

Mycobacterium spp. are one of the few prokaryotes known to produce lipid droplets (LDs), and their production has been linked to aspects of persistent infection by M. tuberculosis. Unfortunately, little is known about LD production in these organisms, including how LDs are formed, their function, or the identity of proteins that associate with them. In this study, an established M. tuberculosis LD protein and a surrogate Mycobacterium host were used as model systems to study the interactions between proteins and LDs in bacteria. Through these studies, we identified a commonly occurring protein motif that is able to facilitate the association of proteins to LDs in prokaryotes.
KEYWORDS: Mycobacterium, Mycobacterium tuberculosis, phage shock protein, PspA, amphipathic helix, latency, lipid droplets, nonreplicating persistence
ABSTRACT
Mycobacterium tuberculosis is a global pathogen of significant medical importance. A key aspect of its life cycle is the ability to enter into an altered physiological state of nonreplicating persistence during latency and resist elimination by the host immune system. One mechanism by which M. tuberculosis facilitates its survival during latency is by producing and metabolizing intracytoplasmic lipid droplets (LDs). LDs are quasi-organelles consisting of a neutral lipid core such as triacylglycerol surrounded by a phospholipid monolayer and proteins. We previously reported that PspA (phage shock protein A) associates with LDs produced in Mycobacterium. In particular, the loss or overproduction of PspA alters LD homeostasis in Mycobacterium smegmatis and attenuates the survival of M. tuberculosis during nonreplicating persistence. Here, M. tuberculosis PspA (PspAMtb) and a ΔpspA M. smegmatis mutant were used as model systems to investigate the mechanism by which PspA associates with LDs and determine if other Mycobacterium proteins associate with LDs using a mechanism similar to that for PspA. Through this work, we established that the amphipathic helix present in the first α-helical domain (H1) of PspA is both necessary and sufficient for the targeting of this protein to LDs. Furthermore, we identified other Mycobacterium proteins that also possess amphipathic helices similar to PspA H1, including a subset that localize to LDs. Altogether, our results indicate that amphipathic helices may be an important mechanism by which proteins target LDs in prokaryotes.
IMPORTANCE Mycobacterium spp. are one of the few prokaryotes known to produce lipid droplets (LDs), and their production has been linked to aspects of persistent infection by M. tuberculosis. Unfortunately, little is known about LD production in these organisms, including how LDs are formed, their function, or the identity of proteins that associate with them. In this study, an established M. tuberculosis LD protein and a surrogate Mycobacterium host were used as model systems to study the interactions between proteins and LDs in bacteria. Through these studies, we identified a commonly occurring protein motif that is able to facilitate the association of proteins to LDs in prokaryotes.
INTRODUCTION
Mycobacterium tuberculosis is an acid-fast bacillus and is responsible for the respiratory disease tuberculosis (TB). In 2016, this bacterium was responsible for ∼10.4 million cases of TB and more than 1.7 million deaths (1), making M. tuberculosis a leading cause of death in the world due to an infectious agent. The World Health Organization currently estimates that 1.7 billion people are latently infected with M. tuberculosis (1). During latency, M. tuberculosis resides within granulomatous lesions in a metabolically altered state of nonreplicating persistence. The ability of M. tuberculosis to establish, maintain, and reactivate from nonreplicating persistence is a critical aspect of its life cycle, contributing to the continued circulation of this pathogen within the human population.
One strategy thought to be utilized by M. tuberculosis to persist during latency is the production of lipid droplets (LDs) which can be used as an energy reservoir under conditions of stress. LDs are quasi-organelles comprised of a neutral lipid core such as triacylglycerol and are surrounded by a phospholipid monolayer and proteins. Several lines of evidence indicate that LDs regulate key aspects of the M. tuberculosis life cycle. Bacilli present in sputum samples derived from TB patients with active disease possess intracytoplasmic LDs (2). In addition, environmental conditions and/or cell types thought to be encountered by M. tuberculosis within the granuloma are capable of inducing the production of LDs within M. tuberculosis and other disease-causing mycobacterial species (3–7). Finally, M. tuberculosis mutants that are defective in their ability to synthesize or hydrolyze triacylglycerol exhibit survival and/or reactivation defects in vitro and in vivo (4–11). These results suggest that proteins involved in LD production, maintenance, and/or mobilization may be rational targets for therapeutic intervention.
While LDs are produced by nearly all eukaryotes, only a limited number of prokaryotes, including species of Mycobacterium and Rhodococcus, have been shown to produce LDs (reviewed in references 12–14). Consequently, much less is known about LDs in bacteria, including their proteomic composition or the features that target proteins to these structures. To date, several hundred proteins have been identified in the LD proteome of Rhodococcus (15, 16). In contrast, only seven proteins thus far have been described in the proteome of LDs from Mycobacterium (17, 18). Five of these proteins are predicted to function in the synthesis and/or hydrolysis of TAG, including Tgs1 (BCG_3153c), Tgs2 (BCG_3794c), BCG_1169c, BCG_1489c, and BCG_1721 (18). Of the other two determinants, Acr (HspX) is a stress-responsive protein that functions as an ATP-independent chaperone (9, 11, 18) and PspA is a member of the phage shock protein A (PspA) family (17). While PspA has been observed on LDs produced in Rhodococcus, its function in these organisms remains unknown (63, 64). In Mycobacterium smegmatis, the loss or overproduction of PspA alters the size distribution of LDs produced by these organisms (17). In M. tuberculosis, the loss or overproduction of PspA attenuates survival during nonreplicating persistence (17). These observations indicate that PspA may play an important role in the physiology and/or persistence of M. tuberculosis within the host during latency.
PspA is a member of an evolutionarily conserved family of proteins (PspA/Vipp1/IM30) that are found in bacteria, algae, and plants (reviewed in reference 19). In most Gram-positive and Gram-negative bacteria, PspA binds to the inner face of the plasma membrane and regulates plasma membrane homeostasis under conditions of cell envelope stress (19–21). In plants, algae, and photosynthetic bacteria, the PspA ortholog, Vipp1 (vesicle-inducing protein in plastids 1), binds to the inner membrane of chloroplasts where it regulates the biogenesis and maintenance of these organelles (22–25). While members of the PspA/Vipp1/IM30 protein family are generally unrelated at the amino acid level, they all possess a largely α-helical secondary structure and are able to form large homo-oligomeric complexes that assume ring- and/or rodlike superstructures (17, 26–30). Furthermore, these proteins share the ability to bind phospholipids and membranes through an amphipathic helix that resides within the N and/or C terminus of these proteins (23, 24, 31).
In this study, PspA from M. tuberculosis (PspAMtb) and a ΔpspA M. smegmatis mutant were used to delineate the proteomic composition of Mycobacterium LDs and begin interrogating the mechanism by which proteins are able to target LDs in prokaryotes. Through these studies, we have determined that the amphipathic helix present within the H1 domain of PspAMtb is both necessary and sufficient for this protein to bind Mycobacterium LDs. Furthermore, amphipathic helices with similarity to the PspAMtb H1 domain are present in other M. smegmatis proteins, including a subset that localize to Mycobacterium LDs. In summary, our data implicate amphipathic helices as an important mechanism by which proteins associate with LDs in prokaryotes.
RESULTS
H1 domain is necessary and sufficient for localization of PspAMtb to LDs.
PspAMtb is largely α-helical and contains five distinct α-helical domains (Fig. 1A) (17). As amphipathic helices are one motif used by eukaryotic proteins to target LDs (32–36), PspAMtb was first analyzed using Heliquest to identify possible amphipathic helices (37). Results from this search indicated the presence of an amphipathic helix in the N-terminal H1 domain of PspAMtb. The 18 residues comprising this motif have a hydrophobic moment (μH) of 0.543 and a mean hydrophobicity (H) of 0.589 (Fig. 1B). This domain is highly conserved in PspA family members (31), including in Mycobacterium species (Fig. 1C). To investigate the importance of this domain for PspAMtb localization to LDs, a variant of PspAMtb lacking the first 22 amino acids which includes the H1 domain (PspAMtbΔH1) was generated (Fig. 1A). This derivative, along with wild-type PspAMtb, was produced in an M. smegmatis ΔpspA strain under conditions promoting LD production. The protein was then localized by Western blotting following subcellular fractionation. Wild-type PspAMtb localized predominantly to LDs (Fig. 1D), similar to that previously reported (17). The higher molecular weight banding pattern observed is due to the presence of SDS-resistant PspAMtb multimers, a characteristic that has been observed previously (17). In contrast, PspAMtbΔH1 was observed exclusively in the cytoplasmic compartment, indicating that the H1 domain is necessary for PspAMtb to localize to LDs (Fig. 1E). MprB and GroES, membrane and cytoplasmic control proteins, respectively, were present in their expected subcellular compartments, confirming the fractionation procedure (Fig. 1D and E). Thus, the H1 domain is necessary for PspAMtb to associate with LDs.
FIG 1.

Helix 1 is necessary and sufficient for PspA localization to lipid droplets. (A) Schematic of Psipred-predicted helices (blue segments; helix 1 in dark blue) of full-length PspAMtb (top) and helix-1-truncated PspAMtb (PspAMtbΔH1) (bottom). (B) Helical wheel diagram of helix 1 comprising residues 5 to 22 of the protein. The hydrophobic face of the amphipathic helix is highlighted in red. Hydrophobicity (H) and mean hydrophobic moment (μH) values are noted. The arrow indicates the angle of the mean hydrophobic moment. (C) The PspA H1 helix (residues 5 to 22) from various mycobacterial species was aligned using Clustal W (DNASTAR; Lasergene, Madison, WI). Amino acid residues that are identical are shaded gray. PspA from the following species were used: Mycobacterium tuberculosis, Mycobacterium smegmatis, Mycobacterium abscessus, Mycobacterium avium, Mycobacterium bovis BCG, Mycobacterium gilvum, Mycobacterium leprae, Mycobacterium marinum, Mycobacterium paratuberculosis, Mycobacterium ulcerans, and Mycobacterium vanbaalenii. (D and E) Western blots showing localization of 3×Flag-PspAMtb (D) and 3×Flag-PspAMtbΔH1 (E) following production in M. smegmatis ΔpspA. PspA derivatives were detected using anti-Flag antibody. Anti-GroES and anti-MprB antibodies were used to detect control proteins. Fraction abbreviations: Cyt, cytosol; M, membrane; LD, lipid droplets. The images shown are representative of experiments performed in triplicate. (F) Distribution of PspAMtb-His (left) and PspAMtbΔH1-His (right) following ultracentrifugation on 10 to 50% continuous sucrose gradients. Fractions were collected from top to bottom of the gradient tubes and are displayed on the x axis. Protein concentration as a percentage of total protein detected for each sample is displayed on the y axis. Arrows indicate the fractions in which molecular weight standards localized in gradients that were run in parallel: ADH, alcohol dehydrogenase (150 kDa); Thy, thyroglobulin (670 kDa); BD, blue dextran (∼2,000 kDa). (G) Negative-stain electron microscopy of PspAMtb-His (left) or PspAMtbΔH1-His (right). Protein was purified from fraction 18 (PspAMtb-His) or 23 (PspAMtbΔH1-His), stained with uranyl acetate, and examined at ×100,000 magnification. White arrowheads indicate spheroid structures, and black arrowheads indicate rodlike superstructures. (H) Schematic of 3×Flag-GFPmut3 constructs with either 1, 3, or no H1 tag(s) cloned at the N terminus. (I) Western blot showing localization of wild-type and mutant GFPmut3 variants following production in M. smegmatis ΔpspA. GFPmut3 variants were detected using anti-Flag antibody. Anti-GroES and anti-MprB antibodies were used to detect control proteins. Fraction abbreviations: Cyt, cytosol; M, membrane; LD, lipid droplets. The images shown are representative of experiments performed in triplicate.
To investigate if the removal of the N-terminal 22 amino acids of PspAMtb alters global protein structure, PspAMtb and PspAMtbΔH1 were produced and purified in Escherichia coli (see Fig. S1 in the supplemental material). The oligomeric nature of the purified proteins was then characterized by analyzing their mobility on a 10 to 50% continuous sucrose gradient alongside protein standards of known mass and by negative-stain electron microscopy (EM). Following ultracentrifugation, wild-type PspAMtb localized predominantly to fraction 18, between thyroglobulin (fraction 14; ∼670 kDa) and blue dextran (fraction 23). In addition, PspAMtb formed spheroid and rodlike superstructures (Fig. 1F and G), similar to that seen previously (17). In contrast, PspAMtbΔH1 localized primarily to fraction 23, similar to blue dextran (>2,000 kDa) (Fig. 1F). When analyzed by negative stain EM, PspAMtbΔH1 also formed spheroids and rodlike superstructures (Fig. 1G). While these results indicate that the removal of the H1 domain does not induce gross structural changes to PspAMtb, the loss of this region does shift the fractionation pattern within the sucrose gradient to the right, possibly reflecting an increased propensity of the PspAMtbΔH1 variant to form larger rodlike superstructures, as indicated by negative-stain EM (Fig. 1F and G).
Finally, to determine if the H1 domain is also sufficient to facilitate the localization of PspAMtb to LDs, the first 22 amino acids of PspAMtb were fused in single copy, or in multiple tandem copies, to the N terminus of green fluorescent protein mutant GFPmut3 (Fig. 1H). The ability of these H1-GFPmut3 chimeras to localize to M. smegmatis LDs was then assessed. While GFPmut3 alone partitioned completely in the cytoplasmic compartment (Fig. 1I), the addition of either one or three copies of the PspAMtb H1 domain to GFPmut3 altered the localization such that a predominant fraction of GFP now localized to LDs (Fig. 1I). Altogether, these results indicate that the H1 domain of PspAMtb is both necessary and sufficient for PspAMtb to localize to LDs.
PspA from Rhodococcus contains an amphipathic helix in its H1 domain and localizes to LDs when produced in M. smegmatis.
PspA has been identified in the LD proteome of Rhodococcus jostii strain RHA1 (PspARho) (16). PspARho is 77% identical to PspAMtb at the primary amino acid level (17); however, other aspects of PspARho, including the mechanism by which it localizes to LDs, remain unknown. To determine if PspARho is α-helical and possesses an amphipathic helix similar to that of PspAMtb, PspARho was analyzed using Psipred and Heliquest (37). PspARho forms five α-helical domains (Fig. 2A), similar to PspAMtb. Furthermore, the H1 domain of PspARho (residues 5 to 22) is predicted to be amphipathic, with μH and H values of 0.528 and 0.532, respectively (Fig. 2B). Interestingly, the H1 domain of PspARho differs from PspAMtb H1 by only two residues (L12M and S17N) (Fig. 2C). To determine if PspARho forms higher order homo-oligomers like PspAMtb, PspARho was produced in E. coli, purified, and analyzed using sucrose gradient ultracentrifugation (Fig. S1). PspARho localized predominantly to fraction 20 between the control proteins thyroglobulin (fraction 13) and blue dextran (fraction 24) (Fig. 2D). These results are similar to those observed for PspAMtb (Fig. 1E). Finally, to determine if PspARho binds Mycobacterium LDs, the protein was produced in M. smegmatis ΔpspA under LD-inducing conditions, and its subcellular localization was assessed as described previously. PspARho was observed predominantly in the LD fraction (Fig. 2E). Importantly, control proteins, including MprB and GroES, localized to their expected compartments (Fig. 2E), confirming the fractionation procedure. Thus, PspARho is largely α-helical, possesses an amphipathic helix in its H1 domain that is nearly identical to that in PspAMtb, and is able to bind LDs when produced in M. smegmatis ΔpspA.
FIG 2.
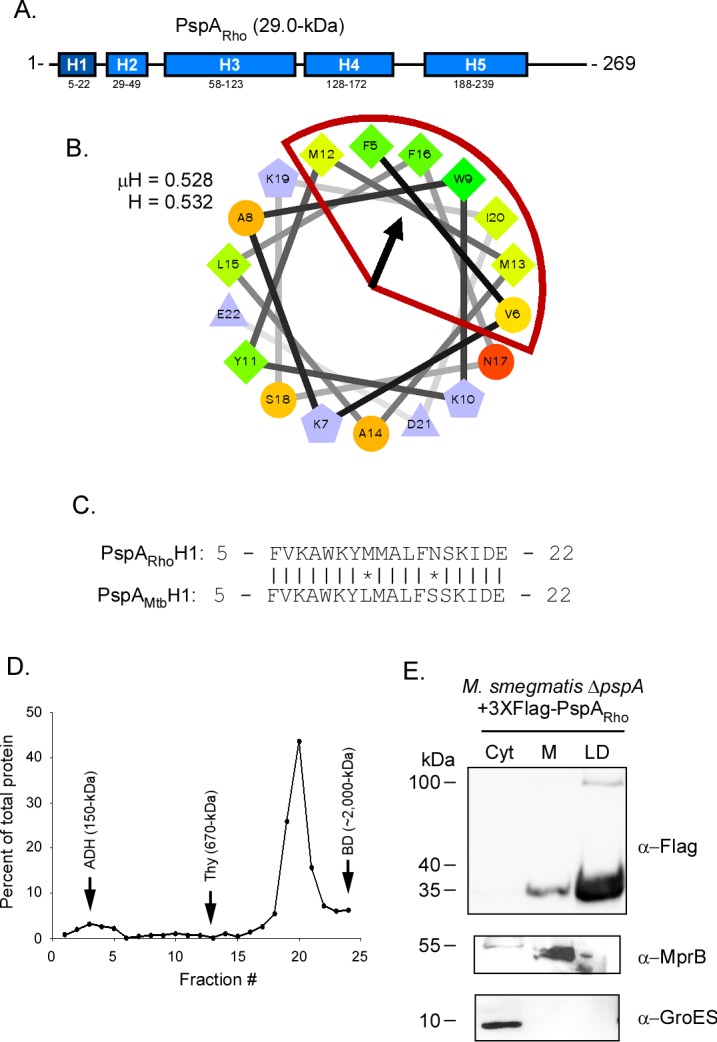
PspA from Rhodococcus localizes to Mycobacterium lipid droplets. (A) Schematic of Psipred-predicted helices of PspARho (blue segments; helix 1 in dark blue). (B) Helical wheel diagram of helix 1 from PspARho comprising residues 5 to 22 of the protein. The hydrophobic face of the amphipathic helix is highlighted in red. Hydrophobicity (H) and mean hydrophobic moment (μH) values are noted. The arrow indicates the angle of the mean hydrophobic moment. (C) Sequence alignment between the H1 domains of PspARho and PspAMtb. Vertical lines, identical residues; *, mismatched residues. (D) Distribution of PspARho-His following ultracentrifugation on 10 to 50% continuous sucrose gradients. Fractions were collected from top to bottom of the gradient tubes and are displayed on the x axis. Protein concentration as a percentage of total protein detected for each sample is displayed on the y axis. Arrows indicate the fractions in which molecular weight standards localized in gradients that were run in parallel: ADH, alcohol dehydrogenase (150 kDa); Thy, thyroglobulin (670 kDa); BD, blue dextran (∼2,000 kDa). (E) Western blot showing localization of 3×Flag-PspARho following production in M. smegmatis ΔpspA. PspARho was detected using anti-Flag antibody. Anti-GroES and anti-MprB antibodies were used to detect control proteins. Fraction abbreviations: Cyt, cytosol; M, membrane; LD, lipid droplets. The images shown are representative of experiments performed in triplicate.
PspA from E. coli associates with LDs when produced in M. smegmatis.
The ability of PspAMtb and PspARho to bind LDs in Mycobacterium may be due to the amphipathic nature of their H1 domains. Alternatively, the ability of these proteins to associate with LDs may be due to the highly conserved residues comprising this domain. To differentiate between these possibilities, PspA from E. coli (PspAEco) was produced in M. smegmatis ΔpspA and its binding to LDs assessed. PspAEco possesses a largely α-helical domain architecture (Fig. 3A) (27, 38, 39) and also contains an amphipathic helix within its H1 domain (also called AHa; residues 2 to 19) (Fig. 3B) (31); however, PspAEco exhibits little to no amino acid sequence identify with PspAMtb and PspARho in the H1 region (Fig. 3C) (17). We first confirmed by sucrose gradient ultracentrifugation that PspAEco produced in E. coli homo-oligomerizes as has been reported previously (27, 28). As expected, PspAEco localized predominantly to fraction 20, between thyroglobulin (fraction 13) and blue dextran (fraction 24) (Fig. 3D; Fig. S1), confirming that this protein forms higher order homo-oligomeric complexes. Interestingly, when PspAEco was produced in M. smegmatis ΔpspA, the protein localized almost exclusively within the LD compartment (Fig. 3E). In contrast, the control proteins MprB and GroES were localized within the membrane and cytoplasmic compartments, respectively, as expected (Fig. 3E). Altogether, these results indicate that the ability of PspA to associate with LDs is likely due to the amphipathic nature of its H1 (AHa) domain rather than the specific amino acid residue(s) that comprises this domain.
FIG 3.

PspA from E. coli localizes to Mycobacterium lipid droplets. (A) Schematic of Psipred-predicted helices of PspAEco (blue segments; helix 1 in dark blue). (B) Helical wheel diagram of helix 1 from PspAEco comprising residues 2 to 19 of the protein. The hydrophobic face of the amphipathic helix is highlighted in red. Hydrophobicity (H) and mean hydrophobic moment (μH) values are noted. The arrow indicates the angle of the mean hydrophobic moment. (C) Sequence alignment of the H1 domain from PspAEco with the H1 domains of PspARho and PspAMtb. Vertical line, identical residues; *, mismatched residues. (D) Distribution of PspAEco-His following ultracentrifugation on 10 to 50% continuous sucrose gradients. Fractions were collected from top to bottom of the gradient tubes and are displayed on the x axis. Protein concentration as a percentage of total protein detected for each sample is displayed on the y axis. Arrows indicate the fractions in which molecular weight standards localized in gradients that were run in parallel: ADH, alcohol dehydrogenase (150 kDa); Thy, thyroglobulin (670 kDa); BD, blue dextran (∼2,000 kDa). (E) Western blot showing localization of 3×Flag-PspAEco following production in M. smegmatis ΔpspA. PspAEco was detected using anti-Flag antibody. Anti-GroES and anti-MprB antibodies were used to detect control proteins. Fraction abbreviations: Cyt, cytosol; M, membrane; LD, lipid droplets. The images shown are representative of experiments performed in triplicate.
Amino acid substitutions within the H1 domain of PspAMtb reduce but do not abrogate binding to LDs.
Recently, it was demonstrated that the localization of amphipathic helices to LDs is largely independent of the H or μH (35). Rather, the binding of amphipathic helices to LDs is more dependent on the number of large hydrophobic residues (I, F, L, M, W, or Y) that are present on the hydrophobic face of the helix (35). In particular, amphipathic helices with 5 or more large hydrophobic residues are able to bind LDs, with the efficiency of binding proportional to the number of large hydrophobic residues present (35). As the PspAMtb H1 domain contains 6 large hydrophobic residues on its hydrophobic face (Fig. 1B), we postulated that the mutation of one or more of these residues may reduce or abrogate the ability of PspAMtb to bind LDs. To test this possibility, select amino acid residues along the hydrophobic or hydrophilic face of the PspAMtb H1 domain were mutated to alanine (Fig. 4A). F5, W9, and F16 were chosen for the mutations, as these residues are on the hydrophobic face of the helix and are positioned nearest the hydrophobic moment (Fig. 4A). K7 and Y11 were also chosen, as they are on the hydrophilic face of the helix and were not expected to contribute substantively to LD binding (Fig. 4A). While individual substitutions at F5 and W9 alone did not alter the distribution pattern of PspAMtb relative to that of the wild-type protein (Fig. 4B and C), a substitution at F16 significantly reduced the percentage of PspAMtb localized to LDs (Fig. 4B and C). Furthermore, PspAMtb variants harboring two (F5A/W9A, F5A/F16A, or W9A/F16A) or three (5FA/W9A/F16A) amino acid substitutions on the hydrophobic face of the H1 helix were also reduced in their ability to bind LDs relative to that of the wild-type PspA (Fig. 4B and C). Interestingly, the level of binding of these variants was similar to that observed for the single F16A mutant even though these mutant proteins possessed 4 or fewer large hydrophobic residues on the hydrophobic face of the helix (Fig. 4B and C). PspAMtb variants carrying substitutions along the hydrophilic face of the H1 domain (K7 or Y11) were also reduced in their ability to bind LDs (Fig. 4B and C), indicating that amino acid residues along the hydrophilic face of PspAMtb may also contribute to LD interactions. Altogether, our results indicate that amino acids on the hydrophobic and hydrophilic faces of the H1 amphipathic helix contribute to LD binding by PspAMtb.
FIG 4.

Impact of H1 amino acid substitutions on PspA and H1-GFP chimera localization. (A, left) Amino acid sequence of the PspAMtb H1 domain (residues 5 to 22). *, residues targeted for alanine mutagenesis. (Right) Helical wheel diagram of the PspAMtb H1 domain. *, mutagenized residues. The arrow indicates the angle of the mean hydrophobic moment. (B) Western blots showing localization of wild-type and mutant variants of 3×Flag-PspAMtb following production in M. smegmatis ΔpspA. PspAMtb was detected using anti-Flag antibody. Anti-GroES and anti-MprB antibodies were used to detect control proteins. Fraction abbreviations: Cyt, cytosol; M, membrane; LD, lipid droplets. Hydrophobicity (H) and mean hydrophobic moment (μH) values are noted for each H1 domain. The images shown are representative of experiments performed in triplicate. (C) Graph indicating the average percentages of proteins present in the lipid droplet fractions relative to the average percentages of each protein present in the cytosolic and membrane fractions. *, P < 0.05 versus wild-type PspAMtb or H1-GFPmut3. Values were derived from experiments performed in triplicate. (D) Western blots showing localization of 3×Flag-H1-GFPmut3 and 3×Flag-H1-GFPmut3 mutant variants following production in M. smegmatis ΔpspA. H1-GFPmut3 derivatives were detected using anti-Flag antibody. Anti-GroES and anti-MprB antibodies were used to detect control proteins. Fraction abbreviations: Cyt, cytosol; M, membrane; LD, lipid droplets. Hydrophobicity (H) and mean hydrophobic moment (μH) values are noted for each H1 domain. The images shown are representative of experiments performed in triplicate.
In PspAEco, substitutions at single hydrophobic residues (F4E or V11E) along the hydrophobic face of the H1 (AHa) domain are sufficient to abrogate binding of this protein to lipids (31). Given our inability to completely abrogate binding of PspAMtb to LDs following a substitution at one or more residues along the hydrophobic face of the H1 helix, we wondered if the propensity for PspAMtb to bind itself may partially mask LD-binding defects. To test this possibility, a subset of the H1 amino acid substitutions were generated in the H1-GFPmut3 chimera, including W9A, W9E, and 5FA/W9A/F16A. The resulting protein variants were then tested for their ability to associate with LDs following production in M. smegmatis ΔpspA. The localization patterns of the generated H1-GFPmut3 chimeras were similar to those observed for their full-length PspAMtb counterparts (Fig. 4C and D), demonstrating that homo-oligomerization of PspAMtb does not regulate H1 binding to LDs.
In summary, our results indicate that the binding of PspAMtb to LDs can be achieved with as few as three large hydrophobic residues along the hydrophobic face of the H1 amphipathic helix. Furthermore, the binding of PspAMtb to LDs is dependent on residues present on both the hydrophobic and hydrophilic faces of the H1 helix but is independent of homo-oligomerization of the protein.
Identification and validation of potential LD-binding amphipathic helices in other mycobacterial proteins.
Six proteins have previously been identified in the LD proteome of Mycobacterium bovis BCG (18). To determine if any of these proteins possess amphipathic helices similar to the H1 domain of PspAMtb, BCG_1169c, BCG_1489c, BCG_1721, HspX (BCG_2050c), Tgs1 (BCG_3153c), and Tgs2 (BCG_3794) were analyzed using Heliquest. Of the six proteins analyzed, four contained helices which satisfied search parameter requirements, including Tgs1, BCG_1721, Tgs2, and BCG_1169c (Fig. 5). Notably, Tgs2 contained two such helices. The numbers of large hydrophobic residues present in the amphipathic helices of these proteins ranged between 4 and 5. Thus, a subset of M. bovis BCG proteins known to bind Mycobacterium LDs possess one or more amphipathic helices with similarity to the H1 domain of PspAMtb.
FIG 5.

LD-associated proteins from M. bovis BCG possess amphipathic helices. Amphipathic helices were identified in BCG_3153 (Tgs1), BCG_1721, BCG_3794 (Tgs2), and BCG_1169c (18) using Heliquest. The length of each protein is shown. The location of amphipathic helices within each sequence is denoted by a blue box. The hydrophobic face of each helix is highlighted in red. The mean hydrophobic moment (μH) and hydrophobicity (H) values are noted. The arrows indicate the angles of the mean hydrophobic moments.
We next investigated whether PspAMtb H1-like amphipathic helices exist in other proteins from M. smegmatis. To investigate this possibility, the complete M. smegmatis proteome was searched using Heliquest. The resulting candidates were then screened to identify candidates that possessed 5 or more large hydrophobic residues along the hydrophobic face of the helix (35, 37). Of the 6,602 proteins analyzed, 330 proteins (∼5% of the total proteome) satisfied these search parameter criteria (see Table S2). Amphipathic helices were positioned throughout the amino acid sequences of the identified proteins, with the numbers of large hydrophobic residues ranging between 5 and 9. Importantly, MSMEG_2695, the M. smegmatis homolog of PspA, was present in the list.
To determine if any of the proteins identified from the Heliquest search were able to bind LDs, LDs were purified from wild-type M. smegmatis and treated with chloroform-methanol to extract proteins, and the resulting proteins were identified by liquid chromatography-tandem mass spectroscopy (LC-MS/MS). For these studies, LDs were isolated from three independent cultures of wild-type M. smegmatis. Each of these biological replicates was further split into technical triplicates prior to their injection into the mass spectrometer. Only proteins in which 3 or more peptides were observed in at least 2 of the 3 technical replicates, and in at least 2 of the 3 biological replicates, were designated as being LD associated. In total, 480 proteins (∼7% of the M. smegmatis proteome) satisfied these criteria (see Table S3). This is similar to the number of LD proteins reported in Rhodococcus jostii RHA1 (16). The 50 top scoring hits from this analysis based on relative protein abundance (average label-free quantification [LFQ]) are shown in Table 1. The identified proteins are predicted to possess diverse biological functions, including biosynthesis, metabolism, protein folding, replication, transcription, and translation. Of note, MSMEG_2695 (PspA), was the second most abundant protein observed on LDs (Table 1). Interestingly, the most abundant protein was MSMEG_0919, a homolog of the heparin-binding hemagglutinin (HbhA) from M. tuberculosis (Table 1) (40). HbhA is an established surface-exposed adhesion and a multifunctional virulence determinant (41–46). MSMEG_0919 is also orthologous to Rhodococcus microorganism lipid droplet small (MLDS), an LD-associated protein that regulates LD size and content (16). In addition, this protein binds to genomic DNA on the surface of LDs, and enhances the protection against genotoxic stress encountered by Rhodococcus (47). Of the 480 proteins identified by LC-MS/MS, 26 (∼5%) were also identified in the Heliquest search as possessing one or more amphipathic helices with characteristics similar to the PspAMtb H1 domain (Table 2). Several of these proteins are predicted to be involved in metabolic processes (Table 2), while others regulate the processes of transcription, translation, and protein degradation (Table 2). Thus, M. smegmatis harbors a total of 22 proteins that possess H1-like amphipathic helices similar to that in PspAMtb and that localize to the surface of LDs.
TABLE 1.
Fifty most abundant proteins in the M. smegmatis LD proteome based on label-free quantification
| UniProt accession no. | Gene | Protein | No. of unique peptides | % coverage | Avg LFQ |
|---|---|---|---|---|---|
| Q3I5Q7 | MSMEG_0919 | HBHA-like protein | 38 | 69.4 | 2.14E+09 |
| A0QVU2 | MSMEG_2695 | 35-kDa protein | 36 | 97.1 | 1.99E+09 |
| A0R5R3 | MSMEG_6282 | KanY protein | 18 | 95.2 | 8.81E+08 |
| A0QR91 | MSMEG_1030 | 4-Hydroxyacetophenone monooxygenase | 50 | 86.8 | 7.22E+08 |
| A0R2B0 | MSMEG_5048 | Membrane protein | 17 | 60.1 | 4.45E+08 |
| A0QPE8 | fadA2 | 3-Ketoacyl coenzyme A thiolase | 27 | 78.5 | 3.91E+08 |
| A0R535 | MSMEG_6049 | Secreted protein | 19 | 75.8 | 3.77E+08 |
| A0QPE7 | fabG | 3-Ketoacyl-ACP reductase | 44 | 96 | 2.88E+08 |
| A0QQU5 | groL1 | 60-kDa chaperonin 1 | 44 | 82.4 | 2.31E+08 |
| A0QRD5 | MSMEG_1077 | Deazaflavin-dependent nitroreductase family protein | 19 | 88.6 | 2.23E+08 |
| A0QSD7 | rpsC | 30S ribosomal protein S3 | 35 | 80 | 2.15E+08 |
| A0QNM6 | MSMEG_0098 | Methyltransferase | 19 | 83.5 | 2.05E+08 |
| A0QZX9 | MSMEG_4188 | Short-chain dehydrogenase | 29 | 91.5 | 2.02E+08 |
| Q9ZHC5 | hup | DNA-binding protein HU homolog | 13 | 39.4 | 1.92E+08 |
| A0QQS8 | MSMEG_0863 | Short-chain dehydrogenase | 23 | 74.3 | 1.57E+08 |
| A0QYS3 | MSMEG_3767 | Acyl coenzyme A synthase | 57 | 76.5 | 1.53E+08 |
| A0R200 | atpD | ATP synthase subunit beta | 33 | 90.7 | 1.52E+08 |
| A0R5I8 | MSMEG_6207 | Polyketide cyclase | 18 | 80.6 | 1.50E+08 |
| A0QWW2 | gapA | Glyceraldehyde-3-phosphate dehydrogenase | 18 | 68.2 | 1.47E+08 |
| A0R1E4 | MSMEG_4722 | Short-chain dehydrogenase | 27 | 93.7 | 1.40E+08 |
| A0QSE0 | rpsQ | 30S ribosomal protein S17 | 19 | 87.8 | 1.38E+08 |
| A0R0W7 | MSMEG_4533 | Sulfate ABC transporter substrate-binding protein | 17 | 66.8 | 1.28E+08 |
| A0QR89 | MSMEG_1028 | Geranylgeranyl reductase | 30 | 79.5 | 1.28E+08 |
| A0QR11 | MSMEG_0946 | NAD-dependent epimerase/dehydratase family protein | 26 | 76.9 | 1.25E+08 |
| A0QW19 | dxs | 1-Deoxy-d-xylulose-5-phosphate synthase | 43 | 89 | 1.22E+08 |
| A0QQ49 | MSMEG_0625 | Peptidase | 21 | 66.4 | 1.22E+08 |
| A0R3D1 | MSMEG_5430 | Oxidoreductase | 18 | 85.9 | 1.15E+08 |
| A0QX80 | MSMEG_3204 | Uncharacterized protein | 13 | 73 | 1.13E+08 |
| A0QTV7 | MSMEG_1981 | Nitroreductase | 14 | 75.3 | 1.09E+08 |
| A0R157 | MSMEG_4632 | Enoyl reductase | 35 | 91.6 | 1.03E+08 |
| A0R723 | MSMEG_6753 | Oxidoreductase, short-chain dehydrogenase/reductase family protein | 17 | 75.4 | 1.01E+08 |
| A0QNZ7 | MSMEG_0220 | Monoglyceride lipase | 18 | 87.5 | 9.79E+07 |
| A0QNF5 | cwsA | Cell wall synthesis protein CwsA | 6 | 57.4 | 9.72E+07 |
| A0QPJ9 | MSMEG_0424 | Hsp20/alpha crystallin family protein | 18 | 84.8 | 9.70E+07 |
| A0R5S1 | MSMEG_6291 | Amino acid dehydrogenase | 25 | 74.8 | 9.55E+07 |
| A0R058 | asnB | Asparagine synthetase | 39 | 73.3 | 9.35E+07 |
| A0QS97 | rpsG | 30S ribosomal protein S7 | 15 | 71.2 | 9.15E+07 |
| A0QTL0 | MSMEG_1882 | Acyltransferase, ws/dgat/mgat subfamily protein | 41 | 88.5 | 8.98E+07 |
| A0QXG0 | MSMEG_3287 | Alpha/beta hydrolase | 17 | 68.3 | 8.96E+07 |
| A0R202 | atpA | ATP synthase subunit alpha | 34 | 65.5 | 8.53E+07 |
| A0QSZ3 | icd2 | Isocitrate dehydrogenase | 40 | 66.1 | 8.36E+07 |
| A0QQY3 | MSMEG_0918 | Transcriptional regulator, XRE family | 14 | 97.2 | 8.17E+07 |
| A0R073 | MSMEG_4284 | Multidrug MFS transporter | 14 | 69.9 | 8.16E+07 |
| A0QS66 | rpoC | DNA-directed RNA polymerase subunit beta | 87 | 76.8 | 8.12E+07 |
| A0QS98 | tuf | Elongation factor Tu | 25 | 86.4 | 7.90E+07 |
| A0R2S3 | MSMEG_5215 | Nitroreductase | 12 | 75.6 | 7.90E+07 |
| A0R5H6 | MSMEG_6195 | Anion transporter | 25 | 67.8 | 7.82E+07 |
| A0QPG9 | MSMEG_0394 | Uncharacterized protein | 11 | 100 | 7.61E+07 |
| A0QR12 | MSMEG_0947 | Acyltransferase | 21 | 74.3 | 7.50E+07 |
| A0QPG2 | MSMEG_0387 | Rmt2 protein | 16 | 79.5 | 7.13E+07 |
TABLE 2.
M. smegmatis LD proteins possessing amphipathic helices like the PspAMtb H1 domain
| UniProt accession no. | Gene | Protein | No. of unique peptides | % coverage | Avg LFQ | Helix position | Helix sequence |
|---|---|---|---|---|---|---|---|
| A0QVU2 | MSMEG_2695 | 35-kDa protein | 36 | 97.1 | 1.99E+09 | 5–23 | FVKAWKYLMALFSSKVDEY |
| A0QWW2 | gapA | Glyceraldehyde-3-phosphate dehydrogenase | 18 | 68.2 | 1.47E+08 | 167–185 | LAKVLNDEFGIVKGLMTTI |
| A0QS66 | rpoC | DNA-directed RNA polymerase subunit beta | 87 | 76.8 | 8.12E+07 | 835–852 | IKSSFREGLTVLEYFINT |
| A0R2B1 | kgd | Multifunctional 2-oxoglutarate metabolism enzyme | 55 | 60.4 | 4.89E+07 | 972–989 | LWEAQFGDFVNGAQSIID |
| A0R203 | atpFH | ATP synthase subunit b-delta | 31 | 67.4 | 4.86E+07 | 198–215 | GLTNLADELASVAKLLLS |
| A0QV46 | MSMEG_2444 | Dienelactone hydrolase family protein | 11 | 51.3 | 3.85E+07 | 211–228 | AATTDAWTRVFAFFDEHL |
| A0QSD8 | rplP | 50S ribosomal protein L16 | 8 | 62.3 | 3.50E+07 | 52–69 | IAINRHIKRGGKVWINIF |
| A0R6J9 | MSMEG_6575 | Beta-lactamase | 22 | 73.8 | 3.11E+07 | 28–45 | RAIEQIWDSVRYWYQSGL |
| A0R7C5 | MSMEG_6859 | Oxidoreductase | 15 | 87.6 | 1.57E+07 | 106–123 | LADWHRTIDVNIGGVLNV |
| A0QW02 | sigA | RNA polymerase sigma factor SigA | 17 | 38.4 | 1.21E+07 | 390–407 | SFTLLQDQLQSVLETLSE |
| A0QT91 | MSMEG_1757 | DEAD/DEAH box helicase | 31 | 30.5 | 9.42E+06 | 1193–1210 | ILETLGHGGAYFFRQLTD |
| A0R3M4 | sucC | Succinate coenzyme A ligase (ADP-forming) subunit beta | 12 | 37.2 | 8.63E+06 | 166–183 | LDAAAVTIQKLWEVFVKE |
| A0R2B5 | MSMEG_5053 | Acetoin dehydrogenase | 18 | 74.1 | 5.88E+06 | 247–264 | KILDVIVRITGSGYQRLF |
| A0R564 | disA | DNA integrity scanning protein DisA | 11 | 37.6 | 5.71E+06 | 346–364 | GIGSMWARHIREGLSLLAE |
| A0R564 | disA | DNA integrity scanning protein DisA | 11 | 37.6 | 5.71E+06 | 318–337 | AHVDLLVRSFGSLQNLLAAS |
| A0R5R4 | MSMEG_6283 | FAD-binding domain protein | 16 | 51.5 | 5.40E+06 | 185–202 | FHSITDLVAAMDRIIETG |
| A0QNJ7 | MSMEG_0067 | Conserved hypothetical proline and alanine rich protein | 13 | 43.7 | 4.66E+06 | 348–365 | GLFQKATRGVLSTISGLV |
| A0QS46 | rplA | 50S ribosomal protein L1 | 8 | 44.3 | 4.20E+06 | 94–117 | IVGSDDLIEKIQGGFLDFDAAIAT |
| A0R574 | clpC1 | ATP-dependent Clp protease, ATP-binding subunit ClpC1 | 20 | 38.2 | 3.67E+06 | 725–743 | DEIIQMVDLMIGRVSNQLK |
| A0R1C6 | MSMEG_4703 | Glycerol-3-phosphate acyltransferase | 21 | 42.2 | 3.04E+06 | 212–229 | EMLDELSTGWSRFSVDLI |
| A0QT70 | MSMEG_1736 | Glycerol-3-phosphate dehydrogenase | 12 | 32.4 | 2.82E+06 | 443–462 | VRHLLDRYGSLIGEVLALAD |
| A0QTW2 | MSMEG_1986 | Tartrate dehydrogenase | 9 | 27.3 | 2.73E+06 | 88–105 | SLWGSLIQFRRHFDQYVN |
| A0QVM7 | infB | Translation initiation factor IF-2 | 10 | 25.6 | 2.68E+06 | 491–508 | SVIYQAIDEIEAALKGML |
| A0R730 | MSMEG_6761 | Glycerol-3-phosphate dehydrogenase | 12 | 28.2 | 2.11E+06 | 435–452 | VAHLLGRYGTLTDELLEI |
| A0QRV5 | MSMEG_1252 | Uncharacterized protein | 28 | 21.9 | 2.01E+06 | 1374–1391 | GMSVRALMDNFGELITQI |
| A0QWV7 | MSMEG_3079 | Nucleotide-binding protein MSMEG_3079/MSMEI_3001 | 8 | 40.9 | 1.57E+06 | 222–240 | GALEFLDTYHRLLDVVIDG |
| A0R4M2 | MSMEG_5884 | 3-Hydroxyisobutyrate dehydrogenase family protein | 5 | 34.4 | 9.87E+05 | 181–198 | IASVIESLGEAMALVGKA |
Finally, to validate the results from computational and LC-MS/MS analyses, the DNA sequence encoding the amphipathic helix present in MSMEG_4703 (residues 212 to 229) (Table 2) (Fig. 6A) was synthesized and cloned in front of GFPmut3. This helix has μH and H values of 0.545 and 0.539, respectively (Fig. 6B). Importantly, there are six large hydrophobic residues on the hydrophobic face of this helix (Fig. 6B). Following construction, the resulting plasmid was introduced into M. smegmatis ΔpspA, and the localization of the chimeric GFPmut3 variant was assessed as described previously. While GFPmut3 localized exclusively to the cytoplasmic compartment (data not shown), the MSMEG_4703 (212 to 229) GFPmut3 chimera was observed in multiple fractions, including the LD compartment (Fig. 6C). In contrast, GroES and MprB were observed in the cytoplasmic and membrane compartments, respectively, confirming the fractionation procedure (Fig. 6C). In summary, the M. smegmatis proteome includes proteins predicted to contain one or more PspAMtb H1-like amphipathic helices. Furthermore, a subset of these proteins is present on M. smegmatis LDs, possibly through interactions facilitated by one or more amphipathic helices.
FIG 6.
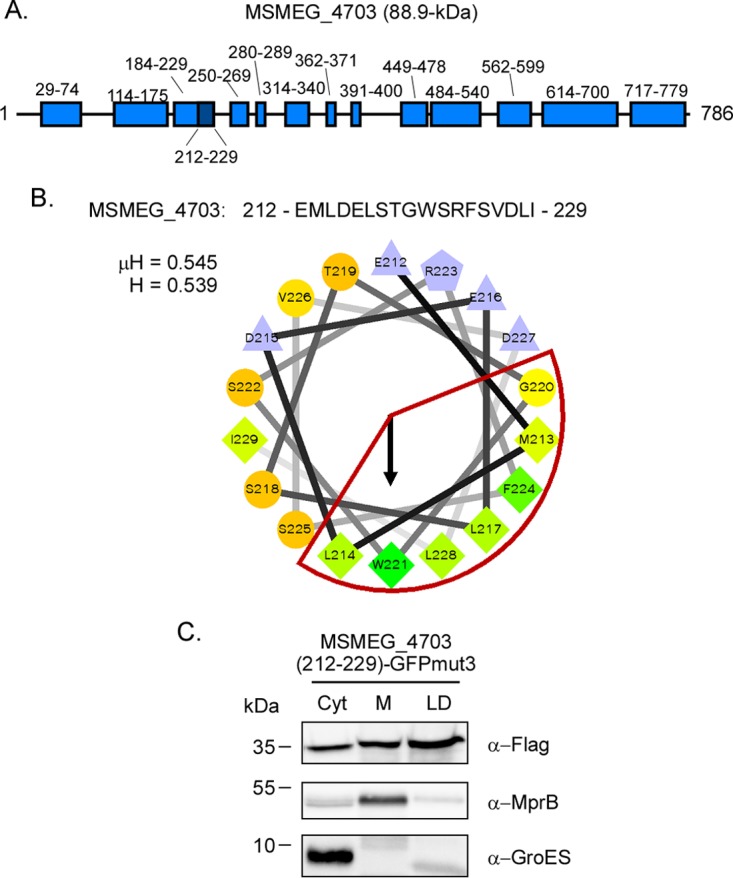
The amphipathic helix from MSMEG_4703 targets GFPmut3 to lipid droplets. (A) Schematic of Psipred-predicted helices of MSMEG_4703. Light blue boxes denote the location of α-helical domains, and the dark blue box denotes the location of the amphipathic helix identified using Heliquest. (B) The amino acid sequence of the amphipathic helix (residues 212 to 229) and the helical wheel diagram of residues 212 to 229. The hydrophobic face of the amphipathic helix is highlighted in red. Hydrophobicity (H) and mean hydrophobic moment (μH) values are noted. The arrow indicates the angle of the mean hydrophobic moment. (C) Western blot showing localization of 3×Flag-MSMEG_4703 (212 to 229) GFPmut3 following production in M. smegmatis ΔpspA. PspAEco was detected using anti-Flag antibody. Anti-GroES and anti-MprB antibodies were used to detect control proteins. Fraction abbreviations: Cyt, cytosol; M, membrane; LD, lipid droplets. The images shown are representative of experiments performed in triplicate.
DISCUSSION
LDs are unique in biology as they represent the only naturally occurring structures utilizing a phospholipid monolayer to separate organic from aqueous phases. Nearly all eukaryotic cells are able to produce LDs, and as a result, much is known about their structure and function within these hosts. However, LD production in bacteria is much less common, and our understanding of prokaryotic LDs is still in its infancy. Recent studies from our laboratory have identified phage shock protein PspA as a constituent of LDs produced by Mycobacterium spp., including M. tuberculosis and M. smegmatis (17). The deletion or overproduction of PspA in these organisms alters the size distribution of LDs and negatively impacts bacterial survival in an in vitro culture model of nonreplicating persistence (17). To begin elucidating the properties of prokaryotic LDs, PspA proteins from M. tuberculosis and an M. smegmatis ΔpspA surrogate host were used as model systems to (i) begin defining the mechanism by which PspA localizes to Mycobacterium LDs and (ii) determine if other Mycobacterium proteins associate with LDs using a similar mechanism. The use of M. smegmatis ΔpspA for these studies was advantageous for several reasons. The isolation of LDs requires the use of aerosol-generating equipment not currently available in our biosafety level 3 laboratory, precluding the use of M. tuberculosis as an LD source. Furthermore, the production of proteins in the ΔpspA mutant background enabled the study of protein-LD interactions independent of interactions between endogenous PspA and cloned PspA variants.
The PspAMtb H1 domain contains 6 large hydrophobic residues (F5, W9, L12, M13, F16, and I20) along the hydrophobic face of the helix and five charged residues (K7, K10, K19, D21, E22) on the hydrophilic face of the helix, resulting in a net charge of +1. The removal of the H1 domain completely abrogated the binding of PspAMtb to LDs. Similarly, cloning of the H1 domain to the N terminus of GFP enabled this otherwise cytoplasmic protein to associate with LDs. An amphipathic helix is present in the N terminus of most if not all PspA/Vipp1 family members that have been characterized to date. Although the N-terminal amphipathic helix present in E. coli PspA (AHa, residues 2 to 19) shares no sequence homology with the PspAMtb H1 domain, PspAEco is capable of associating with LDs when produced in M. smegmatis. Interestingly, only a minor fraction of PspAEco associates with membranes, a predominant site of localization for this protein when produced endogenously in E. coli. While the reasons for this discrepancy are currently unclear, we speculate that the lipid surface area of LDs far exceeds that of the plasma membrane under the conditions utilized in these studies. Alternatively, or in addition to, LDs are expected to possess a much higher stored curvature elastic stress (see below) compared to that of the plasma membrane of unstressed bacteria. In contrast to that of PspAEco, the H1 domain of PspARho differs by only two residues from the H1 domain of PspAMtb (L12M and S17N). PspARho binds LDs when produced in its natural host (16) but is also capable of binding LDs when produced in M. smegmatis. While it remains unclear whether the N-terminal amphipathic helices present in PspAEco and PspARho facilitate their association to LDs, PspAEco variants lacking AHa are deficient in their ability to bind lipids (23, 31, 48). Furthermore, fusion of the AHa domain to the N terminus of GFP promotes the association of this protein with lipid membranes (23). Thus, the N-terminal amphipathic helix present in PspAMtb, and possibly other PspA family members, regulates the binding to Mycobacterium LDs.
Amphipathic helices are a common motif of proteins that interact with lipid membranes. However, amphipathic helices are also an established mechanism by which cytoplasmic proteins associate with the lipid monolayer of LDs produced in eukaryotes (32–36). A recent study indicates that the most important characteristic for an amphipathic helix to bind LDs is the number of large hydrophobic residues (I, F, L, M, W, and Y) that are present on the hydrophobic face of the helix (35). In particular, LD binding is most frequently observed with amphipathic helices possessing at least 5 large hydrophobic residues, with the amount of binding proportional to the number of large hydrophobic residues present (35). In contrast, little if any correlation is observed between LD binding and the net charge or hydrophobic moment of the amphipathic helix (35). The N-terminal amphipathic helices present in PspAMtb, PspAEco, and PspARho each possess 5 or more large hydrophobic residues along their hydrophobic faces. Interestingly, PspAMtb double and triple mutants (F5A/W9A, F5A/F16A, W9A/F16A, and F5A/W9A/F16A) that harbor only 4 or 3 large hydrophobic residues on the hydrophobic face of the H1 helix remain able to bind Mycobacterium LDs. This phenotype is conserved when H1 mutant variants are cloned in front of GFPmut3. Therefore, H1 binding to LDs occurs independently of PspAMtb oligomerization and is able to proceed with only a limited number of large hydrophobic residues relative to other amphipathic helices shown to bind LDs. Interestingly, additional residues along the hydrophilic face of the H1 domain may also contribute to LD binding by PspAMtb. For example, the PspAMtb K7A mutant is reduced in its ability to bind Mycobacterium LDs. A substitution at this residue alters the net charge of the helix from +1 to neutral. Therefore, the K7A substitution may impact LD binding by altering electrostatic interactions between the hydrophilic face of the H1 domain and the negatively charged phospholipids of the LD monolayer. Regardless, our results indicate that the properties of PspAMtb important for its ability to bind LDs are complex and multifaceted and may include interactions that require both faces of the H1 amphipathic helix.
In addition to the H1 amphipathic helix, the ability of PspA family members to bind Mycobacterium LDs may also be related to one or more general characteristics of the phospholipid monolayer surrounding LDs. The H1 domains of PspA and Vipp1 exhibit high affinities for membranes possessing lipid-packing defects (i.e., membranes exhibiting stored curvature elastic [SCE] stress) (23, 24). The spherical conformation of LDs by their very nature induces SCE stress within the phospholipid monolayer due to the space restrictions placed on acyl chains (49). In addition, the neutral lipid core of LDs pokes into the phospholipid monolayer and imparts additional packing defects on the phospholipid acyl chains comprising the LD monolayer (35). Apart from inherent SCE stress, the negatively charged head groups present on the LD phospholipid monolayer are also known to mediate interactions with PspA and Vipp1 (23, 24). As the phospholipid monolayer of bacterial LDs is thought to be derived from the plasma membrane, the M. smegmatis LD phospholipid monolayer is expected to contain several negatively charged lipid species, including phosphatidic acid, phosphatidylserine, phosphatidylglycerol, cardiolipin, and various phosphatidylinositol species (50–52). Thus, the presence of anionic phospholipids and the general lipid packing defects present within the phospholipid monolayer may regulate the localization of PspA family members to LDs.
Apart from PspAMtb, other Mycobacterium proteins may also associate with LDs via amphipathic helices. For example, several of the proteins present on M. bovis BCG LDs contain one or more amphipathic helices with characteristics similar to PspAMtb (18). Furthermore, of the >6,000 proteins predicted to be encoded by the M. smegmatis genome, 330 of these proteins possess PspAMtb-like amphipathic helices that contain five or more large hydrophobic residues along the hydrophobic face. Finally, LC-MS/MS analyses identified a total of ∼480 proteins that are present on LDs isolated from M. smegmatis, and 26 of these possess amphipathic helices similar to the PspAMtb H1 domain. One of these proteins, MSMEG_4703, is a predicted glycerol-3-phosphate acyltransferase. This enzyme functions in the first of two committed steps in the synthesis of phosphatidic acid, a precursor needed to produce both triacylglycerol and anionic phospholipids (53, 54). Cloning of the identified amphipathic helix from MSMEG_4703 (residues 212 to 229) in front of GFPmut3 alters the localization pattern of this protein to now include LDs. The M. tuberculosis homolog of this protein (Rv1551) also contains an amphipathic helix in the same region that differs by only 4 residues from MSMEG_4703 (data not shown). The expression of Rv1551 in E. coli leads to higher levels of several glycerophospholipids within the cell, including phosphatidylglycerol, phosphatidylethanolamine, and cardiolipin (55). Thus, MSMEG_4703/Rv1551 is likely to regulate several aspects of LD biology in Mycobacterium. In addition to MSMEG_4703, several other LD-associated proteins that possess PspAMtb-like amphipathic helices may also regulate the synthesis or metabolism of LDs. These include two predicted glycerol-3-phosphate dehydrogenases (MSMEG_1736/GlpD2 and MSMEG_6761), which are involved in the conversion of glycerol-3-phosphate to dihydroxyacetone and vice versa (56), and glyceraldehyde-3-phosphate dehydrogenase (MSMEG_3084/GapA), which mediates the reversible conversion of glyceraldhehyde-3-phosphate to 3-phospho-d-glyceroyl phosphate. Interestingly, orthologs of these three enzymes have also been identified on LDs produced by R. jostii RHA1 (16), indicating that they, along with PspA and HbhA, may represent part of the core proteome of LDs produced in bacteria.
In summary, our understanding of prokaryotic LDs remains in its infancy. The work described here extends our limited knowledge of these quasi-organelles by defining the proteomic composition of LDs produced in M. smegmatis and by identifying a key structural motif that facilitates the interaction of PspA, as well as other proteins, with LDs produced in Mycobacterium. Going forward, this information may help in the development of novel therapeutics capable of disrupting key LD-protein interactions needed by M. tuberculosis for survival during nonreplicating persistence.
MATERIALS AND METHODS
Bacterial strains, media, and culture conditions.
The strains and plasmids used in this study are described in Table 3. Escherichia coli strains DH5α, Top10, (Thermo Fisher Scientific, Waltham, MA), and NovaBlue (MilliporeSigma, Burlington, MA) were used for cloning. Protein purification was carried out in E. coil strain BL21(DE3) containing plasmid pLysS. All E. coli strains were incubated at 37°C in Luria-Bertani (LB) medium with 100 μg/ml ampicillin, 50 μg/ml kanamycin, 150 μg/ml hygromycin B, and/or 25 μg/ml chloramphenicol for selection, as necessary. Rhodococcus jostii strain RHA1 was kindly provided by Lindsay Eltis (University of British Columbia, Canada) and was cultured at 30°C in LB medium. All Mycobacterium strains are from American Type Culture Collection (ATCC; Manassas, VA) and are derivatives of Mycobacterium tuberculosis H37Rv (ATCC 27294) or Mycobacterium smegmatis mc2155 (ATCC 700084). For general culturing, mycobacteria were incubated at 37°C in Middlebrook 7H9 broth (BD, Franklin Lakes, NJ) with shaking or on Middlebrook 7H10 agar (BD, Franklin Lakes, NJ). Broth and agar media were supplemented with 0.5% glycerol (MilliporeSigma, Burlington, MA), 0.05% Tween 80 (Sigma, St. Louis, MO), and either 10% albumin-dextrose-catalase (ADC; M. smegmatis) or 10% oleic acid-albumin-dextrose-catalase (OADC; M. tuberculosis) (BD, Franklin Lakes, NJ). Mycobacteria were also grown in Sauton's medium (57) at 37°C for 7 days to induce LD production. When needed, Mycobacterium medium was supplemented with 50 μg/ml hygromycin B.
TABLE 3.
Strains and plasmids
| Strain or plasmid | Genotype or descriptiona | Reference or source |
|---|---|---|
| Strains | ||
| E. coli DH5α | F− ϕ80lacZΔM15 Δ(lacZYA-argF)U169 recA1 endA1 hsdR17(rK− mK+) phoA supE44 λ thi-1 byrA96 relA1 | Lab collection |
| E. coli TOP10 | F− mcrA Δ(mrr-hsdRMS-mcrBC) ϕ80lacZΔM15 ΔlacX74 recA1 araD139 Δ(ara-leu)7697 galU galK rpsL str endA1 nupG | Lab collection |
| E. coli BL21(DE3)pLysS | F− ompT hsdSB(rB− mB−) gal dcm (DE3); contains pLysS | Lab collection |
| E. coli NovaBlue Singles | endA1 hsdR17(rK12− mK12+) supE44 thi-1 recA1 gyrA96 relA1 lac F′[proA+B+ lacIqZΔM15::Tn10] (Tetr) | Novagen |
| Rhodococcus jostii RHA1 | Laboratory strain | Gift of Eltis lab |
| M. smegmatis mc2155 | Laboratory strain | ATCC 700084 |
| M. tuberculosis H37Rv | Laboratory strain | ATCC 27294 |
| TCZ2054 | M. smegmatis ΔMSMEG_2695 (ΔpspA) | 17 |
| TCZ2365 | M. smegmatis ΔMSMEG_2695 (ΔpspA) containing pTZ1227 | 17 |
| TCZ2547 | E. coli BL21(DE3) with pLysS containing pTZ1445 | This study |
| TCZ2632 | M. smegmatis ΔMSMEG_2695 (ΔpspA) containing pTZ1477 | This study |
| TCZ2634 | M. smegmatis ΔMSMEG_2695 (ΔpspA) containing pTZ1479 | This study |
| TCZ2642 | E. coli BL21(DE3) with pLysS containing pTZ1482 | This study |
| TCZ2646 | E. coli BL21(DE3) with pLysS containing pTZ1484 | This study |
| TCZ2664 | M. smegmatis ΔMSMEG_2695 (ΔpspA) containing pTZ1494 | This study |
| TCZ2729 | M. smegmatis ΔMSMEG_2695 (ΔpspA) containing pTZ1509 | This study |
| TCZ2731 | M. smegmatis ΔMSMEG_2695 (ΔpspA) containing pTZ1511 | This study |
| TCZ2758 | M. smegmatis ΔMSMEG_2695 (ΔpspA) containing pTZ1534 | This study |
| TCZ2783 | M. smegmatis ΔMSMEG_2695 (ΔpspA) containing pTZ1535 | This study |
| TCZ2784 | M. smegmatis ΔMSMEG_2695 (ΔpspA) containing pTZ1536 | This study |
| TCZ2785 | M. smegmatis ΔMSMEG_2695 (ΔpspA) containing pTZ1537 | This study |
| TCZ2786 | M. smegmatis ΔMSMEG_2695 (ΔpspA) containing pTZ1545 | This study |
| TCZ2787 | M. smegmatis ΔMSMEG_2695 (ΔpspA) containing pTZ1546 | This study |
| TCZ2788 | M. smegmatis ΔMSMEG_2695 (ΔpspA) containing pTZ1547 | This study |
| TCZ2789 | M. smegmatis ΔMSMEG_2695 (ΔpspA) containing pTZ1548 | This study |
| TCZ2790 | M. smegmatis ΔMSMEG_2695 (ΔpspA) containing pTZ1549 | This study |
| TCZ2794 | M. smegmatis ΔMSMEG_2695 (ΔpspA) containing pTZ1550 | This study |
| TCZ2800 | M. smegmatis ΔMSMEG_2695 (ΔpspA) containing pTZ1551 | This study |
| TCZ2801 | M. smegmatis ΔMSMEG_2695 (ΔpspA) containing pTZ1552 | This study |
| TCZ2804 | M. smegmatis ΔMSMEG_2695 (ΔpspA) containing pTZ1555 | This study |
| TCZ2811 | M. smegmatis ΔMSMEG_2695 (ΔpspA) containing pTZ1561 | This study |
| Plasmids | ||
| pCR2.1-TOPO | 3.9-kb plasmid for cloning PCR products; Ampr Kanr | Invitrogen |
| pSTBlue1 | 3.8-kb plasmid for cloning PCR products; Ampr Kanr | Novagen |
| pSE100 | TetR-regulated protein expression vector, Hygr | 59 |
| pET24b | 5.3-kb plasmid for expression in E. coli, Kanr | Novagen |
| pTZ842 | pET24b cloning plasmid with 5′ 3×Flag inserted | 58 |
| pTZ1160 | pSE100 containing 3×Flag-6×His tags flanking the multiple cloning site; Hygr | This study |
| pTZ1167 | pSE100 containing 3×Flag-6×His tags flanking the multiple cloning site, altered cutsites from pTZ1160; Hygr | 58 |
| pTZ1168 | pET-24b containing Rv2744c coding sequence; Kanr | 58 |
| pTZ1227 | pTZ1167 containing Rv2744c (pspA) coding sequence; Hygr | 17 |
| pTZ1445 | pET24b expression vector containing pspAMtbΔH1 | This study |
| pTZ1477 | pTZ1167 containing pspA coding sequence from E. coli BL21; Hygr | This study |
| pTZ1479 | pTZ1167 containing pspA coding sequence from Rhodococcus RHA1; Hygr | This study |
| pTZ1482 | pET24b containing pspAEco | This study |
| pTZ1484 | pET24b containing pspARho | This study |
| pTZ1490 | pspAMtbΔH1 inserted into pTZ842 for addition of 5′ 3×Flag and 3′ 6×His tags | This study |
| pTZ1494 | 3×Flag-pspAMtbΔH1-6×His from pTZ1490 inserted into pSE100 | This study |
| pTZ1509 | pTZ1167 containing GFPmut3 coding sequence; Hygr | This study |
| pTZ1510 | pTZ1167 containing GFPmut3 in-frame for insertion of amphipathic helix | This study |
| pTZ1511 | pTZ1167 containing 3× H1-tagged GFPmut3 coding sequence; Hygr | This study |
| pTZ1514 | pTZ1167 containing HbhA from M. smegmatis (MSMEG_0919) | This study |
| pTZ1526 | pCR2.1 TOPO containing Rv2744c w/NheI and BglII cutsites for mutagenesis/insertion into pTZ1160 | This study |
| pTZ1534 | pTZ1160 containing Rv2744c (pspA) F5A mutant coding sequence; Hygr | This study |
| pTZ1535 | pTZ1160 containing Rv2744c (pspA) W9A mutant coding sequence; Hygr | This study |
| pTZ1536 | pTZ1160 containing Rv2744c (pspA) F5A/W9A double mutant coding sequence; Hygr | This study |
| pTZ1537 | pTZ1160 containing Rv2744c (pspA) F16A mutant coding sequence; Hygr | This study |
| pTZ1539 | pCR2.1-TOPO containing PspA H1 domain fused to GFPmut3 | This study |
| pTZ1545 | pTZ1160 containing Rv2744c (pspA) F5A/F16A double mutant coding sequence; Hygr | This study |
| pTZ1546 | pTZ1160 containing Rv2744c (pspA) F5A/W9A/F16A triple mutant coding sequence; Hygr | This study |
| pTZ1547 | pTZ1160 containing Rv2744c (pspA) W9A/F16A double mutant coding sequence; Hygr | This study |
| pTZ1548 | pTZ1160 containing Rv2744c (pspA) K7A mutant coding sequence; Hygr | This study |
| pTZ1549 | pTZ1160 containing Rv2744c (pspA) Y11A mutant coding sequence; Hygr | This study |
| pTZ1550 | pTZ1167 containing 1× H1-tagged GFPmut3 coding sequence; Hygr | This study |
| pTZ1551 | pTZ1167 containing 1× H1 (W9A) GFPmut3 coding sequence; Hygr | This study |
| pTZ1552 | pTZ1167 containing 1× H1 (W9E) GFPmut3 coding sequence; Hygr | This study |
| pTZ1555 | pTZ1167 containing MSMEG_4703 (residues 212–229) GFPmut3 coding sequence; Hygr | This study |
| pTZ1561 | pTZ1167 containing 1× H1 (F5A/W9A/F16A) GFPmut3 coding sequence; Hygr | This study |
Hygr, hygromycin resistance; Tetr, tetracycline resistance; Kanr, kanamycin resistance; Ampr, ampicillin resistance.
Construction of plasmids.
Primers used in this study are described in Table S1 in the supplemental material and were synthesized by Eurofins Genomic (Louisville, KY). To generate expression constructs, the coding sequences from genes of interest were first amplified by PCR with Platinum Pfx (Thermo Fisher Scientific, Waltham, MA) using primer sets containing engineered restriction endonuclease recognition sequences. Amplified products were then cloned into pCR2.1-TOPO (Thermo Fisher Scientific, Waltham, MA) or pSTBlue-1 (MilliporeSigma, Burlington, MA), and cloned DNA was confirmed by sequencing (MCLAB, San Francisco, CA). The resulting constructs were then digested with the appropriate restriction enzyme(s), and the released product of interest was subcloned into an E. coli or Mycobacterium expression vector. pTZ1227 has been described previously and was the vector used to produce 3×Flag-PspAMtb-6×His in Mycobacterium (17). PspAMtbΔH1 was generated by amplifying M. tuberculosis genomic DNA using primers 2744c_H1FWDNheI and 2744cRev4-NotI. This variant lacks the first 22 amino acids of PspAMtb, which includes the H1 domain. The resulting product was subcloned into pTZ842 (58) to allow incorporation of a 3×Flag epitope at the N terminus and a 6×His epitope tag at the C terminus of PspAMtbΔH1, resulting in pTZ1490. The sequence encoding 3×Flag-PspAMtbΔH1-6×His was then amplified from pTZ1490 using the primers Rv2744cFpSE100PstI and 2744cRpSE100EcoRV, and the resulting product was subcloned into pSE100 for expression in Mycobacterium (59), resulting in pTZ1494. pTZ1168 was used to produce PspAMtb-His in E. coli and has been described previously (58). To produce PspAMtbΔH1-His in E. coli, the sequence encoding PspAMtbΔH1 was amplified from M. tuberculosis H37Rv genomic DNA using primers 2744c_H1FWDNheI and 2744cRev4-NotI and subcloned into pET24b (EMD Millipore, Burlington, MA), resulting in pTZ1445. The GFPmut3 coding sequence was generated by amplifying pMV261-gfpmut3 plasmid DNA (kindly provided by Eric Rubin, Harvard Medical School, Boston, MA) with primers GFPmut3_no_fusion_F and GFPmut3_both_R. The resulting product was then subcloned into pTZ1167, a derivative of pSE100 that carries the coding sequences for 3×Flag and 6×His flanking the multiple cloning site (58), resulting in pTZ1509. To generate chimeras in which the H1 domain from PspAMtb was fused to GFP, gfpmut3 was amplified from pMV261-gfpmut3 plasmid DNA using primers GFPmut3_fusion_F and GFPmut3_both_R. The resulting product was then subcloned into pTZ1167, resulting in pTZ1510. Next, double-stranded DNA encoding three tandemly arranged copies of the H1 domain (the first 22 amino acids of PspAMtb; H11-H12-H13) was chemically synthesized by GeneArt Gene Synthesis (Thermo Fisher Scientific, Waltham, MA). This fragment was designed to incorporate a small linker encoding Ala-Ala-Ser-Ala at the 3′ end of each H1 domain to enhance flexibility. In addition, restriction endonuclease recognition sequences were engineered between and flanking each H1 domain to facilitate downstream cloning procedures. Finally, the fragment encoding 3×H1 was cloned upstream of gfpmut3 in pTZ1510, resulting in pTZ1511. To generate a GFP chimera in which a single copy of the H1 domain was fused to GFP, pTZ1511 was digested with restriction endonucleases to release the fragment encoding H13-GFPmut3. The resulting fragment was then amplified with primers H1_trucation_F and GFPmut3_both_R, and the resulting product was subcloned into pTZ1167, producing pTZ1550. To generate the GFP chimera in which the amphipathic helix present in MSMEG_4703 (amino acids 212 to 229) was fused to gfpmut3, MSMEG_4703_HelixF and MSEMG_4703_HelixR were phosphorylated with T4 DNA kinase, mixed together, and annealed, and the corresponding product was cloned into pTZ1510, resulting in pTZ1555. To generate PspAEco for expression in M. smegmatis, E. coli BL21 chromosomal DNA was amplified with primers F_Eco_pspA_pET24 and R_Eco_pspA_pSE100, and the corresponding product was subcloned into pTZ1167, resulting in pTZ1477. To generate PspARho for expression in M. smegmatis, R. jostii RHA1 chromosomal DNA was amplified with primers F_Rho_pspA_BamHI and R_Rho_pspA_HindIII, and the corresponding product was subcloned into pTZ1167, resulting in pTZ1479. To generate PspAEco-His or PspARho-His for production in E. coli, pspAEco or pspARho was amplified from the chromosomal DNA using primers F_Eco_pspA_pET24 and Eco_R_pET24real or F_Rho_pspA_BamHI and Rho_R_pET24real, respectively. The corresponding products were then subcloned into pET24b, resulting in pTZ1482 (PspAEco-His) and pTZ1484 (PspARho-His). Amino acid substitutions within the H1 domain were generated by site-directed mutagenesis using the Change-IT multiple mutation site-directed mutagenesis kit according to the manufacturer's instructions. The base plasmids used for these reactions were pTZ1526 and pTZ1539. Mutagenic primers were designed to include 15 to 18 bp of sequence homology flanking the codon(s) targeted for mutagenesis. All primers were synthesized to include a 5′ phosphate. The Amp REV primer provided in the mutagenesis kit was used to synthesize the opposite strand. Mutant variants harboring multiple codon substitutions were generated by sequential mutagenesis. Once mutant alleles were generated and nucleotide changes confirmed, the corresponding sequence was amplified from the vector using Rv2744cFWD-NheI and Rv2744c_Rev_BglII (PspAMtb variants) or Rv2744cFWD-NheI and GFPmut3_both_R (H1-GFPmut3 variants). The resulting products were then subcloned into pTZ1160 or pTZ1167, respectively. Using this strategy, the following mutant derivatives were generated: pTZ1534 (PspAMtbF5A), pTZ1535 (PspAMtbW9A), pTZ1536 (PspAMtbF5A/W9A), pTZ1537 (PspAMtbF16A), pTZ1545 (PspAMtbF5A/F16A), pTZ1546 (PspAMtbF5A/W9A/F16A), pTZ1547 (PspAMtbW9A/F16A), pTZ1548 (PspAMtbK7A), pTZ1549 (PspAMtbY11A), pTZ1551 (H1[W9A]-GFPmut3), pTZ1552 (H1[W9E]-GFPmut3), and pTZ1558 (H1[F5A/W9A/F16A]-GFPmut3).
Induction and isolation of Mycobacterium lipid droplets.
Mid-log-phase cultures of M. smegmatis that were grown in 7H9 were washed 3 times in phosphate-buffered saline (PBS) and inoculated in fresh Sauton's medium supplemented with 0.05% Tween 80 at an optical density at 600 nm (OD600) of ∼0.1. The cultures were grown for 1 week at 37°C with shaking, and bacteria were harvested by centrifugation. Bacterial pellets were suspended in 20 ml buffer A (25 mM Tricine, 250 mM sucrose [pH 7.8]) containing 1:1,000 protease inhibitor cocktail (Sigma, St. Louis, MO), and were incubated on ice for at least 30 min. Bacteria were lysed by passage 3 times through a French pressure cell, and the lysate was cleared of large debris by centrifugation. Eight milliliters of cleared lysate was then placed in an SW41 ultracentrifuge tube, and 2 ml of buffer B (20 mM HEPES, 100 mM KCl, 2 mM MgCl2 [pH 7.4]) was carefully layered on top. Samples were centrifuged at ∼100,000 × g for 1 h at 4°C, and LDs were carefully skimmed off the top of the tubes. LDs were washed 3 times in buffer B and stored at −20°C until needed. Cytoplasmic proteins were collected from the middles of the ultracentrifugation tubes and were cleared of contaminants via a second ultracentrifugation step at 100,000 × g and 4°C. Membrane proteins were collected by suspending the pellets from the initial ultracentrifugation step in buffer B with 0.1% Triton X-100 and agitating overnight at 4°C. Cytoplasmic and membrane proteins were stored at −20°C until needed.
SDS-PAGE and Western blotting.
Ten micrograms of total protein was denatured by incubating with 2× SDS loading dye for >10 min at 95°C and resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Proteins were stained with Coomassie brilliant blue (Thermo Fisher Scientific, Waltham, MA) or were transferred onto polyvinylidene difluoride (PVDF) membrane using the Trans-Blot Turbo transfer system (Bio-Rad, Hercules, CA). For Western blotting, membranes were first blocked in TTBS (20 mM Tris-HCl [pH 7.5], 500 mM NaCl, 0.5% Tween 20) containing 5% skim milk for 1 h and then probed with antiserum diluted in TTBS overnight at 4°C. The primary antibodies used included mouse anti-Flag (1:5,000; Sigma, St. Louis, MO), mouse anti-GroES (1:3,000; BEI Resources, NIAID, NIH), and rabbit anti-MprB (1:3,000; Covance, Denver, PA). Membranes were then washed twice for 5 min each in TBS (20 mM Tris-HCl [pH 7.5], 500 mM NaCl) and three times for 10 min each in TTBS before incubating for 2 h at room temperature with goat anti-mouse IgG (1:5,000; Thermo Fisher Scientific, Waltham, MA) or donkey anti-rabbit IgG (1:1,500) secondary antibody conjugated to horseradish peroxidase. Following washing in TBS and TTBS, the blots were then developed using the SuperSignal West Pico chemiluminescent substrate kit (Thermo Fisher Scientific) and visualized on a ChemiDoc Touch imaging system (Bio-Rad, Hercules, CA).
Production and purification of PspAMtb-His, PspAMtbΔH1-His, PspAEco-His, and PspARho-His.
PspA from various bacterial species was produced and purified in E. coli using Ni2+ affinity chromatography. Briefly, E. coli BL21(DE3)pLysS producing PspA-His variants was grown in 1 liter LB medium with selection at 37°C until cells reached mid-exponential phase. Protein expression was then induced by incubation with 1.0 mM IPTG (isopropyl-β-d-thiogalactopyranoside) for 3 h at 37°C. After induction, the cultures were harvested by centrifugation at 8,000 × g at 4°C. The bacterial pellet was suspended in 30 ml binding buffer (50 mM Na2HPO4 [pH 8.0], 300 mM NaCl, 10 mM imidazole) containing 3 μg/ml DNase, 3 μg/ml RNase, and 30 μl protease inhibitor cocktail (all from Sigma, St. Louis, MO) and lysed by passage through a French pressure cell. The total cell lysate was clarified by centrifugation at 25,000 × g for 30 min, rocked with Ni2+-agarose (Qiagen, Venlo, the Netherlands) for 90 min at 4°C, and then processed by batch purification. Bound protein was incubated with binding buffer, followed by incubating twice with wash buffer (50 mM Na2HPO4 [pH 8.0], 300 mM NaCl, 25 mM imidazole). Protein was removed by incubating Ni2+-resin sequentially with 1-ml aliquots of elution buffer (50 mM Na2HPO4 [pH 8.0], 300 mM NaCl, 150 mM imidazole). The collected fractions were then analyzed by SDS-PAGE. Gels were stained with Coomassie brilliant blue to assess the extent of protein induction and enrichment.
Sucrose gradient ultracentrifugation and negative-stain electron microscopy.
The oligomeric status of purified PspA was assessed using sucrose gradient ultracentrifugation. Briefly, 0.5 mg of wild-type PspA, PspA variants, or protein standards (β-amylase, 200 kDa; thyroglobulin, 670 kDa; blue dextran, >2,000 kDa) was loaded onto 10-ml 10 to 50% linear sucrose gradients (Gradient Master; Biocomp Instruments) and resolved using an SW-41 swinging-bucket rotor at 100,000 × g for 16 h and 4°C; 0.5-ml aliquots were then removed sequentially from the top of each gradient, and protein content assessed by measuring A280. The fraction containing the largest amount of protein was then used for negative-stain electron microscopy. Fractions were diluted to 100 μg/ml, adsorbed onto ionized copper/Formvar 400 mesh grids, and imaged on a Hitachi H600 transmission electron microscope (TEM) at an accelerating voltage of 75 kV with a magnification from ×100,000 to ×300,000.
Secondary structure predictions and helical wheel analyses.
Alpha-helical domains were identified using Psipred (http://bioinf.cs.ucl.ac.uk/psipred/) and other secondary structure prediction algorithms. Helical wheel diagrams were generated using the website http://rzlab.ucr.edu/scripts/wheel/wheel.cgi. Residues were colored from most hydrophobic (green) to least hydrophobic (red), with charged residues colored gray. The mean hydrophobic moment (μH) vector of each helix was represented by an arrow, and the magnitude (number) calculated using the equation
where is the vector sum of the hydrophobicity of each residue of helix length, N. In addition, the mean hydrophobicity (H) was also calculated using the equation
where Hi is the hydrophobicity of each residue in helix length N (60). The values for residue hydrophobicities were established by Fauchere et al. (61). Heliquest (http://heliquest.ipmc.cnrs.fr/) was used to screen specific protein determinants, or the complete proteome of M. smegmatis, for potential LD-targeting helices. This online search algorithm slides an 18-residue window of peptides through a user-submitted database, generating helical wheel diagrams of these residues and calculating various helix parameters, including charge (z), mean hydrophobicity, and mean hydrophobic moment. Heliquest searches of the M. bovis BCD LD-associated proteins were conducted using the following parameters: hydrophobicity, μH ranging from 0.40 to 0.75; mean hydrophobic moment, H ranging from 0.40 to 0.60; z, −4 to +4; polar residues, ≥2; uncharged residues (Ser, Thr, Asn, Gln, His), ≥1; no glycine; charged residues, ≤10; no proline at i, i + 3/n − 3, n; no cysteine; no geometric rules; no BlackList. A higher stringency Heliquest search was used to identify PspAMtb-like amphipathic helices present in the M. smegmatis proteome (UniProt). These parameters were based on values from PspAMtb, PspARho, and PspAEco, and included μH ranging from 0.0.467 to 0.546, H ranging from 0.507 to 0.607, z from −4 to +4; polar residues of ≥7, uncharged residues (Ser, Thr, Asn, Gln, and His) of ≥2, no glycine, charged residues of ≤5, no proline at i of i + 3/n − 3, n, no cysteine, no geometric rules, and no BlackList.
Mass spectrometry of the LD proteome.
The protein concentration of LDs was determined using the QuBit protein assay and instrument (Thermo Fisher Scientific, Waltham, MA). One hundred micrograms of LDs was precipitated by chloroform-methanol extraction and suspended in 50 mM ammonium bicarbonate. The samples were reduced with 10 mM dithiothreitol (DTT) for 30 min at 37°C and alkylated with 50 mM iodoacetamide. Excess alkylating agent was removed by the addition of 10 mM DTT. The samples were digested with mass spectrometry-grade Trypsin Gold (Promega, Madison, WI) at an enzyme-to-substrate ratio of 1:50 overnight at 37°C. Digestion reactions were stopped by the addition of trifluoroacetic acid to a final concentration of 0.1%. The samples were then desalted and processed by Zip-Tip C18 columns (MilliporeSigma, Burlington, MA). For each sample, approximately 5 μg of tryptic peptides were loaded via an autosampler (AS-2; Eksigent Technologies Inc.) onto a 100-μm inside diameter (i.d.) trapping column that was hand packed with 2.5 cm of 5-μm 200-Å C18 (Magic C18 AQ) over 6 min in 2% acetonitrile with 0.1% formic acid. Peptides were eluted from the trapping column onto a 75-μm i.d. analytical column that was hand-packed with 10 cm of 3-μm 200-Å C18 (Magic C18 AQ). Elution occurred over 180 min on a 2 to 98% buffer B gradient at a flow rate of 300 nl/min delivered by a NanoLC 2D high-pressure liquid chromatography (HPLC) pump (AB Sciex Eskigent). Buffer A was 0.1% formic acid in H2O and buffer B was 0.1% formic acid in acetonitrile. The HPLC effluent was directly coupled to the nanoelectrospray ionization source of an LTQ-Orbitrap Velos hybrid mass spectrometer (Thermo Fisher Scientific). Precursor ion scans were collected with one internal lock mass at a resolution setting with a resolution of 60,000 for full MS followed by data-dependent and targeted MS/MS acquisitions of the 10 most abundant ions. The normalized collision energy was set to 35. Dynamic exclusion, monoisotopic precursor selection, and predicted ion injection time were enabled. This was done in technical triplicates for each of three independent biological experiments. The LC-MS/MS data were analyzed with MaxQuant software (version 1.2.2.5) (62) using a UniProtKB Mycobacterium smegmatis reference proteome database through MaxQuant's built-in Andromeda search engine. Default parameters were set and protein identification and quantification were performed using the MaxLFQ label-free quantification algorithm previously described. Default parameters in MaxQuant were used wherever applicable. Briefly, variable modifications included protein N-terminal acetylation and oxidized methionine. Carbamidomethylated cysteine was the only fixed modification specified. Full trypsin specificity was selected as digestion mode, and maximum missed cleavages were set to 2. Peptides with lengths of a minimum of 7 amino acids were considered, with both the peptide and protein false discovery rate (FDR) set to 1%. Precursor mass tolerance was set to 20 ppm for the first search and 4.5 ppm for the main search. Product ions were searched with a mass tolerance of 20 ppm. Protein identification required a minimum of three peptides with at least one razor or unique peptide. Label-free quantification was performed using quantities of unique and razor peptides and required a minimum of three peptides. Identified proteins that could be reconstructed from a set of peptides are “grouped” and termed protein groups. The top matched or leading candidate in a protein group is defined as the protein with the greatest number of identified peptides within the group. Protein groups marked as contaminant, reverse, or “identified by site only” in MaxQuant results were discarded.
Statistical analysis of PspAMtb localization.
The abundance of PspAMtb or H1-GFP chimeras was quantified by densitometry using ImageLab (Bio-Rad, Hercules, CA). The relative abundance of protein in the LD fraction, or in the cytoplasm plus membrane fraction, was expressed as a percentage of the total protein signal detected in all three fractions, which was set at 100%. The values shown represent the average percentages of proteins present determined from three independent cultures prepared and processed in parallel. Statistical significance was determined by a two-tailed, unpaired t test with assumed unequal variance.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by a grant from the Potts Foundation to T.C.Z.
We thank Dara Frank, Jenifer Coburn, and Blake Hill for helpful discussions regarding the study.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00240-18.
REFERENCES
- 1.Anonymous. 2017. WHO world tuberculosis report 2017. World Health Organization, Geneva, Switzerland: http://www.who.int/tb/publications/global_report/gtbr2017_main_text.pdf. [Google Scholar]
- 2.Garton NJ, Christensen H, Minnikin DE, Adegbola RA, Barer MR. 2002. Intracellular lipophilic inclusions of mycobacteria in vitro and in sputum. Microbiology 148:2951–2958. doi: 10.1099/00221287-148-10-2951. [DOI] [PubMed] [Google Scholar]
- 3.Caire-Brändli I, Papadopoulos A, Malaga W, Marais D, Canaan S, Thilo L, de Chastellier C. 2014. Reversible lipid accumulation and associated division arrest of Mycobacterium avium in lipoprotein-induced foamy macrophages may resemble key events during latency and reactivation of tuberculosis. Infect Immun 82:476–490. doi: 10.1128/IAI.01196-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Daniel J, Kapoor N, Sirakova T, Sinha R, Kolattukudy P. 2016. The perilipin-like PPE15 protein in Mycobacterium tuberculosis is required for triacylglycerol accumulation under dormancy-inducing conditions. Mol Microbiol 101:784–794. doi: 10.1111/mmi.13422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Daniel J, Maamar H, Deb C, Sirakova TD, Kolattukudy PE. 2011. Mycobacterium tuberculosis uses host triacylglycerol to accumulate lipid droplets and acquires a dormancy-like phenotype in lipid-loaded macrophages. PLoS Pathog 7:e1002093. doi: 10.1371/journal.ppat.1002093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deb C, Lee C-M, Dubey VS, Daniel J, Abomoelak B, Sirakova TD, Pawar S, Rogers L, Kolattukudy PE. 2009. A novel in vitro multiple-stress dormancy model for Mycobacterium tuberculosis generates a lipid-loaded, drug-tolerant, dormant pathogen. PLoS One 4:e6077. doi: 10.1371/journal.pone.0006077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Elamin AA, Stehr M, Spallek R, Rohde M, Singh M. 2011. The Mycobacterium tuberculosis Ag85A is a novel diacylglycerol acyltransferase involved in lipid body formation. Mol Microbiol 81:1577–1592. doi: 10.1111/j.1365-2958.2011.07792.x. [DOI] [PubMed] [Google Scholar]
- 8.Baek SH, Li AH, Sassetti CM. 2011. Metabolic regulation of mycobacterial growth and antibiotic sensitivity. PLoS Biol 9:e1001065. doi: 10.1371/journal.pbio.1001065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Daniel J, Deb C, Dubey VS, Sirakova TD, Abomoelak B, Morbidoni HR, Kolattukudy PE. 2004. Induction of a novel class of diacylglycerol acyltransferases and triacylglycerol accumulation in Mycobacterium tuberculosis as it goes into a dormancy-like state in culture. J Bacteriol 186:5017–5030. doi: 10.1128/JB.186.15.5017-5030.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kapoor N, Pawar S, Sirakova TD, Deb C, Warren WL, Kolattukudy PE. 2013. Human granuloma in vitro model, for TB dormancy and resuscitation. PLoS One 8:e53657. doi: 10.1371/journal.pone.0053657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Low KL, Rao PSS, Shui G, Bendt AK, Pethe K, Dick T, Wenk MR. 2009. Triacylglycerol utilization is required for regrowth of in vitro hypoxic nonreplicating Mycobacterium bovis bacillus Calmette-Guerin. J Bacteriol 191:5037–5043. doi: 10.1128/JB.00530-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Murphy DJ. 2012. The dynamic roles of intracellular lipid droplets: from archaea to mammals. Protoplasma 249:541–585. doi: 10.1007/s00709-011-0329-7. [DOI] [PubMed] [Google Scholar]
- 13.Thiam AR, Farese RV Jr, Walther TC. 2013. The biophysics and cell biology of lipid droplets. Nat Rev Mol Cell Biol 14:775–786. doi: 10.1038/nrm3699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walther TC, Farese RV Jr. 2012. Lipid droplets and cellular lipid metabolism. Annu Rev Biochem 81:687–714. doi: 10.1146/annurev-biochem-061009-102430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ding Y, Zhang S, Yang L, Na H, Zhang P, Zhang H, Wang Y, Chen Y, Yu J, Huo C, Xu S, Garaiova M, Cong Y, Liu P. 2013. Isolating lipid droplets from multiple species. Nat Protoc 8:43–51. doi: 10.1038/nprot.2012.142. [DOI] [PubMed] [Google Scholar]
- 16.Ding Y, Yang L, Zhang S, Wang Y, Du Y, Pu J, Peng G, Chen Y, Zhang H, Yu J, Hang H, Wu P, Yang F, Yang H, Steinbüchel A, Liu P. 2012. Identification of the major functional proteins of prokaryotic lipid droplets. J Lipid Res 53:399–411. doi: 10.1194/jlr.M021899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Armstrong RM, Adams KL, Zilisch JE, Bretl DJ, Sato H, Anderson DM, Zahrt TC. 2016. Rv2744c is a PspA ortholog that regulates lipid droplet homeostasis and nonreplicating persistence in Mycobacterium tuberculosis. J Bacteriol 198:1645–1661. doi: 10.1128/JB.01001-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Low KL, Shui G, Natter K, Yeo WK, Kohlwein SD, Dick T, Rao SPS, Wenk MR. 2010. Lipid droplet-associated proteins are involved in the biosynthesis and hydrolysis of triacylglycerol in Mycobacterium bovis bacillus Calmette-Guerin. J Biol Chem 285:21662–21670. doi: 10.1074/jbc.M110.135731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Manganelli R, Gennaro ML. 2017. Protecting from envelope stress: variations on the phage-shock-protein theme. Trends Microbiol 25:205–216. doi: 10.1016/j.tim.2016.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Joly N, Engl C, Jovanovic G, Huvet M, Toni T, Sheng X, Stumpf MPH, Buck M. 2010. Managing membrane stress: the phage shock protein (Psp) response, from molecular mechanisms to physiology. FEMS Microbiol Rev 34:797–827. doi: 10.1111/j.1574-6976.2010.00240.x. [DOI] [PubMed] [Google Scholar]
- 21.Darwin AJ. 2005. The phage-shock-protein response. Mol Microbiol 57:621–628. doi: 10.1111/j.1365-2958.2005.04694.x. [DOI] [PubMed] [Google Scholar]
- 22.Zhang L, Sakamoto W. 2015. Possible function of VIPP1 in maintaining chloroplast membranes. Biochim Biophys Acta 1847:831–837. doi: 10.1016/j.bbabio.2015.02.013. [DOI] [PubMed] [Google Scholar]
- 23.McDonald C, Jovanovic G, Ces O, Buck M. 2015. Membrane stored curvature elastic stress modulates recruitment of maintenance proteins PspA and Vipp1. mBio 6:e01188-. doi: 10.1128/mBio.01188-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McDonald C, Jovanovic G, Wallace BA, Ces O, Buck M. 2017. Structure and function of PspA and Vipp1 N-terminal peptides: insights into the membrane stress sensing and mitigation. Biochim Biophys Acta 1859:28–39. doi: 10.1016/j.bbamem.2016.10.018. [DOI] [PubMed] [Google Scholar]
- 25.Westphal S, Heins L, Soll J, Vothknecht UC. 2001. Vipp1 deletion mutant of synechocystis: a connection between bacterial phage shock and thylakoid biogenesis? Proc Natl Acad Sci U S A 98:4243–4248. doi: 10.1073/pnas.061501198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gao F, Wang W, Zhang W, Liu C. 2015. α-Helical domains affecting the oligomerization of Vipp1 and its interaction with Hsp70/DnaK in Chlamydomonas. Biochemistry 54:4877–4889. doi: 10.1021/acs.biochem.5b00050. [DOI] [PubMed] [Google Scholar]
- 27.Joly N, Burrows PC, Engl C, Jovanovic G, Buck M. 2009. A lower-order oligomer form of phage shock protein A (PspA) stably associates with the hexameric AAA(+) transcription activator protein PspF for negative regulation. J Mol Biol 394:764–775. doi: 10.1016/j.jmb.2009.09.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Standar K, Mehner D, Osadnik H, Berthelmann F, Hause G, Lünsdorf H, Brüser T. 2008. PspA can form large scaffolds in Escherichia coli. FEBS Lett 582:3585–3589. doi: 10.1016/j.febslet.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 29.Kobayashi R, Suzuki T, Yoshida M. 2007. Escherichia coli phage-shock protein A (PspA) binds to membrane phospholipids and repairs proton leakage of the damaged membranes. Mol Microbiol 66:100–109. doi: 10.1111/j.1365-2958.2007.05893.x. [DOI] [PubMed] [Google Scholar]
- 30.Hankamer BD, Elderkin SL, Buck M, Nield J. 2004. Organization of the AAA(+) adaptor protein PspA is an oligomeric ring. J Biol Chem 279:8862–8866. doi: 10.1074/jbc.M307889200. [DOI] [PubMed] [Google Scholar]
- 31.Jovanovic G, Mehta P, McDonald C, Davidson AC, Uzdavinys P, Ying L, Buck M. 2014. The N-terminal amphipathic helices determine regulatory and effector functions of phage shock protein A (PspA) in Escherichia coli. J Mol Biol 426:1498–1511. doi: 10.1016/j.jmb.2013.12.016. [DOI] [PubMed] [Google Scholar]
- 32.Bouvet S, Golinelli-Cohen MP, Contremoulins V, Jackson CL. 2013. Targeting of the Arf-GEF GBF1 to lipid droplets and Golgi membranes. J Cell Sci 126:4794–4805. doi: 10.1242/jcs.134254. [DOI] [PubMed] [Google Scholar]
- 33.Drin G, Antonny B. 2010. Amphipathic helices and membrane curvature. FEBS Lett 584:1840–1847. doi: 10.1016/j.febslet.2009.10.022. [DOI] [PubMed] [Google Scholar]
- 34.Krahmer N, Guo Y, Wilfling F, Hilger M, Lingrell S, Heger K, Newman HW, Schmidt-Supprian M, Vance DE, Mann M, Farese RV Jr, Walther TC. 2011. Phosphatidylcholine synthesis for lipid droplet expansion is mediated by localized activation of CTP:phosphocholine cytidylyltransferase. Cell Metab 14:504–515. doi: 10.1016/j.cmet.2011.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Prévost C, Sharp ME, Kory N, Lin Q, Voth GA, Farese RV Jr, Walther TC. 2018. Mechanism and determinants of amphipathic helix-containing protein targeting to lipid droplets. Dev Cell 44:73.e4–86.e4. doi: 10.1016/j.devcel.2017.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rowe ER, Mimmack ML, Barbosa AD, Haider A, Isaac I, Ouberai MM, Thiam AR, Patel S, Saudek V, Siniossoglou S, Savage DB. 2016. Conserved amphipathic helices mediate lipid droplet targeting of perilipins 1-3. J Biol Chem 291:6664–6678. doi: 10.1074/jbc.M115.691048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gautier R, Douguet D, Antonny B, Drin G. 2008. HELIQUEST: a web server to screen sequences with specific alpha-helical properties. Bioinformatics 24:2101–2102. doi: 10.1093/bioinformatics/btn392. [DOI] [PubMed] [Google Scholar]
- 38.Dworkin J, Jovanovic G. 2000. The PspA protein of Escherichia coli is a negative regulator of sigma(54)-dependent transcription. J Bacteriol 182:311–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Elderkin S, Bordes P, Jones S, Rappas M, Buck M. 2005. Molecular determinants for PspA-mediated repression of the AAA transcriptional activator PspF. J Bacteriol 187:3238–3248. doi: 10.1128/JB.187.9.3238-3248.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lanfranconi MP, Alvarez HM. 2016. Functional divergence of HBHA from Mycobacterium tuberculosis and its evolutionary relationship with TadA from Rhodococcus opacus. Biochimie 127:241–248. doi: 10.1016/j.biochi.2016.06.002. [DOI] [PubMed] [Google Scholar]
- 41.de Lima CS, Marques MA, Debrie AS, Almeida EC, Silva CA, Brennan PJ, Sarno EN, Menozzi FD, Pessolani MC. 2009. Heparin-binding hemagglutinin (HBHA) of Mycobacterium leprae is expressed during infection and enhances bacterial adherence to epithelial cells. FEMS Microbiol Lett 292:162–169. doi: 10.1111/j.1574-6968.2009.01488.x. [DOI] [PubMed] [Google Scholar]
- 42.Esposito C, Marasco D, Delogu G, Pedone E, Berisio R. 2011. Heparin-binding hemagglutinin HBHA from Mycobacterium tuberculosis affects actin polymerisation. Biochem Biophys Res Commun 410:339–344. doi: 10.1016/j.bbrc.2011.05.159. [DOI] [PubMed] [Google Scholar]
- 43.Lebrun P, Raze D, Fritzinger B, Wieruszeski JM, Biet F, Dose A, Carpentier M, Schwarzer D, Allain F, Lippens G, Locht C. 2012. Differential contribution of the repeats to heparin binding of HBHA, a major adhesin of Mycobacterium tuberculosis. PLoS One 7:e32421. doi: 10.1371/journal.pone.0032421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Menozzi FD, Bischoff R, Fort E, Brennan MJ, Locht C. 1998. Molecular characterization of the mycobacterial heparin-binding hemagglutinin, a mycobacterial adhesin. Proc Natl Acad Sci U S A 95:12625–12630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mueller-Ortiz SL, Wanger AR, Norris SJ. 2001. Mycobacterial protein HbhA binds human complement component C3. Infect Immun 69:7501–7511. doi: 10.1128/IAI.69.12.7501-7511.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pethe K, Aumercier M, Fort E, Gatot C, Locht C, Menozzi FD. 2000. Characterization of the heparin-binding site of the mycobacterial heparin-binding hemagglutinin adhesin. J Biol Chem 275:14273–14280. doi: 10.1074/jbc.275.19.14273. [DOI] [PubMed] [Google Scholar]
- 47.Zhang C, Yang L, Ding Y, Wang Y, Lan L, Ma Q, Chi X, Wei P, Zhao Y, Steinbuchel A, Zhang H, Liu P. 2017. Bacterial lipid droplets bind to DNA via an intermediary protein that enhances survival under stress. Nat Commun 8:15979. doi: 10.1038/ncomms15979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Otters S, Braun P, Hubner J, Wanner G, Vothknecht UC, Chigri F. 2013. The first alpha-helical domain of the vesicle-inducing protein in plastids 1 promotes oligomerization and lipid binding. Planta 237:529–540. doi: 10.1007/s00425-012-1772-1. [DOI] [PubMed] [Google Scholar]
- 49.Bigay J, Antonny B. 2012. Curvature, lipid packing, and electrostatics of membrane organelles: defining cellular territories in determining specificity. Dev Cell 23:886–895. doi: 10.1016/j.devcel.2012.10.009. [DOI] [PubMed] [Google Scholar]
- 50.Bansal-Mutalik R, Nikaido H. 2014. Mycobacterial outer membrane is a lipid bilayer and the inner membrane is unusually rich in diacyl phosphatidylinositol dimannosides. Proc Natl Acad Sci U S A 111:4958–4963. doi: 10.1073/pnas.1403078111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chiaradia L, Lefebvre C, Parra J, Marcoux J, Burlet-Schiltz O, Etienne G, Tropis M, Daffe M. 2017. Dissecting the mycobacterial cell envelope and defining the composition of the native mycomembrane. Sci Rep 7:12807. doi: 10.1038/s41598-017-12718-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Crellin PK, Luo C-Y, Morita YS. 2013. Chapter 6. Metabolism of plasma membrane lipids in mycobacteria and corynebacteria. In Baez RV (ed), Lipid metabolism. InTech, Rijeka, Croatia. doi: 10.5772/52781. [DOI] [Google Scholar]
- 53.Gao Q, Shang Y, Huang W, Wang C. 2013. Glycerol-3-phosphate acyltransferase contributes to triacylglycerol biosynthesis, lipid droplet formation, and host invasion in Metarhizium robertsii. Appl Environ Microbiol 79:7646–7653. doi: 10.1128/AEM.02905-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hayashi JM, Luo CY, Mayfield JA, Hsu T, Fukuda T, Walfield AL, Giffen SR, Leszyk JD, Baer CE, Bennion OT, Madduri A, Shaffer SA, Aldridge BB, Sassetti CM, Sandler SJ, Kinoshita T, Moody DB, Morita YS. 2016. Spatially distinct and metabolically active membrane domain in mycobacteria. Proc Natl Acad Sci U S A 113:5400–5405. doi: 10.1073/pnas.1525165113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Law JD, Daniel J. 2017. The mycobacterial Rv1551 glycerol-3-phosphate acyltransferase enhances phospholipid biosynthesis in cell lysates of Escherichia coli. Microb Pathog 113:269–275. doi: 10.1016/j.micpath.2017.10.050. [DOI] [PubMed] [Google Scholar]
- 56.Pol A, Gross SP, Parton RG. 2014. Review: biogenesis of the multifunctional lipid droplet: lipids, proteins, and sites. J Cell Biol 204:635–646. doi: 10.1083/jcb.201311051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Allen B. 1998. Mycobacteria, p 15–30. In Parish T, Stoker N (ed), Mycobacteria protocols, vol 101 Humana Press, New York, NY. [Google Scholar]
- 58.White MJ, Savaryn JP, Bretl DJ, He H, Penoske RM, Terhune SS, Zahrt TC. 2011. The HtrA-Like serine protease PepD interacts with and modulates the Mycobacterium tuberculosis 35-kDa antigen outer envelope protein. PLoS One 6:e18175. doi: 10.1371/journal.pone.0018175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Guo XV, Monteleone M, Klotzsche M, Kamionka A, Hillen W, Braunstein M, Ehrt S, Schnappinger D. 2007. Silencing essential protein secretion in Mycobacterium smegmatis by using tetracycline repressors. J Bacteriol 189:4614–4623. doi: 10.1128/JB.00216-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Eisenberg D, Weiss RM, Terwilliger TC. 1982. The helical hydrophobic moment: a measure of the amphiphilicity of a helix. Nature 299:371–374. doi: 10.1038/299371a0. [DOI] [PubMed] [Google Scholar]
- 61.Fauchere J, Pliska V. 1983. Hydrophobic parameters pi of amino-acid side chains from the partitioning of N-acetyl-amino-acid amides. Eur J Med Chem 18:369–975. [Google Scholar]
- 62.Cox J, Mann M. 2008. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol 26:1367. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 63.Ding Y, Yang L, Zhang S, Wang Y, Du Y, Pu J, Peng G, Chen Y, Zhang H, Yu J, Hang H, Wu P, Yang F, Yang H, Steinbüchel A, Liu P. 2012. Identification of the major functional proteins of prokaryotic lipid droplets. J Lipid Res 53:399–411. doi: 10.1194/jlr.M021899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen Y, Ding Y, Yang L, Yu J, Liu G, Wang X, Zhang S, Yu D, Song L, Zhang H, Zhang C, Huo L, Huo C, Wang Y, Du Y, Zhang H, Zhang P, Na H, Xu S, Zhu Y, Xie Z, He T, Zhang Y, Wang G, Fan Z, Yang F, Liu H, Wang X, Zhang X, Zhang MQ, Li Y, Steinbüchel A, Fujimoto T, Cichello S, Yu J, Liu P. 2014. Integrated omics study delineates the dynamics of lipid droplets in Rhodococcus opacus PD630. Nucleic Acids Res 42:1052–1064. doi: 10.1093/nar/gkt932. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.