

Disulfide bond formation has a great impact on bacterial pathogenicity. Thus, disulfide-bond-forming proteins represent new targets for the development of antibacterials, since the inhibition of disulfide bond formation would result in the simultaneous loss of the activity of several classes of virulence factors. Here, we identified five candidate proteins encoded by the M. tuberculosis genome as possible substrates of the M. tuberculosis VKOR protein involved in disulfide bond formation. We then reconstituted the mycobacterial disulfide bond formation pathway in E. coli and showed that of the five candidates, only M. tuberculosis DsbA is efficiently oxidized by VKOR in E. coli. We also present evidence for the involvement of VKOR in DsbA oxidation in M. smegmatis.
KEYWORDS: Dsb proteins, VKOR, disulfide bond, mycobacteria, thioredoxin-like protein
ABSTRACT
Disulfide bonds influence the stability and activity of many proteins. In Escherichia coli, the DsbA and DsbB enzymes promote disulfide bond formation. Other bacteria, including the Actinobacteria, use instead of DsbB the enzyme vitamin K epoxide reductase (VKOR), whose gene is found either fused to or in the same operon as a dsbA-like gene. Mycobacterium tuberculosis and other Gram-positive actinobacteria secrete many proteins with even numbers of cysteines to the cell envelope. These organisms have predicted oxidoreductases and VKOR orthologs. These findings indicate that such bacteria likely form disulfide bonds in the cell envelope. The M. tuberculosis vkor gene complements an E. coli dsbB deletion strain, restoring the oxidation of E. coli DsbA. While we have suggested that the dsbA gene linked to the vkor gene may express VKOR's partner in mycobacteria, others have suggested that two other extracytoplasmic oxidoreductases (DsbE or DsbF) may be catalysts of protein disulfide bond formation. However, there is no direct evidence for interactions of VKOR with either DsbA, DsbE, or DsbF. To identify the actual substrate of VKOR, we identified two additional predicted extracytoplasmic DsbA-like proteins using bioinformatics analysis of the M. tuberculosis genome. Using the five potential DsbAs, we attempted to reconstitute disulfide bond pathways in E. coli and in Mycobacterium smegmatis, a close relative of M. tuberculosis. Our results show that only M. tuberculosis DsbA is oxidized by VKOR. Comparison of the properties of dsbA- and vkor-null mutants in M. smegmatis shows parallels to the properties of dsb mutations in E. coli.
IMPORTANCE Disulfide bond formation has a great impact on bacterial pathogenicity. Thus, disulfide-bond-forming proteins represent new targets for the development of antibacterials, since the inhibition of disulfide bond formation would result in the simultaneous loss of the activity of several classes of virulence factors. Here, we identified five candidate proteins encoded by the M. tuberculosis genome as possible substrates of the M. tuberculosis VKOR protein involved in disulfide bond formation. We then reconstituted the mycobacterial disulfide bond formation pathway in E. coli and showed that of the five candidates, only M. tuberculosis DsbA is efficiently oxidized by VKOR in E. coli. We also present evidence for the involvement of VKOR in DsbA oxidation in M. smegmatis.
INTRODUCTION
Disulfide bond formation plays an important role in protein folding and function. In Gram-negative bacteria, the formation of disulfide bonds in proteins is directly catalyzed by enzymes that are localized to the periplasmic space. In Gram-positive bacteria, such enzymes are anchored to the cytoplasmic membrane, with their active domains located in the extracytoplasmic space. So far, all of the many bacterial enzymes identified that promote disulfide bond formation are members of the thioredoxin superfamily, containing the Cys-X-X-Cys active-site motif characteristic of this family. The Escherichia coli pathway consists of the periplasmic enzyme DsbA, which catalyzes the formation of disulfide bonds when its two active-site cysteines are joined in a disulfide bond. DsbA donates its disulfide bond to substrate proteins and in the process becomes reduced. Therefore, a cytoplasmic transmembrane protein, DsbB, oxidizes this reduced DsbA, regenerating DsbA's disulfide bond to start a new round of substrate oxidation. In this oxidative step, electrons are transferred from DsbA to DsbB's cysteines and thence to quinones and, ultimately, to oxygen under aerobic growth conditions (1).
In many bacteria, the enzyme that oxidizes DsbA has been identified as DsbB. However, a bioinformatic analysis based on the genome sequences of nearly 400 bacteria revealed a somewhat different pathway for disulfide bond formation. Bacteria such as Actinobacteria, Cyanobacteria, and some deltaproteobacteria use vitamin K epoxide reductase (VKOR) instead of DsbB, whose gene is found either fused to or in the same operon as a dsbA homolog. Several lines of evidence are consistent with the proposal that VKOR performs the same function as DsbB, even though the two proteins are not homologs. First, the existence of vkor in an operon with or fused to a dsbA homolog in many bacteria is suggestive. Second, we showed previously that when the vkor gene is cloned from Mycobacterium tuberculosis, it complements an E. coli dsbB deletion strain, restoring the oxidation of E. coli DsbA and disulfide bond formation (2). Third, the crystal structure of fused VKOR-DsbA from Synechococcus sp. shows interactions and a disulfide bond joining the two parts of the protein, consistent with a mechanism for the oxidation of DsbA similar to that for DsbB (3). Finally, a detailed comparison of the mechanism of electron transfer between M. tuberculosis VKOR and E. coli DsbA reveals strong parallels (4).
M. tuberculosis causes tuberculosis, which results in 1.5 million deaths every year, and recently, the emergence of strains resistant to multiple antibiotics has posed a great threat to the treatment of the disease. Proteins, including virulence factors as well as other exported proteins, from the M. tuberculosis predicted proteome contain even numbers of cysteines, which suggests that the formation of disulfide bonds might be important for the folding and functioning of these proteins (5, 6). The vkor gene is essential for the growth of M. tuberculosis (7), and the deletion of a vkor homolog in a close relative and oft-used, faster-growing surrogate for M. tuberculosis, Mycobacterium smegmatis, causes a severe growth defect (5). In M. tuberculosis, there are other potential extracytoplasmic thioredoxin homologs that might be the direct catalysts of protein disulfide bond formation and possible substrates of VKOR. These potential candidates include DsbE, DsbF, and the product of Rv2969c (which is located upstream of the vkor gene, Rv2968c [for the reasons mentioned above, the protein product of this gene has been named DsbA]). Both DsbE and DsbF display oxidase activity in vitro (8, 9). DsbA is a membrane-anchored protein and itself contains a structural disulfide bond (10–12). DsbA also shows oxidase activity in vitro and binds in a cysteine-dependent manner to a hexapeptide derived from the periplasmic loop of VKOR (10). Another report shows that the DsbA protein exhibits only isomerase activity in vitro (12). However, none of these proteins have yet to be shown to interact with M. tuberculosis VKOR (MtVKOR).
Thus, here, in addition to the three above-mentioned thioredoxin candidates, we have identified two other likely exported thioredoxin-like proteins in the M. tuberculosis genome by a bioinformatics approach, and we seek to identify the substrate of VKOR by reconstituting a disulfide bond pathway in an E. coli strain missing both its DsbB and DsbA proteins by expressing M. tuberculosis VKOR and each of the M. tuberculosis extracytoplasmic thioredoxin-like candidates. Our results show that only MtDsbA is efficiently oxidized by MtVKOR. Furthermore, the expression of MtDsbA and MtVKOR in M. smegmatis presents additional direct evidence for the involvement of VKOR in DsbA oxidation. A comparison of the properties of dsbA- and vkor-null mutants in M. smegmatis shows parallels to the properties of dsbA and dsbB mutations in E. coli.
RESULTS
Identification of five thioredoxin-like exported proteins of M. tuberculosis as potential substrates of MtVKOR.
We used a bioinformatics approach to predict the DsbA-like candidates in M. tuberculosis (see Materials and Methods). First, we assumed that since E. coli DsbA (EcDsbA) is efficiently oxidized by MtVKOR in E. coli, VKOR's substrate would be a member of the thioredoxin family, as is DsbA (2). Second, we assumed that a candidate DsbA would be a protein exported to the cell envelope since this is the location of other DsbAs and of VKOR's substrate, EcDsbA, when it is functional in E. coli. Therefore, using a hidden Markov model (13), we sought candidate proteins in M. tuberculosis that were members of the thioredoxin family and that exhibited signal sequences or transmembrane components when assessed by the Web-based prediction program Phobius (14), which predicted such proteins to have the active enzymatic portion of the protein localized to the extracytoplasmic space. We identified five proteins in the M. tuberculosis genome that fulfilled these criteria (see Table S1 in the supplemental material): (i) Rv2969c (dsbA product), which has a transmembrane sequence and was shown to be located in the membrane fraction (11), and (ii) Rv0526, Rv0816c, Rv1677, and Rv2878c, which have a signal peptide or transmembrane segment, indicating the export of their thioredoxin domains to the cell envelope. Rv0526 has been annotated as a possible thiroredoxin and was found in a proteomics study to be secreted by cultured M. tuberculosis cells (15). Rv0816c (thiX product) has also been annotated as a thioredoxin family member, and it is conserved in mycobacteria. Rv1677 (dsbF product) is a conserved lipoprotein possibly involved in thiol-disulfide interchange, and it shares high similarity with the C terminus of Rv2878c (dsbE product). The crystal structures of Rv1677 and Rv2878c indicate that both proteins exhibit the typical thioredoxin fold (8, 9).
Reconstitution of the M. tuberculosis disulfide bond formation pathway in E. coli.
Initially, we attempted to reconstitute the disulfide bond formation pathway of M. tuberculosis in E. coli by cloning into a pBAD33 plasmid the predicted gene of each thioredoxin candidate with a myc tag at the carboxy termini. However, with the full arabinose induction of this promoter, neither tagged dsbF nor tagged dsbE was expressed in E. coli at detectable levels by Western blotting using anti-myc antibody. These low expression levels may be due to poor translation or to problems in recognizing possible N-terminal mycobacterial signal sequences properly. Thus, we constructed versions of these genes in which the signal sequences or transmembrane sequences were truncated from the N termini of the proteins and replaced with the E. coli TorT cotranslational signal sequence (16). With these constructs, each thioredoxin homolog should be translocated into the E. coli periplasm and expressed as a soluble periplasmic protein with its signal sequence removed. In addition, the truncated proteins are all fused to a FLAG tag at their C termini. These new constructs were introduced into the pBAD33 plasmid, where the proteins were all expressed by the addition of arabinose to the growth medium.
We first tested the ability of each His-MtVKOR/M. tuberculosis thioredoxin-FLAG pair to restore disulfide bond formation to an E. coli strain deleted for its dsbA and dsbB genes by assessing the motility of such strains. Motility promoted by the flagella of the bacteria is dependent on the protein FlgI, a component of E. coli flagella, which requires disulfide bonds for its stability and activity. Thus, the ΔdsbA ΔdsbB strain is nonmotile. Previously, we have shown that MtVKOR can restore the motility of E. coli ΔdsbB due to its ability to oxidize EcDsbA (2). We proceeded to express both MtVKOR (from a pTrc plasmid, induced with isopropyl-β-d-thiogalactopyranoside [IPTG]) and, independently, each of the five predicted exported thioredoxins in the E. coli ΔdsbA ΔdsbB strain induced by the addition of arabinose (Fig. 1). We found that only MtDsbA, when coexpressed with MtVKOR in a ΔdsbA ΔdsbB mutant, can confer motility similar to that of the control strain coexpressing EcDsbA with MtVKOR (Fig. 1). Motility was dependent on the presence of both mycobacterial proteins. This result is consistent with the suggestion that the MtDsbA protein is the only one of the thioredoxin-like proteins that is an effective substrate of MtVKOR and that can promote disulfide bond formation.
FIG 1.
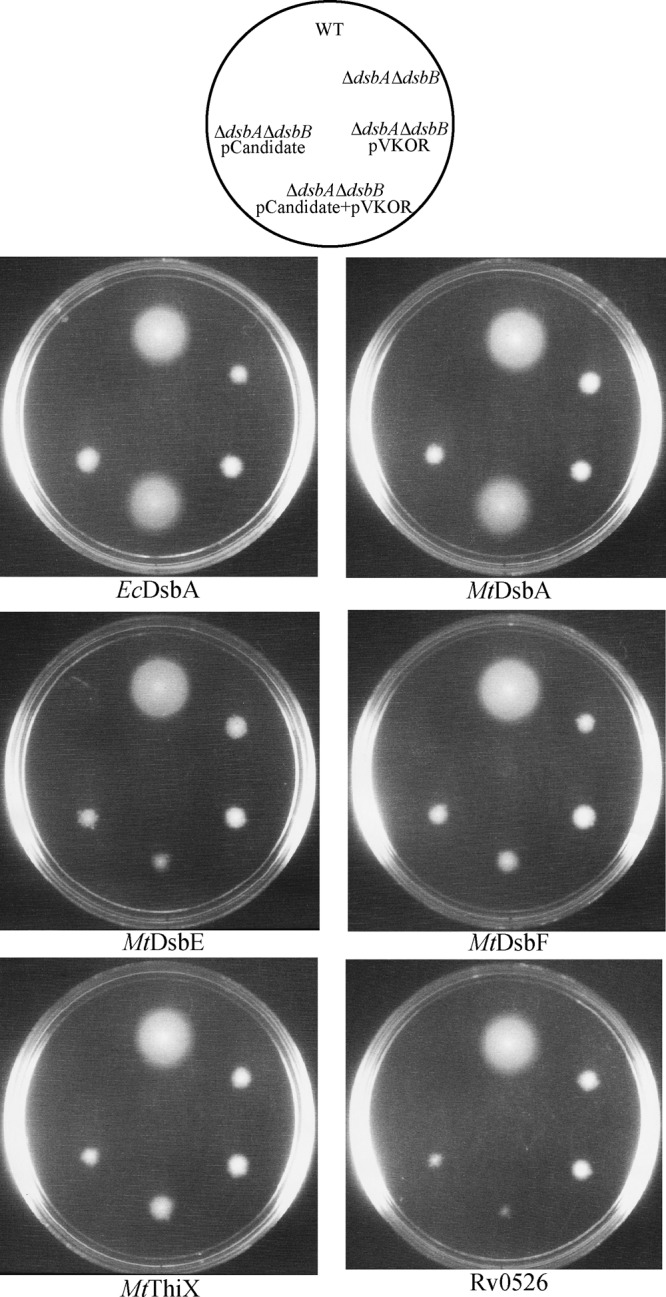
MtDsbA restores motility of the E. coli ΔdsbAB strain when coexpressed with MtVKOR. Shown are data from motility assays to test the five thioredoxin candidates as partners of MtVKOR in their ability to restore disulfide bond formation in E. coli. Strains used were as follows: WT (wild type), NK225 (HK295 with pTrc99a and pMER200); ΔdsbA ΔdsbB strain, NK211 (HK329 with pTrc99a and pMER200); ΔdsbA ΔdsbB pVKOR strain, NK212 (HK329 with pTrc99a-MtVKOR and pMER200); ΔdsbA ΔdsbB pVKOR + pCandidate strains, NK220, NK218, NK214, NK216, NK222, and NK224 (HK329 with pTrc99a-MtVKOR and pMER200 carrying each thioredoxin gene); and ΔdsbA ΔdsbB pCandidate strains, NK219, NK217, NK213, NK215, NK221, and NK223 (HK329 with pTrc99a and pMER200 carrying each thioredoxin). Cells were grown on soft-agar M63 plates (0.3% agar) with 0.2% maltose as the carbon source, 100 μg/ml ampicillin, 10 μg/ml chloramphenicol, 1 mM IPTG to induce the expression of MtVKOR, and 0.2% arabinose to induce the expression of thioredoxins. The plates were incubated at 30°C for 2 days. Images show representative results from at least three independent experiments.
Oxidation of MtDsbA depends on MtVKOR in E. coli.
There are alternative explanations for the negative results observed with four of the five thioredoxin-like proteins in the motility assay. Two of these explanations are (i) that the four thioredoxins are expressed at a much lower level than MtDsbA and (ii) that the four thioredoxins can be oxidized by MtVKOR, but they are unable to oxidize FlgI (see Discussion for an additional alternative explanation). In order to test these explanations, we determined the expression level and the oxidation state of each candidate protein in the presence of the cysteine-alkylating agent α-[3-(3-maleimido-1-oxopropyl)amino]propyl-ω-methoxy polyoxyethylene (Mal-PEG2k) in these strains when coexpressed with MtVKOR. With this agent, one can distinguish between proteins with free cysteines and those with cysteines joined in disulfide bonds via the substantial molecular weight differences that the alkylating agent (Mal-PEG2k) would add to the molecular weight of proteins with free cysteines. These differences are readily observable by immunoblotting.
Our results show that there are no dramatic differences in expression levels of the five M. tuberculosis thioredoxins when fully induced by arabinose (Fig. 2A; see also Fig. S1A in the supplemental material), thus confirming that the lack of motility of the ΔdsbA ΔdsbB strain was not due to a lack of expression of the candidates. In addition, we observed that without the presence of MtVKOR, all six thioredoxins (including EcDsbA) are mostly in the reduced form (Fig. 2A and Fig. S1, lane 2), as expected, since there is no reoxidizing partner in the strain. However, when MtVKOR is coexpressed, MtDsbA and EcDsbA are present mostly in the oxidized form, while the other thioredoxins remain in the reduced state (Fig. 2A and Fig. S1, lane 4). We note that in addition to the two cysteines in the active-site C89XXC92 motif, MtDsbA has two additional cysteines that form a structural disulfide bond (10, 12). Thus, three bands of MtDsbA appear in the Western blot; the middle band is the form of the protein that carries one disulfide bond and two free cysteines (partially oxidized). For this middle band, we cannot determine whether the disulfide bond is formed between the two cysteines in the C89XXC92 motif or is a structural disulfide bond between the non-active-site cysteines (C140-C192). Even in the presence of MtVKOR, not all MtDsbA is fully oxidized, but the appearance of both the fully oxidized form and a form with only one disulfide bond indicates that this VKOR/thioredoxin pair is restoring disulfide bond formation. The degree to which EcDsbA and MtDsbA are oxidized in these experiments may be due to the levels of expression of MtVKOR or due to a decreased efficiency of MtVKOR (2). Three of the other thioredoxin-like proteins exhibit either none or an extremely small amount of the oxidized form in the presence of MtVKOR (Fig. S1, lane 4).
FIG 2.

Oxidation of MtDsbA is dependent on MtVKOR in E. coli. (A) In vivo oxidation states of MtDsbA and EcDsbA with or without MtVKOR in E. coli. MtdsbA and EcdsbA were cloned into the pBAD33 vector and expressed in a truncated form (deletion of its original signal peptide or transmembrane sequence) using an E. coli TorT signal sequence in the ΔdsbA ΔdsbB strain. His-MtVKOR was expressed from the pTrc99a plasmid. Cells were grown in M63 medium with antibiotics and 0.2% arabinose to induce the expression of the FLAG-tagged proteins. A total of 1 mM IPTG was used to induce the expression of His-MtVKOR. Proteins were TCA precipitated and then alkylated with Mal-PEG2k. DsbA proteins were detected with anti-FLAG antibody. The sample without Mal-PEG2k treatment shows the position of the oxidized protein, which is the same as that of the reduced protein with no alkylating agent present. The sample with Mal-PEG2k refers to the position of the protein with reduced cysteines that are alkylated with Mal-PEG2k, which adds to the molecular weight of the protein. red, reduced; ox, oxidized. Blots show representative results from at least two independent experiments. (B) Oxidation states of full-length MtDsbA in the E. coli ΔdsbA ΔdsbB strain. MtDsbA-myc or MtDsbAC92A-myc was cloned with its native transmembrane sequence into pBAD33. His-MtVKOR or His-MtVKORC57A was cloned into pTrc99a. Cell growth and sample preparation were done as described in Materials and Methods. MtDsbA was detected with anti-myc antibody. *, nonspecific band (lane 1) when alkylated at 49 kDa and when not alkylated at 37 kDa. 0, 1, 2, 3, and 4 indicate the numbers of cysteines (Cys) labeled with Mal-PEG2k. The blot shows a representative result from two independent experiments.
Because MtDsbA is the only one of the M. tuberculosis proteins that was oxidized in the presence of MtVKOR, we further investigated the steps in its oxidation. For this, we expressed MtDsbA as a full-length protein with its transmembrane sequence and a C-terminal myc tag in E. coli from a pBAD plasmid. The oxidation of MtDsbA depends on MtVKOR, as shown in Fig. 2B (lanes 6 and 7). Without the expression of MtVKOR, there is reduced (form 4) (Fig. 2B, lane 6) and partially oxidized (form 2) (Fig. 2B, lane 6) MtDsbA, and there is no fully oxidized MtDsbA (form 0) (Fig. 2B, lane 6). With the expression of MtVKOR, the oxidized band of MtDsbA appears (form 0) (Fig. 2B, lane 7), and there is still some amount of partially oxidized (form 2) (Fig. 2B, lane 7) and fully reduced (form 4) (Fig. 2B, lane 7) MtDsbA.
When the active cysteine C57 in MtVKOR, involved in binding to reduced DsbA (4), was mutated to alanine, the amount of fully oxidized MtDsbA decreased (forms 0 and 1) (Fig. 2B, lane 8). Most MtDsbA is found in the fully reduced state (forms 3 and 4) (Fig. 2B, lane 8) or a partially oxidized form (form 2) (Fig. 2B, lane 8). When the second cysteine in the C89XXC92 motif of MtDsbA was mutated to alanine, MtDsbA cysteines were mostly reduced (form 3 compared to the faint form 1) (Fig. 2B, lane 9), suggesting that the structural disulfide bond between C140 and C192 of MtDsbA relies on the catalytic activity of the C89XXC92 disulfide bond of MtDsbA, which is in turn reoxidized by MtVKOR.
Oxidation of MtDsbA depends on MtVKOR in M. smegmatis.
M. smegmatis is a fast-growing, nonpathogenic mycobacterium species widely used as a host to study genes and proteins from M. tuberculosis. Deletion of vkor (MSMEG_2411) in M. smegmatis causes a growth defect that can be complemented by the expression of Mtvkor from a plasmid (5). In order to determine whether MtVKOR affects the oxidation state of MtDsbA, we integrated myc-tagged MtdsbA into a phage L5 attachment site on the chromosome (using the pJEB402 plasmid) (17) of the M. smegmatis Δvkor strain, which expresses Mtvkor from the pTetG plasmid. The expression of MtVKOR was regulated by the Tet promoter and induced with anhydrotetracycline (ATc), while myc-tagged MtDsbA was under the control of a constitutive promoter (17). The M. smegmatis strain with a deletion of vkor grows slowly in 7H9 medium with 1.6 mM cysteine (or oxidized cysteine) (5). When this strain is complemented instead with the pTetG plasmid expressing MtVKOR, the strain grows similarly to the wild type, even without the addition of ATc. Thus, the basal MtVKOR expression level from the pTetG plasmid is sufficient to support the growth of the Δvkor strain.
We determined the redox state of MtDsbA in vivo in M. smegmatis in the presence or absence of MtVKOR using the same cysteine-alkylating agent, Mal-PEG2k. We observed that when MtVKOR expression was induced with ATc in the Δvkor strain (Fig. 3B, lanes 3 and 4), MtDsbA was found fully oxidized, with some amount of reduced MtDsbA (Fig. 3A, lane 4). However, when there is no VKOR in the cell (Fig. 3B, lanes 1 and 2), all the MtDsbA protein was found in the fully reduced state (Fig. 3A, lane 2). Therefore, in the absence of MtVKOR, MtDsbA is not reoxidized in M. smegmatis, which is consistent with our results that indicate that MtDsbA requires MtVKOR to start a new catalytic cycle.
FIG 3.

Oxidation of MtDsbA is dependent on MtVKOR in M. smegmatis. (A and B) Oxidation states of MtDsbA in the M. smegmatis Δvkor strain with pJEB402-MtDsbA-myc inserted into the chromosome and complemented with either the pTetG (lanes 1 and 2) or pTetG-His-MtVKOR (lanes 3 and 4) plasmid. Strains were grown in 7H9 medium (plus 1.6 mM cystine for pTetG or 50 ng/ml of ATc for pTetG-His-MtVKOR), and proteins were TCA precipitated, followed by alkylation with Mal-PEG2k. MtDsbA (A) was detected with anti-myc antibody, and MtVKOR (B) was detected with anti-VKOR antibody. (C) MtDsbA forms a complex with MtVKOR in E. coli. Wild-type MtVKOR (pTrc99a-His-MtVKOR) or the C65A mutant (pTrc99a-His-MtVKORC65A) was coexpressed with MtDsbA or other mycobacterial thioredoxins (pMER200-MtThioredoxin-FLAG) in the E. coli ΔdsbA ΔdsbB strain. Cell lysates were loaded onto a nickel affinity spinning column. The bound proteins were eluted with 0.5 M imidazole, and eluted samples were analyzed by Western blotting with anti-FLAG antibody. Lane 1, coexpression of MtVKOR and MtDsbA; lane 2, expression of MtVKOR only; lanes 3 to 7, coexpression of the MtVKORC65A mutant with the five thioredoxin candidates. (D) MtDsbA forms a complex with MtVKOR in M. smegmatis. Wild-type MtVKOR (pTetG-His-MtVKOR) or the C65A mutant (pTetG-His-MtVKORC65A) was coexpressed with wild-type MtDsbA (pJEB402-MtDsbA-myc) or the MtDsbAC92A mutant (pJEB402-MtDsbAC92A-myc) in M. smegmatis. Samples were treated with anti-myc antibody as described above for panel B. Blots show representative results from two independent experiments.
Only MtDsbA forms a complex with MtVKOR among the five thioredoxin candidates.
Based on previous studies on the structure of cyanobacterial VKOR and the topology of MtVKOR, a model of the oxidation of the disulfide bond formation protein by MtVKOR was established in E. coli (3, 4). During the oxidation of MtDsbA, the first cysteine (C57) of MtVKOR forms an intermolecular disulfide bond with the first cysteine of the C89XXC92 motif of MtDsbA (4). The second cysteine (C65) of MtVKOR then attacks this disulfide bond and resolves the intermediate. Therefore, a C65 mutant of MtVKOR would help to trap the intermediate with MtDsbA by slowing the resolution of the intermediate complex.
To test whether MtDsbA and MtVKOR can interact specifically, we constructed MtVKOR with a C-to-A mutation at residue 65 (MtVKORC65A) and coexpressed it with each of the five thioredoxin candidates in an E. coli ΔdsbA ΔdsbB strain. We then used nickel affinity resin to pull down His-tagged MtVKOR and determine whether the proteins are pulled down together with MtVKORC65A by immunoblotting with anti-myc antibody. As shown in Fig. 3C (lanes 1 and 3), both MtVKOR and MtVKORC65A can form a complex with MtDsbA but not with the other thioredoxin proteins expressed in E. coli (lanes 4 to 7). The size of the DsbA-VKOR complex is around 50 kDa, similar to the predicted size of the two proteins. When the sample is treated with dithiothreitol (DTT), the disulfide bond that links the two proteins is reduced, and the complex is dissociated into two smaller bands (data not shown). Even when the expression levels of the other four thioredoxin candidates were similar to that of MtDsbA (Fig. 2A), no complexes were detected with the MtVKORC65A mutant (Fig. 3C). Hence, MtDsbA is the only protein among the five thioredoxins that can directly interact with MtVKOR in E. coli. Additionally, we looked at the interaction between MtDsbA and MtVKOR in M. smegmatis by coexpressing MtDsbA with the MtVKORC65A mutant. We also used a mutant of the second cysteine in the DsbA active-site motif (MtDsbAC92A), since this cysteine is also important to resolve the intermediate formed between C89 of MtDsbA and C57 of MtVKOR. Either MtDsbAC92A or MtVKORC65A pulled down DsbA with His-tagged VKOR (Fig. 3D, lanes 4 to 6). Thus, our results confirm the specific interaction between mycobacterial VKOR and DsbA in M. smegmatis.
Deletion of the MtDsbA homolog in M. smegmatis causes a growth defect.
In M. tuberculosis, both MtDsbA and MtVKOR are essential for growth (7). A strain of M. smegmatis deleted for vkor (MSMEG_2411), showing a growth defect, is unable to grow on 7H10 medium and forms tiny colonies on rich medium (5). We hypothesize that VKOR is essential for proper growth because the cell cannot form disulfide bonds efficiently in some essential protein(s) required for cell survival. The MtDsbA homolog in M. smegmatis, annotated as MSMEG_2410, is 80% identical to that of M. tuberculosis (see Fig. S2 in the supplemental material) and is located upstream of the vkor gene, forming an operon similar to that of the M. tuberculosis dsbA-vkor genes.
In order to determine whether M. smegmatis DsbA (MsDsbA) is also essential, we made a clear gene deletion in M. smegmatis by using a sacRB method described previously (18). The deletion was made by using a pTetG plasmid that expresses the MsDsbA gene from a Tet promoter. All our attempts to make a deletion without the complementing plasmid failed, which suggests that MsDsbA is also essential for the growth of M. smegmatis on 7H10 medium. The deletion of MsDsbA causes a more severe growth defect than does the deletion of MsVKOR (5), since this strain cannot grow on either 7H10 medium (Fig. 4A, top) or N-Z-Amine (NZ) medium (Fig. 4A, bottom) without inducing the expression of MsDsbA from the plasmid (i.e., it requires the addition of ATc). Unlike the Δvkor strain, the addition of 1.6 mM cysteine did not restore the growth of the ΔdsbA strain. This may be due to the fact that small-molecule oxidants cannot substitute for DsbA, while they can reoxidize DsbA when VKOR is not present.
FIG 4.
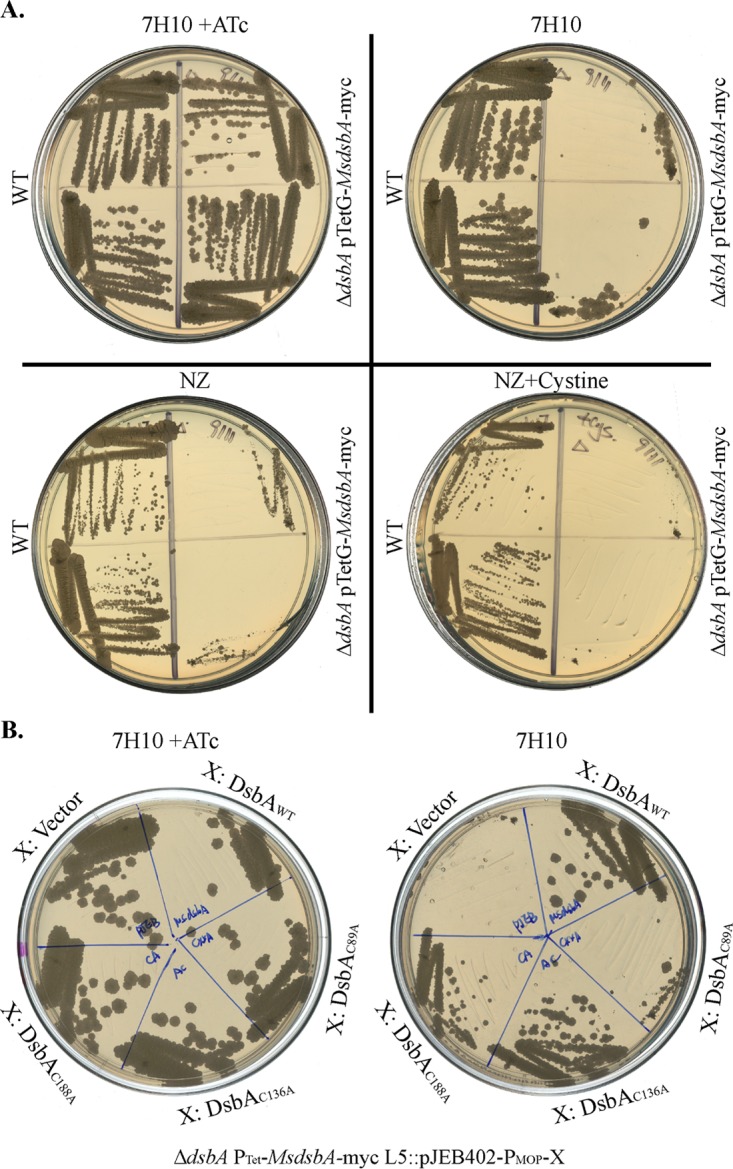
Deletion of the DsbA homolog causes a growth defect in M. smegmatis. (A) Deletion of MsdsbA in M. smegmatis expressing MsDsbA from a plasmid (pTetG-MsDsbA). M. smegmatis wild-type and conditionally lethal ΔdsbA strains were streaked on 7H10 plates with (top left) or without (top right) 100 ng/ml ATc to induce the expression of MsDsbA and with NZ medium (bottom left) or NZ medium supplemented with 1.6 mM cystine (bottom right). Strains were streaked in duplicate onto each plate. (B) Wild-type MsDsbA, MsDsbAC89A (C86XXA mutant), MsDsbAC136A, or MsDsbAC188A (mutants in the structural disulfide bond) was cloned under the control of a constitutive mycobacterial optimized promoter (MOP) present in the pJEB402 plasmid and expressed in the conditionally lethal M. smegmatis ΔdsbA strain with plasmid pTetG-MsDsbA, which requires ATc to induce the expression of MsDsbA in order to grow. Strains were streaked on 7H10 medium plates with 100 ng/ml ATc (left) or without ATc (right). Images show representative results from two independent experiments.
We then determined the essentiality of the four cysteines present in MsDsbA by mutating each residue to alanine. We cloned MsDsbA and its cysteine variants into the pJEB402 plasmid and then integrated the plasmid into the M. smegmatis ΔdsbA strain complemented with pTetGMsdsbA. We tested for growth in the presence or absence of ATc to determine whether the cysteine mutants are able to complement and restore growth. Similarly to MtDsbA, MsDsbA harbors four cysteines (two disulfide bonds), one in the catalytic C86XXC89 motif and one predicted structural disulfide bond between C136 and C188. Disrupting the cysteines of the structural disulfide bond permitted growth of the M. smegmatis conditionally lethal strain, as strains carrying C136 and C188 mutants grew when no ATc was added to the medium (Fig. 4B). However, the C89A mutant did not grow in the absence of ATc (Fig. 4B), suggesting that C86 and C89 are the essential catalytic cysteines of MsDsbA and that the disulfide bond in the active-site motif is essential for the growth of M. smegmatis.
DISCUSSION
M. tuberculosis as well as other Gram-positive actinobacteria, including Corynebacterium and Streptomyces, secrete many proteins with an even number of cysteines to the cell envelope. These organisms have predicted oxidoreductases and VKOR orthologs in their genomes. These properties together indicate that these bacteria likely form disulfide bonds in the cell envelope (2, 6). We have previously shown that the vkor gene from M. tuberculosis restores the oxidation of E. coli DsbA and, thus, disulfide bond formation in an E. coli dsbB deletion strain (2). A recent study has shown that M. tuberculosis VKOR can restore disulfide bond formation in the Gram-positive bacterium Actinomyces oris by interacting with the A. oris DsbA-like protein, MdbA (19). While it has been suggested that DsbA may be the VKOR partner in mycobacteria, no evidence for an interaction between the two proteins was shown (10, 19). In fact, two other oxidoreductases (M. tuberculosis DsbE and DsbF) that exhibit oxidase activity in vitro have been suggested to be the catalysts of protein disulfide bond formation (8, 9). Therefore, in the present study, we investigated the functionality of five candidate M. tuberculosis proteins that belong to the thioredoxin superfamily (DsbA, DsbE, DsbF, ThiX, and Rv0526) and their interactions with M. tuberculosis VKOR in E. coli and in a close relative of M. tuberculosis, M. smegmatis.
When mycobacterial DsbA and VKOR, which are genetically linked in M. tuberculosis, are coexpressed in an E. coli dsbAB double-deletion mutant, they confer motility, a phenotype dependent on a functional disulfide bond pathway (Fig. 1). Motility is not restored when MtVKOR is coexpressed with mycobacterial DsbE, DsbF, ThiX, or Rv0526, consistent with the proposal that mycobacterial DsbA and VKOR are the proteins that act as partners in introducing disulfide bonds into substrate proteins. The failure to confer motility with each of the mycobacterial proteins DsbE, DsbF, ThiX, and Rv0526 when combined with mycobacterial VKOR was not due to poor protein expression. In contrast, the expression of MtDsbA and MtVKOR provided the only combination that resulted in oxidation by MtVKOR in E. coli (Fig. 2A). Consistently, when His-tagged MtVKOR or MtVKORC65A was used to pull down the complex with the thioredoxin candidates in E. coli, only MtDsbA was detected, thus indicating a direct interaction with MtVKOR (Fig. 3C). In addition, the oxidation of MtDsbA was dependent on C57 of MtVKOR, which is involved in binding to reduced DsbA (Fig. 2B). Similarly, the second cysteine in the catalytic C89XXC92 motif of MtDsbA was necessary for the oxidation of MtDsbA (Fig. 2B). While our results show that MtDsbA interacts with MtVKOR, we cannot rule out the possibility that one or more of these thioredoxin candidates are functional, but their functionality in disulfide bond formation does not show up under the several conditions that we have examined. Also, we cannot discard the possibility that these candidates are able to oxidize substrates in M. tuberculosis by a different mechanism and not via MtVKOR, e.g., by small-molecule oxidants.
We have used M. smegmatis as a model for studying the proteins involved in disulfide bond formation of M. tuberculosis. The deletion of vkor (MSMEG_2411) in M. smegmatis complemented with tagged MtDsbA displays only reduced MtDsbA, while the Δvkor strain complemented with MtVKOR shows some MtDsbA in the oxidized state (Fig. 3A), indicating that oxidation of MtDsbA by MtVKOR takes place in M. smegmatis. The reason for the incomplete MtDsbA oxidation may be due to the levels of MtVKOR expression. The interaction between MtDsbA and MtVKOR can also be trapped in M. smegmatis by expressing the MtDsbAC92A mutant or, to a lesser extent, the MtVKORC65A mutant (Fig. 3C). However, wild-type MtVKOR did not pull down MtDsbA in M. smegmatis as it did in E. coli. Perhaps MtVKOR and MtDsbA interact more efficiently with each other in M. smegmatis than in E. coli because the transmembrane segments are still intact when expressed in M. smegmatis. This less efficient interaction between MtDsbA and MtVKOR in E. coli may help to trap MtDsbA in E. coli pulldown assays. Additionally, the vkor deletion in M. smegmatis causes a severe growth defect in rich medium, and this strain does not grow in 7H10 medium unless it is supplemented with cysteine (5). On the other hand, the DsbA homolog of M. smegmatis cannot be deleted unless a second copy of the gene is expressed in the strain (by the addition of ATc); if induction is suspended, the strain is unable to grow, indicating MsDsbA essentiality (Fig. 4A). Expressing the MsDsbAC89A mutant in the conditionally lethal dsbA strain cannot restore growth, while the MsDsbAC136A and MsDsbAC188A mutants complement growth (Fig. 4B). This indicates that MsDsbA catalyzes disulfide bond formation via its C86XXC89 motif. Consistent with M. smegmatis phenotypes, MtVKOR and MtDsbA, but not MtDsbE or MtDsbF, are essential for growth in M. tuberculosis (7). This is in contrast to the E. coli DsbA and DsbB proteins, which are not essential under aerobic growth conditions, although the mutants do not grow anaerobically (20). The reason why these mutants grow aerobically is that the background oxidation of proteins, such as the cell division protein FtsN and the lipopolysaccharide assembly protein LptD, is sufficient to maintain a significant pool of active essential proteins to permit E. coli growth (20, 21). On the other hand, anaerobic growth has no such background oxidation, and E. coli eventually stops growing (20). Thus, altogether, our results demonstrate that MtDsbA is the redox partner of MtVKOR participating in the formation of disulfide bonds in mycobacteria. Our results also indicate that the structural disulfide bond between C140 and C192 of MtDsbA relies on the reoxidation activity of MtDsbA by MtVKOR, as observed by the fully reduced state of cysteines in the MtDsbAC92A mutant (Fig. 2B, lane 9), suggesting that the C140-C192 disulfide bond of MtDsbA is a substrate of the pathway and that it is formed by the catalytic cysteines C89 and C92, which are then reoxidized by MtVKOR cysteines. Our studies together with others allow us to propose a model of the interaction between MtDsbA and MtVKOR (depicted in Fig. 5); in this model, DsbA donates its Cys89-Cys92 disulfide bond to the substrate (e.g., C140-C192 of MtDsbA) and becomes reduced. Reduced Cys89 of DsbA then interacts with Cys57-Cys65 of VKOR, and by thiol-disulfide exchange reactions with Cys139-Cys142 of VKOR, the electrons are shuffled to quinones, releasing oxidized DsbA and VKOR.
FIG 5.

Proposed model of disulfide bond formation in M. tuberculosis. From its C89XXC92 motif, DsbA oxidizes a substrate that requires disulfide bonds to be properly folded and presumably essential for the growth of M. tuberculosis. A substrate of the pathway identified in our studies is the structural disulfide bond (C140-C192) of DsbA itself, which is formed by the catalytic activity of the C89XXC92 motif. Once DsbA transfers its disulfide to substrate proteins, DsbA becomes reduced, and VKOR is required to reoxidize DsbA's cysteines by transferring electrons to quinone. Therefore, the transfer of electrons goes from the substrate to DsbA to VKOR and thence to quinones. Black arrows indicate electron transfer. VK, vitamin K or menaquinone; Cys, cysteine; SH, reduced cysteine; S-S, oxidized cysteines; CW, cell wall; CM, cytoplasmic membrane.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Strains and plasmids used in this study are listed in Table 1. E. coli was grown in NZ medium supplied with suitable antibiotics at 37°C. M. smegmatis mc2155 was grown in either NZ medium supplied with 0.5% Tween 80, 7H9 medium, or 7H10 medium plus oleic acid-albumin-dextrose-catalase (OADC) at 37°C. The antibiotics used were ampicillin at 100 μg/ml, chloramphenicol at 10 μg/ml, kanamycin at 40 μg/ml for E. coli and 20 μg/ml for M. smegmatis, and hygromycin at 100 μg/ml. Mycobacterial genes were cloned by standard molecular biology techniques. The sequences of the identified mycobacterial thioredoxins cloned into pMER200 are given in the supplemental material.
TABLE 1.
Strains and plasmids
| Strain or plasmid | Genotype or description | Reference or source |
|---|---|---|
| Strains | ||
| E. coli | ||
| HK295 | MC1000 Δara714 leu+ | 23 |
| HK329 | HK295 ΔdsbA ΔdsbB | H. Kadokura |
| NK225 | HK295 with pMER200 (Cmr) and pTrc99a (Ampr) | This study |
| NK211 | HK329 with pMER200 (Cmr) and pTrc99a (Ampr) | This study |
| NK212 | HK329 with pMER200 (Cmr) and pRD33 (pTrc99a-6XHis-MtVKOR) (Ampr) | This study |
| NK213 | HK329 with pMER201 (pMER200-MtDsbE-FLAG) (Cmr) and pTrc99a (Ampr) | This study |
| NK214 | HK329 with pMER201 (pMER200-MtDsbE-FLAG) (Cmr) and pRD33 (pTrc99a-6XHis-MtVKOR) (Ampr) | This study |
| NK215 | HK329 with pMER202 (pMER200-MtDsbF-FLAG) (Cmr) and pTrc99a (Ampr) | This study |
| NK216 | HK329 with pMER202 (pMER200-MtDsbF-FLAG) (Cmr) and pRD33 (pTrc99a-6XHis-MtVKOR) (Ampr) | This study |
| NK217 | HK329 with pMER203 (pMER200-MtDsbA-FLAG) (Cmr) and pTrc99a (Ampr) | This study |
| NK218 | HK329 with pMER203 (pMER200-MtDsbA-FLAG) (Cmr) and pRD33 (pTrc99a-6XHis-MtVKOR) (Ampr) | This study |
| NK219 | HK329 with pMER204 (pMER200-EcDsbA-FLAG) (Cmr) and pTrc99a (Ampr) | This study |
| NK220 | HK329 with pMER204 (pMER200-EcDsbA-FLAG) (Cmr) and pRD33 (pTrc99a-6XHis-MtVKOR) (Ampr) | This study |
| NK221 | HK329 with pNK188 (pMER200-MtThiX-FLAG) (Cmr) and pTrc99a (Ampr) | This study |
| NK222 | HK329 with pNK188 (pMER200-MtThiX-FLAG) (Cmr) and pRD33 (pTrc99a-6XHis-MtVKOR) (Ampr) | This study |
| NK223 | HK329 with pNK190 (pMER200-Rv0526-FLAG) (Cmr) and pTrc99a (Ampr) | This study |
| NK224 | HK329 with pNK190 (pMER200-Rv0526-FLAG) (Cmr) and pRD33 (pTrc99a-6XHis-MtVKOR) (Ampr) | This study |
| XW173-1 | HK329 with pXW251 (pBAD33-MtDsbA-myc) (Cmr) and pRD33 (pTrc99a-6Xhis-MtVKOR) (Ampr) | This study |
| XW173-13 | HK329 with pXW264 (pBAD33-MtDsbAC92A-myc) (Cmr) and pRD33 (pTrc99a-6Xhis-MtVKOR) (Ampr) | This study |
| XW173-11 | HK329 with pXW260 (pBAD33-MtDsbAC89A-myc) (Cmr) and pRD33 (pTrc99a-6Xhis-MtVKOR) (Ampr) | This study |
| XW173-15 | HK329 with pXW261 (pBAD33-MtDsbAC140A-myc) (Cmr) and pRD33 (pTrc99a-6Xhis-MtVKOR) (Ampr) | This study |
| XW173-16 | HK329 with pXW262 (pBAD33-MtDsbAC192A-myc) (Cmr) and pRD33 (pTrc99a-6Xhis-MtVKOR) (Ampr) | This study |
| XW173-14 | HK329 with pXW263 (pBAD33-MtDsbAC140A-C192A-myc) (Cmr) and pRD33 (pTrc99a-6Xhis-MtVKOR) (Ampr) | This study |
| XW169-5 | HK329 with pMER201 (pMER200-MtDsbE-FLAG) (Cmr) and pXW169 (pTrc99a-6Xhis-MtVKORC65A) (Ampr) | This study |
| XW169-6 | HK329 with pMER202 (pMER200-MtDsbF-FLAG) (Cmr) and pXW169 (pTrc99a-6Xhis-MtVKORC65A) (Ampr) | This study |
| XW169-1 | HK329 with pMER203 (pMER200-MtDsbA-FLAG, Cmr) and pXW169 (pTrc99a-6Xhis-MtVKORC65A) (Ampr) | This study |
| XW169-9 | HK329 with pNK188 (pMER200-MtThiX-FLAG) (Cmr) and pXW169 (pTrc99a-6Xhis-MtVKORC65A) (Ampr) | This study |
| NK392 | HK329 with pNK190 (pMER200-Rv0526-FLAG) (Cmr) and pXW169 (pTrc99a-6Xhis-MtVKORC65A) (Ampr) | This study |
| M. smegmatis | ||
| NK168 | Mycobacterium smegmatis mc2155 | E. J. Rubin's laboratory collection |
| RD149 | M. smegmatis Δvkor | 5 |
| XW311 | M. smegmatis Δvkor with pTetG (empty) (Hygr) L5::pNK341 (pJEB402-MtDsbA-myc) (Kmr) | This study |
| XW312 | M. smegmatis Δvkor with pNK307 (pTetG-6XHis-MtVKOR) (Hygr) L5::pNK341 (pJEB402-MtDsbA-myc) (Kmr) | This study |
| NK351 | M. smegmatis with pNK307 (pTetG-6XHis-MtVKOR) (Hygr) L5::pNK341 (pJEB402-MtDsbA-myc) (Kmr) | This study |
| NK352 | M. smegmatis with pNK307 (pTetG-6XHis-MtVKOR) (Hygr) L5::pXW307 (pJEB402-MtDsbAC92A-myc) (Kmr) | This study |
| NK354 | M. smegmatis with pNK308 (pTetG-6XHis-MtVKORC65A) (Hygr) L5::pNK341 (pJEB402-MtDsbA-myc) (Kmr) | This study |
| NK355 | M. smegmatis with pNK308 (pTetG-6XHis-MtVKORC65A) (Hygr) L5::pXW307 (pJEB402-MtDsbAC92A-myc) (Kmr) | This study |
| NK317 | M. smegmatis ΔdsbA with pNK252 (pTetG-MsDsbA-myc) (Hygr) | This study |
| NK328 | M. smegmatis ΔdsbA with pNK252 (pTetG-MsDsbA-myc) (Hygr) L5::pJEB402 (Kmr) | This study |
| NK394 | M. smegmatis ΔdsbA with pNK252 (pTetG-MsDsbA-myc) (Hygr) L5::pNK338 (pJEB402-MsDsbA-myc) (Kmr) | This study |
| NK393 | M. smegmatis ΔdsbA with pNK252 (pTetG-MsDsbA-myc) (Hygr) L5::pNK395 (pJEB402-MsDsbAC89A-myc) (Kmr) | This study |
| NK336 | M. smegmatis ΔdsbA with pNK252 (pTetG-MsDsbA-myc) (Hygr) L5::pNK339 (pJEB402-MsDsbAC136A-myc) | This study |
| NK335 | M. smegmatis ΔdsbA with pTetG-MsDsbA-myc) (Hygr) L5::pNK340 (pJEB402-MsDsbAC188A-myc) | This study |
| Plasmids | ||
| pTrc99a | Expression vector; Ampr | 24 |
| pRD33 | pTrc99a-6XHis-MtVKOR (Ampr) | 2 |
| pJEB402 | Chromosomal integration (L5 attachment site) vector, mycobacterial constitutive promoter MOP (Kmr) | 17 |
| pTetG | Mycobacterial expression vector (Hygr) | 25 |
| pMER200 | pBAD33-TorTss-FLAG (Cmr) | This study |
| pMER201 | pMER200-TorTss-MtDsbE-FLAG (Cmr) | This study |
| pMER202 | pMER200-TorTss-MtDsbF-FLAG (Cmr) | This study |
| pMER203 | pMER200-TorTss-MtDsbA-FLAG (Cmr) | This study |
| pMER204 | pMER200-TorTss-EcDsbA-FLAG (Cmr) | This study |
| pNK188 | pMER200-TorTss-MtThiX-FLAG (Cmr) | This study |
| pNK190 | pMER200-TorTss-Rv0526-FLAG (Cmr) | This study |
| pXW251 | pBAD33-MtDsbA-myc (Cmr) | This study |
| pXW264 | pBAD33-MtDsbAC92A-myc (Cmr) | This study |
| pXW260 | pBAD33-MtDsbAC89A-myc (Cmr) | This study |
| pXW261 | pBAD33-MtDsbAC140A-myc (Cmr) | This study |
| pXW262 | pBAD33-MtDsbAC192A-myc (Cmr) | This study |
| pXW263 | pBAD33-MtDsbAC140A-C192A-myc (Cmr) | This study |
| pXW168 | pTrc99a-6XHis-MtVKORC57A (Ampr) | This study |
| pXW169 | pTrc99a-6XHis-MtVKORC65A (Ampr) | This study |
| pNK307 | pTetG-6XHis-MtVKOR (Hygr) | This study |
| pNK308 | pTetG-6XHis-MtVKORC65A (Hygr) | This study |
| pNK341 | pJEB402-MtDsbA-myc (Kmr) | This study |
| pXW307 | pJEB402-MtDsbAC92A-myc (Kmr) | This study |
| pNK252 | pTetG-MsDsbA-myc (Hygr) | This study |
| pNK338 | pJEB402-MsDsbA-myc (Kmr) | This study |
| pNK395 | pJEB402-MsDsbAC89A-myc (Kmr) | This study |
| pNK339 | pJEB402-MsDsbAC136A-myc (Kmr) | This study |
| pNK340 | pJEB402-MsDsbAC188A-myc (Kmr) | This study |
Identifying candidate disulfide bond formation enzymes in M. tuberculosis.
A hidden Markov model of dsbA was constructed based on a few known DsbA protein sequences (13). The HMMsearch program in the HMMER suite took the model and searched for homologs of the model in the M. tuberculosis genome (14). The hits with E values of <10−5 were used to predict their cellular location by Phobius (http://phobius.sbc.su.se/) (14). The ones that were predicted to be a secreted protein or an exported membrane protein were taken as the candidates for disulfide-bond-forming enzymes.
Motility assay.
A motility assay was done on M63 medium soft-agar plates (0.3% agar). The colonies used for the motility assay were picked from a single colony by a pointed toothpick and pricked into a soft-agar plate containing proper antibiotics, 0.5 to 1 mM IPTG (to induce VKOR expression), and 0.2% arabinose (to induce the expression of thioredoxin proteins). The plates were incubated at 30°C for 48 h.
Determining in vivo redox states of the mycobacterial candidates in E. coli.
E. coli cultures were trichloroacetic acid (TCA) precipitated and washed with acetone as described previously (22). Mal-PEG2k {α-[3-(3-maleimido-1-oxopropyl)amino]propyl-ω-methoxy polyoxyethylene; NOF America Corporation} was used as an alkylating reagent to block reduced thiol, adding 2 kDa to the molecular weight of each cysteine. Controls were done by adding DTT (dithiothreitol) and then Mal-PEG2k in order to show the oxidized/nonalkylated (DTT only) or alkylated (DTT and Mal-PEG) protein.
Determining in vivo redox states of MtDsbA in M. smegmatis.
Cultures were started in NZ medium supplemented with 0.5% Tween 80 and either 1.6 mM cysteine for the Δvkor strain or 50 ng/ml of ATc to induce the expression of the Δvkor strain complemented with a plasmid expressing Mtvkor. Cells were grown at 37°C to an optical density at 600 nm (OD600) of 1. Cells were harvested, washed with 7H9 medium plus OADC three times, and resuspended in medium with either 1.6 mM cysteine for the Δvkor strain or 50 ng/ml of ATc to induce the expression of MtVKOR. Cells were continually grown for 4 to 8 h at 37°C. The cells were then pelleted and resuspended in 50 mM Tris–5 mM EDTA (pH 8) with a protease inhibitor. Cell lysis was done by bead beating cells for five times during 30 s with 1-min ice incubation intervals. Lysates were centrifuged for 10 min at 13,000 rpm, and the supernatants were subjected to TCA precipitation and alkylation as described above. MtVKOR antibody was raised in rabbit by using the peptide PMRENRGSQERVGAR (GenScript, USA).
Supplementary Material
ACKNOWLEDGMENT
This work was supported by U.S. National Institute of General Medical Sciences grant GMO41883 (to J.B.).
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00137-18.
REFERENCES
- 1.Kadokura H, Beckwith J. 2010. Mechanisms of oxidative protein folding in the bacterial cell envelope. Antioxid Redox Signal 13:1231–1246. doi: 10.1089/ars.2010.3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dutton RJ, Boyd D, Berkmen M, Beckwith J. 2008. Bacterial species exhibit diversity in their mechanisms and capacity for protein disulfide bond formation. Proc Natl Acad Sci U S A 105:11933–11938. doi: 10.1073/pnas.0804621105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li W, Schulman S, Dutton RJ, Boyd D, Beckwith J, Rapoport TA. 2010. Structure of a bacterial homologue of vitamin K epoxide reductase. Nature 463:507–512. doi: 10.1038/nature08720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang X, Dutton RJ, Beckwith J, Boyd D. 2011. Membrane topology and mutational analysis of Mycobacterium tuberculosis VKOR, a protein involved in disulfide bond formation and a homologue of human vitamin K epoxide reductase. Antioxid Redox Signal 14:1413–1420. doi: 10.1089/ars.2010.3558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dutton RJ, Wayman A, Wei J-R, Rubin EJ, Beckwith J, Boyd D. 2010. Inhibition of bacterial disulfide bond formation by the anticoagulant warfarin. Proc Natl Acad Sci U S A 107:297–301. doi: 10.1073/pnas.0912952107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Daniels R, Mellroth P, Bernsel A, Neiers F, Normark S, Von Heijne G, Henriques-Normark B. 2010. Disulfide bond formation and cysteine exclusion in gram-positive bacteria. J Biol Chem 285:3300–3309. doi: 10.1074/jbc.M109.081398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sassetti CM, Rubin EJ. 2003. Genetic requirements for mycobacterial survival during infection. Proc Natl Acad Sci U S A 100:12989–12994. doi: 10.1073/pnas.2134250100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goulding CW, Apostol MI, Gleiter S, Parseghian A, Bardwell J, Gennaro M, Eisenberg D. 2004. Gram-positive DsbE proteins function differently from gram-negative DsbE homologs: a structure to function analysis of DsbE from Mycobacterium tuberculosis. J Biol Chem 279:3516–3524. doi: 10.1074/jbc.M311833200. [DOI] [PubMed] [Google Scholar]
- 9.Chim N, Riley R, The J, Im S, Segelke B, Lekin T, Yu M, Hung LW, Terwilliger T, Whitelegge JP, Goulding CW. 2010. An extracellular disulfide bond forming protein (DsbF) from Mycobacterium tuberculosis: structural, biochemical, and gene expression analysis. J Mol Biol 396:1211–1226. doi: 10.1016/j.jmb.2009.12.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Premkumar L, Heras B, Duprez W, Walden P, Halili M, Kurth F, Fairlie DP, Martin JL. 2013. Rv2969c, essential for optimal growth in Mycobacterium tuberculosis, is a DsbA-like enzyme that interacts with VKOR-derived peptides and has atypical features of DsbA-like disulfide oxidases. Acta Crystallogr D Biol Crystallogr 69:1981–1994. doi: 10.1107/S0907444913017800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Patarroyo MA, Plaza DF, Ocampo M, Curtidor H, Forero M, Rodriguez LE, Patarroyo ME. 2008. Functional characterization of Mycobacterium tuberculosis Rv2969c membrane protein. Biochem Biophys Res Commun 372:935–940. doi: 10.1016/j.bbrc.2008.05.157. [DOI] [PubMed] [Google Scholar]
- 12.Chim N, Harmston CA, Guzman DJ, Goulding CW. 2013. Structural and biochemical characterization of the essential DsbA-like disulfide bond forming protein from Mycobacterium tuberculosis. BMC Struct Biol 13:23. doi: 10.1186/1472-6807-13-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eddy SR. 1996. Hidden Markov models. Curr Opin Struct Biol 6:361–365. doi: 10.1016/S0959-440X(96)80056-X. [DOI] [PubMed] [Google Scholar]
- 14.Käll L, Krogh A, Sonnhammer ELL. 2004. A combined transmembrane topology and signal peptide prediction method. J Mol Biol 338:1027–1036. doi: 10.1016/j.jmb.2004.03.016. [DOI] [PubMed] [Google Scholar]
- 15.Malen H, Berven FS, Fladmark KE, Wiker HG. 2007. Comprehensive analysis of exported proteins from Mycobacterium tuberculosis H37Rv. Proteomics 7:1702–1718. doi: 10.1002/pmic.200600853. [DOI] [PubMed] [Google Scholar]
- 16.Huber D, Boyd D, Xia Y, Olma MH, Gerstein M, Beckwith J. 2005. Use of thioredoxin as a reporter to identify a subset of Escherichia coli signal sequences that promote signal recognition particle-dependent translocation. J Bacteriol 187:2983–2991. doi: 10.1128/JB.187.9.2983-2991.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee MH, Pascopellat L, Jacobs WR, Hatfull GF. 1991. Site-specific integration of mycobacteriophage L5: integration-proficient vectors for Mycobacterium smegmatis. Proc Natl Acad Sci U S A 88:3111–3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pavelka MS Jr, Jacobs WR Jr. 1999. Comparison of the construction of unmarked deletion mutations in Mycobacterium smegmatis, Mycobacterium bovis bacillus Calmette-Guerin, and Mycobacterium tuberculosis H37Rv by allelic exchange. J Bacteriol 181:4780–4789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Luong TT, Reardon-Robinson ME, Siegel DS, Ton-That H. 2017. Reoxidation of the thiol-disulfide oxidoreductase MdbA by a bacterial vitamin K epoxide reductase in the biofilm-forming actinobacterium Actinomyces oris. J Bacteriol 199:e00817-. doi: 10.1128/JB.00817-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meehan BM, Landeta C, Boyd D, Beckwith J. 2017. The disulfide bond formation pathway is essential for anaerobic growth of Escherichia coli. J Bacteriol 199:e00120-. doi: 10.1128/JB.00120-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meehan BM, Landeta C, Boyd D, Beckwith J. 2017. The essential cell division protein FtsN contains a critical disulfide bond in a non-essential domain. Mol Microbiol 103:413–422. doi: 10.1111/mmi.13565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chng SS, Dutton RJ, Denoncin K, Vertommen D, Collet JF, Kadokura H, Beckwith J. 2012. Overexpression of the rhodanese PspE, a single cysteine-containing protein, restores disulphide bond formation to an Escherichia coli strain lacking DsbA. Mol Microbiol 85:996–1006. doi: 10.1111/j.1365-2958.2012.08157.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kadokura H, Beckwith J. 2002. Four cysteines of the membrane protein DsbB act in concert to oxidize its substrate DsbA. EMBO J 21:2354–2363. doi: 10.1093/emboj/21.10.2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Amann E, Ochs B, Abel K. 1988. Tightly regulated tuc promoter vectors useful for the expression of unfused and fused proteins in Escherichia coli. Gene 69:301–315. doi: 10.1016/0378-1119(88)90440-4. [DOI] [PubMed] [Google Scholar]
- 25.Ehrt S, Guo XV, Hickey CM, Ryou M, Monteleone M, Riley LW, Schnappinger D. 2005. Controlling gene expression in mycobacteria with anhydrotetracycline and Tet repressor. Nucleic Acids Res 33:e21. doi: 10.1093/nar/gni013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.