

Abstract
Biallelic pathogenic variants in PIBF1 have been identified as one of the genetic etiologies of Joubert syndrome. We report a two-years-old girl with global developmental delay, facial dysmorphism, hypotonia, enlarged cystic kidneys, molar tooth sign and thinning of corpus callosum. A novel homozygous 36-bp insertion in PIBF1 (c.1181_1182ins36) was identified by exome sequencing as the likely cause of her condition. This is the second publication demonstrating the cause and effect relationship between PIBF1 and Joubert syndrome.
Keywords: Joubert syndrome, PIBF1, Variant, Exome sequencing
INTRODUCTION
Joubert syndrome is a genetically heterogeneous ciliopathy characterized by intellectual disability, hypotonia, facial dysmorphism, retinal dystrophy, cystic kidney disease and molar tooth sign.1 Pathogenic variants in 37 genes are known to be associated with Joubert syndrome. Recently, using genome wide siRNA knockdown and exome sequencing, Wheway et al. identified recessive mutations in PIBF1 in seven patients with Joubert syndrome 33 (MIM #213300).2 Here, we report a subject with Joubert syndrome harboring a novel biallelic in-frame insertion of 36-bp in exon 9 of PIBF1.
METHODOLOGY
Clinical summary
A two-years-old girl was evaluated for global developmental delay. She was born to consanguineous couple by cesarean section (Fig 1A). She weighed 2.25 kg (−1 SD) at birth. Ventriculomegaly was noted on antenatal ultrasonography at 16 weeks of gestation. She had delayed cry at birth and an episode of seizure on first day of life. She received intensive neonatal care for first six days. She had feeding difficulties in the neonatal period. She could stand with support, followed simple commands and had minimal non-verbal communication skills at age two years. The couple’s first child had succumbed to congenital diaphragmatic hernia.
Fig 1.

Pedigree (A). Proband at age 2 years has frontal prominence, deep set eyes and midface hypoplasia (B, C). Magnetic resonance imaging of brain revealed thickening and lengthening of superior cerebellar peduncle (arrow head shows molar tooth sign, D), prominent cerebrospinal fluid space (asterix, D, E), thinning of corpus callosum (arrow, E), and perisylvian polymicrogyria (black arrow, F).
At age two years, she had a height of 84 cm (1 SD), weight of 8.2 kg (−4 SD) and occipito-frontal circumference of 47 cm (1 SD). She had frontal prominence, deep-set eyes and midface retrusion (Fig 1B, 1C). She had hypotonia and absent deep tendon reflexes. She had an enlarged liver extending 3 cm below right costal margin. She had acute renal failure at two years of age with elevated blood urea (71 mg/dL; normal range: 10–45 mg/dL) and creatinine (3.2 mg/dL; normal range: 0.6–45 mg/dL). Enlarged kidneys with increased cortical echogenicity and blurred cortico-medullary differentiation with mild ascites were noted by ultrasonography. Renal biopsy revealed periglomerular fibrosis, tubular atrophy, luminal hyaline casts and interstitial inflammation suggesting nephronophthisis. Chest radiograph was unremarkable (Fig S1). Micturating cystourethrogram showed grade IV vesicoureteric reflux. Echocardiography returned normal results. Magnetic resonance imaging of brain revealed polymicrogyria in right fronto-parieto-temporal and perisylvian region with thin splenium of corpus callosum and molar tooth sign (Fig 1D, 1E, 1F). Ophthalmology evaluation showed hypermetropia. Urine analysis was unremarkable. Written informed consent was obtained from the participants. This study has the approval of institutional ethics committee.
Mutation analysis
DNA was extracted from leukocytes by conventional phenol-chloroform method. Chromosomal microarray was performed using Affymetrix Cytoscan 750K platform (Santa Clara, CA, US) and analyzed using Chromosome Analysis Suite (ChAS) software package (Affymetrix, USA). A commercially available ciliopathy panel was used to test 29 genes (supplementary note; PIBF1 was not a part of this panel as it was discovered recently). Exome sequencing was done using Illumina’s Nextera Rapid Capture Exome Kit on the Illumina NextSeq Platform (Illumina, San Diego, California, USA). The average coverage depth was 130x, with ~95% of the bases covered at >20x, and a sensitivity of >90%. Data were stored and analyzed using a previously-published automated pipeline, SeqMule v1.2.5.3 The variant call format (.vcf) file was annotated by ANNOVAR v.2016Feb01.4 The strategy used for the variant filtering is described in the Table S1. Candidate variants were validated and segregation analysis was carried out by Sanger sequencing. Genes implicated in the molecular mechanisms characterizing ciliary function and associated with ciliopathies (n=303, Table S4), were included from the SYSCILIA consortium gold standard (SCGSv1).5 Genotyping was carried out using Devyser Compact V3 QF-PCR kit and analyzed by GeneMarker software v.2.4.0. In-silico tools were used to predict the effect of candidate variants. The phenotypic information and the variant details can be accessed at ClinVar (Accession ID- SCV000583465.2)
RESULTS
Clinical findings in the proband suggested the diagnosis of Joubert syndrome. Autosomal recessive inheritance pattern was inferred from the pedigree. Next generation sequencing panel for ciliopathies was non-diagnostic. Only 45 rare variants remained after filtering the exome data for exonic, splicing, and homozygous variants (Table S1 and Table S2). The variant, NM_006346.2, c.1181_1182insGCTTCGCAGATTGAAAACCAACCAAGAAATTGATCA, p.(Gln394_Leu395ins12) in PIBF1 was observed in homozygous state in the proband and parents were heterozygous carriers (Fig 2). This variant in PIBF1 appeared as a discrete and significant increase in the read number on Integrative Genomics Viewer (IGV) (Fig S2).6 This variant is not observed in 1000 Genomes Project, ESP, gnomAD and in our in-house exome data of 357 individuals.7, 8 The variant is predicted to have damaging effects on the protein by PROVEAN (−17.967), PaPI (0.783), SIFT Indels 2 (0.858) and Human Splicing Finder (Alteration of an exonic ESE site).9–12 Father was homozygous for the candidate variants in IFT27, CELSR1 and TTLL6. All the four candidate variants evaluated were in the regions of homozygosity as determined by chromosomal microarray (Table S3). We carried out genotyping by QF-PCR in the proband and the parents to rule out the possibility of sample mix-up as all the three rare candidate variants were observed in homozygous state in the father. The alleles segregated and possibility of sample mix-up was ruled out in the family.
Fig 2.
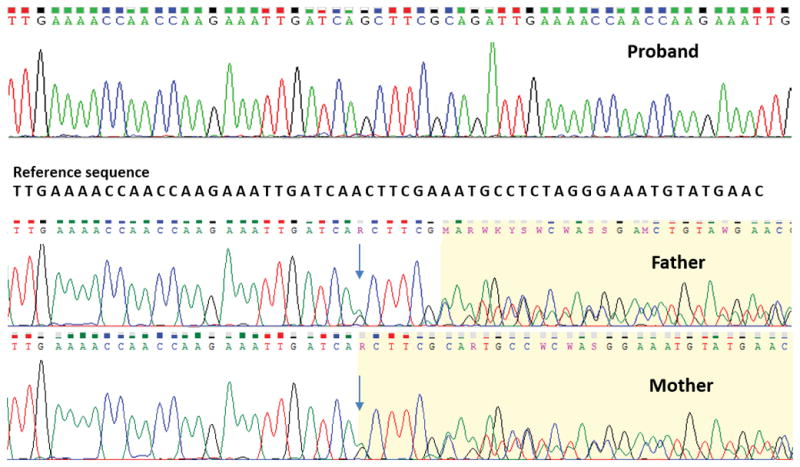
Sanger chromatograms show the variant, c.1181_1182insGCTTCGCAGATTGAAAACCAACCAAGAAATTGATCA in PIBF1 in homozygous state in proband (upper panel) and heterozygous state in father (middle panel) and mother (lower panel).
DISCUSSION
We evaluated a consanguineous family with a two-years-old girl with global developmental delay, facial dysmorphism, hypotonia, polymicrogyria and molar tooth sign on brain imaging. Exome sequencing revealed a novel 36-bp insertion in exon 9 of PIBF1 as the likely cause of the condition. Pathogenic variants in PIBF1 have recently been reported in seven patients from five families including four Hutterite families (Table 1). All the earlier reported patients presented with global developmental delay, hypotonia and ataxia. Ataxia could not be assessed in the proband in our study. Cystic kidney disease and retinal dystrophy were noted in the proband. The characteristic molar tooth sign on brain imaging was observed in five families including ours, while it was absent in two patients in a single family. In addition to this, our patient also had perisylvian polymicrogyria and hypoplastic corpus callosum.2
Table 1.
Summary of clinical features and variants observed in patients with Joubert syndrome 33
| Current study | Wheway et al. 2015 | |||||||
|---|---|---|---|---|---|---|---|---|
| Clinical findings | Proband | UW155-3 | H1-3 | H1-4 | H2-3 | H2-4 | H3-3 | H4-3 |
| Developmental delay | + | + | + | + | + | + | + | + |
| Hypotonia | + | + | + | + | + | + | + | + |
| Ataxia | NA | + | + | + | + | + | + | + |
| Cystic kidney disease | + | NA | NA | NA | NA | NA | NA | NA |
| Retinal degeneration | − | NA | NA | NA | NA | NA | NA | NA |
| Molar tooth sign | + | + | − | − | NA | + | + | + |
| Perisylvian polymicrogyria | + | − | − | − | − | − | − | − |
| Hypoplasia of corpus callosum | + | − | − | − | − | − | − | − |
| Vermian hypoplasia | + | + | + | + | NA | + | + | + |
| Foramen magnum cephalocele | − | NA | − | − | NA | − | − | + |
| cDNA change | c.1181_1182ins36 | c.1214G>A, c.1669delC | c.1910A>C | c.1910A>C | c.1910A>C | c.1910A>C | c.1910A>C | c.1910A>C |
| Protein change | p.(Gln394_Leu395ins12) | p.Arg405Gln, p.Leu557Phefs*18 | p.Asp637Ala | p.Asp637Ala | p.Asp637Ala | p.Asp637Ala | p.Asp637Ala | p.Asp637Ala |
Abbreviations: + present, − absent, NA not available
The gene, PIBF1, encodes a protein that is produced during pregnancy in response to progesterone. PIBF1 is a core component of the human centrosome and is crucial for the accumulation of centriolar satellites, eventually forming the primary cilia.1 Depletion of PIBF1 is known to cause mitotic arrest, misaligned chromosomes, spindle pole fragmentation. A whole-genome siRNA reverse genetics screen for defects in biogenesis and maintenance of the primary cilium, combined with exome sequencing data identified recessive mutations in PIBF1 in seven individuals with Joubert syndrome.2 Also, exogenous expression of human wild-type PIBF1 following siRNA knockdown was found to rescue ciliogenesis in mIMCD3 cells.
The pathogenic variants previously identified in PIBF1 include two missense and a truncating variant (Table 1). The biallelic 36-bp insertion in PIBF1 identified in our proband is predicted to have a damaging effect on the protein by in-silico tools.2 Further analysis for the effect of this variant on protein function could not be done as a suitable template for reliable protein modeling is unavailable at present. Also, reluctance of the family to participate in further functional studies hindered validation of the variant as a loss-of-function allele.
In conclusion, our report validates PIBF1 as a causative gene for Joubert syndrome and expands the clinical and molecular spectrum of the condition. However, identification of additional variants in PIBF1 and further functional validation are warranted for elucidation of the underlying pathogenic mechanism.
Supplementary Material
Acknowledgments
We thank the family who cooperated with evaluation of the children and consented for participation in this study.
FUNDING
This work was supported by National Institutes of Health funded the project titled ‘Genetic Diagnosis of Heritable Neurodevelopmental Disorders in India: Investigating the Use of Whole Exome Sequencing and Genetic Counseling to Address the High Burden of Neurodevelopmental Disorders’ (1R21NS094047-01)
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
References
- 1.Kim K, Lee K, Rhee K. CEP90 is required for the assembly and centrosomal accumulation of centriolar satellites, which is essential for primary cilia formation. PLoS One. 2012;7:e48196. doi: 10.1371/journal.pone.0048196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wheway G, Schmidts M, Mans DA, Szymanska K, Nguyen TM, Racher H, et al. An siRNA-based functional genomics screen for the identification of regulators of ciliogenesis and ciliopathy genes. Nature cell biology. 2015;17:1074–1087. doi: 10.1038/ncb3201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guo Y, Ding X, Shen Y, Lyon GJ, Wang K. SeqMule: automated pipeline for analysis of human exome/genome sequencing data. Scientific reports. 2015;5:14283. doi: 10.1038/srep14283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shukla A, Hebbar M, Srivastava A, Kadavigere R, Upadhyai P, Kanthi A, et al. Homozygous p.(Glu87Lys) variant in ISCA1 is associated with a multiple mitochondrial dysfunctions syndrome. J Hum Genet. 2017 doi: 10.1038/jhg.2017.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van Dam TJ, Wheway G, Slaats GG, Huynen MA, Giles RH. The SYSCILIA gold standard (SCGSv1) of known ciliary components and its applications within a systems biology consortium. Cilia. 2013;2:7. doi: 10.1186/2046-2530-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Briefings in bioinformatics. 2013;14:178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, et al. A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Desmet FO, Hamroun D, Lalande M, Collod-Beroud G, Claustres M, Beroud C. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic acids research. 2009;37:e67. doi: 10.1093/nar/gkp215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Choi Y, Chan AP. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics (Oxford, England) 2015;31:2745–2747. doi: 10.1093/bioinformatics/btv195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Limongelli I, Marini S, Bellazzi R. PaPI: pseudo amino acid composition to score human protein-coding variants. BMC bioinformatics. 2015;16:123. doi: 10.1186/s12859-015-0554-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hu J, Ng PC. SIFT Indel: predictions for the functional effects of amino acid insertions/deletions in proteins. PLoS One. 2013;8:e77940. doi: 10.1371/journal.pone.0077940. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.