

Abstract
Recurrent Laryngeal Neuropathy (RLN) is a highly prevalent and predominantly left‐sided, degenerative disorder of the recurrent laryngeal nerves (RLn) of tall horses, that causes inspiratory stridor at exercise because of intrinsic laryngeal muscle paresis. The associated laryngeal dysfunction and exercise intolerance in athletic horses commonly leads to surgical intervention, retirement or euthanasia with associated financial and welfare implications. Despite speculation, there is a lack of consensus and conflicting evidence supporting the primary classification of RLN, as either a distal (“dying back”) axonopathy or as a primary myelinopathy and as either a (bilateral) mononeuropathy or a polyneuropathy; this uncertainty hinders etiological and pathophysiological research. In this review, we discuss the neuropathological changes and electrophysiological deficits reported in the RLn of affected horses, and the evidence for correct classification of the disorder. In so doing, we summarize and reveal the limitations of much historical research on RLN and propose future directions that might best help identify the etiology and pathophysiology of this enigmatic disorder.
Keywords: axonopathy, bilateral mononeuropathy, horse, larynx, myelinopathy, polyneuropathy, recurrent laryngeal neuropathy
Abbreviations
- RLN
recurrent laryngeal neuropathy
- CMT
Charcot‐Marie‐Tooth disease
- RLn
recurrent laryngeal nerve
- EM
electron microscopy
- EDL
extensor digitorum longus muscle
- PMP22
peripheral myelin protein 22 gene
- MFN2
mitofuscin 2
- Cx‐32
connexin 32
- SARM1
sterile alpha and TIR motif containing 1
- SCG‐10
stathmin‐2
- NMNAT2
nicotinamide nucleotide adenylyltransferase 2
- NAD+
nicotinamide adenine dinucleotide
- ATP
adenosine triphosphate
- CMAP
compound motor action potential
- NCV
nerve conduction velocity.
1. INTRODUCTION
Recurrent laryngeal neuropathy (RLN), is an equine degenerative disorder of the recurrent laryngeal nerves (RLn) of (particularly) tall breeds such as Thoroughbreds and Drafts.1, 2 The disease is characterized by preferential degeneration of the left RLn over the right RLn3, 4 that causes paresis or, in severe cases, paralysis of the left intrinsic laryngeal muscles preventing arytenoid movement, which is vital for enabling unimpeded inspiratory airflow. Recurrent laryngeal neuropathy (RLN) is performance limiting in horses performing at their greatest exertion, in particular, in racehorses.5 Many studies have identified RLN‐associated neuropathological changes in both RLns (left more so than right) in clinically unaffected horses, leading authors to postulate whether the majority of unaffected horses are in fact subclinical cases.3, 4, 6, 7 The mechanisms that lead to the varying severity (from subclinical to severe) in horses are largely unknown, however, the identification of many subclinical cases means that case selection for “unaffected” controls for research is challenging. Amongst the apparent risk factors, horse height is reported as a significant contributor to RLN‐status, with taller horses being at greater risk1, 2; it is unclear whether height itself is the principal risk factor or simply a covariate for nerve length. Males are more commonly affected than females.2, 8 Genetic risk factors are plausible given that the heritability of RLN ranges from 8 to 40% depending on the breed affected9, 10; however, as of yet no specific risk alleles have been reported despite many published studies.1, 11, 12 Indeed, very little is known about the cause of RLN13: the array of etiologies proposed includes mechanical stress14 or ischemic nerve damage,4 pressure damage,4 an infectious etiology,13 vitamin deficiencies,15 and toxic insults.16, 17, 18, 19
RLN is consistently described as a peripheral neuropathy but without a more detailed classification describing the primary defect.20, 21 Conflicting data is available supporting RLN as affecting just the RLn (ie, a mononeuropathy) or whether it affects other peripheral nerves (a polyneuropathy).3, 4, 7, 20, 22, 23, 24 Also unanswered is whether the degeneration begins within the axon or the myelin sheath. Providing answers to these key questions would likely expedite the search for the etiology, and potential therapies by helping focus research. For instance, molecular therapies are in development for similar human peripheral neuropathies, such as the group of peripheral neuropathies known as the Charcot‐Marie‐Tooth (CMT) diseases25 but a key element to developing these therapies is the understanding of where the degeneration begins (ie, in the nerve cell body, axon, synapse, myelin sheath or glia), to enable focused treatment. Potential issues that have hindered research in these areas for RLN include the selection of adequate control groups (as outlined previously), the trend for studies to evaluate subjective histopathological features in unblinded, uncontrolled fashion and that many RLN‐studies are typically statistically unpowered.
This review article discusses the current literature (veterinary and human) pertaining to the pathophysiology underlying RLN, addressing the issues relating to characterizing RLN as an axon‐ versus myelinopathy and a mononeuropathy or polyneuropathy.
2. EQUINE RLn
The RLn contains the longest motor axons in the horse, measuring up to 2.5 m in length in Draft and Thoroughbred horses, with the left nerve being ∼30 cm longer than the right in tall breeds6 (Figure 1). Their cell bodies are situated within the nucleus ambiguus in the brainstem and they project their axons caudally within the vagus nerves before RLn fibers exit and, on the left, traverse medially around the aorta and on the right, medially around the right subclavian artery.26 The nerves then follow an identical bilateral course, moving rostrally, adjacent to, and on either side of the trachea and carotid arteries. Their long axonal course is established during embryonic development: in the growing embryo, the RLn branch from the vagus nerve at the level of the sixth aortic arch, whereby they innervate the developing intrinsic laryngeal muscles. As the neck of the embryo elongates, the larynx migrates rostrally (relative to the aortic arches) resulting in elongation of the RLn axons as they are held distally by the aortic arches.27 At the level of the aorta, the equine RLn is ∼1.5 mm in diameter and the epineurium contains between 6 and 29 perineuronal bundles (fascicles), (generally between 11 and 15; Figure 2).28 Several authors have reported structural variations at the hairpin turn of the left RLn, as it courses around the aorta (eg, Haslam's Anomaly and Renaut bodies) although these appear with the same frequency in horses with and without RLN.4, 28, 29, 30
Figure 1.
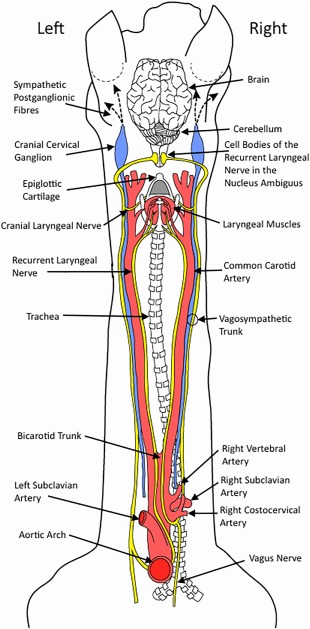
Anatomical representation of the dorsal view of the course taken by the RLn (yellow) in the horse, including both vagosympathetic trunks, vagus nerves, and important vasculature. Both RLn originate in the medulla oblongata where their cell bodies are located within the nucleus ambiguus. The left RLn courses around the aortic arch before innervating the muscles of the larynx and the right RLn courses around the right subclavian artery before it courses rostrally
Figure 2.
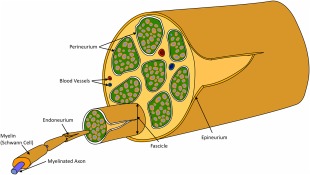
Schematic representative of a peripheral nerve. The main connective tissue structures include the epineurium (surrounding the entire nerve), the perineurium (surrounding each fascicle within the nerve) and the endoneurium (located around an individual axon). The perineurium also contains blood vessels and lymphatics
The RLn provide general somatic efferent innervation to the laryngeal and cranial esophageal muscles26, 31 and are comprised of wide diameter, alpha, myelinated nerve fibers of 12–20 µm diameter.28 Interspersed between the large myelinated fibers are less numerous, narrower myelinated fibers (that are speculated to be beta and gamma fibers) and small unmyelinated fibers.26 A study of 9 mixed‐breed horses under 2 years old with no evidence of laryngeal disease, revealed that the total number of myelinated fibers, mean fiber diameter and percentage of fibers over 9.5 μm in diameter were significantly lower in the left RLn compared with the right (p < 0.05).32 In other species where greater numbers of myelinated nerve fibers have been reported in the right RLn compared with the left (humans, dog, and cats) typically the myelinated nerve fiber diameter is larger in the left RLn, which is believed to compensate for the greater length of the left RLn, thus maintaining comparable conduction properties since wider axons have faster conduction speeds.33, 34, 35, 36, 37 Unequal fiber numbers are not found in all species: indeed equal (or near equal) fiber numbers have also been reported in humans, rats, and giraffes.37, 38 The greater numbers of myelinated fibers in the right RLn might reflect the presence of afferent (sensory) fibers, however the presence of sensory axons within equine RLn has not been investigated. In humans, these sensory axons receive input from the mucosa in the region of the vocal folds, and in cats, the pharyngeal swallow reflex occurs in part, from esophageal pressure increases transmitted through the RLn.39 However, staining for afferent branches in the rat RLn did not show any sensory branches40 suggesting that there are species differences in RLn‐associated sensory pathways.
2.1. The importance of the classification of neuropathies
Historically, human peripheral neuropathies were classified according to the site of the primary pathology—as a myelinopathy or axonopathy—caused by degeneration of the myelin sheath or axon respectively, and by the type of neuron affected: sensory or motor.41 In addition, the involvement of a single nerve or many different nerves in the disease process is described as a mononeuropathy or polyneuropathy, respectively.41 These classifications were critical for accurate diagnoses, and they underpinned all investigations into the diseases' etiopathogenesis. However, with advanced genomics, many inherited neuropathies are now classified by their causative genetic mutations. The genetic era has highlighted that a single disease phenotype can result from mutations in different genes, and further, that subtle differences in a disease phenotype can be explained by different mutations within the same gene.
The characterization of a peripheral neuropathy as either an axonopathy or a myelinopathy is achieved by amalgamating data from nerve conduction velocity (NCV) testing, pathological observations (from nerve biopsy specimen or postmortem examination) and genetic studies.42, 43, 44 Nerve conduction velocities (NCVs) are markedly slowed when there is extensive demyelination, and normal to partially slowed, with reduced compound muscle or sensory action potentials, in an axonopathy.42 In the diagnosis of human CMT disorders, genetic testing has largely superseded nerve biopsy43: when a causative genetic mutation is found, differentiating an axonopathy from a myelinopathy is simplified by the mutated protein's normal localized expression. However, mutation of an axonally localized protein can lead to a disease phenotype typically attributed to a myelinopathy and vice versa (see below). As a genetic cause of RLN has not yet been identified, the disease's classification as an axonopathy or myelinopathy can only be based on pathological and electrophysiological studies.
2.2. Charcot Marie tooth disease complex
The CMT diseases are inherited human peripheral polyneuropathies that affect either sensory or motor axons, or both, and are arranged into categories depending upon their primary site of pathology.43 Where possible, a definitive diagnosis of a specific form of CMT is made after genetic testing, however where a mutation is not found, pathological classifications provide the framework for the diagnosis and importantly, prognostic information.43
Demyelinating forms of CMT are classified as CMT type 1 (median motor NCV <38 m/s), with subgroups labeled according to the underlying genotype; CMT1a is a common form of CMT1 caused by a duplication in peripheral myelin protein 22 gene (PMP22).43 Human PMP22 duplications were discovered after observation that a mouse model (Trembler) with similar neuropathological changes to those found in CMT1a (peripheral hypomyelination) had a point mutation within PMP22,45, 46, 47 highlighting the importance of accurate and detailed histopathological disease descriptions. The axonal forms of CMT are classified as type 2 (median motor NCV > 38 m/s), with dominant mutations in the mitofusin 2 (MFN2) gene being the most common (CMT2a).48
An intermediate form of CMT disease (CMTX1) that produces axonal and myelin‐related pathological changes, as it results from mutations within Connexin‐32 (Cx‐32).49 Cx‐32 protein is expressed by Schwann cells in the peripheral nervous system and is involved in tight junction formation between the myelin lamella at the paranode and Schmidt‐Lantermann incisions.50, 51 CMTX1 accounts for ∼10% of all human CMT cases, and as a result of the paranodal abnormalities, NCVs are usually slower than CMT type 2 (axonal), ranging from 18 to 60 m/s, but not as slow as the hypomyelinated forms of CMT (eg, CMT type 1).42
3. CLASSIFICATION OF RLN
3.1. Neuropathology of RLN
The majority of pathological studies investigating RLN have been conducted by a handful of groups.3, 4, 6, 7, 20, 23, 24, 29, 30, 52, 53, 54, 55, 56 Their research has demonstrated loss of large, alpha myelinated nerve fibers in the left distal RLn (Figure 3), and in some severely affected horses, within the proximal left RLn, and the distal right RLn.3, 4, 7, 20, 23 Consequently, RLN has been classified as a distal axonopathy by some authors.3, 4, 20 However, preferential loss of large myelinated nerve fibers in a distal to proximal manner does not per se indicate either an axonopathy or myelinopathy. For example, distal to proximal axonal degeneration is typically seen with toxic or metabolic “dying‐back” or “central‐peripheral” distal axonopathies (ascending destruction of the distal axon of both central and peripheral nerves).57 Likewise, humans with end‐stage CMT type 1 (with primary demyelination), present primarily with length‐dependent axonal degeneration, with the distal appendages displaying muscle atrophy and the foot deformity, pes cavus.58, 59, 60
Figure 3.
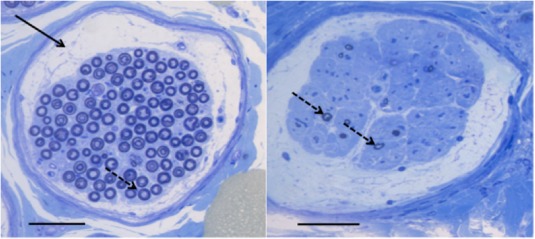
Distal Left RLn individual fascicle sections from a horse with Havemeyer grade I/IV (unaffected) RLN (left) and a horse with Havemeyer grade IV/IV (severely affected) RLN (right). There is obvious loss of large myelinated nerve fibers (dotted arrow) in the affected horse (right) compared with the unaffected horse (left), and also note the increased subperineurial space and endoneurial connective tissue present in the section from the affected horse. A Renaut Body is highlighted by a solid black arrow in the normal horse. Black bar: 50 μm
Clues as to the primary definition of RLN's status as an axonopathy or myelinopathy might then be possible from other pathological features. Common additional pathological changes affecting the distal left RLn in severely affected cases (Havemeyer grade III/IV or IV/IV61) include high numbers of Büngner bands and clusters of regenerating fibers.3, 4 These changes are also found in the proximal left RLn and (less commonly) in the distal right RLn in severely affected horses.3, 4 Büngner bands are formed from Schwann cell aggregates within the original basal lamina of the Schwann cell‐axon unit, and result from proliferation of that Schwann cell after axonal injury41; they are believed to serve as scaffolding to allow for axonal regeneration.62 Regenerating clusters are composed of axonal sprouts that form within the basal lamina as a result of axonal degeneration.41, 62 The presence of both Büngner Bands and regenerating clusters are suggestive of primary axonal degeneration.62
Other common pathological features seen in RLN are onion bulb formation and segmental demyelination/remyelination. The development of these structures cannot immediately be attributed to that of a primary axonopathy or demyelinating disorder. Onion bulbs are classically seen with repeated cycles of demyelination/remyelination: they are composed of concentric layers of remyelinating Schwann cells (that macrophages have failed to clear) before a successive demyelination episode.41 Repeated demyelination/remyelination is most commonly reported in primary myelinopathies41 but does occur in primary axonopathies, (whence they are termed pseudo‐onion bulbs).63, 64 Segmental demyelination/remyelination manifests as inconsistent internodal lengths and myelin thicknesses, typically within the same fiber, and is a common feature of CMT type 1 (demyelinating form).41, 58, 60 However, segmental myelin loss also occurs secondarily in primary axonopathies when there is loss of communication with the axon terminal65, 66; this is a key feature of RLN.20
As both onion bulbs and segmental demyelination/remyelination can occur with either primary myelinopathies or axonopathies, clues as to the primary site of degeneration might be evident from other associated localized pathological changes. In subclinical RLN cases, onion bulbs and regions of demyelination/remyelination occur most commonly at prominent sites of axonal histopathological change.3, 53 This is consistent with secondary demyelination that occurs as a direct result of axonal degeneration.41 These axonal histopathological changes included paranodal axoplasmic outpouchings (see below),53 organelle accumulations within swollen axons and active axonal degeneration at sites of onion bulb formation.3, 7 Specifically, Cahill and Goulden3 remarked that swollen fibers with dense axoplasm and attenuated myelin sheaths were observed in the RLn; often these swollen fibers were at the center of onion bulb formations. Duncan and Hammang53 stated at [paranodal] areas there was evidence of paranodal demyelination and remyelination, and as in all the other similarly swollen axons studied, that there was notable evidence of concentric Schwann cell proliferation leading to typical onion bulb formation. They concluded that the myelin attenuation occurring around axonal spheroids (axonal degeneration) resulted from mechanical slippage of the paranodal myelin.53 The paranode is the main anchorage point of the myelin lamellae to the axon, and mechanical myelin slippage is a well‐recognized consequence of paranodal pathology.67, 68, 69
When sural nerve biopsy specimen from human patients with a demyelinating polyneuropathy were compared to those with a primary axonal polyneuropathy (diagnosed on electrophysiological tests), there was no difference in the number of onion bulbs between the two conditions.64 The authors explained the lack of difference in onion bulb numbers resulted from the fact that onion bulbs identified in patients with the primary axonopathy had been misclassified: they were instead, pseudo‐onion bulbs resulting from axonal degeneration/regeneration.66 The differentiation of pseudo‐onions bulbs from onions bulbs can be difficult, and is typically made by an experienced pathologist who will assimilate the clinical history, results from other diagnostic tests and the type and distribution of the pathology present in a nerve biopsy specimen (sometimes with electron microscopy [EM] and immunohistochemistry); where the concentric Schwann cell processes mainly contain axons or axonal sprouts, this is more indicative of axonal degeneration and regeneration, and these Schwann cell processes are therefore termed pseudo‐onion bulbs.70 As Bosboom et al64 shows, correctly classifying these Schwann Cell proliferations as onion bulbs or as pseudo‐onions relies on knowing the underlying pathological process at work, so it is entirely feasible that the demyelination/remyelination reported in RLN results from axonal pathology and not primary demyelination.
Defining the primary site of disease pathology is likely harder in end‐stage disease tissue; study of tissue from individuals early in the disease course to identify active pathological processes might be more informative. In the RLn from subclinical cases (equivalent to grade II‐IIa61) organelle accumulations were seen in the paranodes (Figure 4) and along some internodal regions3, 4, 7, 53 in the most distal 40 cm of the left RLn and became more numerous in the most distal regions; they were only found in the distal 10 cm of the right RLn. The majority of the paranodal swellings were located proximal to the nearest Node of Ranvier. These swellings were further identified as axoplasmic evaginations that protruded into the myelin sheath, containing mitochondria (normal and degenerate) and dense lamellar bodies (lysosomes).53 Distal to the swellings, the neurofilaments aggregated at the axon center, causing the mitochondria to be displaced peripherally. Duncan and Hammang53 hypothesized that deficits in axonal transport were most likely the cause of the paranodal evaginations, as they occurred at the bracelet of Nageotte, the site of myelin attachment at the paranode which is a point of axolemmal weakness; they speculated that if axonal transportation is perturbed, cargo will accumulate and cause bulging of the axolemma at its weakest point.71 Axonal transportation deficits are typically seen in primary axonopathies; in particular, certain forms of CMT type 2 (2a, 2e, 2f) all exhibit transportation deficits whereas different forms of CMT type 1 (myelinopathies) do not (reviewed by Millecamps and Julien72).
Figure 4.
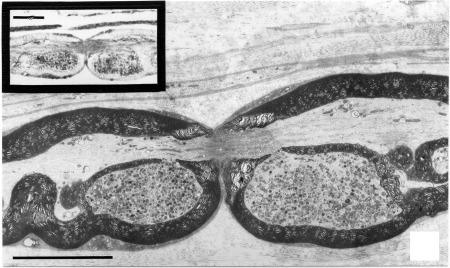
Electron microscopic image from longitudinal sections of the left RLn from a subclinically RLN‐affected horse. The node of Ranvier, with both surrounding paranodes containing large organelle accumulations (mitochondria and lysosomes) within splits to the myelin sheath can be easily appreciated. The image in the upper left corner is an etched section (1 μm) showing positive staining for anti‐cathepsin D in the region of the paranodal evaginations. Cathepsin D is found ubiquitously distributed in lysosomes, confirming their presence within the organelle accumulations. Both black bars correspond to 10 μm. Reprinted by permission from RightsLink: Springer Link, Journal of Neurocytology, Duncan and Hammang53 Copyright 1987
Further evidence that these paranodal evaginations result from pathology arising in the axon can be found when looking for their replication in other diseases. Duncan and Hammang53 commented that the paranodal evaginations appear to be a hallmark of RLN; however, they are almost identically replicated in certain toxic polyneuropathies (acrylamide and 2,5‐hexane‐diol) and were also identified in a rat neuroma study.73, 74, 75, 76 Acrylamide‐ and hexane‐associated polyneuropathies share many other similarities to RLN. They both cause a “dying‐back” central‐peripheral neuropathy, preferentially affecting long motor neurons.77, 78 Pathological studies of these toxic neuropathies identified mitochondria (and neurofilaments) located within the paranodal evaginations, typically located proximal to the node.79 In addition the evaginations precede classical signs of axonal degeneration in acute models of intoxication.71, 74, 76, 80 The exact etiopathogenesis of these toxins has not yet been fully accepted,81, 82 but both bind to microtubule‐associated proteins and neurofilaments,81 disrupting their maintenance and function, leading to long term deficits in the cytoskeleton and axonal transportation. Both anterograde and retrograde axonal transportation deficits have been recorded in acute acrylamide and hexane neurotoxicity.79, 83, 84, 85, 86 Another important observation made from models of acrylamide and hexane neurotoxicity is that these paranodel evaginations disrupt axoplasmic flow leading to defective organelle transportation79, 82 that could contribute to further neurodegeneration. Acrylamide and hexane neurotoxicity are classified as primary axonopathies, which lends support to classification of RLN also as an axonopathy.57, 73, 79, 81, 82
Axonal paranodal evaginations were also detected in the proximal sciatic nerve axons of rats, 1 month after the formation of a neuroma, after crushing and ligation.76 The evaginations contained similar membrane‐bound organelles and dense laminar bodies seen in subclinical RLN cases and in neurons from acrylamide‐ or hexane‐treated animals53, 71, 73, 74, 80; they were most numerous in the large myelinated fibers.41 These findings suggest that loss of trophic support from the muscle could be another important factor leading to generation of these paranodal evaginations,76 or it could simply reflect that disruption to both anterograde and retrograde axonal transportation systems can cause evagination.
Other histopathological features reported to occur in the RLn of subclinical RLN cases are axonal spheroids and axonal atrophy though they have not been quantified.3, 4, 7, 53 Axonal spheroids are the hallmark pathological sign of axonal dystrophy, a process that is distinct from axonal degeneration, and associated with synaptic dysplasia, abnormal axonal regeneration, or an imbalance with axonal transportation.87, 88 The timing of axonal spheroid development can serve as a clue for a primary (before degeneration signs occurring) versus a secondary (development of end‐bulbs after degeneration) axonal transportation deficit.89 End‐bulb formation (where the axon terminal swells) has not been reported specifically in RLN, and with the reported paranodal swellings, it is likely that axonal transportation deficits arise in the RLn (left more than right) before the loss of axonal integrity. Axonal atrophy is detectable in end‐stage axonal degeneration of various types.41 These pathological changes, although associated with many different etiologies, are all interpreted as an axonal pathology, supporting primary axon degeneration in RLN.
4. AXONAL DEGENERATION
For many years, axonal degeneration, studied in humans and mice, was thought simply to encompass Wallerian degeneration and apoptosis via two separate pathways. Wallerian degeneration is seen in the axon distal to a site of injury and involves activation of calpain, resulting from increased axonal calcium influx that leads to orderly degradation of the cytoskeleton and membrane proteins.90 The influx of calcium is hypothesized to result from the reversed action of the Na+‐Ca2+ exchanger as the Na+/K+ ATPase pump fails.91 The axonal debris generated by the proteolysis of the structural proteins and axonal fragmentation is then removed in a coordinated glial response,92 including phagocytosis by infiltrating macrophages.93
In 2002, Raff et al90 proposed a different classification for types of axonal degeneration:
the classical example of Wallerian degeneration of the distal axon after axotomy, whereby the proximal axon and cell body degenerate via apoptosis
dying‐back degeneration of the distal axon following:
chronic insult to the cell body, resulting in the distal axon degeneration extending proximally which can, eventually, result in the death of the cell body
pathology at the axon terminal (eg, removal of a neurotrophic factor), that maintains the cell body's integrity (eg, axonal pruning during metamorphosis, whereby excessive or redundant neurons are refined in the central nervous system during development)
These observations of 3 different axonal degeneration scenarios were first made from in vitro experimental observations and mouse models94 but there was very little understanding of the relevant cellular pathways that contributed to them. Coleman89 expanded upon the consequences of the classical “transecting” lesion inducing Wallerian degeneration, by hypothesizing that one or more focal lesions, not necessarily transecting the axon, trigger degeneration of the whole, distal axon. This would explain why proximal axonal numbers remain nearly normal in many peripheral neurodegenerative diseases.41 Indeed, current evidence suggests that neuronal cell body numbers in the nucleus ambiguous are unaffected in RLN, providing further supportive evidence that RLN is a distal axonopathy.22, 55, 95 Raff et al90 speculated that degeneration of only the distal axon could convey a distinct advantage to neurons if they become disconnected from their target cells: it would allow conservation of neuronal resources enabling the axon to reconnect with its terminal when conditions become more favorable. Coleman surmised that Wallerian degeneration could be the final convergent pathway of axonal degeneration in dying‐back and distal axonopathies,89 which would explain this key pathological feature of RLN.6
Defining RLN as a distal axonopathy and the confirmation experimentally, of deficits in axonal transportation, would provide an opportunity for extrapolation from related research in other species that might help define novel therapeutic targets. In recent years, significant strides have been made towards unraveling the molecular signaling of different forms of axonal degeneration96 (reviewed by Conforti et al97) and identifying pathways that converge on the common Wallerian degeneration pathway, proposed by Coleman89 (Figure 5). A key upstream event is the activation of SARM1, which occurs after axotomy,98 traumatic brain injury,99 mitochondrial potential loss,100 and excitotoxicity,101 that is, both in disease and with injury, leading to MAP kinase pathway initiation102 and SCG10 (Stathmin‐2; neuronal associated growth protein) loss.103 Overexpression of SARM1 alone does not result in axonal degeneration; an additional signal (such as an axotomy) is essential.104 The other key upstream event that initiates axonal degeneration is down regulation of the enzyme NMNAT2 (a nicotinamide mononucleotide adenylyltransferase that undergoes bidirectional axonal transport). NMNAT2 converts nicotinamide mononucleotide to NAD+, and its overexpression in experimental models delays Wallerian degeneration.97 NMNAT2 is also essential for axonal survival and growth.105, 106 The exact mechanism behind the protective effects of NMNAT2 overexpression are undetermined, but it does not appear simply to be mediated through an increase in NAD+; the basal levels of NAD+ are not increased by this overexpression, and supplementation of neurons with NAD+ in vitro, does not protect against degeneration.107, 108, 109 However, SARM1 overexpression does trigger rapid NAD+ consumption,96 supporting the idea that changes in NAD+ concentrations might play a role in this model of axonal degeneration. SCG10 loss accelerates axonal degeneration in a similar fashion to NMNAT2,103 however its protective effects are less pronounced. SARM1 activation and NMNAT2 inactivation both lead to MAPK activation, and in turn, ATP levels are depleted (causing failure of the Na+/K+ ATPase pump), and intracellular calcium concentrations will increase leading to cytoskeletal disassembly.97 The increased calcium concentration activates calpain and in turn, degrades calpastatin, both of which are well‐established events in the latter phases of Wallerian degeneration.110, 111 It is clear that mitochondria play a key role in Wallerian degeneration, but whether their roles are limited to the initiation or downstream execution of axonal degeneration (or both) is not yet established. Axonal degeneration is a key feature of many neurological conditions caused by mutations in mitochondrial proteins (eg, CMT type 2K caused by mutations in GDAP1),112 implying that mitochondrial dysfunction itself can initiate axonal degeneration under certain circumstances.
Figure 5.
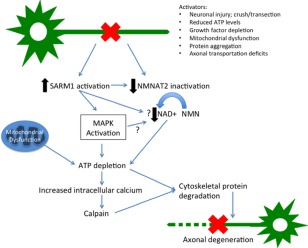
Working model of an integrated axon degeneration signaling cascade. Injury leads to SARM1 activation and NMNAT2 depletion. Energetic failure promotes ionic imbalance including intra‐axonal calcium accumulation, leading to calpain activation and proteolysis of intermediate filaments in the axonal cytoskeleton. Cumulative structural damage leads to irreversible fragmentation of the damaged axon. Arrows with questions marks reflect postulated interactions
Although two key elements in this pathway of axonal degeneration (SARM1 and NMNAT2) have been identified (Figure 5), their exact actions are, as yet, unknown. It will be crucial to identify disease‐specific degeneration pathways associated with distal axon loss to ensure that targeted, efficacious therapies can be developed in RLN.
4.1. Bilateral mononeuropathy or polyneuropathy?
When the severity of RLN is sufficient to cause clinical signs, the typical neuropathological changes identifiable in the left RLn, are also detected (with less severity) in the right6, 20, 23, 55, 56, 113, 114; a disparity that has been explained by the right RLn's shorter length compared with that of the left.21 If true, this feature alone implicates length‐dependency as a key feature in this disease. Although degeneration of the right RLn is consistently reported in pathological studies, development of right arytenoid dysfunction has been reported in only one horse with RLN115 (RLN cases typically present with left laryngeal dysfunction in isolation20, 115, 116). However, these pathological studies confirm the disorder's bilateral occurrence and reveal that the disorder cannot solely be explained by the physical course of the left RLn.
Bilateral degeneration of one pair of peripheral nerves has been reported in humans as a component of the mononeuropathy multiplex syndrome (defined as the isolated and differential involvement of separate nerves) often because of a generalized systemic disorder and most commonly diabetes mellitus.117 Signs of nerve dysfunction relate to the specific nerve affected: frequently they are initially asymmetrical, but they progress to symmetrical nerve dysfunction and then to other nerve groups.41, 117 This suggests that systemic disorders (at least initially) can have localized nerve involvement. Indeed, length dependency is a common feature of many peripheral nerve disorders with a genetic basis: even though defective protein expression is a generalized feature, the signs are most readily apparent in the longest nerves.60, 118 In humans, the longest nerves are to the distal extremities and length dependency is revealed by many of the CMT disorders that have distal sensory loss, or distal neurogenic muscle atrophy (leading to pes cavus deformity).60, 118 Length dependency is also a feature of some acquired neuropathies such as diabetic neuropathy (reviewed by Kazamel and Dyck119).
Determining whether this equine neuropathy affects only the RLn or in addition, other long nerves, has been a key objective in several studies over past decades3, 6, 7, 20, 23, 113 because the answer has major implications for research into the etiopathogenesis. In particular, certain acquired causes (eg, trauma from neck stretch or thoracic vessel pulsation) would be ruled out if other long nerves are affected, whereas toxic or genetic causes might be more likely if the disease is found to be a polyneuropathy. Polyneuropathies typically produce diverse clinical signs according to the nerves that are diseased120: for example, equine polyneuropathies include those with a toxic etiology (such as Australian Stringhalt, lead, and possibly haloxon) result in laryngeal dysfunction, dysphagia, proprioceptive deficits and gait abnormalities.121, 122, 123, 124, 125, 126, 127 Similarly, disorders such as equine motor neuron disease, are associated with widespread, generalized and symmetrical peripheral motor nerve involvement.128
Clinical signs associated with involvement of other nerves is not a consistent feature of RLN, but the sub‐clinical involvement of the right RLn, suggests that other nerves might also have subclinical involvement. Consequently, some researchers investigating RLN's pathology, have included the histological examination of other long peripheral nerves in horses, such as the phrenic, lateral palmar, ulnar, tibial, medial plantar, median, and peroneal nerves.3, 4, 7, 20, 23, 24 Unfortunately, many of these studies were underpowered, unblinded, subjective or poorly controlled. Some authors report a lack of involvement of other nerves in this disorder. For example, Duncan et al4 examined multiple additional peripheral nerves from 2 Thoroughbreds with RLN, but with no significant pathological findings. Hahn et al20 examined median, peroneal and phrenic nerves of 3 horses (a Thoroughbred, Warmblood, and a Clydesdale) with severe RLN, concluding that there were no significant neuropathological findings in nonlaryngeal nerves, even though one of the horses was reported with moderate loss of large myelinated fibers, multiple onion bulbs, and an increased frequency of hypomyelinated internodes in semi‐thin sections of peroneal and median nerves.
In contrast, several reports detail the possible significance of RLN‐associated pathological features in other long, nonlaryngeal nerves.3, 7, 23 Indeed, Duncan et al (14 years after their study described above) examined the deep peroneal nerves of 14 male Draft horse foals, reporting the presence of Büngner bands and onion bulbs in nerves from 6 month old foals.24 The authors made no comment about clinical signs associated with peroneal nerve dysfunction being present in any foal, nor was there any investigation into pathological changes within the peroneal nerve‐innervated muscles.54 Cahill and Goulden examined long limb motor nerves (median, peroneal and tibial) in 2 of 4 male Thoroughbred horses with clinically relevant RLN3, 7, 23, 56 that ranged in age from 2 to 22 years; unfortunately the ages of the 2 cases chosen for limb nerve analysis were not disclosed3, 7, 23 and no control horses were included. The pathology was graded as the number of pathological changes (regenerating clusters, onion bulbs, demyelination/remyelination and myelin debris in the Schwann cell cytoplasm) per fascicle. The 2 cases had occasional (1–2 changes per fascicle) or moderate (5–10 changes per fascicle) pathological changes in the extensor digitorum longus (EDL) peroneal branch, superficial and common peroneal, median and the tibial nerves, by light microscopy.3 EM and teased fiber studies on the peroneal nerves revealed that the type and degree of pathology in the peroneal nerves from RLN‐affected horses was similar to that present in the proximal left and distal right RLn.7, 23
Some authors have additionally, or instead, searched for histopathological, neuropathic changes in selected limb muscles because RLN‐associated intrinsic laryngeal muscle pathology is readily identified.20, 56 Cahill and Goulden56 found no significant histopathological changes in the tibialis cranialis, gastrocnemius or biceps femoris muscles of 4 horses with clinically relevant RLN. In contrast, presumed neurogenic muscle fiber atrophy was detected in the EDL muscle of 3 of the 4 horses, although in only one of these animals was the key feature of neuronal regeneration (seen in RLN), known as fiber type grouping detected.56 It is noteworthy that these apparent EDL muscle changes occurred despite the lack of nerve pathological changes in previous studies of the deep peroneal nerve by the same and other authors3, 23, 56 suggesting either that muscle histopathological assessment for neurogenic change has greater sensitivity than nerve histology, or that the detected muscle changes were not neurogenic in origin.
Conflicting results obtained by these various groups and between examination of nerves and muscles, reveal the problems associated with unblinded, uncontrolled, subjective histopathological assessment. In particular, including greater numbers of height‐, sex‐, and breed‐matched control horses would have enabled more robust conclusions to be drawn, given that each is associated with RLN development.1, 2, 8, 14, 129 Furthermore, typically authors have failed to consider the influence of age, even though age‐related neuronal degenerative changes occur in many peripheral nerves of various species.67, 130, 131, 132, 133 Indeed, neurodegenerative changes reported in the median and peroneal nerves of a horse with clinically relevant RLN, might have been age‐related (the horse was 16 years old)20 rather than associated with underlying disease. In a study looking at the lateral palmar (sensory) nerve in horses free from detectable neuromuscular disease (unknown breeds) horses aged between 5 and 7 years old had evidence of demyelination/remyelination in up to 20% of myelinated nerve fibers.133 Other than the RLn (and the paired phrenic nerves), the longest peripheral nerves in horses are sensory rather than motor; however, no studies have evaluated clinical or histopathological sensory nerve involvement in horses with RLN.
In certain neuropathies in other species, both the peripheral and central nervous systems can be involved.134 For example, in horses with equine motor neuron disease, there is prominent degeneration of the spinal cord ventral horn cell bodies of the peripheral motor nerves.128 Cahill and Goulden22, 55 examined the nucleus ambiguus of clinically affected or subclinically affected RLN horses55 but identified no relevant pathological features. However, a search for central involvement in RLN (ie, in a length‐dependent neuropathy) might most logically be made in the longest central nerve tracts: these would be the sensory proprioceptive tracts from the thoracic limbs (funiculus cuneate) and pelvic limbs (funiculus gracilis), with the latter containing the longest axons. The same authors reported significantly higher numbers of spheroids in the lateral cuneate nuclei (which receives sensory input from the cervicothoracic spinal nerves) in subclinical and clinical RLN cases when compared with the control group,22 but despite evaluating the gracilis nucleus for increased spheroids, none was reported.55 Since spheroid formation is seen with aging, the apparent increased spheroidal numbers in the diseased groups could be explained by the lack of age matching between the control and diseased groups: the horses in the control group were all <3 years old, whereas the diseased group's horses ranged from 2 to 22 years old.135
Perhaps then, RLN represents the localized involvement of two nerves (the left and right RLn) associated with a systemic disorder that has subclinical involvement of other long nerves in only a few (eg, the most severely affected, or tallest) horses. Unfortunately, despite the previous work, no clear conclusion regarding the possible involvement of other nerves in RLN can be made. In particular, the trend for studies to evaluate subjective histopathological features in unblinded, uncontrolled fashion and with low power, means that this crucial question remains unanswered such that RLN cannot yet be defined as bilateral mononeuropathy or a polyneuropathy with certainty.
5. OBJECTIVE FUNCTIONAL TESTING OF THE RLn
Nerve function testing for a suspected neuropathy helps define each tested nerve's involvement but also the type of pathology (axonal or myelin‐based).44 Function is assessed either directly, by examining the speed of conduction (NCV) or, indirectly, via the measure of latency (delay from nerve stimulation to muscle contraction) and through functionality of the innervated structure (ie, the larynx in RLN). Testing of the paired RLn' functionality has included direct measurement of NCV and nerve conduction latency measurements.136, 137, 138, 139 Of these techniques, accurate measurement of the NCV in the RLn (and others) in affected horses might best help define if RLN is an axonopathy or myelinopathy and whether other nerves are also involved. This routine technique is used diagnostically in human peripheral neuropathy patients to help define the precise phenotypic features of the disease (distinguishing between CMT types or sensorimotor peripheral neuropathies for example).140 The characteristic electrophysiological features of an axonopathy include a reduction in the amplitude of a compound motor action potential (CMAP) and a normal NCV, whereas a myelinopathy is characterized by a normal CMAP, with a slowed NCV.44 Conduction blocks have historically been recognized as signifying demyelination, especially segmental demyelination, however more recently this phenomenon has also been detected in distal axonopathies.141 Presumably, conduction blocks could occur in grade 4 RLN horses when the loss of myelinated axons in the left RLn is great enough to stop the propagation of the action potential to the laryngeal muscles. In this situation, pathological evaluation of the RLn would be necessary to differentiate axonal loss from demyelination as the cause for the conduction block. Unfortunately, NCVs are influenced by many factors such as the individual's age, sex, height, and skin temperature: for instance, in humans, every 1 cm increase in height, accounts for a 0.2 m/s decrease in NCV142, 143 and women have slower NCVs in the RLn than men.144 The negative correlation between height and NCV has been attributed to the abrupt tapering of the nerve fiber diameter with increasing nerve length (which is mainly caused by a reduction in the axonal diameter) rather than to any effect on internodal distance.145, 146, 147
Likely, because of practical constraints, very few researchers have evaluated the NCV of the RLn in horses. The latency (using electrolaryngeography) of the RLn in Clydesdale horses with varying grades of RLN was reported by Hawe et al (2001). They reported no significant difference between the Clydesdales' size‐adjusted latency values and their RLN endoscopic grade.138 There was no information included in the study regarding the amplitude of the CMAPs produced. Recurrent laryngeal nerves (RLn) NCVs were reported directly by Cheetham et al137 in horses with normal (Havemeyer grade 1) arytenoid function: the NCV of the left RLn (39.1 ± 4.4 m/s; mean ± SD) was significantly slower than that of the right RLn (50 ± 4.2 m/s). The authors concluded that this data suggests that the primary pathology associated with RLN might be demyelination rather than axonal loss, although significant demyelination has not been reported in horses with (grade 1) normal arytenoid function.3, 4, 7 Finally, a slower left RLn NCV compared with the right was reported in one horse with subclinical RLN by Duncan et al,113 although the velocities were not included.
Steiss et al reported the latency of a CMAP, as a measure of the NCV, in both the RLn in 5 normal horses and 7 ponies. This revealed a longer conduction latency in the proximal left RLn compared with the proximal right nerve (no left‐right differences were detected in the distal RLn).139 However, no correction was made for differences in nerve length in our study, which likely explains the longer latency of the left RLn. Nevertheless, the similar left and right RLn conduction latencies reported in dogs and humans144, 148 were explained by the left side's wider diameter (and therefore faster conducting) axons when compared to the right, compensating for the longer path.37 The left distal RLn of young (<2 year old) horses that lacked histopathological features of RLN, had significantly fewer myelinated nerve fibers, and smaller diameter myelinated nerve fibers than those in the right distal RLn. Furthermore, the proportion of the widest myelinated fibers (>9.5 μm) was also always lower in the left RLn compared with the right.149 These findings imply that the equine RLn and their NCVs are unusual. Nerve conduction velocities (NCVs) in the left and right RLn of horses have been shown to differ between the two nerves in several studies; this has been recapitulated in normal ponies and horses, as well as in RLN cases.113, 137, 138, 139, 150 The interpretation of this finding differs between authors: is RLN simply ubiquitous or does it result from deterioration of ‘normal’ equine anatomy?
Despite this previous research, the correct classification of RLN's primary basis based on objective electrophysiological recordings remains unclear. This might largely because of differences between methodologies, low numbers of animals, and differences in signalment in the animals examined. It also very likely relates to the practical difficulties encountered when performing these sorts of tests in horses.
6. CONCLUSIONS
The majority of the histopathological changes (Büngner bands, regenerating clusters, paranodal evaginations, and spheroids) reported in RLN‐affected horses are associated with primary axonal dysfunction.3, 4, 6, 7, 20, 30, 52, 53, 113, 114 Further, we have discussed how pathological features traditionally associated with primary myelinopathies (ie, onion bulbs and demyelination and remyelination) also occur in primary axonopathies. In RLN, these histopathological changes occur simultaneously with other pathological features that are exclusively reported in axonopathies such as paranodal evaginations (that occur in toxic axonopathies and axotomies) and at sites of axonal degeneration.4, 53 In addition, we have highlighted evidence that suggests axonal transportation deficits might play a role in the etiopathogenesis of RLN.
While there is compelling evidence that RLN affects both the left and (to a lesser extent) right RLn, it is not yet clear whether in some horses, other long nerves are also affected. There is some pathological evidence to suggest subclinical involvement of other long peripheral nerves in cases of RLN (Büngner bands, and onion bulbs in nonlaryngeal peripheral nerves),3, 4, 7, 20, 23, 24 nevertheless these findings are not found in all studies; typically, and unfortunately, most of these studies suffer from the limitations discussed throughout this review article and a comprehensive, objective, blinded study that evaluates other long peripheral nerves, and long spinal tracts of the CNS, with age, height, breed, and sex matched controls is required.
In summary, in our opinion RLN is best currently classified as a distal axonopathy with clear evidence of involvement of both RLn. Further, from a functional and clinical perspective, this disease can best be regarded as a mononeuropathy of the left RLn: however, when considering the etiology and pathophysiology of this disorder, there is insufficient evidence to classify the disease as either a bilateral mononeuropathy or polyneuropathy, a classification that remains fundamentally important for the study of this enigmatic, and highly prevalent equine neurodegenerative disease.
CONFLICT OF INTEREST DECLARATION
Authors declare no conflict of interest.
OFF‐LABEL ANTIMICROBIAL DECLARATION
Authors declare no off‐label use of antimicrobials.
INSTITUTIONAL ANIMAL CARE AND USE COMMITTEE (IACUC) OR OTHER APPROVAL DECLARATION
Ethic approval for all work conducted by Alex Draper, during her PhD was received from the Ethics Committee, Royal Veterinary College. Ref No. 2015 1381.
ACKNOWLEDGMENTS
The authors acknowledge the many discussions had over the course of several years with colleagues that have helped inform content within this review, and in particular, those with Robert Adalbert, Julian Blake, Jon Cheetham, Michael Coleman, Norm Ducharme, Ian Duncan, Rosalind King, Joe Mayhew, Vince Molony, Justin Perkins, Mary Reilly, Ed Robinson, Giampietro Schiavo, and Laura Tulloch. Figures 1 and 2 were kindly generated by Brian Cox. Alex Draper is a PhD scholar at the Royal Veterinary College supported by a grant from the Horserace Betting Levy Board of the United Kingdom. The manuscript was approved by the Royal Veterinary College's research office and assigned the following unique reference number: CSS_01735.
Draper ACE, Piercy RJ. Pathological classification of equine recurrent laryngeal neuropathy. J Vet Intern Med. 2018;32:1397–1409. 10.1111/jvim.15142
Funding information Horserace Betting Levy Board, United Kingdom
REFERENCES
- 1. Boyko AR. Genomic analysis establishes correlation between growth and laryngeal neuropathy in Thoroughbreds. BMC Genomics. 2014;15:259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Goulden BE, Anderson LJ, Cahill JI. Roaring in Clydesdales. N Z Vet J. 1985;33:73–76. [DOI] [PubMed] [Google Scholar]
- 3. Cahill JI, Goulden BE. Equine laryngeal hemiplegia. Part I. A light microscopic study of peripheral nerves. N Z Vet J. 1986;34:161–169. [DOI] [PubMed] [Google Scholar]
- 4. Duncan ID, Griffiths IR, Madrid RE. A light and electron microscopic study of the neuropathy of equine idiopathic laryngeal hemiplegia. Neuropathol Appl Neurobiol. 1978;4:483–501. [DOI] [PubMed] [Google Scholar]
- 5. Taylor SE, Barakzai SZ, Dixon P. Ventriculocordectomy as the sole treatment for recurrent laryngeal neuropathy: long‐term results from ninety‐two horses. Vet Surg. 2006;35:653–657. [DOI] [PubMed] [Google Scholar]
- 6. Duncan ID, Griffiths IR. Pathological changes in equine laryngeal muscles and nerves In: Proceedings of the 19th Annual Convention of the American Association of Equine Practitioners, Atlanta, Georgia. 1973;97–223.
- 7. Cahill JI, Goulden BE. Equine laryngeal hemiplegia. Part II. An electron microscopic study of peripheral nerves. N Z Vet J. 1986;34:161–165. [DOI] [PubMed] [Google Scholar]
- 8. Cook WR. The diagnosis of respiratory unsoundness in the horse. Vet Rec. 1965;77:516–528. [PubMed] [Google Scholar]
- 9. Takayuki IBI, Takeshi M, Seiji H, Hironori O, Nobushige I, Yoshiyuki S. Estimation of heritability of laryngeal hemiplegia in the thoroughbred horse by Gibbs sampling. J Equine Sci. 2003;14:81–86. [Google Scholar]
- 10. Barakzai S. Heritability of Recurrent Laryngeal Neuropathy. In: 4th World Equine Airways Symposium (WEAS 09); 2009; Berne, Switzerland. [Google Scholar]
- 11. Dupuis MC, Zhang Z, Durkin K, Charlier C, Lekeux P, Georges M. Detection of copy number variants in the horse genome and examination of their association with recurrent laryngeal neuropathy. Anim Genet. 2013;44:206–208. [DOI] [PubMed] [Google Scholar]
- 12. Dupuis MC, Zhang Z, Druet T, et al. Results of a haplotype‐based GWAS for recurrent laryngeal neuropathy in the horse. Mamm Genome. 2011;22:613–620. [DOI] [PubMed] [Google Scholar]
- 13. Cahill JI, Goulden BE. The pathogenesis of equine laryngeal hemiplegia–a review. N Z Vet J. 1987;35:82–90. [DOI] [PubMed] [Google Scholar]
- 14. Rooney JR, Delaney FM. An hypothesis on the causation of laryngeal hemiplegia in horses. Equine Vet J. 1970;2:35–37. [Google Scholar]
- 15. Loew FM. Thiamine and equine laryngeal hemiplegia. Vet Rec. 1973;92:372–373. [DOI] [PubMed] [Google Scholar]
- 16. Cavanagh JB. The problems of neurons with long axons. Lancet. 1984;1:1284–1287. [DOI] [PubMed] [Google Scholar]
- 17. Burrows GE. Lead poisoning in the horse. Equine Prac. 1982;4. [Google Scholar]
- 18. Duncan ID, Brook D. Bilateral laryngeal paralysis in the horse. Equine Vet J. 1985;17:228–233. [DOI] [PubMed] [Google Scholar]
- 19. Fleming G. Roaring in horses. London: Balliere, Tindall & Cox; 1889. [Google Scholar]
- 20. Hahn CN, Matiasek K, Dixon PM, Molony V, Rodenacker K, Mayhew IG. Histological and ultrastructural evidence that recurrent laryngeal neuropathy is a bilateral mononeuropathy limited to recurrent laryngeal nerves. Equine Vet J. 2008;40:666–672. [DOI] [PubMed] [Google Scholar]
- 21. Griffiths IR. The pathogenesis of equine laryngeal hemiplegia. Equine Vet J. 1991;23:75–76. [DOI] [PubMed] [Google Scholar]
- 22. Cahill JI, Goulden BE. Further evidence for a central nervous system component in equine laryngeal hemiplegia. N Z Vet J. 1989;37:89–90. [DOI] [PubMed] [Google Scholar]
- 23. Cahill JI, Goulden BE. Equine laryngeal hemiplegia. Part III. A teased fibre study of peripheral nerves. N Z Vet J. 1986;34:181–185. [DOI] [PubMed] [Google Scholar]
- 24. Duncan ID. Determination of the early age of onset of equine recurrent laryngeal neuropathy. 2. Nerve pathology. Acta Neuropathol. 1992;84:316–321. [DOI] [PubMed] [Google Scholar]
- 25. Ekins S, Litterman NK, Arnold RJG, et al. A brief review of recent Charcot‐Marie‐Tooth research and priorities. F1000Res. 2015;4:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. de Lahunta A, Glass E, Kent M. Veterinary Neuroanatomy and Clinical Neurology. St Louis, Missouri, USA: Saunders Elsevier; 2009:134–167. [Google Scholar]
- 27. McGeady TA, Quinn PJ, Fitzpatrick ES, Ryan MT, Kilroy D, Lonergan P. Veterinary Embryology. Oxford: Blackwell Publishing; 2008:120–122. [Google Scholar]
- 28. Mason JE. Laryngeal Hemiplegia: a further look at Haslam's anomaly of the left recurrent nerve. Equine Vet J. 1973;5:150–155. [DOI] [PubMed] [Google Scholar]
- 29. Cahill JI. The Pathology of Laryngeal Hemiplegia in Horses. New Zealand: Massey University; 1985. [Google Scholar]
- 30. Duncan ID. The Pathology of Equine Laryngeal Hemiplegia. United Kingdom: University of Glasgow; 1975. [Google Scholar]
- 31. Dyer KR, Duncan ID. The intraneural distribution of myelinated fibres in the equine recurrent laryngeal nerve. Brain. 1987;110:1531–1543. [DOI] [PubMed] [Google Scholar]
- 32. Lopez‐Plana C, Sautet JY, Ruberte J. Muscular pathology in equine laryngeal neuropathy. Equine Vet J. 1993;25:510–513. [DOI] [PubMed] [Google Scholar]
- 33. Murray JG. Innervation of the intrinsic muscles of the cat's larynx by the recurrent laryngeal nerve: a unimodal nerve. J Physiol. 1957;135:206–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shin T, Rabuzzi DD. Conduction studies of the canine recurrent laryngeal nerve. Laryngoscope. 1971;81:586–596. [DOI] [PubMed] [Google Scholar]
- 35. Tomasch J, Britton WA. A fibre analysis of the laryngeal nerve‐supply in man. Acta Anat (Basel). 1955;23:386–398. [PubMed] [Google Scholar]
- 36. Gacek RR, Lyon MJ. Fiber components of the recurrent laryngeal nerve in the cat. Ann Otol Rhinol Laryngol. 1976;85:460–471. [DOI] [PubMed] [Google Scholar]
- 37. Dahlqvist A, Carlsoo B, Hellstrom S. Fiber components of the recurrent laryngeal nerve of the rat: a study by light and electron microscopy. Anat Rec. 1982;204:365–370. [DOI] [PubMed] [Google Scholar]
- 38. Harrison DF. Fibre size frequency in the recurrent laryngeal nerves of man and giraffe. Acta Otolaryngol. 1981;91:383–389. [DOI] [PubMed] [Google Scholar]
- 39. Lang IM, Medda BK, Jadcherla SR, Shaker R. Characterization and mechanisms of the pharyngeal swallow activated by stimulation of the esophagus. Am J Physiol Gastrointest Liver Physiol. 2016;311:G827–G837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pascual‐Font A, Hernández‐Morato I, McHanwell S, et al. The central projections of the laryngeal nerves in the rat. J Anat. 2011;219:217–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Love S, Louis DN, Ellison DW. Greenfield's Neuropathology. London, UK: Edward Arnold Publishers Ltd; 2008:1609–1725. [Google Scholar]
- 42. Pareyson D, Marchesi C. Diagnosis, natural history, and management of Charcot‐Marie‐Tooth disease. Lancet Neurol. 2009;8:654–667. [DOI] [PubMed] [Google Scholar]
- 43. Rossor AM, Evans MR, Reilly MM. A practical approach to the genetic neuropathies. Pract Neurol. 2015;15:187–198. [DOI] [PubMed] [Google Scholar]
- 44. Kimura J. Electrodiagnosis in diseases of Nerve and Muscle: Principles and Practice. Oxford: Oxford University Press; 2013:74–92. [Google Scholar]
- 45. Matsunami N, Smith B, Ballard L, et al. Peripheral myelin protein‐22 gene maps in the duplication in chromosome 17p11.2 associated with Charcot‐Marie‐Tooth 1A. Nat Genet. 1992;1:176–179. [DOI] [PubMed] [Google Scholar]
- 46. Patel PI, Roa BB, Welcher AA, et al. The gene for the peripheral myelin protein PMP‐22 is a candidate for Charcot‐Marie‐Tooth disease type 1A. Nat Genet. 1992;1:159–165. [DOI] [PubMed] [Google Scholar]
- 47. Timmerman V, Nelis E, Van Hul W, et al. The peripheral myelin protein gene PMP‐22 is contained within the Charcot‐Marie‐Tooth disease type 1A duplication. Nat Genet. 1992;1:171–175. [DOI] [PubMed] [Google Scholar]
- 48. Züchner S, Mersiyanova IV, Muglia M, et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot‐Marie‐Tooth neuropathy type 2A. Nat Genet. 2004;36:449–451. [DOI] [PubMed] [Google Scholar]
- 49. Hahn AF, Ainsworth PJ, Bolton CF, Bilbao JM, Vallat JM. Pathological findings in the x‐linked form of Charcot‐Marie‐Tooth disease: a morphometric and ultrastructural analysis. Acta Neuropathol. 2001;101:129–139. [DOI] [PubMed] [Google Scholar]
- 50. Birouk N, LeGuern E, Maisonobe T, et al. X‐linked Charcot‐Marie‐tooth disease with connexin 32 mutations: clinical and electrophysiologic study. Neurology. 1998;50:1074–1082. [DOI] [PubMed] [Google Scholar]
- 51. Dubourg O. Clinical, electrophysiological and molecular genetic characteristics of 93 patients with X‐linked Charcot‐Marie‐Tooth disease. Brain. 2001;124:1958–1967. [DOI] [PubMed] [Google Scholar]
- 52. Duncan ID, Griffths IR, McQueen A, Baker GO. The pathology of equine laryngeal hemiplegia. Acta Neuropathol. 1974;27:337–348. [DOI] [PubMed] [Google Scholar]
- 53. Duncan ID, Hammang JP. Ultrastructural observations of organelle accumulation in the equine recurrent laryngeal nerve. J Neurocytol. 1987;16:269–280. [DOI] [PubMed] [Google Scholar]
- 54. Harrison GD, Duncan ID, Clayton MK. Determination of the early age of onset of equine recurrent laryngeal neuropathy. 1. Muscle pathology. Acta Neuropathol. 1992;84:307–315. [DOI] [PubMed] [Google Scholar]
- 55. Cahill JI, Goulden BE. Equine laryngeal hemiplegia. Part V. Central nervous system pathology. N Z Vet J. 1986;34:191–193. [DOI] [PubMed] [Google Scholar]
- 56. Cahill JI, Goulden BE. Equine laryngeal hemiplegia. Part IV. Muscle pathology. N Z Vet J. 1986;34:186–190. [DOI] [PubMed] [Google Scholar]
- 57. Cavanagh JB. The ‘dying back' process. A common denominator in many naturally occurring and toxic neuropathies. Arch Pathol Lab Med. 1979;103:659–664. [PubMed] [Google Scholar]
- 58. Kamholz J, Menichella D, Jani A, et al. Charcot‐Marie‐Tooth disease type 1: molecular pathogenesis to gene therapy. Brain. 2000;123:222–233. [DOI] [PubMed] [Google Scholar]
- 59. Scherer S. Axonal pathology in demyelinating diseases. Ann Neurol. 1999;45:6–7. [DOI] [PubMed] [Google Scholar]
- 60. Krajewski KM. Neurological dysfunction and axonal degeneration in Charcot‐Marie‐Tooth disease type 1A. Brain. 2000;123:1516–1527. [DOI] [PubMed] [Google Scholar]
- 61. Dixon PM, Robinson NE, Wade J. Proceedings of a workshop on equine recurrent laryngeal neuropathy Havemeyer Foundation Monograph Series No 11. Newmarket: R&W Publications; 2003. [Google Scholar]
- 62. Vandevelde M, Higgins RJ, Oevermann A. General Neuropathology. West Sussex, UK: Wiley; 2012:1–37. [Google Scholar]
- 63. Griffin JW, Price DL. Demyelination in experimental beta, beta'‐iminodipropionitrile and hexacarbon neuropathies. Evidence for an axonal influence. Lab Invest. 1981;45:130–141. [PubMed] [Google Scholar]
- 64. Bosboom WM. Diagnostic value of sural nerve demyelination in chronic inflammatory demyelinating polyneuropathy. Brain. 2001;124:2427–2438. [DOI] [PubMed] [Google Scholar]
- 65. Dyck PJ, Johnson WJ, Lambert EH, O'Brien PC. Segmental demyelination secondary to axonal degeneration in uremic neuropathy. Mayo Clin Proc. 1971;46:400–431. [PubMed] [Google Scholar]
- 66. Midroni G, Bilbao JM. Biopsy Diagnosis of Peripheral Neuropathies. Boston: Butterworth‐Heinemann; 1995. [Google Scholar]
- 67. Bertram M, Schröder JM. Developmental changes at the node and paranode in human sural nerves: morphometric and fine‐structural evaluation. Cell Tissue Res. 1993;273:499–509. [DOI] [PubMed] [Google Scholar]
- 68. Schroder JM. Developmental and pathological changes at the node and paranode in human sural nerves. Microsc Res Tech. 1996;34:422–435. [DOI] [PubMed] [Google Scholar]
- 69. Schroder JM. Structure of the nodes and paranodes in peripheral nerves, Part I. Introduction. Microsc Res Tech. 1996;34:397–398. [DOI] [PubMed] [Google Scholar]
- 70. Bilbao JM, Schmidt RE. Biopsy Diagnosis of Peripheral Neuropathy. Switzerland: Springer; 2015:144–145. [Google Scholar]
- 71. Jones HB, Cavanagh JB. Distortions of the nodes of Ranvier from axonal distension by filamentous masses in hexacarbon intoxication. J Neurocytol. 1983;12:439–458. [DOI] [PubMed] [Google Scholar]
- 72. Millecamps S, Julien JP. Axonal transport deficits and neurodegenerative diseases. Nat Rev Neurosci. 2013;14:161–176. [DOI] [PubMed] [Google Scholar]
- 73. Jones HB, Cavanagh JB. The evolution of intracellular responses to acrylamide in rat spinal ganglion neurons. Neuropathol Appl Neurobiol. 1984;10:101–121. [DOI] [PubMed] [Google Scholar]
- 74. Brismar T, Hildebrand C, Tegner R. Nodes of Ranvier in acrylamide neuropathy: voltage clamp and electron microscopic analysis of rat sciatic nerve fibres at proximal levels. Brain Res. 1987;423:135–143. [DOI] [PubMed] [Google Scholar]
- 75. Simonati A, Cavanagh JB. Changes in terminal sprout formation in rat sternocostalis muscle during chronic intoxication with 2,5 hexanedione. Muscle Nerve. 1984;7:355–361. [DOI] [PubMed] [Google Scholar]
- 76. Brismar T, Hildebrand C, Berglund S. Nodes of Ranvier above a neuroma in the rat sciatic nerve: voltage clamp analysis and electron microscopy. Brain Res. 1986;378:347–356. [DOI] [PubMed] [Google Scholar]
- 77. Sayre LM, Autilio‐Gambetti L, Gambetti P. Pathogenesis of experimental giant neurofilamentous axonopathies: a unified hypothesis based on chemical modification of neurofilaments. Brain Res. 1985;10:69–83. [DOI] [PubMed] [Google Scholar]
- 78. Cavanagh JB, Bennetts RJ. On the pattern of changes in the rat nervous system produced by 2,5 hexanediol. A topographical study by light microscopy. Brain. 1981;104:297–318. [DOI] [PubMed] [Google Scholar]
- 79. Sickles DW, Stone JD, Friedman MA. Fast axonal transport: a site of acrylamide neurotoxicity? Neurotoxicology. 2002;23:223–251. [DOI] [PubMed] [Google Scholar]
- 80. Jones HB, Cavanagh JB. Cytochemical staining characteristics of peripheral nodes of Ranvier in hexacarbon intoxication. J Neurocytol. 1983;12:459–473. [DOI] [PubMed] [Google Scholar]
- 81. Tshala‐Katumbay D, Monterroso V, Kayton R, Lasarev M, Sabri M, Spencer P. Probing mechanisms of axonopathy. Part II: protein targets of 2,5‐hexanedione, the neurotoxic metabolite of the aliphatic solvent n‐hexane. Toxicol Sci. 2009;107:482–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. LoPachin RM, Gavin T. Toxic neuropathies: mechanistic insights based on a chemical perspective. Neurosci Lett. 2015;596:78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Harris CH, Wrenn RW, Friedman MA, Sickles DW. Acrylamide effects on cAMP‐dependent MAP phosphorylation and microtubule disassembly. Toxicologist. 1994;14:206. [Google Scholar]
- 84. Sickles DW, Brady ST, Testino A, Friedman MA, Wrenn RW. Direct effect of the neurotoxicant acrylamide on kinesin‐based microtubule motility. J Neurosci Res. 1996;46:7–17. [DOI] [PubMed] [Google Scholar]
- 85. Stone JD, Peterson AP, Eyer J, Oblak TG, Sickles DW. Axonal neurofilaments are nonessential elements of toxicant‐induced reductions in fast axonal transport: video‐enhanced differential interference microscopy in peripheral nervous system axons. Toxicol Appl Pharmacol. 1999;161:50–58. [DOI] [PubMed] [Google Scholar]
- 86. Martenson CH, Sheetz MP, Graham DG. A quantitation of fast axonal transport in cultured chick dorsal root ganglion (DRG) explants following exposure to acrylamide (ACR), methacrylamide (M‐ACR) and cyanide. Toxicologist. 1992;12:191. [Google Scholar]
- 87. Schmidt RE. Neuropathology of human sympathetic autonomic ganglia. Microsc Res Tech. 1996;35:107–121. [DOI] [PubMed] [Google Scholar]
- 88. Schmidt RE. Synaptic dysplasia in sympathetic autonomic ganglia. J Neurocytol. 1996;25:777–791. [DOI] [PubMed] [Google Scholar]
- 89. Coleman M. Axon degeneration mechanisms: commonality amid diversity. Nat Rev Neurosci. 2005;6:889–898. [DOI] [PubMed] [Google Scholar]
- 90. Raff MC, Whitmore AV, Finn JT. Axonal self‐destruction and neurodegeneration. Science. 2002;296:868–871. [DOI] [PubMed] [Google Scholar]
- 91. Yang H, He X, Yang J, et al. Activation of cAMP‐response element‐binding protein is positively regulated by PKA and calcium‐sensitive calcineurin and negatively by PKC in insect. Insect Biochem Mol Biol. 2013;43:1028–1036. [DOI] [PubMed] [Google Scholar]
- 92. Kurant E. Keeping the CNS clear: glial phagocytic functions in Drosophila. Glia. 2011;59:1304–1311. [DOI] [PubMed] [Google Scholar]
- 93. Yang J, Weimer RM, Kallop D, et al. Regulation of axon degeneration after injury and in development by the endogenous calpain inhibitor calpastatin. Neuron. 2013;80:1175–1189. [DOI] [PubMed] [Google Scholar]
- 94. Finn JT, Weil M, Archer F, Siman R, Srinivasan A, Raff MC. Evidence that Wallerian degeneration and localized axon degeneration induced by local neurotrophin deprivation do not involve caspases. J Neurosci. 2000;20:1333–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Hackett SM. The equine nucleus ambiguus: Myotopic and neurotopic representations of motor and sensory components of the recurrent laryngeal nerve. Ithaca, NY, USA: Cornell University; 2000. [Google Scholar]
- 96. Gerdts J, Summers DW, Milbrandt J, DiAntonio A. Axon self‐destruction: new links among SARM1, MAPKs, and NAD+ metabolism. Neuron. 2016;89:449–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Conforti L, Gilley J, Coleman MP. Wallerian degeneration: an emerging axon death pathway linking injury and disease. Nat Rev Neurosci. 2014;15:394–409. [DOI] [PubMed] [Google Scholar]
- 98. Osterloh JM, Yang J, Rooney TM, et al. dSarm/Sarm1 is required for activation of an injury‐induced axon death pathway. Science. 2012;337:481–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Henninger N, Bouley J, Sikoglu EM, et al. Attenuated traumatic axonal injury and improved functional outcome after traumatic brain injury in mice lacking Sarm1. Brain. 2016;139:1094–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Summers DW, DiAntonio A, Milbrandt J. Mitochondrial dysfunction induces Sarm1‐dependent cell death in sensory neurons. J Neurosci. 2014;34:9338–9350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Massoll C, Mando W, Chintala SK. Excitotoxicity upregulates SARM1 protein expression and promotes Wallerian‐like degeneration of retinal ganglion cells and their axons. Invest Ophthalmol Vis Sci. 2013;54:2771–2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Yang J, Wu Z, Renier N, et al. Pathological axonal death through a MAPK cascade that triggers a local energy deficit. Cell. 2015;160:161–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Shin JE, Miller BR, Babetto E, et al. SCG10 is a JNK target in the axonal degeneration pathway. Proc Natl Acad Sci USA. 2012;109:E3696–E3705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Gerdts J, Summers DW, Sasaki Y, DiAntonio A, Milbrandt J. Sarm1‐mediated axon degeneration requires both SAM and TIR interactions. J Neurosci. 2013;33:13569–13580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Gilley J, Coleman MP. Endogenous NMNAT2 is an essential survival factor for maintenance of healthy axons. PLoS Biol. 2010;8:e1000300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Gilley J, Adalbert R, Yu G, Coleman MP. Rescue of peripheral and CNS axon defects in mice lacking NMNAT2. J Neurosci. 2013;33:13410–13424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Marangoni M, Adalbert R, Janeckova L, et al. Age‐related axonal swellings precede other neuropathological hallmarks in a knock‐in mouse model of Huntington's disease. Neurobiol Aging. 2014;35:2382–2393. [DOI] [PubMed] [Google Scholar]
- 108. Mack TG, Reiner M, Beirowski B, et al. Wallerian degeneration of injured axons and synapses is delayed by a Ube4b/Nmnat chimeric gene. Nat Neurosci. 2001;4:1199–1206. [DOI] [PubMed] [Google Scholar]
- 109. Araki T, Sasaki Y, Milbrandt J. Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science. 2004;305:1010–1013. [DOI] [PubMed] [Google Scholar]
- 110. Ma M, Ferguson TA, Schoch KM, et al. Calpains mediate axonal cytoskeleton disintegration during Wallerian degeneration. Neurobiol Dis. 2013;56:34–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. George EB, Glass JD, Griffin JW. Axotomy‐induced axonal degeneration is mediated by calcium influx through ion‐specific channels. J Neurosci. 1995;15:6445–6452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Vallat J‐M, Ouvrier RA, Pollard JD, et al. Histopathological findings in hereditary motor and sensory neuropathy of axonal type with onset in early childhood associated with mitofusin 2 mutations. J Neuropathol Exp Neurol. 2008;67:1097–1102. [DOI] [PubMed] [Google Scholar]
- 113. Duncan ID, Baker GJ, Heffron CJ, Griffiths IR. A correlation of the endoscopic and pathological changes in subclinical pathology of the horse's larynx. Equine Vet J. 1977;9:220–225. [DOI] [PubMed] [Google Scholar]
- 114. Duncan ID, Reifenrath P, Jackson KF, Clayton M. Preferential denervation of the adductor muscles of the equine larynx. II: nerve pathology. Equine Vet J. 1991;23:99–103. [DOI] [PubMed] [Google Scholar]
- 115. Dixon PM, McGorum BC, Railton DI, et al. Clinical and endoscopic evidence of progression in 152 cases of equine recurrent laryngeal neuropathy (RLN). Equine Vet J. 2010;34:29–34. [DOI] [PubMed] [Google Scholar]
- 116. Dixon PM, McGorum BC, Railton DI, et al. Laryngeal paralysis: a study of 375 cases in a mixed‐breed population of horses. Equine Vet J. 2010;33:452–458. [DOI] [PubMed] [Google Scholar]
- 117. Bilbao JM, Schmidt RE. Biopsy Diagnosis of Peripheral Neuropathy. Switzerland: Springer International; 2015:246–247. [Google Scholar]
- 118. Scherer SS, Wrabetz L. Molecular mechanisms of inherited demyelinating neuropathies. Glia. 2008;56:1578–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Kazamel M, Dyck PJ. Sensory manifestations of diabetic neuropathies: anatomical and clinical correlations. Prosthet Orthot Int. 2015;39:7–16. [DOI] [PubMed] [Google Scholar]
- 120. Faissler D, Jurina K, Cauzinille L, Gaschen F, Adama F, Jaggy A. In: Platt SR, ed. Peripheral Nervous System and Musculature, in Small Animal Neurology: An Illustrated Text. Hannover: Schlutersche; 2010. [Google Scholar]
- 121. Aronson AL. Lead poisoning in cattle and horses following long‐term exposure to lead. Am J Vet Res. 1972;33:627–629. [PubMed] [Google Scholar]
- 122. Liu ZP. Lead poisoning combined with cadmium in sheep and horses in the vicinity of non‐ferrous metal smelters. Sci Total Environ. 2003;309:117–126. [DOI] [PubMed] [Google Scholar]
- 123. Sojka JE, Hope W, Pearson D. Lead toxicosis in 2 horses: similarity to equine degenerative lower motor neuron disease. J Vet Intern Med. 1996;10:420–423. [DOI] [PubMed] [Google Scholar]
- 124. Willoughby R, Macdonald E, Mcsherry B, Brown G. The interaction of toxic amounts of lead and zinc fed to young growing horses. Vet Rec. 1972;91:382–383. [DOI] [PubMed] [Google Scholar]
- 125. Willoughby RA, MacDonald E, McSherry BJ, Brown G. Lead and zinc poisoning and the interaction between Pb and Zn poisoning in the foal. Can J Comp Med. 1972;36:348–359. [PMC free article] [PubMed] [Google Scholar]
- 126. Huntington PJ, Jeffcott LB, Friend SC, Luff AR, Finkelstein DI, Flynn RJ. Australian Stringhalt–epidemiological, clinical and neurological investigations. Equine Vet J. 1989;21:266–273. [DOI] [PubMed] [Google Scholar]
- 127. Rose RJ, Hartley WJ, Baker W. Laryngeal paralysis in Arabian foals associated with oral haloxon administration. Equine Vet J. 1981;13:171–176. [DOI] [PubMed] [Google Scholar]
- 128. Step DL, Cummings JF, de Lahunta A, et al. Motor neuron degeneration in a horse. J Am Vet Med Assoc. 1993;202:86–88. [PubMed] [Google Scholar]
- 129. Brakenhoff J, Holcombe S, Hauptman J, Smith HK, Nickels FA, Caron JP. The prevalence of laryngeal disease in a large population of competition draft horses. Vet Surg. 2006;35:579–583. [DOI] [PubMed] [Google Scholar]
- 130. Griffiths IR, Duncan ID. Age changes in the dorsal and ventral lumbar nerve roots of dogs. Acta Neuropathol. 1975;32:75–85. [DOI] [PubMed] [Google Scholar]
- 131. Arnold N, Harriman DG. The incidence of abnormality in control human peripheral nerves studied by single axon dissection. J Neurol Neurosurg Psychiatry. 1970;33:55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Dyck PJ, Karnes J, O'Brien P, Nukada H, Lais A, Low P. Spatial pattern of nerve fiber abnormality indicative of pathologic mechanism. Am J Pathol. 1984;117:225–238. [PMC free article] [PubMed] [Google Scholar]
- 133. Wheeler SJ. Structure and function of equine peripheral nerves: morphology, morphometry and clinical electrophysiology. London: Royal Veterinary College, University of London; 1988:209. [Google Scholar]
- 134. Boillée S, Vande Velde C, Cleveland DW. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron. 2006;52:39–59. [DOI] [PubMed] [Google Scholar]
- 135. Beech J. Neuroaxonal dystrophy of the accessory cuneate nucleus in horses. Vet Pathol. 1984;21:384–393. [DOI] [PubMed] [Google Scholar]
- 136. Kenny M, Cercone M, Rawlinson JJ, et al. Transoesophageal ultrasound and computer tomographic assessment of the equine cricoarytenoid dorsalis muscle: Relationship between muscle geometry and exercising laryngeal function. Equine Vet J. 2017;49:395–400. [DOI] [PubMed] [Google Scholar]
- 137. Cheetham J, Pigott JJ, Leal MT, Soderholm LV, Mitchell LM, Ducharme NG. Nerve conduction in horses with normal arytenoid function. In: Proceedings of the 47th British Equine Veterinary Association Congress; 2008; Liverpool, UK. [Google Scholar]
- 138. Hawe C, Dixon PM, Mayhew IG. A study of an electrodiagnostic technique for the evaluation of equine recurrent laryngeal neuropathy. Equine Vet J. 2010;33:459–465. [DOI] [PubMed] [Google Scholar]
- 139. Steiss JE, Marshall AE, Humburg JM. Electromyographic evaluation of conduction time of the recurrent laryngeal nerve: findings in clinically normal horses and ponies. Equine Vet J. 1989;21:218–220. [DOI] [PubMed] [Google Scholar]
- 140. Bilbao J, Schmidt RE. Biopsy Diagnosis of Peripheral Neuropathy. London: Springer; 2015:7. [Google Scholar]
- 141. Kokubun N, Nishibayashi M, Uncini A, et al. Conduction block in acute motor axonal neuropathy. Brain. 2010;133:2897–2908. [DOI] [PubMed] [Google Scholar]
- 142. Blythe LL, Engel HN Jr., Rowe KE. Comparison of sensory nerve conduction velocities in horses versus ponies. Am J Vet Res. 1988;49:2138–2142. [PubMed] [Google Scholar]
- 143. Stetson DS, Albers JW, Silverstein BA, Wolfe RA. Effects of age, sex, and anthropometric factors on nerve conduction measures. Muscle Nerve. 1992;15:1095–1104. [DOI] [PubMed] [Google Scholar]
- 144. J. Kim S, G. Lee D, Kwon JY. Development of a nerve conduction technique for the recurrent laryngeal nerve. Laryngoscope. 2014;124:2779–2784. [DOI] [PubMed] [Google Scholar]
- 145. Campbell WW Jr., Ward LC, Swift TR. Nerve conduction velocity varies inversely with height. Muscle Nerve. 1981;4:520–523. [DOI] [PubMed] [Google Scholar]
- 146. Simpson AH, Gillingwater TH, Anderson H, et al. Effect of limb lengthening on internodal length and conduction velocity of peripheral nerve. J Neurosci. 2013;33:4536–4539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Williams PL, Wendell‐Smith CP. Some additional parametric variations between peripheral nerve fibre populations. J Anat. 1971;109:505–526. [PMC free article] [PubMed] [Google Scholar]
- 148. Steiss JE, Marshall AE. Electromyographic evaluation of conduction time and velocity of the recurrent laryngeal nerves of clinically normal dogs. Am J Vet Res. 1988;49:1533–1536. [PubMed] [Google Scholar]
- 149. Lopez JM, Alvarez‐Uria M. A morphometric and fine structural study of the myoepithelial cells in the hamster Harderian gland. J Submicrosc Cytol Pathol. 1993;25:223–232. [PubMed] [Google Scholar]
- 150. Cook WR, Thalhammer JG. Electrodiagnostic Test for the Objective Grading of Recurrent Laryngeal Neuropathy in the Horse In: Proceedings of the Annual Convention of the American Association of Equine Practitioners; 1991; AAEP. [Google Scholar]