

Bacterial meningitis is a serious global health problem, and one of the major causative organisms is Neisseria meningitidis, which is also a common commensal in the upper respiratory tract of healthy humans. In bacteria, numerous loci involved in biosynthesis of surface-exposed antigenic structures that are involved in the interaction between bacteria and host are frequently subjected to homologous recombination and phase variation. These mechanisms are well described in Neisseria, and phase variation provides the ability to change these structures reversibly in response to the environment. Protein glycosylation systems are becoming widely identified in bacteria, and yet little is known about the mechanisms and evolutionary forces influencing glycan composition during carriage and disease.
KEYWORDS: Neisseria meningitidis, carriage, whole-genome sequencing, O-linked protein glycosylation, glycan diversity, microheterogeneity
ABSTRACT
Species within the genus Neisseria display significant glycan diversity associated with the O-linked protein glycosylation (pgl) systems due to phase variation and polymorphic genes and gene content. The aim of this study was to examine in detail the pgl genotype and glycosylation phenotype in meningococcal isolates and the changes occurring during short-term asymptomatic carriage. Paired meningococcal isolates derived from 50 asymptomatic meningococcal carriers, taken about 2 months apart, were analyzed with whole-genome sequencing. The O-linked protein glycosylation genes were characterized in detail using the Genome Comparator tool at the https://pubmlst.org/ database. Immunoblotting with glycan-specific antibodies (Abs) was used to investigate the protein glycosylation phenotype. All major pgl locus polymorphisms identified in Neisseria meningitidis to date were present in our isolate collection, with the variable presence of pglG and pglH, both in combination with either pglB or pglB2. We identified significant changes and diversity in the pgl genotype and/or glycan phenotype in 96% of the paired isolates. There was also a high degree of glycan microheterogeneity, in which different variants of glycan structures were found at a given glycoprotein. The main mechanism responsible for the observed differences was phase-variable expression of the involved glycosyltransferases and the O-acetyltransferase. To our knowledge, this is the first characterization of the pgl genotype and glycosylation phenotype in a larger strain collection. This report thus provides important insight into glycan diversity in N. meningitidis and into the phase variability changes that influence the expressed glycoform repertoire during meningococcal carriage.
IMPORTANCE Bacterial meningitis is a serious global health problem, and one of the major causative organisms is Neisseria meningitidis, which is also a common commensal in the upper respiratory tract of healthy humans. In bacteria, numerous loci involved in biosynthesis of surface-exposed antigenic structures that are involved in the interaction between bacteria and host are frequently subjected to homologous recombination and phase variation. These mechanisms are well described in Neisseria, and phase variation provides the ability to change these structures reversibly in response to the environment. Protein glycosylation systems are becoming widely identified in bacteria, and yet little is known about the mechanisms and evolutionary forces influencing glycan composition during carriage and disease.
INTRODUCTION
Neisseria meningitidis is a Gram-negative bacterium colonizing the human oropharynx, normally without causing disease. The carriage prevalence has been shown to range widely (from 3% to 20%) and is influenced by factors such as geography, age, crowded societies, contact with a case, and the epidemic/endemic situation (1). Meningococci may occasionally invade the bloodstream and progress to meningococcal disease with severe meningitis and septicemia.
Bacteria covalently attach different glycan structures to diverse molecules such as lipids and proteins. N. meningitidis exhibits a general O-linked glycosylation system in which several surface-exposed and periplasmic proteins are glycosylated. The major glycoprotein, PilE, is the building subunit of pili, which are important virulence factors. In addition, the meningococci produce lipopolysaccharides (LPS), peptidoglycan, and capsular polysaccharide. These molecules are synthesized by distinct but similar pathways where glycosyltransferases perform the sequential addition of monosaccharides onto a lipid carrier.
N. meningitidis isolates can be classified into 12 different serogroups based on their polysaccharide capsule. The majority of disease cases is caused by the members of serogroups A, B, C, W, X, and Y (2, 3). Some strains of N. meningitidis, especially those isolated from the oropharynx of healthy carriers, can also be rendered noncapsulated through regulation of the capsule production, either due to phase variation of the capsule synthesis genes (4) or due to inactivation of genes in the capsule gene cluster (cps) (5). Other strains lack the genes for capsule synthesis, modification, and transport (cps operon) (6). In these strains, the cps operon is replaced by a noncoding region (cnl region) that resembles that of other neisserial species, such as Neisseria gonorrhoeae and Neisseria lactamica. The capsule is believed to represent an advantage in invasive disease by helping the bacteria to evade complement-mediated and phagocytic killing by the host (2), but cases where noncapsulated meningococci have caused disease have also been reported (7–10). On the other hand, loss of capsule seems to enhance colonization (11).
Neisseria species express a broad spectrum of O-linked protein glycosylation (12–14). The glycoproteins identified in N. gonorrhoeae are predicted to be lipoproteins or transmembrane proteins localized in the periplasm or cell surface (13, 15). The closely related N. meningitidis species is predicted to have a similar glycoprotein repertoire. Neisseria species display high glycoform variability (12, 13, 16–18), and a simplified overview of the glycosylation pathway and the glycosyltransferases that are involved is shown in Fig. 1. The pgl core locus products function in the synthesis of undecaprenyldiphosphate (UndPP) monosaccharides (PglB/PglB2, PglC, and PglD) and in the translocation to the periplasm (PglF). PglB is a bifunctional protein with an acetyltransferase domain and a phosphoglycosyltransferase domain responsible for synthesis of N,N′-diacetylbacillosamine (diNAcBac). The variant bifunctional PglB2 protein has an ATP grasp domain responsible for synthesis of glyceramido-acetamido trideoxyhexose (GATDH). The N-terminal phosphoglycosyltransferase domains in PglB and PglB2 are, however, identical. PglA is a galactosyltransferase that acts on diNAcBac and GATDH, whereas PglE is a galactosyltransferase that elongates the PglA-generated disaccharide to a trisaccharide. PglH is a glucosyltransferase that acts on both diNAcBac and GATDH to generate glucose (Glc)-containing disaccharides. Recently, a PglH2 variant that acts on both diNAcBac and GATDH to generate GlcNAc-containing disaccharides was identified (19). In addition, PglG was characterized as a glycosyltransferase that elaborates the undecaprenyl diphosphate-linked disaccharide in Neisseria elongata subsp. glycolytica with di-N-acetyl hexuronic acid (20), but no PglG activity has been detected in pathogenic Neisseria species. The neisserial glycoforms can be further modified via O-acetylation mediated by the acetyltransferase PglI to further increase neisserial glycan diversity (see Fig. S1 in the supplemental material). To date, evidence has shown that more than 30 different glycoforms can be synthesized by combinations of glycosyltransferases and the O-acetylase within neisserial species (21).
FIG 1.
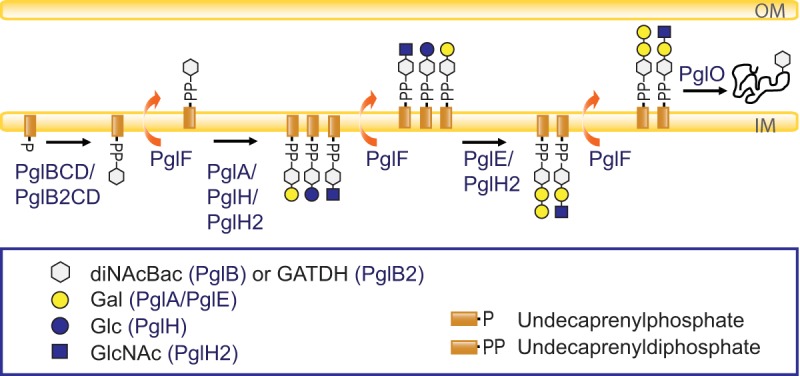
Simplified overview of the O-linked protein glycosylation pathway in Neisseria. The current model of the broad-spectrum O-linked glycosylation pathway expressed by species within the genus Neisseria is shown. OM, outer membrane; IM, inner membrane.
Whole-genome sequencing (WGS) of Neisseria isolates has revealed extensive genomic variation both in gene sequence diversity and in gene content, as a consequence of homologous recombination (22). This is also the case for pgl genes. First, there are two gross polymorphisms at the pgl loci, namely, the variable presence of pglG and pglH and the mutually exclusive presence of pglB and pglB2. The variable presence of two open reading frames (ORFs) in the pgl locus includes a putative glycosyltransferase gene, pglG, in addition to a glucosyltransferase-expressing gene, pglH. Strains lacking these two ORFs retain the first 40 bp of pglG and the last 100 bp of pglH. The pglB2 allele is found among commensal neisserial species and N. meningitidis strains but not among N. gonorrhoeae strains. Second, pglA, pglE, and pglI are absent in a significant number of commensal species, except N. lactamica. Third, polymorphisms also exist at the gene level as described for pglH and pglH2, where only one nonsynonymous mutation is accountable for the glycoform switch from Glc to GlcNAc (19). Fourth, several pgl genes contain mononucleotide or polynucleotide repeat tracts subjected to phase variation and, thus, to reversible on/off expression. Changes in the number of repeats arise by slipped-strand mispairing during DNA replication. The pgl genes have the phase-variable tract within their coding region; consequently, an off configuration results in a nonfunctional, truncated protein due to frameshifting of the correct reading frame. Differential on/off expression can then facilitate the adaptation of bacteria to fluctuations in the environment.
Genetic variation is believed to facilitate escape of the adaptive immune response. A recent study detected homologous recombination within the pgl loci through genomic analysis of 100 African serogroup A isolates representing the clonal replacement of hypervirulent meningococcal clone sequence type 7 (ST-7) by the ST-2859 descendant clone. It was suggested that this recombination event emphasizes the role of protein glycosylation diversity in immune evasion (23). It has also been shown that N. meningitidis isolates that express the antigenic invariable class II pilin display additional pilin glycosylation sites, which supports a model where these strains evade the immune system by changing their sugar structure rather than their pilin structure (24).
The isolates analyzed in this study were collected as part of a longitudinal carriage study in Ethiopia in 2014 (25). A group of individuals identified as asymptomatic carriers of N. meningitidis in a cross-sectional study were followed weekly for asymptomatic carriage in a 2-month period. Paired meningococcal carriage isolates collected 6 to 9 weeks apart from 50 individuals were analyzed by WGS. A high degree of within-host genetic changes was revealed, with genes involved in glycosylation being among those showing the highest degree of genetic variation. All these genes showed phase-variable mononucleotide differences resulting in differential on/off expression within the paired isolates. This observation led us to further characterize each of the pgl genes, as well as how their genetic variability alters the protein glycan repertoire expressed within these paired isolates.
RESULTS
Major polymorphism in pgl gene content within a small geographic area.
The whole genomes of paired N. meningitidis isolates collected approximately 2 months apart from the same asymptomatic carrier were shown to have a high degree of within-host genetic changes (25). As three genes involved in the O-linked protein glycosylation system were identified among the genes that differed in ≥30% of paired isolates, we have here extended the analysis to include all pgl genes in these 100 meningococcal carriage isolates belonging to 10 STs.
The pgl gene compositions represented in our strain collection are shown in Fig. 2. There are two known major polymorphisms within the pgl locus. First, the monosaccharide can be either diNAcBac or GATDH, depending on the presence of pglB or pglB2 alleles, respectively. Within our collection, 70 isolates, belonging to ST-35, ST-53, ST-175, ST-192, and ST-198, carried the pglB allele and thus expressed diNAcBac glycoforms, and 30 isolates, belonging to ST-11, ST-2880, ST-11372, ST-11595, and ST-11597, carried the pglB2 allele variant responsible for GATDH glycoforms (Table 1). A second form of polymorphism at the pgl locus involves the variable presence of two linked ORFs, pglG together with the pglH or the pglH2 variant. Both diNAcBac and GATDH glycoforms can be further extended by addition of Glc or GlcNAc, as determined by the presence of pglH or pglH2 variant alleles, respectively. In our collection, pglH was present in 86 isolates and pglH2 in only three ST-53 isolates, while the linked pglG and pglH ORFs were absent in all 11 isolates belonging to ST-175, ST-198, or ST-11372 (Table 1 and Fig. 2).
FIG 2.

Protein glycosylation gene variants in meningococcal carriage isolates. An overview of the pgl genes present in the collection is shown. There are two major polymorphisms documented in the pgl locus of strains of Neisseria. The configuration associated with pglG and pglH represents the ancestral form, and the other corresponds to the deleted form. Ancestral and deleted forms are found in combination with both pglB and pglB2 allele variants.
TABLE 1.
Major polymorphism in the pgl locus
| Isolate category | No. of isolates with indicated status with respect toa: |
|
|---|---|---|
| pglH and pglH2 | pglB and pglB2 | |
| Gene absent | 11 | 0 |
| Allele variant | 89 (86 + 3) | 100 (70 + 30) |
| Total | 100 | 100 |
Values in parentheses represent numbers of isolates with the indicated status with respect to the genes listed in the column headings.
The pgl locus composition was conserved within the different STs, except for ST-53, where both pglH and pglH2 variants were observed, as shown in Fig. 2. The ST-53 isolates were previously found to form two different subclusters by core genome multilocus sequence typing (cgMLST), Bayesian analysis of population structure (BAPS), and single nucleotide polymorphism (SNP) analysis (25), and our analysis revealed that one subcluster had pglH while the other had the pglH2 variant allele. In addition, pglI was absent in both ST-53 subclusters, eliminating the possibility of O-acetylation of the different glycoforms synthesized in this sequence type.
Allelic variation in the pgl genes within the paired isolates.
We compared the allelic differences in each of the pgl genes within the paired isolates (n = 46) (Table 2), excluding four pairs of isolates that previously were shown to belong to different strains. Three individuals (individuals 47, 48, and 50) carried strains with different STs at the two time points, and a fourth individual (individual 49) clearly carried different strains belonging to the same ST, ST-53 (25) (see Table S1 in the supplemental material). The three pairs with a change of ST differed in all pgl genes. In individual 49, all pgl genes but pglC and pglD differed between the two isolates. In fact, different gene compositions were observed in the isolates consecutively colonizing these four individuals, individual 47 (with or without pglG and pglH), individual 48 (with or without pglI, pglB, and pglB2), individual 49 (with pglH and pglH2) and individual 50 (with pglB and pglB2) (Table S1).
TABLE 2.
Overview of genetic changes in pgl genes in paired meningococcal carriage isolatesa
| NEIS no.b | Gene | Gene product | PV tract | % of pairsc with genetic differences (n = 46) | Mechanism(s) and proportion(s) of pairsd with genetic differences |
||||
|---|---|---|---|---|---|---|---|---|---|
| All STs (n = 46) | ST-11 (n = 5) | ST-53 (n = 4) | ST-192 (n = 23) | ST-2880 (n = 7) | |||||
| NEIS0568 | pglE | Glycosyltransferase | CAACAAAe | 60 | PV, 60% | PV, 100% | PV, 50% | PV, 50% | PV, 71% |
| NEIS0400 | pglH/H2 | Glycosyltransferase | Poly(C) | 44 | PV, 44 %; PM, 4% | PV, 60% | No changef | PV, 70%; PM,g,h 9% | PV, 14% |
| NEIS0380 | pglI | O-Acetyltransferase | Poly(G) | 41 | PV, 41 %; PM, 9% | PV, 60%; PM,h 40% | Gene absent | PV, 52% | PV, 43%; D,g,h 43% |
| NEIS0401 | pglG | Putative glycosyltransferase | Poly(C) | 33 | PV, 30 %; R, 4%; PM, 2% | PV, 40%; R, 20% | PV, 50% | PV, 70%; R,h 9%; PM,h 4% | No change |
| NEIS0213 | pglA | Glycosyltransferase | Poly(G) | 22 | PV, 20%; PM, 4%; R, 2%; D, 2% | PV, 20%; D,h 20% | No change | PV, 13% | PV, 43%; PM,g,h 29% |
| NEIS0402 | pglF | Flippase | 11 | PM, 11% | PM, 20% | PM,g 75% | No change | No change | |
| NEIS0539 | pglO | Oligosaccharyltransferase | 9 | PM, 7%; R, 2% | No change | PM, 25% | PM,g 9%; R, 4.5% | No change | |
| NEIS0399/NEIS2838 | pglB/B2 | N-Acetyltransferase/phospho-glycosyltransferase | No change | No change | No change | No change | No change | No change | |
| NEIS0397 | pglC | Aminotransferase | No change | No change | No change | No change | No change | No change | |
| NEIS0396 | pglD | Dehydratase | No change | No change | No change | No change | No change | No change | |
D, deletion; PM, point mutation; PV, phase variation; R, recombination.
The annotated genes are accessible in Genome Comparator (https://pubmlst.org/).
Data represent pairs with any genetic differences. Pairs from individuals carrying different strains at the two time points were excluded from the analysis.
In some pairs, two or more mechanisms of genetic change were observed.
ST-192 and ST-198 have different repeats as detailed in Table S5.
No PV tract.
The same in all pairs, probably due to recombination.
Close to PV tract.
The sequence was incomplete for one or both of the isolates of the pair for some genes, and those genes were therefore excluded from the analysis. Alignment and comparison of all pgl gene sequences were performed, and the mechanisms involved in the genetic differences observed were assigned as point mutations, deletions, or homologous recombinations. Table 2 shows that the genetic differences between pairs ranged from 60% to no change and that phase variation was the most frequent mechanism. Moreover, all phase-variable pgl genes showed a high degree of allelic differences due to their heptanucleotide (pglE) or mononucleotide (pglA, pglH, pglG, pglI) repeat tract variations. The heptanucleotide variation in pglE was the most extensive, with variation in 60% of the pairs. The levels of variation between pairs in the other phase-variable genes ranged from 20% to 44%. The numbers of repeats also differed between the pgl genes, where pglE had 9 to 59 heptanucleotide repeats, whereas all the other pgl genes had shorter mononucleotide repeats (8 to 19 cytosines or guanosines) (Fig. 3). The largest switches within pairs were observed in the pglE gene of individuals 8, 23, and 34.
FIG 3.

Phase variation of pgl genes within paired isolates. The variations in the lengths of the phase-variable tracts between all paired isolates (n = 46) are represented by a line for each of the pgl genes. The poly(C) tract in pglG ranges between 8 Cs and 15 Cs for all individuals, with the largest change seen in individuals 6 and 7. The poly(C) tract in pglH/H2 ranges between 9 Cs and 16 Cs for all individuals, with the largest change seen in individual 33. Individuals 28, 29, 32, 34, and 42 lacked the poly(C) tract in pglH (which has only three cytosines in this position). The poly(G) tract in pglA ranges between 9 Gs and 15 Gs, with the largest change seen in individual 2. The poly(G) tract in pgI ranges between 9 Gs and 19 Gs for all individuals. The heptanucleotide phase-variable tract in pglE has between 9 and 59 repeats, with the largest change seen in individuals 8, 23, and 34. The line between the two isolates is horizontal for identical repeat numbers. Pairs where the sequence(s) of one or both isolates was incomplete are indicated at the bottom, along the x axis. Note that a missing line for pglG, pglH, or pglI within an individual indicates the absence of the gene.
The pgl core locus genes pglB/B2, pglC, and pglD, responsible for synthesis of monosaccharide, were the most highly conserved pgl genes, with no genetic changes between the paired isolates (Table 2).
Polymorphisms within and adjacent to phase-variable repeat tracts.
In addition to the phase variation mechanism accountable for many of the genetic differences between pairs, there were additional polymorphisms (mechanism assigned as point mutation, deletion, or homologous recombination) observed around the phase-variable regions of pglG, pglH, pglI, and pglA (Table 2).
There were some differences between and within STs adjacent to the polyguanosine [poly(G)] tract in pglA, while the flanking sequences were highly similar (see Fig. S2 in the supplemental material). One of the nine isolates had a deletion of 13 nucleotides just prior to the poly(G) tract in ST-11, and this deletion was not observed in isolates from any of the other STs. Both ST-35 isolates had a guanosine (G)-to-adenine (A) point mutation in the middle of the poly(G) tract, thus interrupting the mononucleotide repeat of the pglA gene and presumably resulting in constitutive PglA expression. Two subjects (individuals 15 and 44) carrying ST-2880 isolates had a two-nucleotide deletion upstream of the poly(G) tract at the second time point.
In ST-2880 isolates, we observed an AGG deletion 3′ to the poly(G) tract of pglI for both time points for individuals 15 and 17 and at the second time point for individuals 2, 5, and 10 (Fig. S3).
The pglH locus showed polymorphisms between STs along the whole gene (Fig. S4). A 5′ point mutation (guanosine to cytosine) adjacent to the polycytosine [poly(C)] tract was found in individuals 20, 22, and 33 carrying ST-192 isolates.
The pglE gene contained the heptanucleotide CAACAAA repeat for most isolates, except the ST-198 isolates and some of the isolates within ST-192 (Fig. S5). Most of the ST-192 isolates (46/48) had a longer sequence of CAACAAA repeats together with one or two repeat variants (CAATAAA) included in the same tract, possibly representing a mechanism that stabilizes the phase-variable region. One ST-192 isolate had a CACCCGA repeat sequence variant. In ST-198, there was also an alternative heptanucleotide repeat unit, CACCCAA.
Homologous recombination in pgl genes.
Several studies has shown that the pgl locus is subject to high genetic variation due to homologous recombination. Although the isolates used in this study were sampled only 6 to 9 weeks apart, homologous recombination was observed in a variety of genes. In individual 40, the pglO gene had two nucleotide substitutions within a 26-bp region (Fig. S6), and in individuals 7 and 46, blocks of nucleotides 3′ to the poly(C) region were changed in the pglG gene (Fig. S7).
Some of the changes observed in close proximity to the phase-variable tracts were probably also due to homologous recombination events since they were identical in different isolate pairs. An identical two-nucleotide difference was observed 5′ in pglA of ST-2880 isolates in individuals 15 and 44. In contrast, a two-nucleotide insertion in the pglA gene was seen 3′ in ST-11372 isolates of individual 26 (Fig. S2). We also observed a two-nucleotide insertion in pglH 5′ to the poly(C) in individuals 20, 22, and 33 carrying ST-192 isolates (Fig. S4) and in pglI for individuals 2, 5, and 10 (ST-2880 isolates) and 11 (ST-11 isolates) (Fig. S3). Altogether, these data indicate that the phase-variable pgl gene regions are subjected to both phase variability and genetic variation likely introduced by homologous recombination. Furthermore, we observed the same point mutation within the pglF gene in three of four paired ST-53 isolates and within pglO in four (9%) of paired ST-192 isolates. All these paired isolates within STs had the exact same variation between the two sampling points, thus probably excluding the possibility of a random insertion/deletion.
Phase variation of pgl genes and their impact on protein expression.
The pgl genes contain mononucleotide or polynucleotide repeat tracts within their coding regions. A change in the number of repeats is reversible, and these genes can therefore change between functional protein expression (on configuration) and nonfunctional protein expression because of premature stop codons and truncation in the noncoding reading frame (off configuration). For all phase-variable genes, sequence alignments and assessments of the homopolymeric tracts were performed (Fig. 3), and the status of each phase-variable gene (on/off configuration) is summarized based both on paired isolates (Fig. 4A; pairs, n = 46) and on all isolates (Fig. 4B; isolates, n = 100). This analysis revealed that all isolates, except two ST-35 isolates, had poly(G) tracts in pglA and that the lengths ranged between 9 and 15 Gs. pglA genes without poly(G) tracts are constitutively in an on configuration. For pglA, 33 isolates had the on configuration while 59 isolates were off; furthermore, both isolates were on in 12 pairs, both isolates were off in 24 pairs, and a change of poly(G) tract length resulted in a switch in on/off status in five pairs. The pglA gene was turned off in two of these individuals, while the pglA gene was turned on in three individuals.
FIG 4.

Phase-variable pgl genes. Overviews of the status of phase-variable pgl genes (on/off configurations) for paired isolates (n = 46) (A) and individual isolates (n = 100) (B). The distributions of absent genes, incomplete sequences, on configurations, and off configurations are shown for pglA, pglE, pglI, pglG, and pglH. In addition, pglA and pglH have some alleles without the phase-variable tract and these are constitutively on (No PV tract) (i.e., no phase variation tract).
All of the pglE alleles contained heptanucleotide repeat tracts with various lengths. We observed between 9 and 59 repeat units. However, the Illumina technology is not capable of covering these long repetitive sequences; therefore, we could not determine the on/off status of this gene by WGS. The pglE gene was therefore PCR amplified and analyzed by Sanger sequencing in all isolates. Comparing the Sanger and Illumina MiSeq sequencing data, the results were consistent only for the shorter pglE repeat tracts (17% of the isolates). The Sanger analysis revealed that for pglE, 41 isolates (10 pairs) had the on configuration while 56 (16 pairs) were off. Among the paired isolates, 27 had a change of heptanucleotide tract length and 18 of these resulted in a switch in on/off status. Interestingly, the pglE genes were switched from on to off in 6 individuals, whereas this gene was switched from off to on in 12 individuals.
The pglI gene was absent in 11 isolates, all belonging to ST-53. The pglI alleles were all phase variable, containing poly(G) tracts with 9 to 19 Gs. In this collection, 35 isolates (10 pairs) had the pglI gene switched on and 53 (20 pairs) had this gene switched off, and there was a change in the on/off configuration in 10 of the paired isolates. Here, we observed equal numbers of changes of configuration in the two directions.
The pglG and pglH insertion was variably present in the isolates as detailed above. Although pglG is not shown to be functional in meningococci, the phase-variable poly(C) tract consists of 8 to 15 cytosines (Cs) and was presumably in the on and off configurations in 55 and 20 isolates (20 and 5 pairs), respectively. Fourteen of the paired isolates had a change of poly(G) tract length, and 7 of these resulted in a switch in predicted on/off status. The pglG gene was turned off in five of these individuals, and the pglG gene was turned on in two individuals. The pglH gene lacked the phase-variable tract in 14 isolates. These were isolates within ST-53, ST-11595, and ST-11597. Of the 75 strains that had the homopolymeric poly(C) tract (9 to 16 Cs), there were changes in 20 of the pairs; 31 isolates (12 pairs) had pglH on and 43 (12 pairs) had pglH off. We observed an on-to-off switch in 16 of the pairs, and there were equal numbers of changes of configuration in the two directions.
The glycosylation phenotype revealed a high degree of microheterogeneity.
All isolates were screened for the protein glycosylation phenotype using immunoblotting with glycan-specific antibodies. For strains carrying pglB, we used glycan-specific monoclonal antibodies (MAbs) (npg1, npg2, and npg3), polyclonal antibody pDAb2, and the lectin wheat germ agglutinin (WGA) which together recognize all known diNAcBac-based glycoforms. The strains with pglB2 could not be analyzed in such detail, as only pDAb2/WGA and npg3 can recognize both diNAcBac- and GATDH-based pglH/H2 disaccharides and trisaccharides, respectively. From the immunoblots, we assigned and scored reactivity for each strain as no reactivity (score of 0), low reactivity (score of 1), moderate reactivity (score of 2), or high reactivity (score of 3). We scored no reactivity (0) when the level of reactivity was comparable to that seen with the glycosylation null mutant and high reactivity (3) when it was comparable to that seen with the positive glycosylation mutants that have previously been well characterized (12, 19). We also summarized the scoring for all immunoblots to compare the overall levels of observed glycosylation in these strains. An overview of all phenotypic glycosylation results is shown in Table S1 (see also Fig. 6) together with the genotypic data for these isolates.
FIG 6.

Core genome MLST, pgl genotype, and protein glycosylation phenotype. An overview of phylogenetic relationships together with genotypic and phenotypic results for this strain collection is presented. The genotypic data include the allelic numbers assigned by the PubMLST genome comparator protein glycosylation typing scheme and the on/off configuration of the phase-variable genes. The color codes are shown in the figure legend. Not determined, incomplete sequence, analysis not performed, or not possible to evaluate. All details presented in this figure are listed in Table S1.
Analysis and interpretation of the glycosylation phenotype can be complicated, and as an example, we present all details from the immunoblots with selected pairs of representative ST-192 isolates in Fig. 5. ST-192 isolates have the pglB allele, and immunoblotting showed variable reactivity with all antibodies investigated. Overall, the ST-192 isolates displayed a high degree of glycan microheterogeneity, as most isolates (67%) reacted with two or more antibodies (Table S1). As shown in Fig. 5, changes in glycosylation patterns of the paired isolates were observed between the two different time points as a consequence of phase variation in individuals 23, 40, 33, 24, 25, and 35. For individual 23, an off-to-on configuration switch in both pglA and pglH changed the synthesis from mainly diNAcBac (npg1) to both pglA disaccharide diNAcBac-Gal (npg2) and pglH disaccharide diNAcBac-Glc (pDAb2). In isolates from individual 40, pglH was turned off and the synthesis changed from diNAcBac-Glc (pDAb2) to diNAcBac (npg1). For individual 33, pglA was turned on and pglH off and synthesis subsequently changed from mainly diNAcBac-Glc (pDAb2) to diNAcBac-Gal (npg2). In individual 24, both pglE and pglH were turned on and both diNAcBac-Gal-Gal (npg3) and diNAcBac-Glc (pDAb2) appeared at a later time point. In individual 25, pglA was turned off and a change from diNAcBac-Gal (npg2) to diNAcBac (npg1) was subsequently seen. In addition, pglE was turned on. However, since pglA was off in this isolate, there were only small amounts of the trisaccharide diNAcBac-Gal-Gal (npg3). For individual 35, an on-to-off configuration switch in pglI increased the detection of pglA disaccharide diNAcBac-Gal (npg2), pglH disaccharide diNAcBac-Glc (pDAb2), and trisaccharide diNAcBac-Gal-Gal (npg3). Forty percent of the ST-192 isolates had npg2 reactivity although pglA was in an off configuration. This result can be explained by the presence of some degree of expression through transcriptional slippage such that a small amount of transcript was made in an on configuration and thus translated into a small amount of functional PglA proteins. This phenomenon has previously been observed in pglA alleles from other strains (18). Although they were less abundant, we also observed npg3 expression in 33% and 6% of isolates that had pglE off and pglH off, respectively (Table S1).
FIG 5.

Protein glycosylation in ST-192 isolates. Data represent the reactivity of endogenous glycoproteins following immunoblotting with the glycan-specific monoclonal antibodies npg1 (specific for diNAcBAc) (A), npg2 (specific for diNAcBAc-Gal) (B), and npg3 (specific for both diNAcBac and GATDH trisaccharides) (C) and pDAb2 polyclonal antibody (specific for diNAcBac-Glc) (D). The strains used were as follows: lane 1, KS104 (pglC; negative control with no glycan synthesized); lane 2, positive glycan controls, including KS141 (N400 pglA) (A), KS100 (N400) (B), KS142 (N400 pglEon) (C), and KS966 (pglA pglI lct::pglH2SK-03-1035) (D); lanes 3 to 18, N. meningitidis ST-192 isolates from time points A and B as indicated. The pglA, pglE, pglH, and pglI genotype configurations for each isolate are shown below the immunoblots.
Although half of the isolates had pglH on, only five of these had pDAb2 reactivity (Fig. 5). In particular, isolates 45B, 1A, and 1B had no competing glycosyltransferases for PglH and were therefore expected to synthesize PglH disaccharide. The overall glycosylation level appeared to be low, suggesting either that there was impaired glycosylation in these isolates or that the glycoforms expressed were not recognized in our analysis. Further genetic, phenotypic, and structural characterization is needed to be able to understand the discrepancies.
Correlation between pgl genotype and glycosylation phenotype.
There was not a complete correlation between glycosylation genotype and phenotype in the ST-192 isolates described above (Fig. 5) or in the other isolates (summarized in Fig. 6 and Table S1). Figure 6 contains phylogeny data together with genotypic and phenotypic results for this strain collection. The STs are indicated by different colors and are in agreement with the clustering of the cgMLST tree. The genotypic data include the allelic numbers assigned by the PubMLST genome comparator typing scheme and the on/off configuration of the phase-variable genes. The alleles are colored with the same code as the STs of the dominant allele within the subgroup, while all other alleles are randomly colored. By using this approach, the color patterns demonstrated that the alleles were highly associated with the STs and cgMLST. Another striking observation is the number of allele variants in the phase-variable genes (pglA, pglI, pglE, pglG, pglH). This was predominantly due to variations in repeat numbers rather than to other nucleotide differences.
The O-linked glycosylation system is at present characterized to such an extent that we can make justified hypotheses concerning the nature of the molecular mechanisms behind most divergences. The phenotypic data include the scoring of antibody reactivity as described above.
In addition, we have interpreted the genotypic and phenotypic data and added table columns corresponding to glycoform (diNAcBac or GATDH based), microheterogeneity (two or more glycoforms present), phase-variable slippage (expression from the off configuration), and correlation between genotypic and phenotypic data. Microheterogeneity was observed in 67% of the isolates and transcriptional slippage in 49% of the isolates with diNAcBAc glycoforms. In examining the level of correlation between genotypic and phenotypic data, we included transcriptional slippage as an accepted mechanism involved in this system and found correlations in 54% of the diNAcBac-synthesizing isolates. The remaining isolates had very low (level 0 to 2) reactivity with the glycan-specific antibodies and/or glycosyltransferases that did not show any detectable activity. The lack of typing reagents for GADTH makes it difficult to determine the levels of microheterogeneity, transcriptional slippage, and correlation in the remaining isolates.
DISCUSSION
WGS analysis revealed a high degree of within-host genetic changes in meningococcal isolates taken 6 to 9 weeks apart, and phase-variable pgl genes were among the genes showing the highest degree of genetic variation (25). This study was intended to unravel the degree of direct conformity between the pgl genotype and the glycosylation phenotype, as well as the level of glycan complexity expressed by this system. That has, to our knowledge, been done for only a very few Neisseria strains. Our isolate collection included all known pgl locus variants identified to date. As the events leading to the major pgl locus variants are believed to have occurred in one location and then spread (26), finding all known variants in a small geographic area in a rural/semirural part of Ethiopia therefore underlines the global spread of the meningococcus and its genes. In addition, the correlation between the pgl genotype and protein glycosylation is complicated by allele polymorphisms and phase variation. Together, these observations pointed to a more thorough investigation of the pgl genotype and protein glycosylation within these isolates.
Analyzing paired carriage isolates gives insight into genetic evolution within the host in short time periods. Genetic and phenotypic changes in the meningococcus have been shown to happen over a very short time in vivo, directly impacting the adaptation of the bacterium to evade the host immune system and to increase its virulence (27). The accidental human passage of a meningococcal strain acquired in the laboratory showed that both pglA and pglI were turned off in the patient whereas they were on in the parental strain. Minor variation in sequence repeat tract leads to entirely altered glycoform expression, resulting in that case in a change from diNAcBac-Gal-AcGal to diNAcBac-Glc expression after human passage of the isolate. Those authors proposed that this change, among others, could confer an advantage to the bacteria to escape the immune system (27).
Phase variation was the predominant mechanism accounting for the genetic differences observed between pairs for all phase-variable pgl genes. The phase variation in pglE was the most extensive, with variation in 60% of the pairs compared to 20% to 44% of the pairs for the other pgl loci. The expansions and extensions were also more extensive in pglE than in the other pgl loci. The heptanucleotide repeat and the longer phase-variable tracts presumably make pglE more unstable, as phase variation rates has been believed to increase proportionally with the tract length. The frequencies of phase variation in the pgl genes, in vitro and in vivo, have not been studied. However, for other phase-variable genes in the meningococcus, such as hemoglobin-binding protein genes hpuAB and hmbR, the in vitro switching rates seen under conditions of growth on agar plates ranged from ∼10−6 to 10−2 CFU−1 (28) and also differed between different serogroups (29). In our study, we did not investigate phase variation during culturing and therefore cannot exclude the possibility that some of the changes observed happened in vitro during processing of the samples. Other possibilities are that multiple phase variants exist in vivo and that the changes do not represent differences over time but rather represent natural selection in vivo of the dominant bacteria colonizing the host. Phase variation during isolation and culture has been investigated in hpuAB and hmbR, using contemporaneously acquired clinical specimens (blood/cerebrospinal fluid), and a protocol for determining in vivo phase variation status has been developed (30). Using such a method to investigate the pgl genes would be highly interesting.
Recombination events were found within paired meningococcal carriage isolates in pglG, pglA, and pglO. In addition, several paired isolates within one ST had exactly the same variation between the two sampling points in pglA, pglH, pglI, and pglF, which was thus likely introduced by homologous recombination. Several studies have detected homologous recombination within the pgl loci, emphasizing a role of protein glycosylation diversity in immune evasion (23, 31). Krauland et al. compared genomes of serogroup Y ST-23 clonal complex early and late strain types and hypothesized that emergence of a late strain type was primarily due to antigenic changes that allowed escape from population immunity. They found that genes exhibiting differences included antigenic outer membrane protein genes; genes involved in iron acquisition and uptake; and genes involved in pilus structure, function, and glycosylation (31). However, the phenotypic outcomes of the observed genetic pgl gene differences were not investigated in any of those studies.
Information on gene content, polymorphisms, and phase variation is crucial to be able to predict glycosylation phenotype and glycan diversity. To date, we have known that a meningococcal strain has the capacity to express between 7 and 21 different glycans. Protein glycosylation genotype and phenotype correlations have been difficult within this system because glycans are not directly encoded by the genome but are enzymatic products that are somewhat more difficult to predict. This is further complicated by the fact that Neisseria exhibits a high degree of polymorphism both at the gene content level and with regard to polymorphic glycosyltransferases. In addition, the neisserial protein glycans can be subject to the phenomenon of microheterogeneity (12, 17), in which variant glycan structures are found at specific attachment sites of a given glycoprotein. Even though the study was somewhat hampered by the lack of a complete set of antibodies to recognize all GADTH glycoforms, we were able to study the glycan diversity. ST-11 isolates in our collection express antigenic invariable class II pilin. Gault et al. suggested that N. meningitidis isolates with a class II pilin display additional pilin glycosylation sites and proposed a model where these strains evade the immune system by changing their sugar structure rather than their pilin structure (24). We were not able to investigate the glycosylation phenotype in ST-11 isolates in detail here since they synthesize GATDH glycoforms, but the genotype indicates that these isolates have the capacity to express up to 21 different glycans. Additional pilin glycosylation sites would therefore also increase the magnitude of microheterogeneity in ST-11 isolates.
Extensive acetylation complexity and microheterogeneity of pgl glycoform O-acetylation by PglI were recently characterized using mass spectrometry (21). In this study, we screened the isolates using immunoblotting, and the complexed acetylation pattern therefore cannot be depicted. The functional implications of pgl oligosaccharide O-acetylation are currently unknown, but the O-acetylation has been shown to alter pgl glycan antigenicity. These studies involved both immunoblotting and immunogold labeling with glycoform-specific MAbs, and the altered reactivity was suggested to have been caused by the acetyl group masking the recognition epitope present on sugar moieties (12). This can be difficult to evaluate in single isolates, as the levels of glycosylation also differ due to the level of activity expressed by each of the involved glycosyltransferases. For individual 35, however, the paired isolates shared the same pgl genotype, with the exception of pglI, and we observed an overall increase in reactivity at time point B that can be explained only by the pglI off configuration. In ST-53 isolates, the number of possible glycoforms is only five since isolates within this sequence type do not have the pglI gene responsible for O-acetylation of the glycans. The majority of pathogenic Neisseria isolates and some commensal Neisseria species (N. lactamica, N. mucosa, and N. polysaccharea) have the pglI gene, while it is missing in the related distal commensal Neisseria species such as N. cinerea, N. flavescens, N. subflava, N. sicca, and N. elongata (32). Interestingly, the two species of pathogenic Neisseria have multiple, dedicated O-acetylation systems targeting phosphatidylglycerol (PG), lipooligosaccharides (LOS), and, in the case of N. meningitidis, capsule polysaccharides (CPS) (33–35). The pglI gene is also predicted to be subject to phase-variable expression in meningococci and N. lactamica but not in gonococci and other commensal species (32).
The number of genes with allelic differences between isolates from different individuals within the same ST was about twice as high as the number of differences between paired isolates from the same carrier (25). A recent study looking at within-host evolution by genomic comparison of throat and blood strain pairs from four patients with meningococcal disease also found changes in pilE, modA, and pglI (36), which were also among the most frequently changed genes in our previous study. Those authors hypothesized that genomic variants arise during carriage and that invasive variants occasionally emerge and cause disease (36). The role of glycosylation in meningococcal pathogenesis has not been resolved, and thus, neither has the influence of phase variation in pgl genes on disease and possibly on vaccine immune responses. Further characterization employing genetic manipulations and mass spectrometry as used in earlier studies (17, 19) will most likely further add to the data showing high glycan variability predicted from the genotype. We suspect that such an outcome is likely since a high number of strains showed low reactivity to the glycan-specific antibodies and since only a restricted number of N. meningitidis strains have been investigated for the glycosylation phenotype thus far. Either new functions assigned to already-characterized glycosyltransferases or novel glycosyltransferases might conceivably be identified. Another plausible explanation for the low level of glycosylation is the competition for substrate between the glycosyltransferases (PglA/PglH or PglH/PglE competition) as previously documented (17, 21).
Here we have shown that glycoform diversity is caused by pgl gene variation, polymorphism, and phase variation and that the glycoproteins can be subject to the phenomenon of microheterogeneity. Taking the data together, the complexity of the O-linked protein glycosylation system requires further studies to understand how N. meningitidis utilizes the high variation in pgl gene content to produce high glycoform diversity and to evade the human immune response in both carriage and disease.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
The isolates were obtained from a cross-sectional meningococcal carriage study conducted among healthy 1- to 29-year-old individuals in the Arba Minch area in southern Ethiopia in 2014 (37), where a subgroup of individuals identified as asymptomatic carriers were followed using repeated (weekly) collection of oropharyngeal samples over a period of 9 weeks as previously described (25). All control bacterial strains used in this study were N. gonorrhoeae (see Table S2 in the supplemental material). Isolates were grown overnight on Colombia agar base plates (Oxoid Ltd., Basingstoke, United Kingdom) containing 5% defibrinated horse blood (TCS Biosciences Ltd., Buckingham, United Kingdom) at 37°C in an atmosphere of 5% CO2.
Next-generation sequencing.
Genomic DNA was extracted using an automated MagNAPure isolation station and a MagNAPure 96 DNA and viral neuraminidase (NA) small-volume kit (Roche, Basel, Switzerland), according to the manufacturer's instructions. The sequencing libraries were made using a Kapa HyperPlus kit (Kapa Biosystems, Wilmington, MA); sequencing (500 cycles) was performed on an Illumina MiSeq platform with MiSeq Reagent kits (v2; Illumina Inc., San Diego, CA) with 250-bp paired-end run modes; and the reads were subsequently trimmed, filtered, and assembled as previously described (25).
Sanger sequencing.
The pglE gene in all isolates was PCR amplified using primers BP009 (TCGARAGTTATTTGGAAGCGTGT) and BP010 (TATTMCACGCCGAACCCGGA). The PCR products were sequenced using a BigDye Terminator v1.1 cycle sequencing kit and an ABI 3730 DNA analyzer (Applied Biosystems) according to the manufacturer's recommendation.
Genome comparison, pgl genotype, and phylogenetic analyses.
Genomes were uploaded to and are available from the PubMLST.org database (http://pubmlst.org/neisseria/) (Table S1), which is served by the Bacterial Isolate Genome Sequence Database (BIGSdb) platform (38). For pgl allelic comparisons, gene-by-gene analyses were performed using the Genome Comparator tool and the glycosylation scheme within the PubMLST website. For core genome MLST analysis, the 1,605 loci defined as the core genome in the database (N. meningitidis cgMLST v1.0) were used (39). Incomplete loci were removed from individual pairs prior to calculations. Distance matrices based on the allelic differences were created using the Neighbor-net method (40). To visualize the phylogenetic network involving all 100 isolates from the 50 individuals, SplitsTree4 v4.14.4 was used (41).
For determinations of the mechanisms of differences between paired isolates, sequences were uploaded, aligned, and compared manually in MEGA7 (42). Mechanisms were classified as representing point mutations where only a single nucleotide difference was present, as recombinations where multiple nucleotide differences were present in the same area, and as phase variations where various lengths of repeat nucleotides were seen, and deletions or insertions were determined in relation to the majority of isolates. In cases where alleles were incomplete or missing in BIGSdb, missed gene sequences were retrieved to some extent in performing BLAST searches (43). For determination of the on/off configuration for the phase-variable genes, the sequenced were translated and compared manually. To confirm that the observed differences in isolate pairs around the pgl locus were supported by the raw data rather than being artifacts introduced by spurious assemblies, we mapped raw FASTQ reads back to previously produced contigs using BWA (44), converted the output to the BAM format (45), and manually inspected the pileup of reads around the observed differences using the program artemis v.16.0.11 (46). A read was considered to span a repeat tract if it contiguously spanned the entire tract and was anchored on each side of the repeat region with at least five nonambiguous, nonrepetitive nucleotides. The repeat number of a region was called by simple majority, i.e., was supported by at least 50% of all reads spanning the repeat tract. Additionally, a minimum of two reads had to span the repeat tract.
SDS-PAGE and immunoblotting.
The procedures used for SDS-PAGE and immunoblotting have been described previously (47). Whole-cell lysates were prepared from equivalent numbers of cells by heating cell suspensions at 65°C for 10 min in SDS sample loading buffer. Immunoreactive proteins were detected by immunoblotting by using glycan-specific monoclonal antibodies npg1, npg2, and npg3 (12); the rabbit polyclonal antibody pDAb2 (17); and an alkaline phosphatase-coupled goat anti-rabbit secondary antibody (Sigma). To identify glycoproteins bearing a terminal GlcNAc, alkaline phosphatase-conjugated succinylated wheat germ agglutinin (sWGA) lectin (EY Labs) was used as described previously (19, 48).
Data availability.
The sequences generated and analyzed during the current study are available at the pubMLST.org website (see identification numbers in Table S1).
Supplementary Material
ACKNOWLEDGMENT
We highly appreciate the generous contribution of glycan-specific antibodies and pgl strains by Mike Koomey. We are very grateful to Ingerid Ørjansen Kirkeleite and Nadia Debech for performing the sequencing analysis at the Norwegian Institute of Public Health (NIPH). We thank all the study participants and the study staff in Ethiopia for being part of the study and for sampling.
This project was supported by the Research Council of Norway (grant 220829 to D.A.C.). This publication made use of the Neisseria Multi Locus Sequence Typing website (https://pubmlst.org/neisseria/) developed by Keith Jolley and sited at the University of Oxford (38). The development of this site has been funded by the Wellcome Trust and European Union.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00794-17.
For a commentary on this article, see https://doi.org/10.1128/JB.00316-18.
REFERENCES
- 1.Trotter CL, Greenwood BM. 2007. Meningococcal carriage in the African meningitis belt. Lancet Infect Dis 7:797–803. doi: 10.1016/S1473-3099(07)70288-8. [DOI] [PubMed] [Google Scholar]
- 2.Rosenstein NE, Perkins BA, Stephens DS, Popovic T, Hughes JM. 2001. Meningococcal disease. N Engl J Med 344:1378–1388. doi: 10.1056/NEJM200105033441807. [DOI] [PubMed] [Google Scholar]
- 3.Stephens DS, Greenwood B, Brandtzaeg P. 2007. Epidemic meningitis, meningococcaemia, and Neisseria meningitidis. Lancet 369:2196–2210. doi: 10.1016/S0140-6736(07)61016-2. [DOI] [PubMed] [Google Scholar]
- 4.Hammerschmidt S, Muller A, Sillmann H, Muhlenhoff M, Borrow R, Fox A, van Putten J, Zollinger WD, Gerardy-Schahn R, Frosch M. 1996. Capsule phase variation in Neisseria meningitidis serogroup B by slipped-strand mispairing in the polysialyltransferase gene (siaD): correlation with bacterial invasion and the outbreak of meningococcal disease. Mol Microbiol 20:1211–1220. doi: 10.1111/j.1365-2958.1996.tb02641.x. [DOI] [PubMed] [Google Scholar]
- 5.Dolan-Livengood JM, Miller YK, Martin LE, Urwin R, Stephens DS. 2003. Genetic basis for nongroupable Neisseria meningitidis. J Infect Dis 187:1616–1628. doi: 10.1086/374740. [DOI] [PubMed] [Google Scholar]
- 6.Claus H, Maiden MC, Maag R, Frosch M, Vogel U. 2002. Many carried meningococci lack the genes required for capsule synthesis and transport. Microbiology 148:1813–1819. doi: 10.1099/00221287-148-6-1813. [DOI] [PubMed] [Google Scholar]
- 7.Hoang LM, Thomas E, Tyler S, Pollard AJ, Stephens G, Gustafson L, McNabb A, Pocock I, Tsang R, Tan R. 2005. Rapid and fatal meningococcal disease due to a strain of Neisseria meningitidis containing the capsule null locus. Clin Infect Dis 40:e38–e42. doi: 10.1086/427875. [DOI] [PubMed] [Google Scholar]
- 8.Xu Z, Zhu B, Xu L, Gao Y, Shao Z. 2015. First case of Neisseria meningitidis capsule null locus infection in China. Infect Dis (Lond) 47:591–592. doi: 10.3109/00365548.2015.1010228. [DOI] [PubMed] [Google Scholar]
- 9.Vogel U, Claus H, von Muller L, Bunjes D, Elias J, Frosch M. 2004. Bacteremia in an immunocompromised patient caused by a commensal Neisseria meningitidis strain harboring the capsule null locus (cnl). J Clin Microbiol 42:2898–2901. doi: 10.1128/JCM.42.7.2898-2901.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Findlow H, Vogel U, Mueller JE, Curry A, Njanpop-Lafourcade BM, Claus H, Gray SJ, Yaro S, Traore Y, Sangare L, Nicolas P, Gessner BD, Borrow R. 2007. Three cases of invasive meningococcal disease caused by a capsule null locus strain circulating among healthy carriers in Burkina Faso. J Infect Dis 195:1071–1077. doi: 10.1086/512084. [DOI] [PubMed] [Google Scholar]
- 11.Tzeng YL, Thomas J, Stephens DS. 2016. Regulation of capsule in Neisseria meningitidis. Crit Rev Microbiol 42:759–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Børud B, Aas FE, Vik A, Winther-Larsen HC, Egge-Jacobsen W, Koomey M. 2010. Genetic, structural, and antigenic analyses of glycan diversity in the O-linked protein glycosylation systems of human Neisseria species. J Bacteriol 192:2816–2829. doi: 10.1128/JB.00101-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vik A, Aas FE, Anonsen JH, Bilsborough S, Schneider A, Egge-Jacobsen W, Koomey M. 2009. Broad spectrum O-linked protein glycosylation in the human pathogen Neisseria gonorrhoeae. Proc Natl Acad Sci U S A 106:4447–4452. doi: 10.1073/pnas.0809504106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ku SC, Schulz BL, Power PM, Jennings MP. 2009. The pilin O-glycosylation pathway of pathogenic Neisseria is a general system that glycosylates AniA, an outer membrane nitrite reductase. Biochem Biophys Res Commun 378:84–89. doi: 10.1016/j.bbrc.2008.11.025. [DOI] [PubMed] [Google Scholar]
- 15.Anonsen JH, Vik A, Egge-Jacobsen W, Koomey M. 2012. An extended spectrum of target proteins and modification sites in the general O-linked protein glycosylation system in Neisseria gonorrhoeae. J Proteome Res 11:5781–5793. doi: 10.1021/pr300584x. [DOI] [PubMed] [Google Scholar]
- 16.Aas FE, Vik A, Vedde J, Koomey M, Egge-Jacobsen W. 2007. Neisseria gonorrhoeae O-linked pilin glycosylation: functional analyses define both the biosynthetic pathway and glycan structure. Mol Microbiol 65:607–624. doi: 10.1111/j.1365-2958.2007.05806.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Børud B, Viburiene R, Hartley MD, Paulsen BS, Egge-Jacobsen W, Imperiali B, Koomey M. 2011. Genetic and molecular analyses reveal an evolutionary trajectory for glycan synthesis in a bacterial protein glycosylation system. Proc Natl Acad Sci U S A 108:9643–9648. doi: 10.1073/pnas.1103321108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Johannessen C, Koomey M, Børud B. 2012. Hypomorphic glycosyltransferase alleles and recoding at contingency loci influence glycan microheterogeneity in the protein glycosylation system of Neisseria species. J Bacteriol 194:5034–5043. doi: 10.1128/JB.00950-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Børud B, Anonsen JH, Viburiene R, Cohen EH, Samuelsen AB, Koomey M. 2014. Extended glycan diversity in a bacterial protein glycosylation system linked to allelic polymorphisms and minimal genetic alterations in a glycosyltransferase gene. Mol Microbiol 94:688–699. doi: 10.1111/mmi.12789. [DOI] [PubMed] [Google Scholar]
- 20.Anonsen JH, Vik A, Børud B, Viburiene R, Aas FE, Kidd SW, Aspholm M, Koomey M. 2016. Characterization of a unique tetrasaccharide and distinct glycoproteome in the O-linked protein glycosylation system of Neisseria elongata subsp. glycolytica. J Bacteriol 198:256–267. doi: 10.1128/JB.00620-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Anonsen JH, Børud B, Vik A, Viburiene R, Koomey M. 1 September 2017. Structural and genetic analyses of glycan O-acetylation in a bacterial protein glycosylation system: evidence for differential effects on glycan chain length. Glycobiology doi: 10.1093/glycob/cwx032. [DOI] [PubMed] [Google Scholar]
- 22.Kong Y, Ma JH, Warren K, Tsang RS, Low DE, Jamieson FB, Alexander DC, Hao W. 2013. Homologous recombination drives both sequence diversity and gene content variation in Neisseria meningitidis. Genome Biol Evol 5:1611–1627. doi: 10.1093/gbe/evt116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lamelas A, Harris SR, Roltgen K, Dangy JP, Hauser J, Kingsley RA, Connor TR, Sie A, Hodgson A, Dougan G, Parkhill J, Bentley SD, Pluschke G. 2014. Emergence of a new epidemic Neisseria meningitidis serogroup A clone in the African meningitis belt: high-resolution picture of genomic changes that mediate immune evasion. mBio 5:e01974-14. doi: 10.1128/mBio.01974-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gault J, Ferber M, Machata S, Imhaus AF, Malosse C, Charles-Orszag A, Millien C, Bouvier G, Bardiaux B, Pehau-Arnaudet G, Klinge K, Podglajen I, Ploy MC, Seifert HS, Nilges M, Chamot-Rooke J, Dumenil G. 2015. Neisseria meningitidis type IV pili composed of sequence invariable pilins are masked by multisite glycosylation. PLoS Pathog 11:e1005162. doi: 10.1371/journal.ppat.1005162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bårnes GK, Brynildsrud OB, Børud B, Workalemahu B, Kristiansen PA, Beyene D, Aseffa A, Caugant DA. 2017. Whole genome sequencing reveals within-host genetic changes in paired meningococcal carriage isolates from Ethiopia. BMC Genomics 18:407. doi: 10.1186/s12864-017-3806-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kahler CM, Martin LE, Tzeng YL, Miller YK, Sharkey K, Stephens DS, Davies JK. 2001. Polymorphisms in pilin glycosylation locus of Neisseria meningitidis expressing class II pili. Infect Immun 69:3597–3604. doi: 10.1128/IAI.69.6.3597-3604.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Omer H, Rose G, Jolley KA, Frapy E, Zahar JR, Maiden MC, Bentley SD, Tinsley CR, Nassif X, Bille E. 2011. Genotypic and phenotypic modifications of Neisseria meningitidis after an accidental human passage. PLoS One 6:e17145. doi: 10.1371/journal.pone.0017145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Richardson AR, Stojiljkovic I. 2001. Mismatch repair and the regulation of phase variation in Neisseria meningitidis. Mol Microbiol 40:645–655. doi: 10.1046/j.1365-2958.2001.02408.x. [DOI] [PubMed] [Google Scholar]
- 29.Richardson AR, Stojiljkovic I. 1999. HmbR, a hemoglobin-binding outer membrane protein of Neisseria meningitidis, undergoes phase variation. J Bacteriol 181:2067–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lucidarme J, Findlow J, Chan H, Feavers IM, Gray SJ, Kaczmarski EB, Parkhill J, Bai X, Borrow R, Bayliss CD. 2013. The distribution and 'in vivo' phase variation status of haemoglobin receptors in invasive meningococcal serogroup B disease: genotypic and phenotypic analysis. PLoS One 8:e76932. doi: 10.1371/journal.pone.0076932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krauland MG, Dunning Hotopp JC, Riley DR, Daugherty SC, Marsh JW, Messonnier NE, Mayer LW, Tettelin H, Harrison LH. 2012. Whole genome sequencing to investigate the emergence of clonal complex 23 Neisseria meningitidis serogroup Y disease in the United States. PLoS One 7:e35699. doi: 10.1371/journal.pone.0035699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marri PR, Paniscus M, Weyand NJ, Rendon MA, Calton CM, Hernandez DR, Higashi DL, Sodergren E, Weinstock GM, Rounsley SD, So M. 2010. Genome sequencing reveals widespread virulence gene exchange among human Neisseria species. PLoS One 5:e11835. doi: 10.1371/journal.pone.0011835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Antignac A, Rousselle JC, Namane A, Labigne A, Taha MK, Boneca IG. 2003. Detailed structural analysis of the peptidoglycan of the human pathogen Neisseria meningitidis. J Biol Chem 278:31521–31528. doi: 10.1074/jbc.M304749200. [DOI] [PubMed] [Google Scholar]
- 34.Gudlavalleti SK, Datta AK, Tzeng YL, Noble C, Carlson RW, Stephens DS. 2004. The Neisseria meningitidis serogroup A capsular polysaccharide O-3 and O-4 acetyltransferase. J Biol Chem 279:42765–42773. doi: 10.1074/jbc.M313552200. [DOI] [PubMed] [Google Scholar]
- 35.Kahler CM, Lyons-Schindler S, Choudhury B, Glushka J, Carlson RW, Stephens DS. 2006. O-Acetylation of the terminal N-acetylglucosamine of the lipooligosaccharide inner core in Neisseria meningitidis. Influence on inner core structure and assembly. J Biol Chem 281:19939–19948. [DOI] [PubMed] [Google Scholar]
- 36.Klughammer J, Dittrich M, Blom J, Mitesser V, Vogel U, Frosch M, Goesmann A, Muller T, Schoen C. 2017. Comparative genome sequencing reveals within-host genetic changes in Neisseria meningitidis during invasive disease. PLoS One 12:e0169892. doi: 10.1371/journal.pone.0169892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bårnes GK, Kristiansen PA, Beyene D, Workalemahu B, Fissiha P, Merdekios B, Bohlin J, Preziosi MP, Aseffa A, Caugant DA. 2016. Prevalence and epidemiology of meningococcal carriage in southern Ethiopia prior to implementation of MenAfriVac, a conjugate vaccine. BMC Infect Dis 16:639. doi: 10.1186/s12879-016-1975-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jolley KA, Maiden MC. 2010. BIGSdb: scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics 11:595. doi: 10.1186/1471-2105-11-595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bratcher HB, Corton C, Jolley KA, Parkhill J, Maiden MC. 2014. A gene-by-gene population genomics platform: de novo assembly, annotation and genealogical analysis of 108 representative Neisseria meningitidis genomes. BMC Genomics 15:1138. doi: 10.1186/1471-2164-15-1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bryant D, Moulton V. 2004. Neighbor-net: an agglomerative method for the construction of phylogenetic networks. Mol Biol Evol 21:255–265. doi: 10.1093/molbev/msh018. [DOI] [PubMed] [Google Scholar]
- 41.Huson DH, Bryant D. 2006. Application of phylogenetic networks in evolutionary studies. Mol Biol Evol 23:254–267. doi: 10.1093/molbev/msj030. [DOI] [PubMed] [Google Scholar]
- 42.Kumar S, Stecher G, Tamura K. 2016. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Boratyn GM, Camacho C, Cooper PS, Coulouris G, Fong A, Ma N, Madden TL, Matten WT, McGinnis SD, Merezhuk Y, Raytselis Y, Sayers EW, Tao T, Ye J, Zaretskaya I. 2013. BLAST: a more efficient report with usability improvements. Nucleic Acids Res 41:W29–W33. doi: 10.1093/nar/gkt282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li H. 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv arXiv:1303.3997 [q-bio.GN]. http://arXiv.org/abs/1207.3907v2.
- 45.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R; 1000 Genome Project Data Processing Subgroup . 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, Rajandream MA, Barrell B. 2000. Artemis: sequence visualization and annotation. Bioinformatics 16:944–945. doi: 10.1093/bioinformatics/16.10.944. [DOI] [PubMed] [Google Scholar]
- 47.Freitag NE, Seifert HS, Koomey M. 1995. Characterization of the pilF-pilD pilus-assembly locus of Neisseria gonorrhoeae. Mol Microbiol 16:575–586. doi: 10.1111/j.1365-2958.1995.tb02420.x. [DOI] [PubMed] [Google Scholar]
- 48.Zachara NE, Vosseller K, Hart GW. 2011. Detection and analysis of proteins modified by O-linked N-acetylglucosamine. Curr Protoc Protein Sci Chapter 17:Unit 17.6. doi: 10.1002/0471142727.mb1706s95. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The sequences generated and analyzed during the current study are available at the pubMLST.org website (see identification numbers in Table S1).