

Abstract
Compelling epidemiological evidence shows a strong positive correlation of obesity with thyroid cancer. In vivo studies have provided molecular evidence that high-fat-diet–induced obesity promotes thyroid cancer progression by aberrantly activating leptin-JAK2-STAT3 signaling in a mouse model of thyroid cancer (ThrbPV/PVPten+/− mice). The ThrbPV/PVPten+/− mouse expresses a dominantly negative thyroid hormone receptor β (denoted as PV) and a deletion of one single allele of the Pten gene. The ThrbPV/PVPten+/− mouse spontaneously develops follicular thyroid cancer, which allows its use as a preclinical mouse model to test potential therapeutics. We recently showed that inhibition of STAT3 activity by a specific inhibitor markedly delays thyroid cancer progression in high-fat-diet–induced obese ThrbPV/PVPten+/− mice (HFD-ThrbPV/PVPten+/− mice). Further, metformin, a widely used antidiabetic drug, blocks invasion and metastasis, but not thyroid tumor growth in HFD-ThrbPV/PVPten+/− mice. To improve efficacy in reducing thyroid tumor growth, we treated HFD-ThrbPV/PVPten+/− with JQ1, a potent inhibitor of the activity of bromodomain and extraterminal domain (BET), and with metformin. We found that the combined treatment synergistically suppressed thyroid tumor growth by attenuating STAT3 and ERK signaling, resulting in decreased anti-apoptotic key regulators such as Mcl-1, Bcl-2, and sruvivin and increased pro-apoptotic regulators such as Bim, BAD and cleave caspase 3. Furthermore, combined treatment of JQ1 and metformin reduced cMyc protein levels to suppress vascular invasion, anaplasia, and lung metastasis. These findings indicate that combined treatment is more effective than metformin alone and suggest a novel treatment modality for obesity-activated thyroid cancer.
INTRODUCTION
Thyroid cancer is the most common endocrine malignancy. It consists of an array of different histologic and biologic types, but the most common are differentiated thyroid carcinomas (DTCs). While the overall survival of patients with DTCs is generally better than many other cancers, recent studies have indicated an increasing incidence of DTCs worldwide in the past decades (Chen, et al. 2009; Pathak, et al. 2013). The increased incidence has been attributed to the widespread use of ultrasonography that has led to detection of micro-carcinomas (tumors with diameter less than 1 cm) [Thyroid Cancer Facts and Figures. National Cancer Institute Surveillance, Epidemiology, and End Results Program. (2015), available from: http://seer.cancer.gov/statfacts/html/thyro.html]. However, other studies have reported that the increased incidence is not limited solely to micro-carcinomas, tumors with other sizes are also increasing (Blomberg, et al. 2012; Chen et al. 2009; Lim, et al. 2017). These observations raise the possibility that other risk factors could also contribute to the increased incidence of thyroid cancer.
One risk factor that has been supported by epidemiological studies is obesity. Extensive analyses reported a positive association of obesity with thyroid cancer (Han, et al. 2013; Kitahara, et al. 2016; Kitahara, et al. 2011; Ma, et al. 2015; Renehan, et al. 2008). Studies by Meta-analysis showed that the risk of thyroid cancer was 25% greater in overweight and 55% greater in obese individuals than their normal-weight peers (Schmid, et al. 2015). A large pooled analysis of 22 prospective studies showed that for each 5-unit increase in the body mass index (BMI), young-adult BMI, adult BMI and waist circumference, were associated with 6%, 13%, 7%, and 3% greater risk of thyroid cancer, respectively (Kitahara et al. 2016). Studies have also suggested that overweight and obesity are significantly associated with more aggressive clinicopathological features in patients with thyroid cancer (Choi, et al. 2015; Kim, et al. 2013a).
The compelling positive association of obesity with thyroid cancer has prompted the use of mouse models of thyroid cancer (ThrbPV/PVPten+/− mice) (Guigon, et al. 2009) to test the hypothesis that obesity could promote thyroid cancer progression. The ThrbPV/PVPten+/− mouse expresses a potent dominantly negative thyroid hormone receptor β (TRβPV) and a deletion of one allele of the Pten gene (phosphatase and tensin homologue deleted from chromosome 10) (Guigon et al. 2009). Pten deficiency further exacerbates the overly activated PI3K/AKT signaling found in ThrbPV/PV mice (Guigon et al. 2009). The Pten haplodeficiency introduced into ThrbPV/PV mice (Furuya, et al. 2006) results in rapid development of aggressive cancer phenotype with decreased survival and increased distant metastasis, making it useful for preclinical studies (Guigon, et al. 2010; Guigon et al. 2009). Using ThrbPV/PVPten+/− mice, Kim et al. found that diet-induced obesity increases tumor growth and promotes anaplastic transformation in thyroid cancer (Kim, et al. 2013b). Further molecular analyses indicated that the obesity-activated aggressive pathological progression is driven by activation of the leptin-JAK2-STAT3 signaling pathway (Kim et al. 2013b). These findings led to studies targeting the STAT3 signaling to block the obesity-activated cancer progression. Remarkably, by using a STAT3 specific inhibitor, S3I-201, Park et al. found that the thyroid tumor growth was inhibited, survival was increased, and lung metastasis was blocked (Park, et al. 2016b).
These preclinical studies provided supporting evidence that ThrbPV/PVPten+/− mice could be used to test other potential therapeutics to prevent obesity-activated thyroid cancer. Accordingly, we tested metformin (1,1-dimethylbiguanide hydrochloride) as a potential therapeutic (Park, et al. 2016a). Metformin is the most commonly prescribed antihyperglycemic drug for treatment of type II diabetes patients (Crandall, et al. 2008; Pierotti, et al. 2013). In addition to its use in patients with diabetes, metformin has been associated with decreased cancer incidence and mortality in mammary, lung, colon, endometrial, and thyroid cancer (Anisimov, et al. 2005; Ben Sahra, et al. 2010; Cantrell, et al. 2010; Hosono, et al. 2010; Klubo-Gwiezdzinska, et al. 2012; Memmott, et al. 2010; Rotella, et al. 2006). Using ThrbPV/PVPten+/− mice, we found that metformin markedly delays thyroid cancer progression by blocking vascular invasion and anaplasia, via deactivation of STAT3-ERK-vimentin and fibronectin-integrin signaling pathways in HFD-ThrbPV/PVPten+/− mice (Park et al. 2016a). However, metformin has no apparent effect on thyroid tumor growth, suggesting that the tumor growth and invasion/metastasis of thyroid cancer could be regulated by different pathways. We therefore searched for another potential therapeutic that could suppress tumor growth and block metastasis when used in combined treatment with metformin.
We tested the efficacy of JQ1 in the combined treatment with metformin to assess the antitumor effects in HFD-ThrbPV/PVPten+/− mice. JQ1 is an inhibitor of the activity of the bromodomain-containing protein 4 (BRD4), a member of the bromodomain and extraterminal domain (BET) family proteins (Filippakopoulos, et al. 2012). JQ1 competes with BRD4 for binding to acetyl-lysines on the chromatin to repress transcription of target genes (Filippakopoulos, et al. 2010). JQ1 has been reported to exhibit inhibitory effects on hematological malignant cancers, glioblastoma, lung and prostate cancers (Asangani, et al. 2014; Lockwood, et al. 2012; Shimamura, et al. 2013). JQ1 has also been shown to suppress cell proliferation and tumor growth of both differentiated and undifferentiated thyroid cancer cell lines (Gao, et al. 2016; Mio, et al. 2016). Recently, we reported that JQ1 markedly decreased thyroid tumor growth and prolonged survival of a mouse model of anaplastic thyroid cancer (ATC, ThrbPV/PVKrasG12D mice). In ThrbPV/PVKrasG12D mice, JQ1 suppresses the cMyc transcription via interference with BRD4 functions to block cMyc tumor promoting activity (Zhu, et al. 2017). Recent reports suggested that JQ1 shows synergistic anticancer effect in combination with other agents, such as HDAC inhibitors (Borbely, et al. 2015) and rapamycin (Lee, et al. 2015). However, the antitumor effects of JQ1 combined with metformin are currently unknown.
The aim of the present study was to evaluate the antitumor effect of JQ1 in combined treatment with metformin on the obesity-activated thyroid cancer of HFD-ThrbPV/PVPten+/− mice. We found that the combined treatment of JQ1 plus metformin synergistically decreased thyroid tumor growth, but treatment with a single agent did not. Furthermore, JQ1 together with metformin blocked occurrence of vascular invasion and anaplasia, and decreased lung metastasis mediated by reducing the cMyc protein levels. These results suggest that metformin combined with JQ1 could be tested as an effective treatment modality for obesity-activated thyroid cancer.
MATERIALS AND METHODS
Mice and treatment with metformin and JQ1
Animal protocols for care and handling in the present study were approved by the National Cancer Institute Animal Care and Use Committee. Generation of ThrbPV/PVPten+/− mice was described in previous studies (Guigon et al. 2009; Kim et al. 2013b). The HFD (60% calories from fat) was purchased from Research Diets (New Brunswick, NJ). The mice were administered HFD diet from the age of 6 weeks until the age of 20 weeks (the end of study). JQ1 was dissolved in DMSO to make a 100 mg/ml stock and, before use, was diluted with 10% β-cyclodextrin (cat# H107, Sigma-Aldrich, St. Louis, MO). JQ1 or vehicle was administrated by intraperitoneal injection 5 days in a week at a dose of 50 mg/kg/mouse from age 17 weeks until the end of study. Metformin (cat#M1566, Spectrum, Gardena, CA) was diluted in drinking water (0.5 mg/ml) and the solution was changed weekly as described previously (Park et al. 2016a). Metformin was administrated from the age of 6 weeks until the study ended at 20 weeks. The weight of the mice was measured weekly. The mice were euthanized at the end point or when they became moribund with rapid weight loss, hunched posture, and labored breathing. After mice were euthanized, the thyroids and lungs were collected for weighing, histologic analysis, and biochemical studies.
Western blot analysis
The Western blot analysis from thyroid tissues was carried as described previously (Park et al. 2016a). Thirty μg of protein sample was loaded and separated for Western blot analysis. Primary antibodies for p-STAT3 (1:500 dilution), total-STAT3 (1:1000 dilution), p-ERK (1:2000 dilution), total-ERK (1:2000 dilution), vimentin (1:1000 dilution), E-cadherin (1:1000 dilution), Bim (1:1000 dilution), BAD (1:1000 dilution), and GAPDH (1:5000 dilution) were purchased from Cell Signaling Technology (Danvers, MA). Antibodies for Mcl-1 (1:500 dilution), Bcl-2 (1:2000 dilution), MMP9 (1:500 dilution), Fibronectin (1:500 dilution), and N-cadherin (1:500 dilution) were purchased from Santa Cruz Biotechnology (Dallas, TX). cMyc antibody (1:5000 dilution) was a gift from Dr. Ira Pastan, NCI. Band intensities were quantified by using NIH IMAGE software (Image J 1.48v).
Histopathologic analysis
Thyroid gland and lung tissues were fixed in 10% neutral-buffered formalin (NBF, approximately 4% formaldehyde) (Sigma-Aldrich, St. Louis, MO) and subsequently embedded in paraffin. Five-μm thickness sections were stained with hematoxylin and eosin (HistoServ, Germantown, MD). For thyroids, morphologic evidence of hyperplasia, capsular invasion, and vascular invasion was routinely examined in that single section.
Immunohistochemistry (IHC)
Cleaved caspase 3 expression was evaluated by immunohistochemical staining of paraffin-embedded thyroid tumor sections. Primary antibody for the cleaved caspase 3 was purchased from R&D systems (Minneapolis, MN). Immunohistochemistry (IHC) was conducted similarly as described (Zhu, et al. 2014). A primary antibody (1:200 dilution) was incubated with tissue section overnight at 4 C. Peroxidase activity from the secondary antibody was developed with diaminobenzidine (DAB). And the sections were counterstained with hematoxylin. Relative positive cell ratio was quantified by using NIH IMAGE software.
Statistical analysis
All statistical analyses and the graphs were performed and generated using GraphPad Prism version 6.0 (GraphPad Software, La Jolla, CA). P<0.05 is considered statistically significant. The Kaplan–Meier method with log-rank test was used to compare survival in each treatment group. All data are expressed as mean ± SEM.
RESULTS
Combined treatment of metformin and JQ1 increases survival rate and decreases thyroid tumor growth in HFD-ThrbPV/PVPten+/− mice
Previously, we reported that in HFD-ThrbPV/PVPten+/− mice metformin decreases the occurrence of invasion and metastasis, but not thyroid tumor growth (Park et al. 2016a). We therefore sought another potential therapeutic that could inhibit thyroid tumor growth. We chose JQ1, which was shown to suppress tumor growth effectively in anaplastic thyroid cancer (Zhu et al. 2017). We therefore evaluated the effect of treatment with metformin combined with JQ1 on the survival, body weight, and thyroid weight of HFD-ThrbPV/PVPten+/− mice. Figure 1A shows that the percent survival rates of single treatment, with either JQ1 (open squares; 67%) or metformin (closed circles; 64%), though trended to longer survival as compared with controls, but were not significantly different from that of the controls (open circles; 40%). However, the combined treatment increased the survival to 82% (closed squares). While % survival of mice treated with JQ1 or metformin only trended toward improved survival as compared with mice treated with vehicle, no statically significant differences were found. However, the difference in the survival between mice treated with combined agents or with vehicle was significant (p=0.0472). These results indicate that combination treatment of metformin and JQ1 led to improved survival of HFD-ThrbPV/PVPten+/− mice.
Figure 1. Effects of metformin, JQ1, and combined treatment on survival and thyroid tumor growth of HFD-ThrbPV/PVPten+/− mice.
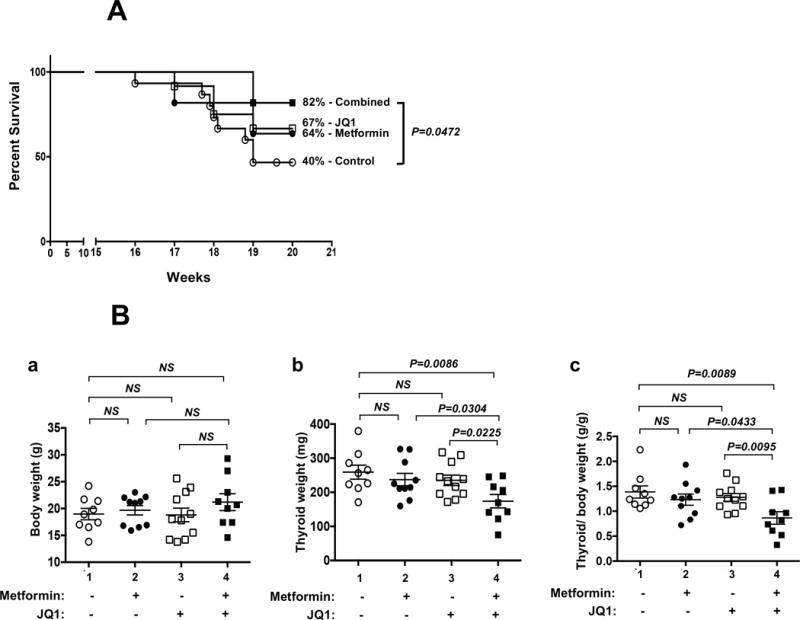
(A) Survival curves for HFD-ThrbPV/PVPten+/− mice treated with vehicle control (open circle, n=15), metformin (closed circles, n=11), JQ1 (open square; n=12), or combination treatment of metformin with JQ1 (closed square, n=11). Mice were treated high fat diet (HFD) and metformin (0.5 mg/kg/mouse) from the age of 6 weeks until the age of 20 weeks (the study’s end). JQ1 (50 mg/kg/mouse) was administrated i.p. 5 times a week from age 17 weeks to the end of study (20 weeks). Survival data are shown as a Kaplan-Meier plot and analyzed by log-rank test. The difference in survival was significant between mice between combined treatment and mice treated with vehicle (p=0.0473); but was not significant between mice-treated with vehicle and mice treated with metformin or JQ1. (B) Body weight (panel a) and thyroid weight (panel b) and ratios of thyroid weight to total body weight (panel c) were compared for the mice treated with vehicle, metformin only, JQ1 only, and a combination of metformin with JQ1 (n=9-11). Values are means ± SEM. The p values are indicated. NS= not significant.
We next examined whether the body weight of HFD-ThrbPV/PVPten+/− mice could be affected by metformin, JQ1, or the combined treatment. Figure 1B-a shows that the body weight of HFD-ThrbPV/PVPten+/− mice was not significantly affected by either single treatment or the combined treatment. The tumor weight was not affected by single treatment with either metformin (237.3±18.3 mg; n=10, Figure 1B-b, data cluster #2) or JQ1 (235.3±15.35 mg, n=11, data cluster #3) as compared with vehicle-treated HFD-ThrbPV/PVPten+/− mice (259±20.55 mg, n=9, data cluster #1). However, combined treatment significantly lowered the tumor weight by ~33% (173.9±19.66 mg, n=9, data cluster #4). We further took into consideration the variability in the individual weight of mice. Using the thyroid tumor: body weight ratios, we found a similar reduction in thyroid tumor weight by combined treatment results (Figure 1B-c, compare data cluster 4 versus 1). These results indicate that JQ1 in combination with metformin synergistically decreased thyroid tumor growth of obesity-activated thyroid cancer.
Inhibition of tumor growth by combined treatment of metformin plus JQ1 in HFD-ThrbPV/PVPten+/− mice is mediated by suppressing STAT3 and ERK signaling pathways
We have previously shown that activation of leptin-JAK2-STAT3 signaling increases thyroid tumor growth in HFD-ThrbPV/PVPten+/− mice (Kim et al. 2013b). We therefore tested the hypothesis that the inhibition of tumor growth by the combined treatment of JQ1 plus metformin was via the attenuation of STAT3 signaling. Western blot analysis showed that the level of p-STAT3 (Y705) in the thyroid tumors of HFD-ThrbPV/PVPten+/− mice was lowered by metformin treatment (Figure 2A-I-a, lanes 4-6). The protein levels of p-STAT3 (Y705) protein levels were markedly decreased in tumors of HFD-ThrbPV/PVPten+/− mice treated with JQ1 (panel a, lanes 7-9) or further with combined treatment (panel a, lanes 10-12). Quantitative analysis indicated that treatment of HFD-ThrbPV/PVPten+/− mice with metformin or JQ1 decreased the ratios of p-STAT3 to total STAT3 by 36% and 52%, respectively (Figure 2A-II-a, bars 2 and 3, respectively). The combined treatment further decreased the ratios of p-STAT3 to total STAT3 by 61% (Figure 2A-II-a, compare bar 4 to bar 1). These data indicated that activity of STAT3 was synergistically suppressed by metformin and JQ1 combined treatment.
Figure 2. Metformin, JQ1, and combined treatment inhibit STAT3 and ERK1/2 activation in the thyroid tumors of HFD-ThrbPV/PV Pten+/− mice.
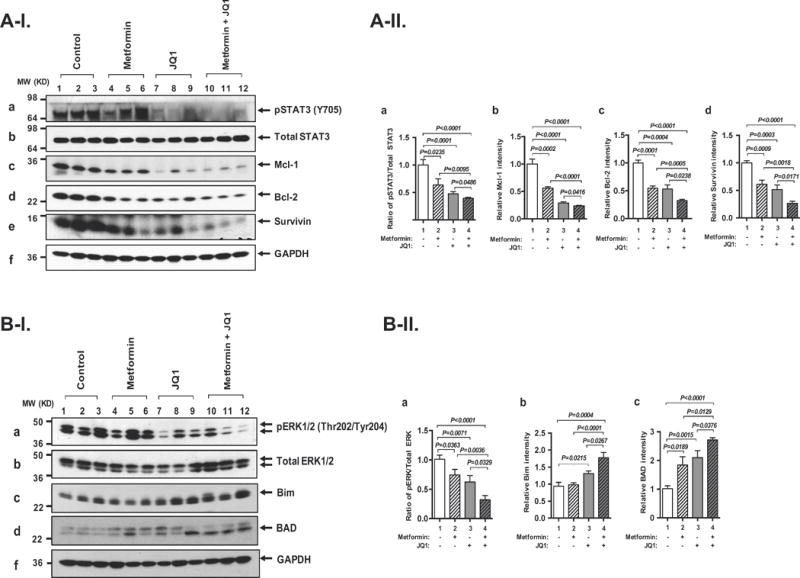
(A-I) Western blot analysis for p-STAT3 (Y705) (panel a), total STAT3 (panel b), Mcl-1 (panel c), Bcl-2 (panel d), survivin (panel e), and GAPDH (panel f) in the thyroids of HFD-ThrbPV/PV Pten+/− mice treated with control, metformin, JQ1, or combination treatment, respectively. GAPDH used as a loading control (n=3-6 for each group). (A-II) The band intensities from western blot analysis detected in (A-I) were quantified by Image J analysis. Ratios of p-STAT3 versus total STAT3 (panel a), Mcl-1 (panel b), Bcl-2 (panel c), survivin (panel d). All band intensities were normalized using GAPDH as a loading control. Values are shown as means ± SEM. The p values are indicated. (B-I). Western blot analysis for p-ERK1/2 (Thr202/Tyr204) (panel a), total ERK1/2 (panel b), Bim (panel c), BAD (panel d), and GAPDH (panel e) in the thyroids of HFD-ThrbPV/PV Pten+/− mice treated with control, metformin, JQ1, or combination treatment, respectively. GAPDH used as a loading control (n=3-6 for each group). Values are shown as means ± SEM. (B-II). The band intensities from western blot analysis detected in (B-I) were quantified by Image J analysis. Ratios of p-ERK1/2 versus total ERK1/2 (panel a), Bim (panel b), and BAD (panel c) are indicated. All band intensities were normalized using GAPDH as a loading control. Values are shown as means ± SEM. The p values are indicated. (C-I). Immuno-histochemical analysis for cleaved caspase 3 was carried out in the thyroid sections from the control (panel b), metformin (panel d), JQ1 (panel f), and combined treatment (panel h) of HFD-ThrbPV/PV Pten+/− mice. The representative positively stained cells are marked by arrows. The negative control panels using IgG are shown in the corresponding panels (panel a, c, e, and g). (C-II). The cleaved caspase 3-positively stained cells were quantified as percentage of cleaved caspase 3-positive cells versus total cells. The p values are indicated.
To further evaluate the functional consequences of attenuated STAT3 activity by single or combined treatment, we ascertained the expression of STAT3 downstream target genes critical for apoptosis at the protein levels. Mcl-1 (myeloid cell leukemia-1) and Bcl-2 (B-cell lymphoma 2), critical for cell survival (anti-apoptotic), are STAT3 downstream target genes (Alvarez, et al. 2005; Bhattacharya, et al. 2005). Western blot analysis showed that metformin treatment decreased Mcl-1 protein level (Figure 2A-I-c, lanes 4-6) 45% (Figure 2B-b, bar 2) as compared with vehicle control (Figure 2A-I-c, lanes 1-3). However, JQ1 or the combined treatment further lowered the Mcl-1 protein level (Figure 2A-I-c, lanes 7-9 and 10-12, respectively) to 71% and 76% (Figure 2A-II-b, bars 3 and 4, respectively). Similarly, metformin or JQ1 decreased Bcl-2 protein levels (Figure 2A-I-d, lanes 4-6 and 7-9, respectively) 45% and 47%, respectively (Figure 2A-II-c, bars 2 and 3) as compared with vehicle control (Figure 2A-I-d, lanes 1-3). The combined treatment further lowered the Bcl-2 protein levels (Figure 2A-I-d, lanes 10-12) 68% (Figure 2A-II-c, bar 4). Survivin, a member of the inhibitor of apoptosis family, is a protein that functions to inhibit caspase activation, thereby leading to negative regulation of apoptosis or programmed cell death. Disruption of the survivin induction pathways leads to an increase in apoptosis and to decrease tumor growth (Mita, et al. 2008). We found that metformin or JQ1 treatment decreased the survivin protein levels (Figure 2A-I-e, lanes 4-6 and 7-9, respectively) 39% and 48%, respectively (Figure 2A-II-c-d, bar 2). The combined treatment further decreased survivin protein levels 72% (Figure 2A-I-e; Figure 2A-II-d, bar 4). These results indicated that metformin and JQ1combined treatment synergistically suppressed STAT3-Mcl-1/Bcl-2/survivin signaling to promote apoptosis of thyroid tumor cells of HFD-ThrbPV/PV Pten+/− mice.
In addition to the STAT3 signaling, leptin could act to increase cancer cell proliferation via an extracellular signal-regulated kinase (ERK) signaling pathway (Saxena, et al. 2007). We found relatively high protein levels of phosphorylated ERK (p-ERK) in the vehicle-treated mice (Figure 2B-I-a, lanes 1-3). The protein levels of p-ERK protein was decreased by metformin, JQ1 or combined treatment (Figure 2B-I-a, lanes 4-6, 7-9, 10-12, respectively). The total ERK was not changed under any of the conditions (Figure 2B-I-b). Figure 2B-II-a shows that ratios of the p-ERK and total ERK were decreased 27%, 39%, and 69%, respectively, by metformin, JQ1 or the combined treatment, indicating the synergistic inhibition of p-ERK activity by combined treatment. Bcl-2-like protein 11, commonly called Bim acting to promote apoptosis (Gillings, et al. 2009), was reported to be a downstream effector of ERK signaling (Takezawa, et al. 2011). Accordingly, we assessed Bim protein levels in mutant mice treated with metformin, JQ1, or the combined treatment. Figure 2B-I-c shows that metformin had no apparent effect on the Bim protein level (Figure 2B-I-c, lanes 4-6 versus lanes 1-3), while JQ1 and the combined treatment elevated Bim protein levels (lanes 7-9 and 10-12, respectively). The quantitative data showed that JQ1 or combined treatment increased 1.3 and 1.8-fold of Bim protein levels, respectively (Figure 2B-II-b). We further analyzed the effects of three different treatments on another pro-apoptotic regulator BAD, which is also a downstream effector of ERK signaling (Lu and Xu 2006). We found that metformin, JQ1 or the combined treatment elevated the BAD proteins levels (Figure 2B-I-d) by 1.8-fold, 2.1-fold and 2.7-fold, respectively (Figure 2B-II-c). The suppression of anti-apoptotic regulators (see Figure 2A) and the increased pro-apoptotic activators (see Figure 2B) prompted us to assess the cleaved caspase 3 activity by examination the protein levels in the tumor cells using immune-histochemical analysis. As shown in Figure 2C, increased cleaved caspase 3 protein abundance was apparent in panel f and h. Quantitative analysis shows 3.3-fold and 7.7-fold increased cleaved caspase 3 protein levels were found for tumor cells from HFD-ThrbPV/PVPten+/− mice treated with JQ1 or metformin and JQ1, respectively (Bars 3 and 4, Figure 2C-II, respectively). While cleaved caspase 3 protein abundance trended slightly higher in tumor cells from mice treated with metformin. The increases were not significantly different from the vehicle control (compare bar 2 to bar 1, Figure 2C-II). Taken together, these results indicated that JQ1 and the combined treatment suppressed the STAT3 and ERK signaling pathway to synergistically promote apoptosis of thyroid tumor cells in HFD-ThrbPV/PV Pten+/− mice.
Combined treatment of JQ1 plus metformin suppresses development of thyroid anaplastic foci and metastasis in HFD-ThrbPV/PVPten+/− mice
To assess the effect of single or combined treatment on thyroid cancer progression, we analyzed histopathological features of thyroid tumors of HFD-ThrbPV/PVPten+/− mice with four different treatments. As shown in Figure 3A-I-a, all mice exhibited hyperplasia that was not affected by any treatment. However, while all vehicle-treated mice developed capsular invasion (100%) (Figure 3A-I-b), treatment with metformin alone and JQ1 alone led to a reduction in the frequency of occurrence by 38% and 75%, respectively. Remarkably, capsular invasion was not detected in HFD-ThrbPV/PVPten+/− mice with the combined treatment at the same age (Figure 3A-I-b, bar 4). The occurrence of vascular invasion was decreased further to 64% in the metformin-treated group (Figure 3A-I-c, bar 2), and was blocked in the JQ1-treated and combined-treatment groups (Figure 3A-I-c, bars 3 and 4). Moreover, no occurrence of anaplasia was detected in either the JQ1-treated or the combined-treatment group (Figure 3A-I-d, bars 3 and 4).
Figure 3. Combined treatment of metformin and JQ1 synergistically decreases thyroid tumor progression and lung metastasis in HFD-ThrbPV/PV Pten+/− mice.
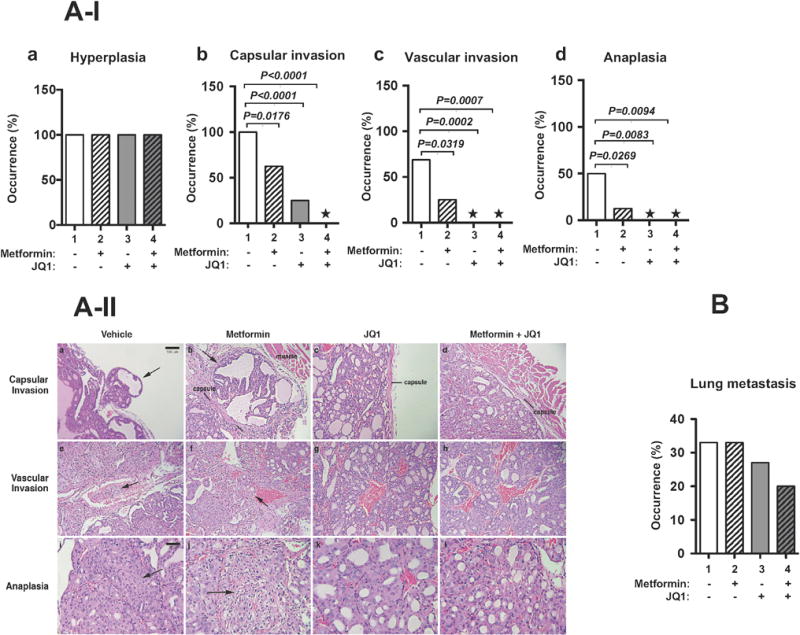
(A-I) The prevalence of hyperplasia (panel a), capsular invasion (panel b), vascular invasion (panel c), and anaplasia (panel d) in the thyroid tumors of HFD-ThrbPV/PVPten+/− mice treated with vehicle, metformin, JQ1, or combination treatment is shown as percentage of occurrence. * Represents no occurrence (n=10-16). P values were obtained by Fischer’s exact test. (A-II). Capsular invasion is shown in samples from vehicle (panel a) and metformin treatment alone (panel b) (arrows), but is absent in samples from JQ1 alone (panel c) and combined treatment of JQ1 plus metformin (panel d), showing intact capsules at the margins of tumors (panels c and d). Vascular invasion is demonstrated in vehicle- (panel e) and metformin-treated tumors (panel f) (arrows show tumor cells in vascular lumens). No tumor cells are seen in vascular spaces in JQ1 (panel g) or combined treatment (panel h). Anaplastic foci (arrows) are evident in vehicle (panel i) and metformin alone treatment (panel j), but no anaplastic foci are seen in JQ1 (panel k) or combined treatment (l). Magnification: panels a–h = ×50; i–l = ×100). (B). The trend in the occurrence of lung metastasis in HFD-ThrbPV/PVPten+/− mice after treatment with vehicle, metformin, JQ1, and combination treatment is shown (n=9-11).
Figure 3A-II shows the representative pathological features in the tumors of HFD-ThrbPV/PVPten+/− mice with four different treatments. The vehicle-treated tumors (controls) consistently showed capsular invasion in all samples (Figure 3A-II-a, indicated by arrows), vascular invasion in the majority (Figure 3A-II-e), and anaplasia in approximately by 50% (Figure 3A-II-i; also Figure 3A-I-d, bar 1). Treatment with metformin alone showed reduction in these features (Figure 3A-II-b and -f), but no significant difference in the frequency of anaplasia (Figure 3A-II-j). JQ1 treatment alone had a more profound effect, with substantial reduction in detection of capsular invasion (Figure 3A-II-c) and essentially no detectable vascular invasion or anaplasia (Figure 3A-II-g and -k). The combined treatment of metformin and JQ1 showed essentially no capsular (Figure 3A-II-d) or vascular invasion (Figure 3A-II-h) and no anaplastic features (Figure 3A-II-l) (please see also Figure 3A-I-d, * in bar 4 indicates no anaplasia was detected).
We further assessed the effect of single or combined treatment on the occurrence of lung metastasis. While our sample number (n=9-11) was not sufficiently large to meaningful analyze the data statistically, Figure 3B shows the trend in the occurrence of lung metastasis. About 33% of vehicle-treated HFD-ThrbPV/PVPten+/− mice developed lung metastasis (Figure 3B, bar 1). No effect of metformin on the occurrence of lung metastasis on HFD-ThrbPV/PVPten+/− mice was detected (Figure 3B, bar 2). However, JQ1 treatment trended to show a ~18% reduction in the occurrence of lung metastasis (Figure 3B, bar 3) as compared with that of vehicle-treated mutant mice. Combined treatment trended a decrease in the frequency of occurrence of lung metastasis (~40%, Figure 3B, bar 4). Taken together, these data suggested that combined treatment of JQ1 and metformin was most effective in the inhibition of thyroid cancer progression.
To understand how the combined treatment inhibited anaplastic progression of thyroid tumors of HFD-ThrbPV/PVPten+/− mice, we evaluated key regulators of epithelial–mesenchymal transition (EMT). EMT is a process by which epithelial cells lose their cell polarity and cell-cell adhesion, and gain migratory and invasion properties to become mesenchymal-type cells. Accordingly, we analyzed how the key regulators of EMT were affected by the single or double treatment. We evaluated cMyc first because it is aberrantly highly expressed in human anaplastic thyroid cancer cells (Enomoto, et al. 2017) and anaplastic thyroid cancer in a mouse model (Zhu et al. 2014). cMyc is known to induce EMT in cancer cells (Cho, et al. 2010; Cowling and Cole 2007; Yin, et al. 2017). Western blot analysis showed that cMyc was highly expressed in the thyroid tumors of vehicle-treated HFD-ThrbPV/PVPten+/− mice (Figure 4A-a, lanes 1-3). Metformin decreased cMyc protein levels in the tumors of HFD-ThrbPV/PV Pten+/− mice (Figure 4A-a, lanes 4-6 versus 1-3). JQ1 or the combined treatment further lowered the cMyc protein levels in the thyroid tumors (Figure 4A-a, lanes 7-9 and 10-12, respectively). Quantitative analysis of the band intensities shows that cMyc protein levels were decreased 55%, 68% and 78% by metformin, JQ1 and the combined treatment, respectively. We next examined the effectors of EMT affected by the single or double treatment of HFD-ThrbPV/PV Pten+/− mice. Matrix metalloproteinase 9 (MMP9) is one of the most important members of the MMPs, known to degrade the extracellular matrix and basement membrane to increase migration, invasion, metastasis, and angiogenesis in cancers (Dragutinovic, et al. 2009). MMP9 is directly regulated by STAT3 signaling (Kamran, et al. 2013). We found that MMP9 was decreased by metformin treatment (Figure 4A-b, lanes 4-6 versus lanes 1-3). Remarkably, JQ1 treatment or combined treatment nearly totally inhibited the expression of MMP9 protein levels (Figure 4A-b, lanes 7-9 and 10-12, respectively; see also the quantitative data in Figure 4B-b, bars 3 and 4).
Figure 4. Metformin, JQ1, and combination treatment decrease the expression of key regulators of EMT in the thyroid tumors of HFD-ThrbPV/PV Pten+/− mice.
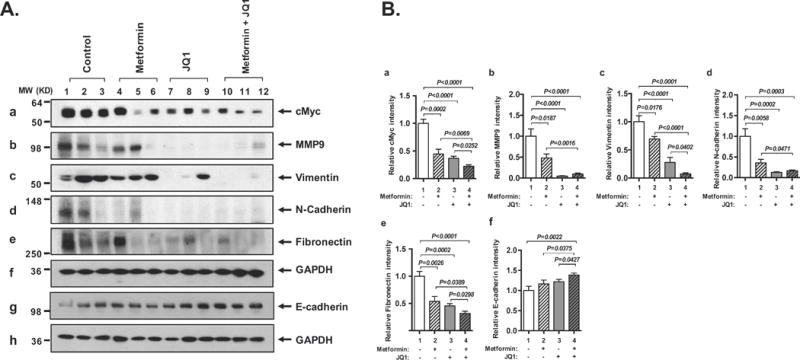
(A) Western blot analysis for cMyc (panel a), MMP9 (panel b), vimentin (panel c), N-cadherin (panel d), fibronectin (panel e), E-cadherin (panel g), and GAPDH (panel f and h) in the thyroid tumors of HFD-ThrbPV/PV Pten+/− mice treated with vehicle, metformin, JQ1, and combination treatment. GAPDH used as a loading control (n=3 for each group). (B) The band intensities from Western blot analysis detected in (A) were quantified by ImageJ analysis. Relative abundance of cMyc (panel a), MMP9 (panel b), vimentin (panel c), N-cadherin (panel d), and fibronectin (panel e), and E-cadherin (panel f) were calculated using the GAPDH as loading control. The p values are indicated.
We next evaluated whether the regulation of vimentin and N-cadherin was responsive to single or combination treatment. Vimentin is a main component of the intermediate filament family (Satelli and Li 2011). N-cadherin is a member of the cadherin family that is mainly located in neural tissue and striated muscle (Pyo, et al. 2007). These major cytoskeletal components are highly expressed in mesenchymal cells. As shown in Figure 4A, expression of vimentin (panel c) and N-cadherin (panel d) was decreased by metformin (lanes 4-6), JQ1 (lanes 7-9), or combination treatment (lanes 10-12) as compared with vehicle treatment (lanes 1-3). Quantitative data showed that vimentin was decreased 31%, 72% and 93%, by metformin, JQ1 and the combined treatment, respectively (Figure 4B, panel c). N-cadherin was decreased 64% by metformin and 83-87% by JQ1 and the combined treatment (Figure 4B, panel d). We further examined the expression of fibronectin. Fibronectin is a high molecular weight protein of the extracellular matrix that binds to membrane-spanning receptor proteins (Pankov and Yamada 2002). As shown in Figure 4A, expression of fibronectin (panel e) was clearly decreased by metformin (lanes 4-6), JQ1 (lanes 7-9), and combination treatment (lanes 10-12) as compared with vehicle treatment (lanes 1-3; also see quantitative data shown in panel e, Figure 4B). The effects of metformin, JQ1 and the combined treatment on the E-cadherin was also evaluated. E-cadherin is a tumor suppressor gene, critical for the formation and maintenance of adherent junctions in areas of epithelial cell-cell contact. Loss of E-cadherin-mediated-adhesion characterizes the transition from benign lesions to invasive, metastatic cancer (Canel, et al. 2013; Kim, et al. 2016; Onder, et al. 2008). As shown in Figure 4A (panel g), while some increases of E-cadherin levels were detected in tumors from mice treated with metformin (lanes 4-6; see also Figure 4B-f), JQ1 (lanes 7-9; Figure 4B-f), a more pronounced increase of E-cadherin protein levels was found by the combined treatment (lanes 10-12; Figure 4B-f, bar 4). Taken together of the elevated E-cadherin and suppressed MMP9, vimentin, N-cadherin, and fibronectin proteins indicated the combined treatment of metformin with JQ1 was most effective to inhibit EMT to suppress thyroid cancer progression of HFD-ThrbPV/PVPten+/− mice (see also Figure 5).
Figure 5. A proposed model to account for the actions by which metformin and JQ1 together inhibit tumor progression in HFD-ThrbPV/PV Pten+/− mice.
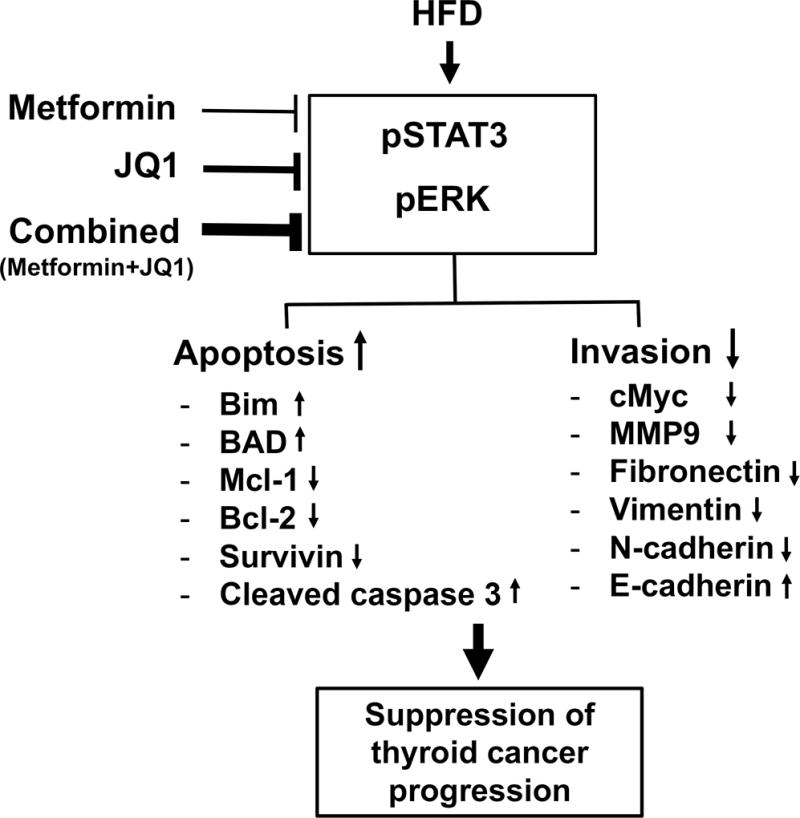
HFD induces the aberrant activation of STAT3 and ERK signaling to promote thyroid cancer progression in HFD-ThrbPV/PV Pten+/− mice. JQ1 and metformin together act to increase the tumor cell apoptosis by elevating the protein levels of pro-apoptotic regulators Bim and BAD and by suppressing the protein levels of anti-apoptotic regulators (Mcl1, Bcl2 and survivin), resulting in increased cleaved caspase 3 activity. Combined treatment of JQ1 and metformin also act to suppress invasion and metastasis by decreasing EMT key regulators (cMyc, MMP9, fibronectin, vimentin, N-cadherin) and increasing E-cadherin protein levels. By acting on these two pathways, JQ1 and metformin together suppress obesity-activated thyroid cancer progression. The extent in the thickness of the lines in the inhibition of pSTAT3 and pERK signaling schematically represents the degree of suppression.
Discussion
While the impact of obesity on cardiovascular disease, diabetes, and stroke is well documented and extensively studied, the effects of obesity on cancer have just begun to be unraveled. The positive association of obesity with thyroid cancer demonstrated by the epidemiological studies has promoted studies aimed at acquiring molecular evidence to demonstrate a direct cause-effect relationship. Indeed, using preclinical mouse models of thyroid cancer, we uncovered that high-fat-diet–induced obesity elevates serum leptin levels to propel JAK2-STAT3 signaling to potentiate thyroid carcinogenesis (Kim et al. 2013b). That STAT3 was a potential molecular target for obesity-activated thyroid cancer was validated by the findings that a STAT3 specific inhibitor markedly inhibits thyroid tumor growth, prolongs survival, reduces invasion, and blocks lung metastasis of HFD-ThrbPV/PVPten+/− mice (Park et al. 2016b). Intriguingly, metformin treatment attenuates signaling pathways, thus leading to the suppression of invasion and metastasis, but does not affect tumor growth (Park et al. 2016a).
To be able to arrest tumor growth concurrently with blocking invasion and metastasis, we chose JQ1 to combine with metformin for treatment. We found that metformin, a widely used antidiabetic drug, when used together with JQ1, was a more effective treatment, not only to inhibit tumor growth, but also to suppress invasion and metastasis of obesity-activated thyroid carcinogenesis. The present study has identified a novel combined treatment modality that could possibly be used to treat thyroid cancer potentiated by obesity. Toward this end, it would be of importance to point out while the combined treatment of JQ1 and metformin was most effective in the suppression of tumor cell invasion, as shown by the concerted changes of cMyc, MMP9, and EMT regulators (see Figure 5). However, the extent of the suppression of tumor cell invasion potential was not linearly correlated with decreases in the prevalence of lung metastasis. This observation is consistent with Stephen Paget’s original “seed and soil” hypothesis, which still holds true today (Paget 1989). The hypothesis is that tumor metastasis is the product of favorable interactions between metastatic tumor cells (the “seed”) and their organ microenvironment (the “soil”). The present studies showed that while combined treatment virtually totally blocked capsular and vascular invasion (see Figure 3A-c and d) of primary tumor cells, the occurrence of lung metastasis was only partially reduced (~ 40%, see Figure 3B). These findings suggested that the therapeutic intervention would be more effective in aiming to make the lung microenvironment less favorable for the metastatic thyroid tumor cells to lodge and to proliferate. For future studies, the HFD-ThrbPV/PVPten+/− mouse could be used a model for elucidating cellular regulators that facilitate the interaction of metastatic tumor cells with the lung microenvironment, thereby identifying potential targets for prevention and better treatment of metastatic thyroid cancer associated with obesity.
The rationale behind our choosing JQ1 to combine with metformin as a treatment was based on findings that JQ1 suppresses thyroid tumor growth in a mouse model of anaplastic thyroid cancer (ATC) (ThrbPV/PVKrasG12D). In that ATC mouse model, the molecular pathways by which JQ1 inhibits tumor growth in ThrbPV/PVKrasG12D mice are mainly via suppression of cMyc expression and its downstream cell cycle regulators, but JQ1 has no effects on the apoptosis pathways (Enomoto et al. 2017). In the ThrbPV/PVKrasG12D mice, the antitumor effects of JQ1 single agent or combined with metformin was mainly via the cMyc-mediated alterations in the expression of apoptotic key regulators to enhance tumor cell apoptosis, but JQ1 has no major effect on the cell cycle regulators (data not shown). In these two thyroid cancer mouse models, both of the mutant mice express the oncogenic TRβPV, but the second mutations differ. In ThrbPV/PVPten+/− mice, the haplodeficiency of the Pten gene aggressively drives the PI3K-AKT signaling, whereas in ThrbPV/PVKrasG12D mice, the KrasG12D mutation propels the downstream MEK signaling. JQ1 is known to preferentially regulate the transcription program of cMyc through super-enhancer on the chromatin (Loven, et al. 2013). However, the present findings from HFD-ThrbPV/PVPten+/− mice and previous studies from ThrbPV/PVKrasG12D mice (Zhu et al. 2017) clearly show distinct molecular pathways downstream of cMyc. Therefore, the antitumor effects of JQ1 would depend on the cellular context due to the type of mutations in a particular cancer. At present, it is unknown how cMyc differentially cross-talks with PTEN-PI3K-AKT signaling (as in ThrbPV/PVPten+/− mice, a model of follicular thyroid cancer) or with MEK signaling (as in ThrbPV/PVKrasG12D mice, a model of anaplastic thyroid cancer) to inhibit thyroid tumor growth in these different mouse models of thyroid cancer. Future studies to identify the cross-talk partners in the network with cMyc would help understanding of how JQ1 could collaborate with other therapeutics in suppressing thyroid tumor growth. To this end, the cell-based approach with the choice of appropriate cancer cell lines would be useful both to dissect how network partners interact at the cellular level and to elucidate the molecular mechanisms underlying the outcome of the cross talks.
The ThrbPV/PVPten+/− mouse represents a unique mouse model of follicular thyroid cancer in that a mutation of thyroid hormone receptor β (TRβPV) activates PI3K activity via direct protein-protein interaction with PI3K-regulatory subunit p85, to markedly increase PI3K kinase activity (Furuya et al. 2006). Together with a deletion of one allele of the Pten gene, PI3K activity is further activated via the loss of the negative regulation of PI3K activity by PTEN (Guigon et al. 2009). The functional consequences of over-activation of PI3K-AKT in the ThrbPV/PVPten+/− mouse is relevant in human thyroid cancer. Studies have reported that genetic alterations in the PI3K-AKT pathway play a critical role in thyroid tumorigenesis and progression (Hou, et al. 2007; Wang, et al. 2007; Xing 2010). Furthermore, association of PTEN methylation with the activating genetic alterations in the PI3K-AKT pathway has been found in human thyroid tumors (Hou, et al. 2008). The activation of PI3K-AKT signaling mediated by the TRβPV mutation accompanied by the deletion of one allele of the Pten gene in ThrbPV/PVPten+/− mouse is phenotypically reminiscent of the silencing of PTEN associated with activating PI3K-AKT pathway reported by Hou et al in human thyroid cancer. In both cases, the negatively regulating activity of PI3K-AKT signaling by PTEN is lost leading to further over-activation of the PI3K-AKT pathway. Thus, while homozygous mutations of the thyroid hormone receptor β have yet to be identified in thyroid cancer patients, the functional consequences of the TRβPV mutation in the over-activation of PI3K-AKT pathways demonstrated in ThrbPV/PVPten+/− mouse is useful as a model to dissect the altered signaling pathways and serve as a model for testing potential molecular targets.
Several recent meta-analyses have shown that the use of metformin in diabetic patients is associated with a lower risk for cancer (Decensi, et al. 2010; Noto, et al. 2012). In particular, metformin has been shown to decrease thyroid cancer risks (Tseng 2014). Further, metformin treatment is associated with a higher remission rate in diabetic patients with thyroid cancer (Klubo-Gwiezdzinska, et al. 2013). These findings are consistent with the present results from our mouse models of thyroid cancer, suggesting that metformin could be a potential therapeutic for obesity-activated thyroid cancer.
Recent studies have indicated that combined treatment is more beneficial than a single agent in the treatment of thyroid cancer (Iyer, et al. 2018; Subbiah, et al. 2018). For example, although effective treatment of anaplastic thyroid cancer is very limited, sixteen patients with BRAFV600E mutated anaplastic thyroid cancer have shown robust clinical response when treated with dabrafenib (a BRAF inhibitor) and trametinib (a MEK inhibitor) (Subbiah et al. 2018). In line with these clinical findings, the present study showed that metformin in combination with JQ1 is more effective than metformin alone. Recently, several second-generation JQ1 analogs (BET inhibitors) are being tested in clinical trials for use in treating solid tumors, such as NUT midline carcinoma, lung cancer, colorectal cancer, neuroblastoma, triple negative breast cancer, castration-resistant prostate cancer and hepatocellular carcinoma, and melanoma (https://clinicaltrials.gov/ct2/show/NCT01587703; https://clinicaltrials.gov/ct2/show/NCT02369029). Metformin is effective, safe, and affordable and could be easily available to combine with other therapeutics to treat obesity-activated thyroid cancer. The present preclinical findings provide a basis for future clinical trials for obesity-activated thyroid cancer in which metformin treatment is combined with second-generation JQ1 analogs. Further studies are warranted to explore other therapeutics as a combined treatment with metformin for obesity-activated thyroid cancer. Our mouse models of thyroid cancer could be used for such preclinical testing of other therapeutics.
Acknowledgments
Funding:
The present research was supported by the Intramural Research Program at the Center for Cancer Research, National Cancer Institute, National Institutes of Health.
Footnotes
Declaration of Interest: Authors have nothing to declare
Author contributions:
Sheue-yann Cheng: Conceptualization, validation of data, review, writing and editing the paper, funding acquisition, investigation, resources, project administration, supervision
Sunmi Park: data curation and analysis, methology, investigation, formal analysis, and writing the first draft
Mark Willingham: analysis of pathology and histology, methodology
Jun Qi: Provided reagents and reviewing of data
References
- Alvarez JV, Febbo PG, Ramaswamy S, Loda M, Richardson A, Frank DA. Identification of a genetic signature of activated signal transducer and activator of transcription 3 in human tumors. Cancer Res. 2005;65:5054–5062. doi: 10.1158/0008-5472.CAN-04-4281. [DOI] [PubMed] [Google Scholar]
- Anisimov VN, Berstein LM, Egormin PA, Piskunova TS, Popovich IG, Zabezhinski MA, Kovalenko IG, Poroshina TE, Semenchenko AV, Provinciali M, et al. Effect of metformin on life span and on the development of spontaneous mammary tumors in HER-2/neu transgenic mice. Exp Gerontol. 2005;40:685–693. doi: 10.1016/j.exger.2005.07.007. [DOI] [PubMed] [Google Scholar]
- Asangani IA, Dommeti VL, Wang X, Malik R, Cieslik M, Yang R, Escara-Wilke J, Wilder-Romans K, Dhanireddy S, Engelke C, et al. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature. 2014;510:278–282. doi: 10.1038/nature13229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben Sahra I, Le Marchand-Brustel Y, Tanti JF, Bost F. Metformin in cancer therapy: a new perspective for an old antidiabetic drug? Mol Cancer Ther. 2010;9:1092–1099. doi: 10.1158/1535-7163.MCT-09-1186. [DOI] [PubMed] [Google Scholar]
- Bhattacharya S, Ray RM, Johnson LR. STAT3-mediated transcription of Bcl-2, Mcl-1 and c-IAP2 prevents apoptosis in polyamine-depleted cells. Biochem J. 2005;392:335–344. doi: 10.1042/BJ20050465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomberg M, Feldt-Rasmussen U, Andersen KK, Kjaer SK. Thyroid cancer in Denmark 1943–2008 before and after iodine supplementation. Int J Cancer. 2012;131:2360–2366. doi: 10.1002/ijc.27497. [DOI] [PubMed] [Google Scholar]
- Borbely G, Haldosen LA, Dahlman-Wright K, Zhao C. Induction of USP17 by combining BET and HDAC inhibitors in breast cancer cells. Oncotarget. 2015;6:33623–33635. doi: 10.18632/oncotarget.5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canel M, Serrels A, Frame MC, Brunton VG. E-cadherin-integrin crosstalk in cancer invasion and metastasis. J Cell Sci. 2013;126:393–401. doi: 10.1242/jcs.100115. [DOI] [PubMed] [Google Scholar]
- Cantrell LA, Zhou C, Mendivil A, Malloy KM, Gehrig PA, Bae-Jump VL. Metformin is a potent inhibitor of endometrial cancer cell proliferation--implications for a novel treatment strategy. Gynecol Oncol. 2010;116:92–98. doi: 10.1016/j.ygyno.2009.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen AY, Jemal A, Ward EM. Increasing incidence of differentiated thyroid cancer in the United States, 1988–2005. Cancer. 2009;115:3801–3807. doi: 10.1002/cncr.24416. [DOI] [PubMed] [Google Scholar]
- Cho KB, Cho MK, Lee WY, Kang KW. Overexpression of c-myc induces epithelial mesenchymal transition in mammary epithelial cells. Cancer Lett. 2010;293:230–239. doi: 10.1016/j.canlet.2010.01.013. [DOI] [PubMed] [Google Scholar]
- Choi JS, Kim EK, Moon HJ, Kwak JY. Higher body mass index may be a predictor of extrathyroidal extension in patients with papillary thyroid microcarcinoma. Endocrine. 2015;48:264–271. doi: 10.1007/s12020-014-0293-z. [DOI] [PubMed] [Google Scholar]
- Cowling VH, Cole MD. E-cadherin repression contributes to c-Myc-induced epithelial cell transformation. Oncogene. 2007;26:3582–3586. doi: 10.1038/sj.onc.1210132. [DOI] [PubMed] [Google Scholar]
- Crandall JP, Knowler WC, Kahn SE, Marrero D, Florez JC, Bray GA, Haffner SM, Hoskin M, Nathan DM, Diabetes Prevention Program, Research G. The prevention of type 2 diabetes. Nat Clin Pract Endocrinol Metab. 2008;4:382–393. doi: 10.1038/ncpendmet0843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decensi A, Puntoni M, Goodwin P, Cazzaniga M, Gennari A, Bonanni B, Gandini S. Metformin and cancer risk in diabetic patients: a systematic review and meta-analysis. Cancer Prev Res (Phila) 2010;3:1451–1461. doi: 10.1158/1940-6207.CAPR-10-0157. [DOI] [PubMed] [Google Scholar]
- Dragutinovic V, Izrael-Zivkovic L, Radovanovic N. Relation of matrix metalloproteinase-9 to different stages of tumors in the serum of gastric cancer. Dig Dis Sci. 2009;54:1203–1207. doi: 10.1007/s10620-008-0472-y. [DOI] [PubMed] [Google Scholar]
- Enomoto K, Zhu X, Park S, Zhao L, Zhu YJ, Willingham MC, Qi J, Copland JA, Meltzer P, Cheng SY. Targeting MYC as a Therapeutic Intervention for Anaplastic Thyroid Cancer. J Clin Endocrinol Metab. 2017;102:2268–2280. doi: 10.1210/jc.2016-3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P, Picaud S, Mangos M, Keates T, Lambert JP, Barsyte-Lovejoy D, Felletar I, Volkmer R, Muller S, Pawson T, et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell. 2012;149:214–231. doi: 10.1016/j.cell.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, et al. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuya F, Hanover JA, Cheng SY. Activation of phosphatidylinositol 3-kinase signaling by a mutant thyroid hormone beta receptor. Proc Natl Acad Sci U S A. 2006;103:1780–1785. doi: 10.1073/pnas.0510849103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Wu X, Zhang X, Hua W, Zhang Y, Maimaiti Y, Gao Z, Zhang Y. Inhibition of BRD4 suppresses tumor growth and enhances iodine uptake in thyroid cancer. Biochem Biophys Res Commun. 2016;469:679–685. doi: 10.1016/j.bbrc.2015.12.008. [DOI] [PubMed] [Google Scholar]
- Gillings AS, Balmanno K, Wiggins CM, Johnson M, Cook SJ. Apoptosis and autophagy: BIM as a mediator of tumour cell death in response to oncogene-targeted therapeutics. FEBS J. 2009;276:6050–6062. doi: 10.1111/j.1742-4658.2009.07329.x. [DOI] [PubMed] [Google Scholar]
- Guigon CJ, Fozzatti L, Lu C, Willingham MC, Cheng SY. Inhibition of mTORC1 signaling reduces tumor growth but does not prevent cancer progression in a mouse model of thyroid cancer. Carcinogenesis. 2010;31:1284–1291. doi: 10.1093/carcin/bgq059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guigon CJ, Zhao L, Willingham MC, Cheng SY. PTEN deficiency accelerates tumour progression in a mouse model of thyroid cancer. Oncogene. 2009;28:509–517. doi: 10.1038/onc.2008.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han JM, Kim TY, Jeon MJ, Yim JH, Kim WG, Song DE, Hong SJ, Bae SJ, Kim HK, Shin MH, et al. Obesity is a risk factor for thyroid cancer in a large, ultrasonographically screened population. Eur J Endocrinol. 2013;168:879–886. doi: 10.1530/EJE-13-0065. [DOI] [PubMed] [Google Scholar]
- Hosono K, Endo H, Takahashi H, Sugiyama M, Uchiyama T, Suzuki K, Nozaki Y, Yoneda K, Fujita K, Yoneda M, et al. Metformin suppresses azoxymethane-induced colorectal aberrant crypt foci by activating AMP-activated protein kinase. Mol Carcinog. 2010;49:662–671. doi: 10.1002/mc.20637. [DOI] [PubMed] [Google Scholar]
- Hou P, Ji M, Xing M. Association of PTEN gene methylation with genetic alterations in the phosphatidylinositol 3-kinase/AKT signaling pathway in thyroid tumors. Cancer. 2008;113:2440–2447. doi: 10.1002/cncr.23869. [DOI] [PubMed] [Google Scholar]
- Hou P, Liu D, Shan Y, Hu S, Studeman K, Condouris S, Wang Y, Trink A, El-Naggar AK, Tallini G, et al. Genetic alterations and their relationship in the phosphatidylinositol 3-kinase/Akt pathway in thyroid cancer. Clin Cancer Res. 2007;13:1161–1170. doi: 10.1158/1078-0432.CCR-06-1125. [DOI] [PubMed] [Google Scholar]
- Iyer PC, Dadu R, Ferrarotto R, Busaidy NL, Habra MA, Zafereo M, Gross N, Hess KR, Gule-Monroe M, Williams MD, et al. Real-World Experience with Targeted Therapy for the Treatment of Anaplastic Thyroid Carcinoma. Thyroid. 2018;28:79–87. doi: 10.1089/thy.2017.0285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamran MZ, Patil P, Gude RP. Role of STAT3 in cancer metastasis and translational advances. Biomed Res Int. 2013;2013:421821. doi: 10.1155/2013/421821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Kim NK, Choi JH, Sohn SY, Kim SW, Jin SM, Jang HW, Suh S, Min YK, Chung JH, et al. Associations between body mass index and clinico-pathological characteristics of papillary thyroid cancer. Clin Endocrinol (Oxf) 2013a;78:134–140. doi: 10.1111/j.1365-2265.2012.04506.x. [DOI] [PubMed] [Google Scholar]
- Kim SA, Inamura K, Yamauchi M, Nishihara R, Mima K, Sukawa Y, Li T, Yasunari M, Morikawa T, Fitzgerald KC, et al. Loss of CDH1 (E-cadherin) expression is associated with infiltrative tumour growth and lymph node metastasis. Br J Cancer. 2016;114:199–206. doi: 10.1038/bjc.2015.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim WG, Park JW, Willingham MC, Cheng SY. Diet-induced obesity increases tumor growth and promotes anaplastic change in thyroid cancer in a mouse model. Endocrinology. 2013b;154:2936–2947. doi: 10.1210/en.2013-1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitahara CM, McCullough ML, Franceschi S, Rinaldi S, Wolk A, Neta G, Olov Adami H, Anderson K, Andreotti G, Beane Freeman LE, et al. Anthropometric Factors and Thyroid Cancer Risk by Histological Subtype: Pooled Analysis of 22 Prospective Studies. Thyroid. 2016;26:306–318. doi: 10.1089/thy.2015.0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitahara CM, Platz EA, Freeman LE, Hsing AW, Linet MS, Park Y, Schairer C, Schatzkin A, Shikany JM, Berrington de Gonzalez A. Obesity and thyroid cancer risk among U.S. men and women: a pooled analysis of five prospective studies. Cancer Epidemiol Biomarkers Prev. 2011;20:464–472. doi: 10.1158/1055-9965.EPI-10-1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klubo-Gwiezdzinska J, Costello J, Jr, Patel A, Bauer A, Jensen K, Mete M, Burman KD, Wartofsky L, Vasko V. Treatment with metformin is associated with higher remission rate in diabetic patients with thyroid cancer. J Clin Endocrinol Metab. 2013;98:3269–3279. doi: 10.1210/jc.2012-3799. [DOI] [PubMed] [Google Scholar]
- Klubo-Gwiezdzinska J, Jensen K, Costello J, Patel A, Hoperia V, Bauer A, Burman KD, Wartofsky L, Vasko V. Metformin inhibits growth and decreases resistance to anoikis in medullary thyroid cancer cells. Endocr Relat Cancer. 2012;19:447–456. doi: 10.1530/ERC-12-0046. [DOI] [PubMed] [Google Scholar]
- Lee DH, Qi J, Bradner JE, Said JW, Doan NB, Forscher C, Yang H, Koeffler HP. Synergistic effect of JQ1 and rapamycin for treatment of human osteosarcoma. Int J Cancer. 2015;136:2055–2064. doi: 10.1002/ijc.29269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim H, Devesa SS, Sosa JA, Check D, Kitahara CM. Trends in Thyroid Cancer Incidence and Mortality in the United States, 1974–2013. JAMA. 2017;317:1338–1348. doi: 10.1001/jama.2017.2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockwood WW, Zejnullahu K, Bradner JE, Varmus H. Sensitivity of human lung adenocarcinoma cell lines to targeted inhibition of BET epigenetic signaling proteins. Proc Natl Acad Sci U S A. 2012;109:19408–19413. doi: 10.1073/pnas.1216363109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loven J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, Young RA. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153:320–334. doi: 10.1016/j.cell.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z, Xu S. ERK1/2 MAP kinases in cell survival and apoptosis. IUBMB Life. 2006;58:621–631. doi: 10.1080/15216540600957438. [DOI] [PubMed] [Google Scholar]
- Ma J, Huang M, Wang L, Ye W, Tong Y, Wang H. Obesity and risk of thyroid cancer: evidence from a meta-analysis of 21 observational studies. Med Sci Monit. 2015;21:283–291. doi: 10.12659/MSM.892035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Memmott RM, Mercado JR, Maier CR, Kawabata S, Fox SD, Dennis PA. Metformin prevents tobacco carcinogen--induced lung tumorigenesis. Cancer Prev Res (Phila) 2010;3:1066–1076. doi: 10.1158/1940-6207.CAPR-10-0055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mio C, Lavarone E, Conzatti K, Baldan F, Toffoletto B, Puppin C, Filetti S, Durante C, Russo D, Orlacchio A, et al. MCM5 as a target of BET inhibitors in thyroid cancer cells. Endocr Relat Cancer. 2016;23:335–347. doi: 10.1530/ERC-15-0322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mita AC, Mita MM, Nawrocki ST, Giles FJ. Survivin: key regulator of mitosis and apoptosis and novel target for cancer therapeutics. Clin Cancer Res. 2008;14:5000–5005. doi: 10.1158/1078-0432.CCR-08-0746. [DOI] [PubMed] [Google Scholar]
- Noto H, Goto A, Tsujimoto T, Noda M. Cancer risk in diabetic patients treated with metformin: a systematic review and meta-analysis. PLoS One. 2012;7:e33411. doi: 10.1371/journal.pone.0033411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onder TT, Gupta PB, Mani SA, Yang J, Lander ES, Weinberg RA. Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res. 2008;68:3645–3654. doi: 10.1158/0008-5472.CAN-07-2938. [DOI] [PubMed] [Google Scholar]
- Paget S. The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev. 1989;8:98–101. [PubMed] [Google Scholar]
- Pankov R, Yamada KM. Fibronectin at a glance. J Cell Sci. 2002;115:3861–3863. doi: 10.1242/jcs.00059. [DOI] [PubMed] [Google Scholar]
- Park J, Kim WG, Zhao L, Enomoto K, Willingham M, Cheng SY. Metformin blocks progression of obesity-activated thyroid cancer in a mouse model. Oncotarget. 2016a;7:34832–34844. doi: 10.18632/oncotarget.8989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JW, Han CR, Zhao L, Willingham MC, Cheng SY. Inhibition of STAT3 activity delays obesity-induced thyroid carcinogenesis in a mouse model. Endocr Relat Cancer. 2016b;23:53–63. doi: 10.1530/ERC-15-0417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathak KA, Leslie WD, Klonisch TC, Nason RW. The changing face of thyroid cancer in a population-based cohort. Cancer Med. 2013;2:537–544. doi: 10.1002/cam4.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierotti MA, Berrino F, Gariboldi M, Melani C, Mogavero A, Negri T, Pasanisi P, Pilotti S. Targeting metabolism for cancer treatment and prevention: metformin, an old drug with multi-faceted effects. Oncogene. 2013;32:1475–1487. doi: 10.1038/onc.2012.181. [DOI] [PubMed] [Google Scholar]
- Pyo SW, Hashimoto M, Kim YS, Kim CH, Lee SH, Johnson KR, Wheelock MJ, Park JU. Expression of E-cadherin, P-cadherin and N-cadherin in oral squamous cell carcinoma: correlation with the clinicopathologic features and patient outcome. J Craniomaxillofac Surg. 2007;35:1–9. doi: 10.1016/j.jcms.2006.11.004. [DOI] [PubMed] [Google Scholar]
- Renehan AG, Tyson M, Egger M, Heller RF, Zwahlen M. Body-mass index and incidence of cancer: a systematic review and meta-analysis of prospective observational studies. Lancet. 2008;371:569–578. doi: 10.1016/S0140-6736(08)60269-X. [DOI] [PubMed] [Google Scholar]
- Rotella CM, Monami M, Mannucci E. Metformin beyond diabetes: new life for an old drug. Curr Diabetes Rev. 2006;2:307–315. doi: 10.2174/157339906777950651. [DOI] [PubMed] [Google Scholar]
- Satelli A, Li S. Vimentin in cancer and its potential as a molecular target for cancer therapy. Cell Mol Life Sci. 2011;68:3033–3046. doi: 10.1007/s00018-011-0735-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena NK, Sharma D, Ding X, Lin S, Marra F, Merlin D, Anania FA. Concomitant activation of the JAK/STAT, PI3K/AKT, and ERK signaling is involved in leptin-mediated promotion of invasion and migration of hepatocellular carcinoma cells. Cancer Res. 2007;67:2497–2507. doi: 10.1158/0008-5472.CAN-06-3075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid D, Ricci C, Behrens G, Leitzmann MF. Adiposity and risk of thyroid cancer: a systematic review and meta-analysis. Obes Rev. 2015;16:1042–1054. doi: 10.1111/obr.12321. [DOI] [PubMed] [Google Scholar]
- Shimamura T, Chen Z, Soucheray M, Carretero J, Kikuchi E, Tchaicha JH, Gao Y, Cheng KA, Cohoon TJ, Qi J, et al. Efficacy of BET bromodomain inhibition in Kras-mutant non-small cell lung cancer. Clin Cancer Res. 2013;19:6183–6192. doi: 10.1158/1078-0432.CCR-12-3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbiah V, Kreitman RJ, Wainberg ZA, Cho JY, Schellens JHM, Soria JC, Wen PY, Zielinski C, Cabanillas ME, Urbanowitz G, et al. Dabrafenib and Trametinib Treatment in Patients With Locally Advanced or Metastatic BRAF V600-Mutant Anaplastic Thyroid Cancer. J Clin Oncol. 2018;36:7–13. doi: 10.1200/JCO.2017.73.6785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takezawa K, Okamoto I, Nishio K, Janne PA, Nakagawa K. Role of ERK-BIM and STAT3-survivin signaling pathways in ALK inhibitor-induced apoptosis in EML4-ALK-positive lung cancer. Clin Cancer Res. 2011;17:2140–2148. doi: 10.1158/1078-0432.CCR-10-2798. [DOI] [PubMed] [Google Scholar]
- Tseng CH. Metformin reduces thyroid cancer risk in Taiwanese patients with type 2 diabetes. PLoS One. 2014;9:e109852. doi: 10.1371/journal.pone.0109852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Hou P, Yu H, Wang W, Ji M, Zhao S, Yan S, Sun X, Liu D, Shi B, et al. High prevalence and mutual exclusivity of genetic alterations in the phosphatidylinositol-3-kinase/akt pathway in thyroid tumors. J Clin Endocrinol Metab. 2007;92:2387–2390. doi: 10.1210/jc.2006-2019. [DOI] [PubMed] [Google Scholar]
- Xing M. Genetic alterations in the phosphatidylinositol-3 kinase/Akt pathway in thyroid cancer. Thyroid. 2010;20:697–706. doi: 10.1089/thy.2010.1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin S, Cheryan VT, Xu L, Rishi AK, Reddy KB. Myc mediates cancer stem-like cells and EMT changes in triple negative breast cancers cells. PLoS One. 2017;12:e0183578. doi: 10.1371/journal.pone.0183578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Enomoto K, Zhao L, Zhu YJ, Willingham MC, Meltzer P, Qi J, Cheng SY. Bromodomain and Extraterminal Protein Inhibitor JQ1 Suppresses Thyroid Tumor Growth in a Mouse Model. Clin Cancer Res. 2017;23:430–440. doi: 10.1158/1078-0432.CCR-16-0914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Zhao L, Park JW, Willingham MC, Cheng SY. Synergistic signaling of KRAS and thyroid hormone receptor beta mutants promotes undifferentiated thyroid cancer through MYC up-regulation. Neoplasia. 2014;16:757–769. doi: 10.1016/j.neo.2014.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]