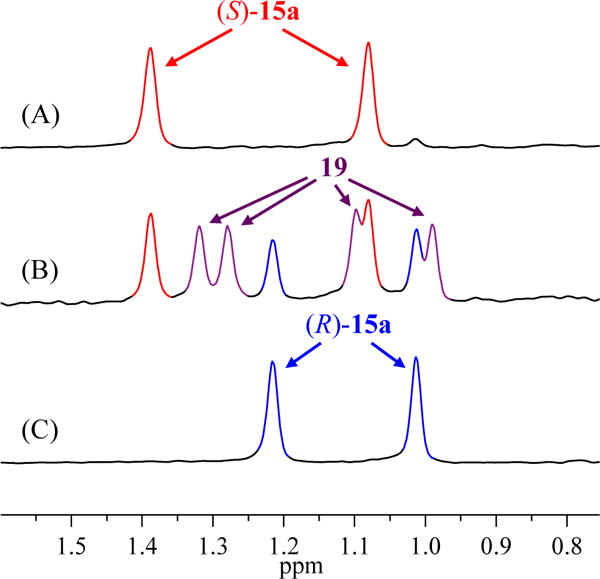
6Li NMR spectra of 0.10 M solutions containing equimolar alkoxide 11 and enolate 15a in 0.11 M THF/toluene at −80 °C: (A) (S)-15a; (B) 1:1 (S)-15a and (R)-15a (1:1); (C) (R)-15a.
Figure 6.

Job plot showing the relative integration of the 6Li resonances of (S)-15a (red), (R)-15a (blue), and 19 (purple) versus the measured mole fraction27 of (R)-1a [χ(R)-1a] monitored with 6Li NMR spectroscopy in 0.11 M THF/toluene at −80 °C. The total enolate and alkoxide titer are constant (0.10 M).
Of course, 2:2 mixed tetramer (S)-15a is necessarily unsolvated in toluene,28 and studies with THF suggested that (S)-15a does not accept coordinating solvents either. Using a control experiment of increasing importance in our laboratory, we added pyridine to toluene solutions to probe lithium ion solvation.22 The coordination of pyridine to a 6Li nucleus causes a marked (>1.0 ppm) downfield chemical shift of any resonance bearing a coordinated solvent.29,30,31 When 1.0 M pyridine was added to (S)-15a, a new species appeared corresponding to a 3:1 mixed aggregate (vide infra), but it had no measurable effect on the resonances of (S)-15a.
The assignment of (S)-15a was completed with the gamut of two-dimensional NMR spectroscopies (Supporting Information). We also examined the efficacy of DFT calculations for distinguishing the five possible stereoisomers of 2:2 mixed aggregates consistent with the 6Li spectroscopic data (one isomer was inconsistent) and obtained the results in Chart 2. The value of 2.2 kcal/mol for the observed form indicated a discrepancy between theory and experiment.
Chart 2.

Computed energies of isomeric 2:2 mixed tetramers (relative kcal/mol).
Spectroscopic data of (R)-15a derived from alkoxide 11 and antipodal enolate (R)-1a were in every respect analogous to those of (S)-15a (Supporting Information). Calculations also failed to show the preference for (R)-15a relative to the isomer analogous to 22 but only by 0.1 kcal/mol.
The solvent-dependent formation of 3:1 mixed aggregates (S)-16a was observed with a THF titration of a 3:1 mixture of alkoxide 11 and enolate (S)-1a (eq 5 and Figure 7). In neat toluene, only mixed (S)-15a and homoaggregated alkoxide tetramer 14 were observed (Figure 7A). During incremental increases in THF concentration, 3:1 mixed tetramer (S)-16a emerged (Figure 7C) and became the sole observable mixed aggregate in neat THF (Figure 7F). The existence of a single enolate subunit within (S)-16a, although not in doubt, was confirmed by using a 3:1 mixture of alkoxide and enolate with equal proportions of (S)-1a and (R)-1a, which yielded (S)-16a and (R)-16a to the exclusion of other species. The existence of two rather than three chelated alkoxides was shown using [15N]11, which afforded two 6Li doublets and a 6Li singlet accompanied by two triplets (1:1:1) and a singlet in the 15N NMR spectrum.
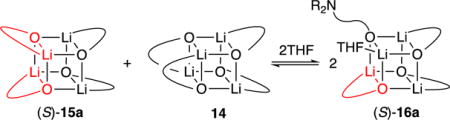 |
(5) |
Figure 7.

6Li spectra of a 3:1 mixture of alkoxide 11 (0.075 M) and (S)-1a (0.025 M) with various concentrations of THF in toluene at −80 °C showing the appearance of THF-solvated aggregate (S)-16a. Concentrations of THF in toluene are (A) 0.0 M, (B) 1.0 M, (C) 3.0 M, (D) 6.0 M, (E) 9.0 M, and (F) neat THF.
Scheme 8.

Stereochemical model I (wrong).
A coordinated THF was confirmed using several strategies. The addition of 4.0 M pyridine to a THF solution of (S)-16a at –80 °C resulted in downfield shifts of the four resonances to various extents. The shifts were small because the degree of pyridine coordination was low. However, at –105 °C, four resonances corresponding to the pyridine solvate were observed in the slow-exchange limit.22 In a second experiment, the addition of 2.0 equiv hexamethylphosphoramide (HMPA) per lithium to a THF solution afforded (S)-25a for which the most upfield resonance appeared as a doublet (0.26 ppm, JLi–P = 4.3 Hz) owing to 6Li –31P coupling, but no change in the four 6Li chemical shifts was observed.32

Characterizing (S)-16a proved a profoundly challenging resolution problem. Fortunately, the full complement of two-dimensional NMR spectroscopies (vide supra) applied to isostructural HMPA solvate (S)-25a provided a three-dimensional picture that included the preferred orientation of the unchelated alkoxide (Supporting Information). Once again, we exploited DFT to examine the 11 possible isomers of (S)-16a (Chart 3). In this instance, the computations predicted the observed form to be the most stable. Applying the spectroscopic analysis described above to (R)-1a provided the structure of (R)-16a. The computations for the 11 isomers of (R)-16a solvated by THF matched the spectroscopic results (Supporting Information).
Chart 3.

Isomers of 3:1 mixed tetramer (S)-16a.
Mixtures of caranolate alkoxide 11 and valine-derived enolates (R)-1b or (S)-1b revealed completely analogous results to those described above: 2:2 mixed aggregates (R)-15b and (S)-15b formed exclusively in neat toluene or in toluene with low concentrations of THF, whereas (R)-16b and (S)-16b were the sole observable mixed aggregates in neat THF. Differences in reactivity and selectivity are noted below.
Structures of ephedrate-derived mixed tetramers
The results of studies using ephedrate-derived alkoxide 8 showed notable departures from those obtained with the carane series. The valine-derived enolates, the b series, gave the cleanest results, and they are presented emblematically (Chart 4). Enolate-rich mixtures of enolate (R)-1b or (S)-1b and alkoxide 8 in toluene yielded no 1:3 (enolate-rich) mixed tetramer (see Figure 8B). Equimolar mixtures afforded 2:2 mixed tetramer (S)-36b or (R)-36b exclusively (see Figure 8C). Alkoxide-rich mixtures with enolate (S)-1b in neat THF afforded complex mixtures. In toluene, however, 3:1 mixed tetramer (S)-37b was produced as a single isomer, albeit not quantitatively (Figure 8D). Neither (S)-36b nor (S)-37b was influenced by added pyridine, which indicated that they are fully chelated. Mixed aggregate (S)-36b was characterized with the full complement of two-dimensional NMR spectroscopies (without 15N labeling). Computations also supported the assignment of (S)-36b (Supporting Information). The stereochemistry of 3:1 mixed tetramer (S)-37b, by contrast, was tentatively distinguished from two other stereoisomers using only computations and should be viewed with some caution. Plotting aggregate concentration versus mole fraction of alkoxide 8 afforded a Job plot (Figure 9) that is almost unrecognizable compared with the statistical analog (see Figure 3). The curves in Figures 3 and 9 both derive from the standard parametric fitting protocol used for any tetramer ensemble.
Chart 4.
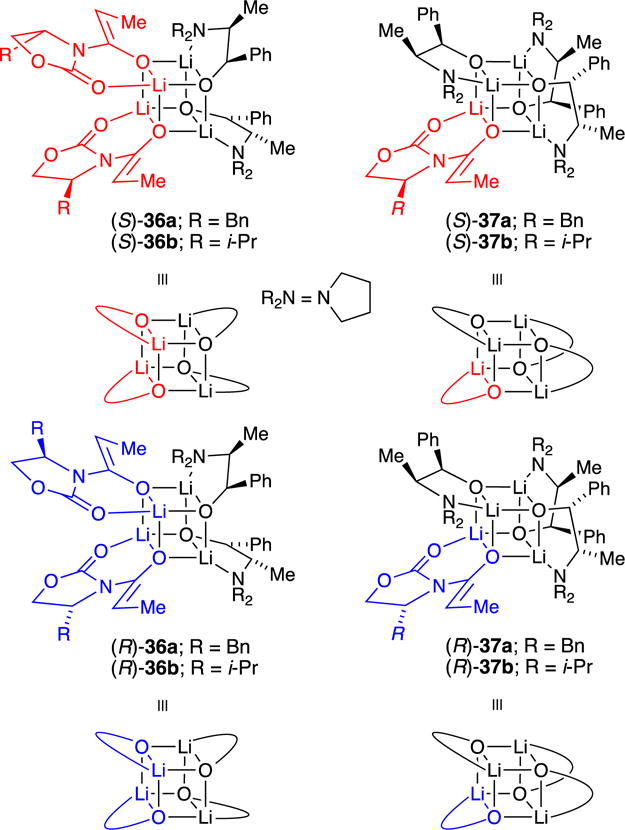
Mixed aggregates ephedrate 8 with (S)- and (R)-16a.
Figure 8.

6Li NMR spectra of 0.10 M solutions of (S)-1b and alkoxide 8 in 0.11 M THF/toluene at −80 °C with 0.11 M [ 6Li]LDA. The measured mole fractions27 of alkoxide 8 (χROLi) are 0.00, 0.18, 0.50, 0.69, and 1.00 for A−E, respectively.
Figure 9.

Job plot showing the relative integration of the 6Li resonances versus the measured mole fraction27 (χROLi) of 8 at a 0.10 M constant total titer of (S)-1b and 8 in 0.11 M THF/toluene at −80 °C.
Equimolar mixtures of 8 and the phenylalanine-derived (S)-1a and (R)-1a enolates in toluene cleanly afforded fully characterized 2:2 mixed aggregates (S)-36a and (R)-36a. Alkoxide-rich mixtures in toluene afforded familiar patterns consistent with (S)-37a and (R)-37a along with other isomers. In neat THF, equimolar and alkoxide-rich samples afforded complex mixtures.
Aldol Additions: Stereoselectivity
Table 1 summarizes a number of aldol additions to i-PrCHO. The aggregate column lists the dominant aggregated form of enolates (S)-1a, (R)-1a, (S)-1b, or (R)-1b observed under the conditions of the aldol addition. (S) and (R) refer to the two antipodes of the Evans enolates that produce what were denoted “matched” and “mismatched” results. Entries 1–2 and 7–8 provide the results in the absence of amino alkoxide additives, as reported previously.8 The poor results in entries 2 and 8 derive not from forcing conditions causing retroaldol additions, as described previously, but rather from slow partial conversion at –78 °C. The low (50%) yields in entries 13, 14, 17, and 18 stem from the stalling of the reaction after the consumption of one enolate subunit within the 2:2 mixed aggregates. Table 2 summarizes a limited survey of aldol selectivities (Scheme 5) using optimal 3:1 mixed aggregate (S)-16b.
Table 1.
Stereoselectivity of aldol additions using chiral amino alkoxides and antipodes of Evans enolates (Chart 5).a
| Entry | Lithium alkoxide | Aggregate | syn/syn (2/38) |
syn/anti ([2+38]/39) |
Yield (%) |
|---|---|---|---|---|---|
| 1 | — | 3a/4a | 12:1 | 16:1 | 91 |
| 2 | — | 5a | 1:1 | 5:1 | <20 |
| 3 | 11 | (S)-15a | 6:1 | 7:1 | 54 |
| 4 | 11 | (R)-15a | 6:1 | 7:1 | 56 |
| 5 | 11 | (S)-16a | 18:1 | 50:1 | 90 |
| 6 | 11 | (R)-16a | 2:1 | 5:1 | 88 |
| 7 | — | 3b/4b | 5:1 | 4:1 | 90 |
| 8 | — | 5b | 1:5 | 3:1 | <20 |
| 9 | 11 | (S)-15b | 50:1 | 15:1 | 48 |
| 10 | 11 | (R)-15b | 50:1 | 14:1 | 45 |
| 11 | 11 | (S)-16b | 30:1 | >100:1 | 92 |
| 12 | 11 | (R)-16b | 1:1 | 6:1 | 88 |
| 13 | 8 | (S)-36a | 10:1 | 16:1 | 55 |
| 14 | 8 | (R)-36a | 10:1 | 14:1 | 50 |
| 15 | 8 | (S)-37a | 10:1 | 25:1 | 85 |
| 16 | 8 | (R)-37a | 10:1 | 8:1 | 78 |
| 17 | 8 | (S)-36b | 9:1 | 13:1 | 52 |
| 18 | 8 | (R)-36b | 9:1 | 13:1 | 55 |
| 19 | 8 | (S)-37b | 4:1 | 3:1 | 80 |
| 20 | 8 | (R)-37b | 9:1 | 4:1 | 79 |
Reaction in neat tetrahydrofuran/–78 °C for 30 min after aging the enolate at 25 °C for 10 min; 2 equiv of electrophile.
Table 2.
Stereoselectivity of selected aldol additions.
| Entry | RC(=O)R′ | Lithium alkoxide | syn/syn | syn/anti | Yield (%) |
|---|---|---|---|---|---|
| 1 | PhC(=O)Ha | — | 1:3 | 1:12 | 70 |
| 2 | PhC(=O)Ha | 11 | >50:1 | 2:1 | 82 |
| 3 | t-BuC(=O)Hb | — | >50:1 | >50:1 | 90 |
| 4 | t-BuC(=O)Hb | 11 | >50:1 | >50:1 | 92 |
| 5 | MeC(=O)CCHb | — | 3:1 | >50:1 | 80 |
| 6 | MeC(=O)CCHb | 11 | 7:1 | >50:1 | 85 |
| 7 | i-PrC(=O)Me | — | — | — | <5 |
| 8 | i-PrC(=O)Me | 11 | — | — | <5 |
Literature compound.1b
Characterized as described in Supporting Information.
Scheme 5.

Aldol additions to various aldehydes and ketones showing amplification.
Aldol Additions: Rate Studies
Aside from the usual technical challenges of carrying out organolithium reaction kinetics, several acute problems arose during this study in particular. The rates of the aldol addition with i-PrCHO were too fast to monitor at –78 °C.33 Given the choice of a lower temperature or a less reactive substrate, we turned to pivaldehyde (t-BuCHO; eq 6).
Rate studies under conditions favoring 2:2 mixed tetramer (S)-15a or (S)-15b were thwarted by odd (non-first-order) substrate losses and product formation. By contrast, the rate studies under conditions affording 3:1 mixed tetramer were far more satisfying. The existence of a single enolate subunit within the mixed aggregate may have helped. Monitoring the aldol addition by following the formation of aldolate (S)-44a (1788 cm–1) rather than the embedded absorbance of t-BuCHO (1724 cm–1) showed growth consistent with34 pseudo-first-order behavior with aldehyde in deficiency and second-order behavior when the mixed aggregate and t-BuCHO were used in 1:1 proportions (Figure 10). Plotting pseudo-first-order rate constants (kobsd) versus aldehyde concentration shows concentration-independent values of kobsd (Figure 11), confirming the first-order dependence. Plotting kobsd versus mixed aggregate concentration (Figure 12) and the concentration of excess alkoxide (Figure 13) shows approximate first-order and zeroth-order dependencies as expected (vide infra).
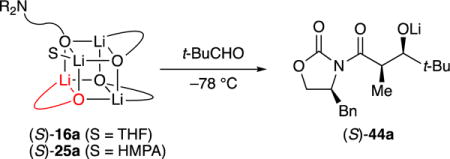 |
(6) |
Figure 10.
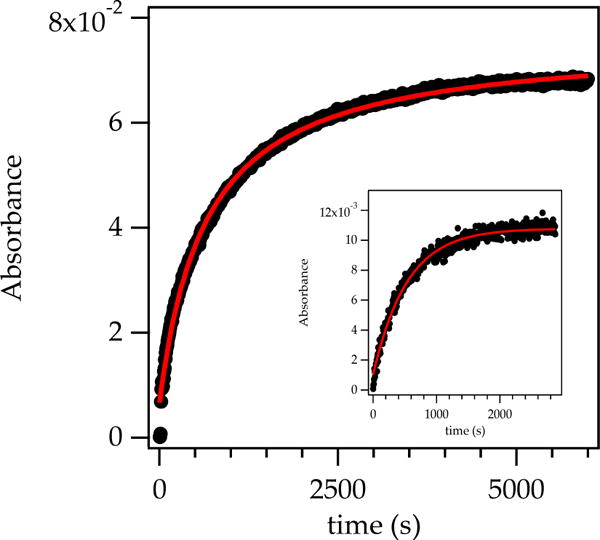
Formation of aldolate (S)-44a (1788 cm–1) carried out using mixed tetramer (S)-16a (0.10 M) at –78 °C under second-order conditions [0.10 M pivaldehyde (t-BuCHO)] and pseudo-first-order conditions (0.010 M, inset). The curves represent fits to f(x) = a – a/(1 + bx) and f(x) = (a–1)e–bx, respectively.
Figure 11.

Plot of pseudo-first-order rate constants (kobsd) versus t-BuCHO concentration in neat THF at −78 °C [0.10 M (S)-16a, neat THF]. The curve depicts an unweighted least-squares fit to the function f(x) = ax + b: a = (–1.3 ± 2.3) × 10–2; b = (2 ± 2) × 10–3.
Figure 12.

Plot of pseudo-first-order rate constants (kobsd) versus (S)-16a concentration at 0.0050 M t-BuCHO in neat THF at −78 °C. The data were fit to y = axn such that n = 1.14 ± 02.
Figure 13.

Plot of pseudo-first-order rate constants (kobsd) versus the concentration of 11 in neat THF at −78 °C [0.10 M (S)-16a, neat THF]. The curve depicts an unweighted least-squares fit to the function f(x) = ax + b: a = (1.94 ± 1.8) × 10–3; b = (1.31 ± 0.11) × 10–3.
Determining the all-important reaction order in THF would require the monitoring of rates over a range of solvent concentrations. However, even modest decreases in THF concentration from 12 M (neat THF) pushed the reactants from exclusive 3:1 mixed tetramer (S)-15a to the 2:2 mixed aggregates (eq 4), irreparably undermining the rate studies. Accordingly, we compromised yet again by using the isostructural HMPA solvate (S)-25a. The free (uncoordinated) HMPA concentration was varied over a 10-fold range (0.025–0.15 M) without a measurable change in structure. It was notable at the outset that, despite the enormous binding affinity of HMPA relative to THF,35 the rates of additions in HMPA/toluene and neat THF were nearly indistinguishable. Despite the possibility of obtaining a number of uninformative outcomes that can result from the addition of such an invasive ligand, we obtained the one result that lent itself to literal interpretation unfettered by qualifiers: the reaction was zeroth-order in HMPA (Figure 14), which showed that the aldol proceeds via a mechanism that requires neither association nor dissociation of the sole coordinated solvent on the 3:1 mixed tetramer framework in the rate-limiting step. Using t-BuCDO afforded a kH/kD of 0.77 ± 0.016, which is consistent with a rate-limiting aldol addition. A rate-limiting complexation would afford a kH/kD of 1.0.
Figure 14.

Plot of pseudo-first-order rate constants (kobsd) versus hexamethylphosphoramide concentration in neat THF at −78 °C [0.10 M (S)-16a, neat THF]. The curve depicts an unweighted least-squares fit to the function f(x) = ax + b: a = (6.4 ± 10.6) × 10–4; b = (1.2 ± 0.09) × 10–3.
Altogether, the rate data implicate the idealized rate law36,37 in eq 7 and the generic mechanism in eq 8, which are discussed below. We note here that the lack of deaggregation and solvent dissociation evidenced by the zeroth-order dependencies is a critical observation.
| (7) |
| (8) |
Discussion
The work described herein falls into three categories: (1) characterization of ROLi-lithium enolate mixed tetramers; (2) development of stereoselective aldol additions of mixed tetramers; and (3) mechanistic study and determination of a stereochemical model for aldol addition.
Structures
The structural studies are summarized using the shorthand mnemonic described in Scheme 6 with color-coded loops for the S (red) and R (blue) enantiomeric series of Evans enolates. Detailed depictions of the tetrameric mixed aggregates are found in Chart 1. The a and b series correspond to the phenylalanine- and valine-derived oxazolidinones, respectively. The characterizations required a range of tactics and strategies. Job plots show the gross aggregation states and stoichiometries of the mixed tetramers. Figure 9 offers the most unusual nonstatistical example we have encountered to date. The presence or absence of chelation was confirmed with 15N labeling of the amino alkoxide ([15N]11). Coordination by external ligands was probed using pyridine as a 6Li chemical shift reagent and HMPA to show specific solvent-lithium contacts through 6Li–31P coupling.32 The stereochemical assignments stem from the application of a battery of two-dimensional spectroscopic methods complemented by DFT computations. Several mixed aggregates received less scrutiny but are secure nonetheless.
Scheme 6.

Summary of 2:2 and 3:1 mixed aggregates.
The various topologies of the D2d-symmetric core of enolate tetramer 5 and the S4-symmetric cores of alkoxide tetramers 13 and 14 conflict en route to the formation of mixed tetramers: the enolate topology dominates, but a structural mess was a distinct possibility. Alkoxides 8 and 11, which form tetrameric cubes 13 and 14 (respectively), combine with both antipodes of the Evans enolates—homotetramers (S)-5a, (R)-5a, (S)-5b, (R)-5b—to form exclusively 2:2 mixed aggregates 15 in toluene, irrespective of stoichiometry. Mixtures containing excess alkoxide 11 cleanly afford (S)-16 or (R)-16 as a single stereoisomer but only in neat THF. The solvent concentration dependence can be traced to the preferential solvation of a single corner of the cube with accompanying scission of one of three amino alkoxide chelates. The stereocontrolled assembly of mixed tetramers 15 and 16 is remarkable given the enormous number of possible isomers (see Charts 2 and 3) consistent with the spectroscopic symmetries (many more if all possibilities are considered). Lithium enolates with excess ephedrate-derived alkoxide 8 received less scrutiny primarily because the structural control of the 3:1 mixed tetramers was poor. The absence of 1:3 enolate-rich mixed tetramers under any conditions is surprising, particularly given that (a) the enolate-like rather than alkoxide-like topologies of the resulting mixed aggregates dominate, and (b) simple enolates afford enolate-rich 1:3 mixed aggregates but not 3:1 alkoxide-rich mixed aggregates.
Stereoselective Aldol Additions
Our goal at the outset was to exploit amino alkoxide-containing mixed tetramers to amplify the inherent 12:1 (syn/syn) selectivity observed for aldol addition using lithiated Evans enolates (see Table 1, entry 1). Once again, we had numerous reasons for pessimism. The credible selectivity for unmodified Evans enolates was obtained from kinetically formed enolate dimers, which readily form fleeting monomers (see Scheme 2). The homotetrameric enolate 5 in THF formed by aging is decidedly unreactive, undergoing aldol addition via monomer stereorandomly and only under forcing conditions (see Table 1, entry 2). A glimmer of hope was found in contemporaneous studies of Weinreb enolates in which fully chelated tetramers showed evidence of coordinating external ligands to afford five-coordinate lithium owing to the geometric constraints of five-membered chelates.22 We hoped the aldehyde might find its way into the mixed tetramer core.
We found two lines of evidence that mixed tetramers react without deaggregation: (1) the reactions were fast at –78 °C even in neat toluene, which contrasts with homotetramer 5, and (2) the 2:2 mixed tetramers (15) markedly altered the stereoselectivity, albeit eroding the selectivity regardless of which enolate antipode was paired with alkoxide 11 (see Table 1, cf. entry 1 and entries 3 and 4). Fortunately, 3:1 mixed aggregates (S)-16 and (R)-16 showed the matched–mismatched influence we sought (Scheme 7). The S series (matched) amplified the aldol selectivity and eliminated low levels of the anti adduct (see Table 1, entries 5 and 11), whereas the R series (mismatched) eroded the selectivities (entries 6 and 12). When measured in kilocalories per mole, the stereochemical effects are small, but the measurable amplification is a satisfying proof of principle nonetheless. Ephedrate-derived alkoxide 8 showed inferior structural control and a lack of stereochemical influence (see Table 1, entries 13–20) that could be construed as evidence of a monomer-based aldol addition. That the aldol additions are fast at –78 °C does not exclude this possibility.
Scheme 7.

Stereoselectivity from matched and mismatched enolate antipodes (see Table 1).
The optimized case, mixed tetramer (S)-16b derived from (S)-valine and alkoxide 11, showed amplification of a few select aldol additions (see Table 2). This amplification is real but modest at best. The addition to benzaldehyde (entries 1 and 2) shows a mechanistically significant alkoxide-mediated reversal to produce synthetically unimportant overall stereoselectivity.
Stereochemical Model I: the Wrong One
We were tempted to invoke the THF-solvated lithium nucleus in 3:1 mixed tetramers (16) as the centerpiece of a stereochemical model (Scheme 8). Imagine the substitution of the lone THF by aldehyde to give fleeting intermediate 45, which then undergoes facile aldol addition via transition structure 46. The first problem is that this aldol addition via (S)-45a predicts that the aldol should be (S)-38a, the wrong product. A simple corrective action is to assume equilibrium between observable mixed tetramer (S)-16 and fleeting isomer (S)-32a, which is calculated to be 5.5 kcal/mol less stable. Substitution via aldehyde and aldol addition would give the correct aldol adduct 47 as the amplified form, a textbook example of the Curtin–Hammett principle. Alas, the computations predict transition structure (S)-46a to be less favorable than (S)-45a. In essence, the model incorrectly predicts an isomeric mismatch. This prediction is not a fundamental problem because computations can be wrong; we noted examples in the results section. A similar analysis using mixed tetramer (R)-16a derived from enolate (R)-1a should display amplified stereocontrol rather than the observed eroded (mismatched) stereoselectivity. The entire model is backwards. There is, however, a more fundamental flaw.
Model I is pedagogically useful (easily envisioned) but incompatible with the rate data. To understand this incompatibility, we must digress. Detailed rate studies showed a first-order dependence on the less-reactive t-BuCHO and mixed tetramer (S)-16a. A zeroth-order dependence on alkoxide confirmed tetramer-based rather than monomer-based aldol addition. Had the reaction proceeded via monomer-based transition structure 6 (see Scheme 2), the rate law would reflect the dissociation of the three alkoxide subunits (eq 9) as inverse three-fourths order in alkoxide 11. However, a zeroth-order was observed. Moreover, substituting the coordinated THF on mixed tetramer 16 with a strongly bound HMPA should inhibit a reaction requiring the substitution of the coordinated solvent: HMPA demonstrably binds but imparts no detectable change in reaction rate or stereochemistry. An affiliated zeroth-order in solvent (HMPA in the rate studies) shows that the solvent is not substituted by the aldehyde en route to the aldol addition. Model I is fatally flawed.
| (9) |
| (10) |
Stereochemical Model II: the Right One?
The non-dissociative pathway mandated by the rate studies requires that (S)-16a react either directly using an ill-defined coordination site or via chelate scission. Although spectroscopic and computational studies of Weinreb amides suggest that five-membered chelates leave open the possibility of trigonal–bipyramidal (five-coordinate) lithiums on cubic tetramers,22 computational evidence lends no support for a five-coordinate lithium in the highly congested 3:1 mixed tetramer (S)-16. Instead, we invoke a second chelate scission with aldehyde coordination (Scheme 9).
Scheme 9.
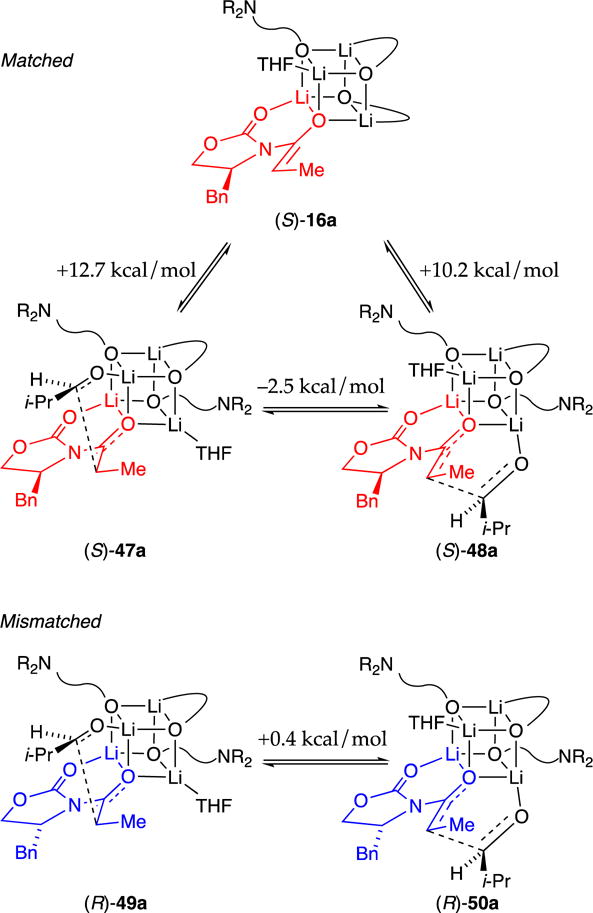
Stereochemical model II showing matched and mismatched selectivities.
Model II correctly predicts amplification in the S series (matched) and eroded selectivities in the R series (mismatched). The absolute activation energies of model II are comparable to those in model I for both the matched and mismatched pairings. We must confess to having had no clue at the outset that our preferred model would involve a mono-chelated mixed tetramer, which is somewhat disruptive to our thinking in that more tightly bound (fully chelated) mixed aggregates would be potentially more predictive and possibly impart even greater selectivity.
The story took a huge detour when we investigated the influence of 2:2 mixed tetramers. Members of the phenylalanine-derived a series were poorly selective regardless of the choice of enantiomeric pairing of (R)-15a or (S)-15a, but there was no evidence of match–mismatch pairing. Valine-derived (S)-15b and (R)-15b also react rapidly at –78 °C. Yields of approximately 50% can be traced to stalling after the consumption of one of the two enolate subunits. Remarkably, (R)-15b and (S)-15b elicit strikingly amplified selectivities, affording 50:1 syn/syn selectivities accompanied by low levels of one anti isomer (eq 11). Moreover, the aldol selectivities are independent of the enantiomeric pairings. The conundrum is that mixed aggregates (R)-15b and (S)-15b promote highly selective alkylation from the face opposite the isopropyl substituent despite strikingly complementary trajectories. The absence of mechanistic data owing to odd curvatures would not stop us from proposing a stereochemical model, but we do not have a particularly satisfying one.
 |
(11) |
The amplification regardless of chiral pairing—that both aggregates act as matched—suggests that the influence of the alkoxide auxiliaries specific to the 2:2 mixed aggregates has nothing to do with chirality. This conclusion prompted a last series of experiments. Equimolar mixtures of achiral alkoxide 51 and enolate (S)-1b cleanly afforded a single 2:2 mixed aggregate shown by the full complement of NMR spectroscopies to be 52. The preference for 52 with the isopropyl moieties proximate to each other rather than 53 with the isopropyl moieties proximate to the alkoxides shows an inherent preference uninfluenced by relative alkoxide stereochemistry. We did indeed modify the selectivity (eq 12) but not favorably.
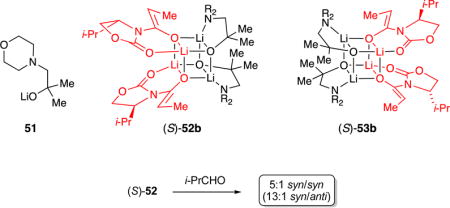 |
(12) |
Conclusions
Our effort to exploit the DuPont and Merck chiral amino alkoxides to impart stereocontrol of enolate reactions is a good proof of principle. Controlling 2:2 and 3:1 mixed tetramer structures and achieving direct reaction without intervening deaggregations are considerable steps. Evidence of matched and mismatched modified selectivities for an aldol addition confirms that we can control selectivity through aggregate structures. It goes without saying that 100:1 selectivities would have been great, but the proof of principle is clear. Our wish list, however, is extensive. Reversing the enolate configuration rather than the alkoxide configuration nicely illustrates the matching and mismatching of chiral subunits but has obvious limitations in a goal to control stereochemistry. An amino alkoxide for which both antipodes are readily available is important. This problem is familiar to those who work with sparteine as a chiral ligand.38 Surprisingly, the 2:2 mixed aggregates show considerable amplification that was fully independent of the relative chiralities of the subunits. This outcome, in theory, solves the chirality problem for the specific study in question, but it begs a fundamental question: How?
We had the notion that 2:2 mixed aggregates could offer a remarkably simple, albeit ironic, solution by amplifying selectivities using achiral alkoxides, or elaborate lithium chloride surrogates if you will. The idea that using even simple achiral amino alkoxides to modify organolithium reactivity has been exploited most notably by Caubère.39 That these achiral compounds could influence stereocontrol is a provocative idea that has been validated: selectivities were amplified but, in a single attempt, the effects were small. There is room for improvement.
Of course, imparting 99% stereoselectivity on reactions of achiral chelating enolates, whether oxazolidinone-based or not, would be a considerable advance. In the long run, however, a universal alkoxide available in both antipodes that can amplify stereoselectivity in a range of chelating enolates would also be meritorious and attainable.
Experimental
Reagents and Solvents
THF, toluene, and pyridine were distilled from solutions containing sodium benzophenone ketyl. LDA, [ 6Li]LDA, [ 6Li,15N]LDA, lithium hexamethyldisilazide (LiHMDS), [ 6Li]LiHMDS, and [ 6Li,15N]LiHMDS were prepared as described previously. Solutions of LDA and LiHMDS were titrated for active base by using a literature method.40 Air- and moisture-sensitive materials were manipulated under argon using standard glove box, vacuum line, and syringe techniques. The Evans enolate precursors were purchased, and amino alcohols were obtained from Merck and Bristol-Myers Squibb (formerly DuPont Pharmaceuticals).
NMR Spectroscopy
Individual stock solutions of substrates and LDA or LiHMDS were prepared at room temperature. An NMR tube under vacuum was flame-dried on a Schlenk line and allowed to return to room temperature. It was then backfilled with argon and placed in a –78 °C dry ice/acetone bath. Appropriate amounts of LDA or LiHMDS (1.1 equiv), amino alcohol, and oxazolidinone enolate were added sequentially via syringe. The tubes were sealed under partial vacuum and vortexed on a vortex mixer for 5 s. They could then be stored for days in a freezer at –86 °C. Each sample routinely contained 0.10 M total substrate with a 0.01 M excess of LDA or LiHMDS. (The excess base could not be observed in samples after aging as shown by 6Li and 15N NMR spectroscopies.) Standard 6Li and 13C NMR spectra were recorded on a 500 MHz spectrometer at 73.57 and 125.79 MHz, respectively. The 6Li resonances were referenced to 0.30 M [ 6Li]LiCl/methanol at –80 °C (0.0 ppm).
IR Spectroscopic Analyses
IR spectra were recorded with an in situ IR spectrometer fitted with a 30-bounce, silicon-tipped probe. The spectra were acquired in 16 scans at a gain of 1 and a resolution of 4 cm−1. A representative reaction was carried out as follows: The IR probe was inserted through a nylon adapter and O-ring seal into an oven-dried, cylindrical flask fitted with a magnetic stir bar and a T-joint. The T-joint was capped with a septum for injections and a nitrogen line. After evacuation under full vacuum, heating, and flushing with nitrogen, the flask was charged with LDA (56 mg, 0.525 mmol) or LiHMDS (88 mg, 0.525 mmol) in THF, cooled in a dry ice/acetone bath prepared with fresh acetone, and charged with (S)-1a (29 mg, 0.25 mmol) and 11 (90 mg, 0.75 mmol) in THF. The total volume was brought to 4.9 mL with THF. After a background spectrum was recorded, t-BuCHO (0.050 mmol) as a 0.25 M stock solution in THF was added with stirring. IR spectra were recorded every 6 s with monitoring of the absorbance at 1788 cm−1 over the course of the reaction.
Preparative Scale Addition of i-PrCHO
LiHMDS (4.11 g, 24.6 mmol) was weighed out in a 100 mL round-bottom flask inside a glove box, and the flask was sealed with a septum. After transferring the flask outside the glove box, the lithium base was dissolved in 40 mL of freshly distilled THF. The solution was cooled to –78 °C in a dry ice/acetone bath. A mixture of (S)-1b (1.11 g, 6.0 mmol) and 11 (4.43 g, 18.5 mmol) in 20 mL of THF was added dropwise to the cooled solution. The mixture was warmed to 25 °C for 10 min, recooled to –78 °C, and allowed to stir for 15 min. Then, 1.10 mL of freshly distilled i-PrCHO (0.87 g, 12.0 mmol) in 2.0 mL of THF was added to the reaction mixture. After 30 min of stirring at –78 °C, the reaction was quenched with 10 mL concentrated HCl. The solution was warmed to room temperature and extracted with three 100 mL portions of diethyl ether. The organic extracts were washed with 100 mL brine, dried over sodium sulfate, and concentrated in vacuo. The crude product was analyzed with 1H and 13C NMR. After flash chromatography with 20% ethyl acetate in hexanes, 1.42 g of product was obtained (92% yield). (S)-2b: 1H NMR (599 MHz, CDCl3) δ 4.43 (ddd, J = 8.4, 4.0, 3.1 Hz, 1H), 4.25 (dd, J = 9.2, 8.4 Hz, 1H), 4.18 (dd, J = 9.2, 3.1 Hz, 1H), 4.03 (qd, J = 7.0, 3.2 Hz, 1H), 3.53 (dd, J = 8.2, 3.2 Hz, 1H), 2.45 (s, 1H), 2.31 (ddq, J = 10.9, 6.9, 3.9, 3.5 Hz, 1H), 1.67 (dp, J = 8.2, 6.7 Hz, 1H), 1.11 (d, J = 7.0 Hz, 3H), 0.99 (d, J = 6.6 Hz, 3H), 0.90 (d, J = 6.8 Hz, 3H), 0.88 (d, J = 7.0 Hz, 3H), 0.85 (d, J = 6.9 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 177.4, 153.7, 77.2, 63.3, 58.5, 39.9, 31.2, 28.5, 19.1, 19.0, 18.0, 14.7, 9.8. Direct analysis in real-time mass spectrometry m/z: calculated for C13H22NO3 (M+H–H2O)+ 240.15942, found 240.15923. The minor isomers were observed using NMR spectroscopy with comparison to independently prepared authentic samples (Supporting Information).
Supplementary Material
Chart 5.

Aldol adducts from mixed aggregates.
Acknowledgments
We thank the National Institutes of Health (GM077167) for support.
Footnotes
Supporting Information: NMR spectroscopic, kinetic, and computational data. This material is available free of charge via the Internet at http://pubs.acs.org.
ORCID
David B. Collum 0000-0001-6065-1655
Janis Jermaks 0000-0002-4828-7210
Notes
The authors declare no competing financial interests.
References and Footnotes
- 1.(a) Evans DA, Bartroli J, Shih TL. J Am Chem Soc. 1981;103:2127. [Google Scholar]; (b) Evans DA, Takacs JM, McGee LR, Ennis MD, Mathre DJ, Bartroli J. Pure Appl Chem. 1981;53:1109. [Google Scholar]
- 2.(a) Ager DJ, Prakash I, Schaad DR. Chem Rev. 1996;96:835. doi: 10.1021/cr9500038. [DOI] [PubMed] [Google Scholar]; (b) Wu G, Huang M. Chem Rev. 2006;106:2596. doi: 10.1021/cr040694k. [DOI] [PubMed] [Google Scholar]; (c) Farina V, Reeves JT, Senanayake CH, Song JJ. Chem Rev. 2006;106:2734. doi: 10.1021/cr040700c. [DOI] [PubMed] [Google Scholar]; (d) Evans DA, Shaw JT. Ĺactualité Chimique. 2003:35. [Google Scholar]; (e) Lin GQ, Li YM, Chan ASC. Principles and Applications of Asymmetric Synthesis, Wiley & Sons: New York. 2001;135 [Google Scholar]; (f) Ager DJ, Prakash I, Schaad DR. Aldrichim Acta. 1997;30:3. [Google Scholar]
- 3.(a) Evans DA, Ennis MD, Mathre DJ. J Am Chem Soc. 1982;104:1737. [Google Scholar]; (b) Kitajima H, Nakamura M, Tamakawa H, Goto N. Bioorg Med Chem Lett. 2000;10:2453. doi: 10.1016/s0960-894x(00)00491-1. [DOI] [PubMed] [Google Scholar]; (c) Bull SD, Davies SG, Nicholson RL, Sanganee HJ, Smith AD. Org Biomol Chem. 2003;1:2886. doi: 10.1039/b305623f. [DOI] [PubMed] [Google Scholar]; (d) Hara A, Morimoto R, Ishikawa Y, Nishiyama S. Org Lett. 2011;15:4036. doi: 10.1021/ol201547q. [DOI] [PubMed] [Google Scholar]; (e) Li L, Ding J, Goa L, Han F. Org Biomol Chem. 2015;13:1133. doi: 10.1039/c4ob01963f. [DOI] [PubMed] [Google Scholar]
- 4.(a) Evans DA, Wu LD, Wiener JJM, Johnson JS, Ripin DHB, Tedrow JS. J Org Chem. 1999;64:6411. [Google Scholar]; (b) Crimmins MT, Emmitte KA, Katz JD. Org Lett. 2000;14:2165. doi: 10.1021/ol006091m. [DOI] [PubMed] [Google Scholar]; (c) Cases M, Gonzalez-Lopez de Turiso F, Hadjisoteriou MS, Pattenden G. Org Biomol Chem. 2005;3:2786. doi: 10.1039/b504545b. [DOI] [PubMed] [Google Scholar]; (d) Green R, Merritt AT, Bull SD. Chem Commun. 2008:508. doi: 10.1039/b713966g. [DOI] [PubMed] [Google Scholar]; (e) McLeod MC, Wilson ZE, Brimble MA. J Org Chem. 2012;77:400. doi: 10.1021/jo201988m. [DOI] [PubMed] [Google Scholar]
- 5.(a) Mahrwald R, editor. Modern Aldol Reactions. 1 and 2. Wiley-VCH; Weinheim: 2004. [Google Scholar]; (b) Palomo C, Oiarbide M, Garcia JM. Chem Soc Rev. 2004;33:65. doi: 10.1039/b202901d. [DOI] [PubMed] [Google Scholar]; (c) Franklin AS, Paterson I. Contemp Org Syn. 1994;1:317. [Google Scholar]; (d) Baiget J, Cosp A, Galvez E, Gomez-Pinal L, Romea P, Urpi F. Tetrahedron. 2008:5637. [Google Scholar]; (e) Velazques F, Olivio H. Curr Org Chem. 2002;6:303. [Google Scholar]
- 6.(a) Singer RA, Ragan JA, Bowles P, Chisowa E, Conway BG, Cordi EM, Leeman KR, Letendre LJ, Sieser JE, Sluggett GW, Stanchina CL, Strohmeyer H, Blunt J, Taylor S, Byrne C, Lynch D, Mullane S, O’Sullivan MM, Whelan M. Org Process Res Dev. 2014;18:26. [Google Scholar]; (b) Fürstner A, Bouchez LC, Morency L, Funel J, Liepins V, Porée F, Gilmour R, Laurich D, Beaufils F, Tamiya M. Chem Euro J. 2009;15:3983. doi: 10.1002/chem.200802067. [DOI] [PubMed] [Google Scholar]; (c) Theurer M, Fischer P, Baro A, Nguyen GS, Kourist R, Bornscheuer U, Laschat S. Tetrahedron. 2010;66:3814. [Google Scholar]; (d) Peters R, Althaus M, Diolez C, Rolland A, Manginot E, Veyrat M. J Org Chem. 2006;71:7583. doi: 10.1021/jo060928v. [DOI] [PubMed] [Google Scholar]; (e) Liu W, Sheppeck JE, II, Colby DA, Huang H, Nairn AC, Chamberlin AR. Bioorg Med Chem Lett. 2003;13:1597. doi: 10.1016/s0960-894x(03)00105-7. [DOI] [PubMed] [Google Scholar]; (f) Ojida A, Yamano T, Taya N, Tasaka A. Org Lett. 2002;4:3051. doi: 10.1021/ol0263189. [DOI] [PubMed] [Google Scholar]; (g) Bartroli J, Turmo E, Belloc J, Forn J. J Org Chem. 1995;60:3000. [Google Scholar]
- 7.(a) Sobahi TR. Oriental J Chem. 2004;20:17. [Google Scholar]; (b) Bonner MP, Thornton ER. J Am Chem Soc. 1991;113:1299. [Google Scholar]; (c) Pridgen LN, Abdel-Magid AF, Lantos I, Shilcrat S, Eggleston DS. J Org Chem. 1993;58:5107. [Google Scholar]; (d) Abdel-Magid A, Pridgen LN, Eggleston DS, Lantos I. J Am Chem Soc. 1986;108:4595. [Google Scholar]; (e) Pridgen LN, Abdel-Magid A, Lantos I. Tett Lett. 1989;30:5539. [Google Scholar]; (f) Banks MR, Blake AJ, Cadogan JIG, Dawson IM, Gaur S, Gosney I, Gould RO, Grant KJ, Hodgson PKG. J Chem Soc, Chem Commun. 1993:1146. [Google Scholar]; (g) Xue C, Voss ME, Nelson DJ, Duan JJW, Cherney RJ, Jacobson IC, He X, Roderick J, Chen L, Corbett RL, Wang L, Meyer DT, Kennedy K, DeGrado WF, Hardman KD, Teleha CA, Jaffee BD, Liu R, Copeland RA, Covington MB, Christ DD, Trzaskos JM, Newton RC, Magolda RL, Wexler RR, Decicco CP. J Med Chem. 2001;44:2636. doi: 10.1021/jm010127e. [DOI] [PubMed] [Google Scholar]; (h) Jacobsen IC, Reddy GP. Tetrahdron Lett. 1996;37:8263. [Google Scholar]
- 8.Tallmadge EH, Jermaks J, Collum DB. J Am Chem Soc. 2016;138:345. doi: 10.1021/jacs.5b10980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tallmadge EH, Collum DB. J Am Chem Soc. 2015;137:13087. doi: 10.1021/jacs.5b08207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.March J. Advanced Organic Chemistry. 4th. Wiley; New York: 1980. p. 959. [Google Scholar]
- 11.(a) Seebach D. Angew Chem, Int Ed Engl. 1988;27:1624. [Google Scholar]; (b) Seebach D. Proceedings of the Robert A Welch Foundation Conferences on Chemistry and Biochemistry. Wiley; New York: 1984. p. 93. [Google Scholar]
- 12.For reviews of structures and reactivities of organolithium mixed aggregates, see:; Harrison-Marchand A, Mongin F. Chem Rev. 2013;113:7470. doi: 10.1021/cr300295w. [DOI] [PubMed] [Google Scholar]; Luderer MR, Bailey WF, Luderer MR, Fair JD, Dancer RJ, Sommer MB. Tetrahedron: Asymmetry. 2009;20:981. [Google Scholar]
- 13.(a) Grabowski EJJ. Reflections on Process Research. In: Abdel-Magid AF, Ragan JA, editors. Chemical Process Research: The Art of Practical Organic Synthesis. American Chemical Society, Washington D.C.; USA: 2004. pp. 1–21. [Google Scholar]; (b) Huffman MA, Yasuda N, DeCamp AE, Grabowski EJJ. J Org Chem. 1995;60:1590. [Google Scholar]; (c) Grabowski EJJ. Chirality. 2005;17:S249. doi: 10.1002/chir.20143. [DOI] [PubMed] [Google Scholar]
- 14.(a) Thompson A, Corley EG, Huntington MF, Grabowski EJJ, Remenar JF, Collum DB. J Am Chem Soc. 1998;120:2028. [Google Scholar]; (b) Xu F, Reamer RA, Tillyer R, Cummins JM, Grabowski EJJ, Reider PJ, Collum DB, Huffman JC. J Am Chem Soc. 2000;122:11212. [Google Scholar]; (c) Thompson AS, Corley EG, Huntington MF, Grabowski EJJ. Tetrahedron Lett. 1995;36:8937. [Google Scholar]
- 15.(a) Pierce ME, Parsons RLJr, Radesca LA, Lo YS, Silverman S, Moore JR, Islam Q, Choudhury A, Fortunak JMD, Nguyen D, Luo C, Morgan SJ, Davis WP, Confalone PN, Chen CY, Tillyer RD, Frey L, Tan LS, Xu F, Zhao D, Thompson AS, Corley EG, Grabowski EJJ, Reamer R, Reider PJ. J Org Chem. 1998;63:8536. [Google Scholar]; (b) Huffman MA, Yasuda N, DeCamp AE, Grabowski EJJ. J Org Chem. 1995;60:1590. [Google Scholar]; (c) Kauffman GS, Harris GD, Dorow RL, Stone BRP, Parsons RLJr, Pesti JA, Magnus NA, Fortunak JM, Confalone PN, Nugent WA. Org Lett. 2000;2:3119. doi: 10.1021/ol006321x. [DOI] [PubMed] [Google Scholar]
- 16.(a) Parsons RLJr, Fortunak JM, Dorow RL, Harris GD, Kauffman GS, Nugent WA, Winemiller MD, Briggs TF, Xiang B, Collum DB. J Am Chem Soc. 2001;123:9135. doi: 10.1021/ja0105616. [DOI] [PubMed] [Google Scholar]; (b) Briggs TF, Winemiller MD, Collum DB, Parsons RLJr, Davulcu AK, Harris GD, Fortunak JD, Confalone PN. J Am Chem Soc. 2004;126:5427. doi: 10.1021/ja0305813. [DOI] [PubMed] [Google Scholar]; (c) Sun X, Winemiller MD, Xiang B, Collum DB. J Am Chem Soc. 2001;123:8039. doi: 10.1021/ja010136c. [DOI] [PubMed] [Google Scholar]
- 17.Parsons RL., Jr Curr Opin Drug Discovery Dev. 2000;3:783. [PubMed] [Google Scholar]
- 18.Bruneau AM, Collum DB. J Am Chem Soc. 2014;136:2885. doi: 10.1021/ja412210d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.For a review on aggregation and cooperative effects on the aldol additions of lithium enolates, see:; Larranaga O, de Cozar A, Bickelhaupt FM, Zangi R, Cossio FP. Chem Eur J. 2013;19:13761. doi: 10.1002/chem.201301597. [DOI] [PubMed] [Google Scholar]
- 20.Renny JS, Tomasevich LL, Tallmadge EH, Collum DB. Angew Chem, Int Ed. 2013;52:11998. doi: 10.1002/anie.201304157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Job P. Ann Chim. 1928;9:113. [Google Scholar]
- 22.(a) Liou LR, McNeil AJ, Ramírez A, Toombes GES, Gruver JM, Collum DB. J Am Chem Soc. 2008;130:4859. doi: 10.1021/ja7100642. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Houghton MJ, Collum DB. J Org Chem. 2016;81:11057. doi: 10.1021/acs.joc.6b02067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.After surveying a subset of the community, we have chosen to refer to (LiX)n and (LiX)m(LiX’)n as “homoaggregate” and “heteroaggregate,” respectively, and reserve the term “mixed aggregate” for (LiX)m(LiY)n.
- 24.(a) Friebolin H. Basic One- and Two-Dimensional NMR Spectroscopy, Wiley VCH: Weinheim. 2010 [Google Scholar]; (b) Claridge TDW. High-Resolution NMR Techniques in Organic Chemistry, 2nd Edition, Elsevier: Amsterdam. 2009 [Google Scholar]
- 25.Gaussian 09, Revision A.02, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox, Gaussian, Inc., Wallingford CT, 2016.
- 26.Phenomena such as “memory of chirality” could owe selectivity to slow aggregate exchange.; Kawabata T, Fuji K. Topics in Stereochemistry. 2003;23:175. [Google Scholar]
- 27.The measured mole fraction—the mole fraction within only the ensemble of interest—rather intended mole fraction of the enolates added to the samples eliminates the distorting effects of impurities.
- 28.For an example and leading references to the influence of aromatic hydrocarbons on organolithium structures and reactivities, see:; Reyes-Rodríguez GJ, Algera RF, Collum DB. J Am Chem Soc. 2017;139:1233. doi: 10.1021/jacs.6b11354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jackman and co-workers described extensive studies of phenolates in pyridine.30
- 30.(a) Jackman LM, DeBrosse CW. J Am Chem Soc. 1983;105:4177. [Google Scholar]; (c) Jackman LM, Smith BD. J Am Chem Soc. 1988;110:3829. doi: 10.1021/ja00226a021. [DOI] [PubMed] [Google Scholar]; (b) Jackman LM, Chen X. J Am Chem Soc. 1997;119:8681. [Google Scholar]; (c) Jackman LM, Petrei MM, Smith BD. J Am Chem Soc. 1991;113:3451. [Google Scholar]
- 31.(a) MacDougall DJ, Noll BC, Kennedy AR, Henderson KW. J Chem Soc Dalton Trans. 2006;15:1875. doi: 10.1039/b515011f. [DOI] [PubMed] [Google Scholar]; (b) Boyle TJ, Pedrotty DM, Alam TM, Vick SC, Rodriguez MA. Inorg Chem. 2000;39:5133. doi: 10.1021/ic000432a. [DOI] [PubMed] [Google Scholar]
- 32.(a) Reich HJ. Chem Rev. 2013;113:7130. doi: 10.1021/cr400187u. [DOI] [PubMed] [Google Scholar]; (b) Reich HJ. J Org Chem. 2012;77:5471. doi: 10.1021/jo3005155. [DOI] [PubMed] [Google Scholar]
- 33.Review of in situ IR spectroscopy:; Rein AJ, Donahue SM, Pavlosky MA. Curr Opin Drug Discovery Dev. 2000;3:734. [PubMed] [Google Scholar]
- 34.One must be cautious in evaluating the quality of the fits to first-order formation of product [f(x) = (a–1)e–bx] and second-order formation of product (f(x) = a – a/(1 + bx).)
- 35.HMPA has been estimated to bind 300 times more strongly than THF in one case.; Reich HJ, Kulicke KJ. J Am Chem Soc. 1996;118:273. [Google Scholar]
- 36.We define the idealized rate law as that obtained by rounding the observed reaction orders to the nearest rational order.
- 37.The rate law provides the stoichiometry of the transition structure relative to that of the reactants:; (a) Edwards JO, Greene EF, Ross J. J Chem Educ. 1968;45:381. [Google Scholar]; (b) Collum DB, McNeil AJ, Ramírez A. Angew Chem, Int Ed. 2007;46:3002. doi: 10.1002/anie.200603038. [DOI] [PubMed] [Google Scholar]; (c) Meek SJ, Pitman CL, Miller AJM. J Chem Educ. 2015;93:275. [Google Scholar]
- 38.Smith BT, Wendt JA, Aubé J. Org Lett. 2002;4:2577. doi: 10.1021/ol026230v. [DOI] [PubMed] [Google Scholar]
- 39.Caubère P. Chem Rev. 1993;93:2317. [Google Scholar]; Choppin S, Gros P, Fort Y. Org Lett. 2000;2:803. doi: 10.1021/ol005538o. [DOI] [PubMed] [Google Scholar]
- 40.Kofron WG, Baclawski LM. J Org Chem. 1976;41:1879. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.