

Abstract
With the demonstration of depositing biomolecules (single or multiple types) onto biomimetic ES fibrous matrices in arbitrary shape, pattern and dosage using the inkjet printing-based technology, we present a cost-effective and high-throughput platform potentially for organizing various cells on an ECM-like matrix and screening biomolecules for stem cell differentiation and therapeutics for cancer therapy.
Keywords: biomolecule, electrospinning, micropattern, cell pattern, cell function
Graphical abstract
In vivo tissue development involves a plethora of interactions between cells and their extracellular surroundings via cascaded molecular recognitions[1], which subsequently regulate intracellular signal transduction pathways for cellular behaviors and functions[2]. For example, during wound healing fibroblast-to-myofibroblast differentiation, essential for prompt wound closure and granulation tissue formation, requires the presence of platelet-derived growth factor (PDGF) for fibroblast chemotaxis, and activated transforming growth factor (TGF)-β for new extracellular matrix (ECM) synthesis in the wound site[3]. As such, it will be of great benefit for the temporary exogenous template to partially if not fully recapitulate these vital regulatory events in biomaterial-guided tissue formation. The advances of biomaterial science and microfabrication technology enable the development of biomimetic matrices that partially recapture some key physicochemical characteristics of the natural cell-growing environment in a given tissue, e.g. micro- or nano-fibrous scaffolds, biological gel systems (e.g., collagen gel, fibrin gel and matrigel), and so on[4]. However, limited by the fabrication processes, such ECM-like analogs often miss the unique temporal and spatial complexity of cells and biomolecules compared to their natural counterparts, which deliver regulatory cues for cell differentiation and tissue morphogenesis[5]. Furthermore, establishment of the capability to spatially control the distribution of cells and relevant biomolecules on an ECM-like substrate in vitro would not only facilitate mechanistic understanding of cell-cell and cell-biomolecule interactions, but also provide an avenue to correlate cell phenotypic expression with various surfaces in a high throughput manner[6], help to identify effective tissue-engineering scaffolds and develop biofunctional chips[5, 7]. Tremendous efforts have been made in recent years to establish such capabilities, especially with the assistance of Micro Electro-Mechanical Systems (MEMS), for patterning biological agents onto planar surfaces such as glass and hydrogel surface[8–10]. In the case of cell patterning, three strategies can be taken: dip-pen nanolithography (DPN)[11], micro- or nano- contact printing (µCP or nCP)[12, 13] and bio-ink printing (BIP)[14, 15]. Compared to the former two, exhibiting high printing resolution (sub 50 nm resolution, for details see the review[16]), BIP, a computer-aided technique, exhibits broader application potential for biochips, biosensors, DNA arrays, and delivery of active proteins[17–21]. Among all the BIP methods, inkjet printing (IP), the most popular one for its accuracy, versatility and cost/time effectiveness, allows high-speed patterning with foreseeable utility in high throughput[22]. Typically, aqueous solutions of biomolecules[23, 24] or cell suspensions [25–27] are loaded in the cartridges and then printed onto the 2D substrates following computer-designed patterns. Despite its capability of depositing multiple cell types to emulate natural tissue organization, direct cell printing faces immediate challenges in maintaining cell viability and preferred phenotype especially upon a prolonged exposure to the printing “ink” (in most cases optimal for printing but not for cells) and experiencing shear stress from printing nozzles[28, 29]. Biomolecule-guided cell patterning offers another means to control cell morphology[30], organization[31] and functions[32]. However, this strategy has yet to be proven for its multi-cell micropatterning capabilities. Furthermore, the intrinsic attributes of substrates may also influence the adhesion of proteins and consequently cells. Increasing evidence demonstrates the superiority of ECM-like fibrous matrices in maintaining cell phenotype[33]. In this regard, the combination of BIP with ECM-like substrates would be of great benefit by means of creating biomimetic microenvironment with spatiotemporal cues to guide tissue formation. Considering the noncontact nature of inkjet printing, the final shape of the printed patterns significantly depends on the diffusion of “bioink” across the substrate. Herein, we first correlated the printing output (e.g., resolution, repeatability, and shape) on electrospun (ES) fibrous matrices with several key parameters (e.g., the physicochemical properties of fibrous matrices, printing drop size (DS), and inter-drop distance (DD)) using a piezoelectric printer (Figure 1A–C and Figure S1, Supporting Information). Using the established platform, we studied the growth factor-induced and dose-dependent fibroblast-to-myofibroblast differentiation and cancer-stroma interaction. All these demonstrations prove that BIP is effective to pattern multiple types of cells on the ES matrices and the combined IP/ES matrices provide a robust biomimetic platform to study cell-cell and cell-matrix interactions, and ultimately facilitate the advances of tissue engineering and regenerative medicine.
Figure 1.

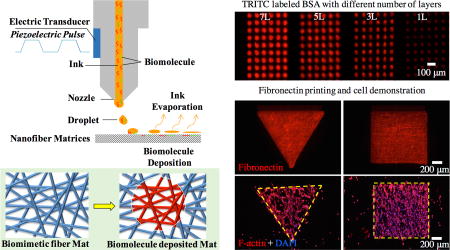
A) Schematic illustration on the inkjet printing setup for ES matrices. The piezoelectric inkjet design allows pushing the “biomolecule ink” drops out of the cartridge reservoir by electrical charge-generated pressure and then depositing onto the nanofiber matrices. B) The key parameters including matrix surface wettability, fiber organization and temperature influence the printing resolution. C) Schematic illustration of the biomimetic fibrous matrices before and after biomolecule printing. D) Fluorescence images of six different ES fibrous matrices [PCL, 3PCL:1collagen (3P1C), 1PCL:1collagen (2P2C), 1PCL:3collagen (1P3C), collagen (Coll) and coverglass (Glass)] printed with TRITC-BSA using 90 µm drop distance (DS) and 10pL drop size (DS). E) Contact angle measurement of various ES matrices (n≥10). F) Quantitative measurement of individual printed drop area on all ES matrices using 6pL and 10pL DS (n≥10). G) Fluorescence images of FITC-BSA printed onto (i) isotropic (Iso) and (ii) anisotropic (Aniso) nanofiber matrix surfaces and quantitative measurement of individual printed drop area (i’) and the dimensions (ii’) (n≥35, *p<0.05). In (ii), dashed double arrow indicates the fiber orientation and white double arrow indicates the printing direction. L and S indicate the measurement of the length as shown in (ii’). H) Fluorescence images of TRITC-BSA printed onto the nanofiber matrices with 1, 3, 5, 7 and 10 repeated printing layers: (i) top view and (ii) transverse view. (iii) Quantitative measurement of the fluorescence intensity per printing area shows a linear increase with the number of layers (n≥20). All scale bars: 100 µm.
ES fibrous matrices have shown great promise in promoting tissue formation as a result of their morphological and dimensional similarity to native ECM fibers and fibril bundles[34–36]. When printing aqueous biomolecule solutions onto such fibrous matrices, the matrix surface properties such as wettability and fiber orientation would influence the printing resolution. To elaborate the correlation, ES fibrous matrices fabricated from either polycaprolactone (PCL) (8%, w/v), or type I collagen (8%, w/v), or their blends at different ratios [3:1(3P1C), 1:1 (2P2C) or 1:3 (1P3C)] were used for biomolecule printing. By tuning the electrospinning conditions, the obtained ES matrices all exhibited similar fiber organization (isotropic) and comparable average fiber diameter (835 nm ± 332 nm, data not shown). Increasing the hydrophilic collagen amount in ES fibers could offset the hydrophobicity of PCL and yielded ES matrices with improved wettability as shown by decreased contact angle (Figure 1E). For those fibers with collagen higher than 50% (w/w), i.e., 2P2C, 1P3C and pure collagen, the ES matrices become hydrophilic. To better visualize the region of interest (ROI), the “ink” solution containing a fluorescently labeled model biomolecule (i.e., TRITC-conjugated bovine serum albumin (TRITC-BSA), 1% w/v) was printed onto various fibrous matrices (n=5) using a piezoelectric inkjet printer (Dimatix Materials Printer DMP-2800) following the input AutoCAD patterns with 90-µm DD (sufficient to avoid the inter-drop overlap). Upon water evaporation, the remaining biomolecules of the “ink” attached to the ES matrices and formed a nearly round dot (Figure 1D and Figure S2, Supporting Information). The area of each round dot of ROI (n=20) was measured using ImageJ (NIH, version 2.0.0). With the “ink” DS of either 6pL or 10 pL, the dot area showed a similar correlation with the wettability of ES matrices, i.e., the area increases over the increase of wettability (Figure 1D E F). The most hydrophobic surface, PCL ES matrices, yielded the smallest dot area, 25 ± 10 µm in diameter, using 6pL DS. Interestingly, the dot area printed on a glass coverslip, which had a contact angle similar to 1P3C ES matrices, was much smaller than that on 1P3C matrices and even slightly smaller than that of PCL matrices (Figure 1F). This observation suggests that besides surface wettability, surface roughness (glass vs. ES matrices), especially the capillary effect from the inter-fiber channels of ES matrices[37], would facilitate the diffusion of the printed “ink” solution and ultimately increase the area of resulting patterns. Similarly, varying fiber arrangement (e.g., randomly oriented or isotropic vs. directionally oriented or anisotropic) within ES matrices would change the organization of inter-fiber channels, which leads to differential solution dispersion and consequently influences the resulting patterns. Indeed, deposition of FITC-conjugated BSA drops (1% w/v, 10pL DS) onto PCL ES matrices with either isotropic or anisotropic fiber organization showed different shapes, i.e., round dots vs. oval dots (Figure 1G-i and ii). Measurement of the dot area by ImageJ revealed that the average area for both shapes was comparable while on the anisotropic ES matrices the long axis of oval dots followed the fiber orientation, resulting from the elevated spread along the fiber orientation (Figure 1G-i’ and ii’). By controlling the voltage of piezoelectricity, it is possible to tune the size of DS between 5 and 10pL. A nearly linear correspondence between DS and the printed dot area was observed on individual ES matrices (Figure S3, Supporting Information). The fluorescence intensity of each dot on the same matrices was also measured and it was found that the dot-to-dot variation was minimal (within 8%) and the fluorescence intensity within the dot was rather uniform, indicating a high printing accuracy and repeatability (Figure S4, Supporting Information). The printing accuracy was further demonstrated by repeated deposition of biomolecules onto the same location of the ROI upon multiple printing cycles (1 to 10). Despite an increase of lateral length due to solution dispersion (Figure 1H-i and ii), the fluorescence intensity of the printed area was a linear function of the printing cycles (Figure 1H-iii). Furthermore, the printing accuracy was also verified by using two different biomolecule models (TRITC-BSA and FITC-BSA) along with the intention to print multiple biomolecules. Once again, the biomolecules could be deposited on the ES matrices in appositional, segregated or overlayed manner (Figure S5, Supporting Information). Examination of the printed region by scanning electronic microscopy (SEM, Figure S6, Supporting Information) showed that ES matrices maintained their original fiber morphology and dimensions, except those of pure collagen fibers, fusing into a continuous film, which might result from their high water-swelling capacity and low mechanical stability[38]. During printing, the ambient temperature would also affect the printing outcomes. Higher temperature could facilitate solvent evaporation and limit solution diffusion for a smaller area. However, to ensure the bioactivity of printed molecules, room temperature was adopted for all the printings.
In the endeavor to explore printing versatility and accuracy, we printed fibronectin (FN) onto isotropic PCL ES matrices following the patterns with designated shapes (circle, rectangle, triangle and square) and sizes (50µm, 100µm, 200µm, 400µm, 800µm for the first three shapes and 70µm, 140µm, 280µm, 560µm, 1120µm for the squares). As shown in Figure 2 and Figure S7 (Supporting Information), the resulting shapes and dimensions closely followed the designed ones with a high fidelity (>99.8% in relation to the design). Besides, the effect of patterned FN on cell adhesion and spreading was studied by culturing mouse smooth muscle cells (MOVAS) on these PCL ES matrices. MOVAS cells have a high affinity to FNs[39] and preferably adhered to the patterned FN regions (Figure 2A and Figure S7, Supporting Information). Similarly, cultured breast cancer cells (MDA-MB-231) on the ES matrices with an array of 200-µm round FN dots yielded the identical cell organization despite a few cells off the patterns (Figure 2B). MCF-7 breast cancer cells and mouse pancreatic endothelial cells (MS-1) were also used to demonstrate the formation of 100-µm dot arrays and blood vessel like patterns, respectively (Figure S8A, B, Supporting Information). However, cultured MS-1 cells on the array of smaller FN dots (25µm) showed that individual MS-1 cells could stick to one dot, but many (>50%) also reached out to the neighboring dots (Figure S8C), indicating that 25µm might be the smallest size required for MS-1 cell spreading. To better correlate cell attachment and spreading with the dimensions of printed patterns, MOVAS and MS-1 were respectively cultured on the printed FN with various patterns and dimensions (Figure 2C and 2D). Both types of cells attached to the printed FN regions after 1-day culture. Upon closer examination, we did notice that reducing dimensions down to 50 µm led to non-compliance of cells, especially MOVAS, to the printed patterns. Considering that cell spreading, i.e., footprint on the surface is directly determined by cell type and cell number, we studied the contribution of cell seeding density to cell patterning accuracy. A density of 3.5 million cells/mL, which was calculated based on the full coverage of 22 mm × 22 mm coverslips by non-spread MOVAS cells (~20µm in diameter) with intercellular separation of 10 µm, and a diluted density of 1.0 million cells/mL was adopted to seed MOVAS onto 50-µm FN lines on PCL ES matrices. At the maximum seeding density, typically 3 to 4 cells attached across the 50-µm FN line, while at the diluted density only 1 to 2 cells were seen. Further culturing revealed that cells at the maximum seeding density not only fully occupied the 50-µm FN lines but also grew out to form much wider cell lines, approximately 100µm in width (Figure S9, Supporting Information). In contrast, the diluted seeding density enabled the formation of 50-µm cell lines similar to FN lines. With the diluted seeding density, we further studied how MOVAS cells responded to various FN patterns (i.e., circle, triangle, square and rectangle) with 50µm dimension. Surprisingly, except the rectangular ones, all others exhibited the incapability of confining MOVAS cells within the patterned FN (Figure 2C), probably due to the limited FN area, not enough for more than three spreading cells, and the biocompatibility of PCL ES matrices for cell spreading. During the initial cell seeding, cells were temporarily confined to the FN area, as a result of selective adhesion of MOVAS to the highly adhesive FN; however, upon further culturing cells migrated out of the FN domains without any preferred orientation. This observation differs from those with cell-repellent surfaces[40], in which cell growth was limited to the adhesive molecule-printed regions. Unlike MOVAS cells, a majority of MS-1 cells, with a cobblestone-like morphology could better remain within all the 50-µm FN patterns (Figure 2D) likely because of their smaller spreading size.
Figure 2.

A) Fluorescence images of fibronectin (FN) printed onto PCL nanofiber matrices after staining with four different shapes: (i) rectangle, (ii) circle, (iii) triangle and (iv) square and their corresponding cell patterns after staining with Phalloidin (F-actin) (i’–iv’). All scale bars: 200 µm. B) Fluorescence images of eight 200-µm dotted MDA-MB-231 cell array with zoomed-in highlight of one cell colony. Scale bar: 200 µm unless stated otherwise. C) Fluorescence images of MOVAS cell patterns with three different sizes. All the shapes are indicated by dashed yellow lines. Scale bar: 50 µm. D) Fluorescence images of MS-1 cell patterns with three different sizes. All the shapes are indicated by dashed yellow lines. Scale bar: 50 µm.
Recognizing the essence of spatially distributed growth factors in regulating cellular activities and functions in native tissues, it becomes desirable to develop the capabilities of fabricating ECM-like surfaces with patterned growth factors[15, 41], allowing for the study of cell-to-biomolecule responses in physiologically relevant settings. For example, during wound healing the elevation of contractile myofibroblasts involves three key local events: 1) accumulation of active transforming growth factor (TGF)-β1, 2) presence of specialized ECM proteins like fibronectin, and 3) high extracellular stress[42, 43]. To better unveil such intertwined regulations with a particular interest in the dose-dependent induction of myofibroblastic differentiation by TGF-β1, TGF-β1 and FN were sequentially deposited onto the surface of PCL fibrous matrices (Figure 3A). With precisely controlled printing of multiple layers of TGF-β1 (100ng/mL solution) to the same location, it was possible to modulate the TGF-β1 amount deposited on the fibrous matrices (Figure 3B). To verify the printing accuracy, TRITC-BSA solution was printed onto the PCL fibrous matrices using 90DD and 10pL DS (Figure 1H-i). With repeated printings, the resulting area proportionally increased for the first 5 layers and then reached a plateau (Figure S10, Supporting Information). A high reproducibility was achieved for all the printed arrays with a negligible dot-to-dot deviation (less than 3%). Examination of the cross-sections of all the TRITC-BSA printed matrices (Figure 1H-ii) revealed that TRITC-BSA remained in the superficial zone of the nanofibrous matrices except for lateral expansion. When large round dots (ϕ = 500 µm) of TRITC-BSA were printed onto the fibrous matrices using 10pL DS and 20µm DD, as shown in Figure 3C, the fluorescence intensity was rather uniform across the dot. Following the same setup, 500-µm round dots of TGF-β1 with varying layers (1, 3, 7 and 10) followed by one layer of cell-adhesive FN (Figure 3D) were printed on the PCL fibrous matrices. Upon seeding with human normal dermal fibroblasts (at passage 4), the cells preferably attached to the TGF-β1/FN dots and became confluent prior to their isotropic migration out of the printed dots. TGF-β1 was known for its ability to induce the fibroblast-to-myofibroblast differentiation[44]. However, it was unclear whether TGF-β1 retained bioactivity after inkjet printing. Thus, cells cultured in the media containing 0 or 10ng/mL exogenous TGF-β1 were included as negative (NC) or positive controls (PC). After culturing for 3 days, cells were immunostained for α-smooth muscle actin (α-SMA), a marker for myofibroblastic differentiation. As shown in Figure 3E, F and Figure S11 (Supporting Information), the number of α-SMA positive cells increased with the increase of TGF-β1 layers and the TGF-β1 concentration in the media. Quantification of α-SMA positive cells normalized by the total number of cells in the field (Figure 3G) showed a dose-dependent differentiation of fibroblasts and 10-layer TGF-β1 dots led to approximately 42±3% myofibroblastic differentiation, comparable to PC (41±2%) (Figure 3G). Different from the ubiquitous differentiation induced by media-containing TGF-β1, myofibroblastic differentiation on PCL fibrous matrices with printed TGF-β1 confined mainly to TGF-β1 dotted area (Figure 3E), suggesting that TGF-β1-regulated fibroblast-to-myofibroblast is a localized event. The result also indicates that TGF-β1 printed on the PCL nanofibers remains biologically active. In a separate experiment to determine the stability of printed biomolecules on PCL ES matrices, we found that FNs were still detectable even after one-week incubation in media (Figure S12, Supporting Information), implying a high affinity of biomolecules to ES matrices. The capability of spatially controlling cellular activities on ECM-like substrates via localized biomolecules, similar to native circumstances, provides not only an avenue to study cell-biomolecule interactions in a physiologically relevant microenvironment, but also an effective approach to identify the regulatory cocktail for lineage differentiation of stem cells[45]. This information is particularly valuable for high-throughput and cost-effective screening with limited amount of biomolecules (e.g., only 0.177 ng TGF-β1 was needed to print 36 of 500-µm 10-layer TGF-β1 dots on one 22mm×22mm coverslip in comparison to 20ng per coverslip in PC group, Figure 3B).
Figure 3.

A) Schematic illustration on the sequential printing of TGF-β1 and fibronectin (FN) on the nanofiber matrices and the seeding with fibroblasts. B) Correlation of TGF-β1 amount with the layers printed onto the nanofiber matrices, negative control (NC) and positive control (PC). C) Fluorescence images of the 500-µm TGF-β1 dots printed onto PCL nanofiber matrices with various printing layers after staining. The fluorescence intensity increases over the repeated printing. D) Fluorescence images of TGF-β1 (red, 5 layers) and FN (green, 1 layer) sequentially printed onto the PCL nanofiber matrices. Top (i) and transverse view (ii) reveal the overlay of both biomolecules. E) Culture of human dermal fibroblasts (passage 4) on the PCL nanofiber matrices with printed TGF-β1/FN for 3 days. Cells predominantly attached to the 500-µm dots, indicated by dashed white circles. Fluorescence images of fibroblasts immunostained for α-SMA (red), F-actin (green) and DAPI (blue). F) Zoomed-in fluorescence images of the cells located within the printed TGF-β1/FN and immunostained for α-SMA (red) and DAPI (blue). G) Quantitative analysis of α-SMA positive cells, i.e., α-SMA positive cells normalized by the total cell number in each image (n≥5, ns: Not significant, *p<0.05, ***p<0.001). All scale bars: 200 µm.
Cumulative evidence has highlighted the pliability of cell phenotype and its adaption to the residing microenvironment, primarily involving cell-cell, cell-matrix, cell-biomolecule and cell-physical stimuli interactions[25]. In tumor development, the mutual regulations between tumor cells and their stroma, mainly composed of fibroblasts, adipocytes, immune cells and blood vessels besides ECM[46, 47], would modify the surrounding stroma accordingly with cancer-prone attributes and lead to the cell phenotypic alteration, e.g., transformation and metastasis[48, 49], which consequently contributes to the heterogeneity of tumorigenesis. The typical in vitro co-culture model, i.e., mixing different cell types together, can neither represent the spatial architecture of native tumor-stroma interactions, nor capture temporospatial tumor progression. In contrast, the established inkjet printing technology may offer a unique opportunity to organize various cells on an ECM-like matrix following the native scenario of tumor-stroma. To this end, PCL fibrous matrices with 200-µm FN dots were first seeded with breast cancer cells (MCF-7) and then seeded with adipose-derived stromal cells (ASC, passage 4, 5,000 cells per scaffold) (Figure 4A). As seen in Figure 4B, MCF-7 cells attached to the FN dots while ASCs occupied the open space between FN dots, partially replicating the in vivo circumstances. Upon culture for designated periods, cells were fluorescently stained for α-SMA, E-cadherin, F-actin and nuclei (DAPI). With the extension of culture, more α-SMA positive ASCs were seen around the MCF-7 clusters (Figure 4B) and by 3 weeks about 70% of them became α-SMA positive (Figure 4C) in contrast to less than 5% for ASC alone controls (Figure 4B-iv). In vivo, cancer-associated fibroblasts (CAFs), a mixture of fibroblasts and myofibroblasts, are often observed in the stroma of tumors. The increase of myofibroblast population is intimately associated with the malignancy development of a tumor[50]. In human tumor xenograft models, CAFs isolated from tumor sites were able to promote tumor growth in comparison to those from non-tumors sites[51]. Increasing evidence suggests that cancer cells not only initiate myofibroblastic differentiation of stromal cells but also maintain their activated phenotype in vivo[52]. Indeed, we observed the persistence of α-SMA positive cells for up to the designated experimental time (4 weeks) (Figure 4D-iv). Interestingly, co-culture of MCF-7 with ASCs could help keep MCF-7 cells in clusters and better detained to FN patterned areas, distinct from those cultures with MCF-7 cells alone, which migrated out of the original clusters as early as two weeks (Figure S13, Supporting Information). However, this circumstance became different for those co-cultures with a lower ASC seeding density (1,000 cells per scaffold). Immunostaining for E-cadherin revealed that MCF-7 cells formed the adherent junctions in the beginning of the culture and then gradually became scattered and infiltrated the ASC-dominated regions in parallel with the appearance of α-SMA positive cells (Figure 4D). By 4 weeks, a majority of MCF-7 cells lost their intercellular junctions and assumed an elongated morphology (Figure 4D-iv), indicating the cells undergoing an epithelial-mesenchymal transition and acquiring a metastatic phenotype[53].
Figure 4.

A) Schematic illustration on the experimental design for studying cancer-stromal cell interaction. 200-µm dots of FNs are printed onto the nanofiber matrices for selective attachment of cancer cells (MCF-7) to form cancer clusters and the remaining area is seeded with human adipose stromal cells (ASCs). Cancer cells would recruit the stromal cells by differentiating them into α-SMA positive cells. B) Co-culture of MCF-7 cells and ASCs (high seeding density) on the PCL nanofiber matrices with printed 200-µm FN dots, onto which MCF-7 cells primarily attached and formed the colony as indicated by dashed white circles. Fluorescence images of the cells immunostainned for α-SMA (red), F-actin (green) and DAPI (blue) after culturing for 1 week (i), 2 weeks (ii), 3 weeks (iii). The culture of ASCs without MCF-7 was included as controls (iv). C) Quantitative analysis of α-SMA positive cells normalized by the total stromal cell number in each image (n≥5, ***p<0.001). Co-culture of MCF-7 cells and ASCs (low seeding density) on the PCL nanofiber matrices with printed 100-µm FN dots, onto which MCF-7 cells primarily attached and formed the colony as indicated by dashed white circles. Fluorescence images of the cells immunostainned for α-SMA (red), E-Cadherin (green) and DAPI (blue) after culturing for 1 week (i), 2 weeks (ii), 3 weeks (iii) and 4 weeks (iv). White arrows indicate those cancer cells migrated out of the cell colony. Scale bars: 100 µm.
Apart from the recruitment of stromal cells into CAFs, cancer cells also secrete multiple angiogenic factors such as basic fibroblast growth factor (b-FGF), platelet-derived growth factor (PDGF), and vascular endothelial growth factor (VEGF), to promote tumor angiogenesis for nutrition supply toward neoplastic growth[54–57]. In fact, tumor angiogenesis has received a particular attention in therapy by means of starving tumors via diminishing blood supply[58]. Thus, setup of a cost-effective in vitro model that can recapitulate tumor angiogenesis would be highly appreciated for fundamental understanding of cancer/vasculature inter-regulation and development of therapeutics for cancer therapy. In alignment with such a demand, we undertook a simplified yet physiologically based strategy by 1) patterning endothelial cells (MS-1) into microvessel-like lines, 2) laying down the suspension of MCF-7/collagen gel (0.5mg/mL) onto 200µm FN/LN dots with defined distances (500 and 700 µm) from the endothelial line, and then 3) seeding ASCs to the remaining area of PCL fibrous matrices (Figure 5A). Figure 5B illustrated the formation of 100µm endothelial line (i) and the attachment of ASCs to the area between endothelial line and cancer clusters (ii). After 1 and 2 weeks in culture, cells were immunostained for platelet-endothelial cell adhesion molecule-1 (PECAM-1, or CD31), a marker for endothelial cells, to visualize endothelial sprouts. The co-culture was stained with TRITC-conjugated Phalloidin (F-actin) and DAPI (nuclei) (Figure 5C and 5D). The presence of MCF-7 cells in the co-culture promoted endothelial sprouting from the endothelial line toward the MCF-7 clusters. Compared to cultures without MCF-7 cells, endothelial sprouting in the presence of MCF-7 cells could reach as far as 700 µm to the edge of the MCF-7 clusters after two weeks (Figure 5E). Closer examination revealed that ASCs elongated perpendicular to the endothelial line with a direct connection with MCF-7 clusters, and endothelial sprouting closely followed the orientation of ASCs (Figure S14, Supporting Information), suggesting that activated ASCs (or CAFs) may serve as the “railways” to guide tumor angiogenesis. Evidence has demonstrated the possible involvement of cancer-secreted PDGF and CAF-expressed PDGF receptors in angiogenesis[59, 60]. Further studies are needed to elaborate the effects of ASCs/CAFs on endothelial sprouting and angiogenesis, especially its directional guidance in combination with secretory factors from cancer cells, markedly different from that disorganized angiogenesis without ASCs (Figure S15, Supporting Information). Clearly, this co-culture model offers a fast and simple way to monitor the endothelial sprouting process.
Figure 5.

A) Schematic illustration on the experimental design for studying cancer-induced angiogenesis. Cancer cells (MCF-7) suspended in collagen gel are dropwise seeded onto the printed laminin (LN)-FN 200-µm dots to form the cancer cluster, and endothelial cells (ECs, MS-1) are seeded onto the printed 100-µm FN line to form the microvessel-like structure on the nanofiber matrices. The remaining area is seeded with ASCs. Factors secreted by cancer cells would encourage the EC sprouting toward cancer clusters. B) Demonstration on the formation of MS-1 cell line (i) and with the presence of ASCs (ii) on PCL nanofiber matrices. MS-1 cells were immunostained for CD31 (green) and all the cells were stained for F-actin (red). C–D) Fluorescence images of the cells cultured for 1 week and 2 weeks. Immunostaining for CD31 reveals pronounced sprouting of MS-1 from its initial lines (dashed purple lines) with the presence of MCF-7 (dashed white circles) compared to the control without MCF-7 (i vs. ii). D–I’) indicates the cancer colonies based on DAPI staining. E) Quantitative analysis of the length of EC sprouting after 1 week and 2 weeks (n≥7, ***p<0.001). Scale bars: 100 µm.
In summary, the established capability, i.e., readily depositing biomolecules (single or multiple types) onto biomimetic ES fibrous matrices with arbitrary shape, pattern and dosage using the inkjet printing-based technology, not only provides a strategy to fabricate multifunctional ECM-like interfaces for medical devices, but also offers the capability of spatially controlling the organization of various cells on ES matrices for fundamental studies (e.g., cell-cell, cell-biomolecule and cell-matrix interactions), and for applications in high-throughput screening of various biomolecules for stem cell differentiation and therapeutics for cancer therapy. With extensive study on the effect of various parameters (e.g., solution drop size, inter-drop distance and physicochemical properties of ES matrices), it becomes possible to achieve high printing resolution (ϕ=25 ± 10µm dot), accuracy (>99.8%) and reproducibility (<8% in variation), which is applicable to either single or multiple biomolecule printing and repeated printing. Importantly, the printed biomolecules remain on the ES matrices for as long as one week in the media. We have successfully demonstrated the utility of such biomolecule-printed ES matrices for studying the dose-dependent fibroblast-to-myofibroblast differentiation induced by TGF-β1, the mutual regulations of breast cancer cells (MCF-7) and stromal cells (ASCs), as well as tumor angiogenesis. Different from a smooth surface, deposition of biomolecules (e.g., FNs) directly onto ECM fibril-like surface can lead to the formation of biomolecule fibrous network, which is generally difficult to develop in vitro but essential to achieve biofunctions similar to in vivo conditions[61]. In combination with the flexibility to modulate the physicochemical properties of ES matrices (e.g., composition, fiber diameter, stiffness), the biomolecule-printed ES matrices can maximally recapitulate the multifunctionality of native ECM for desirable cell phenotype and subsequently functional tissue formation. In recognition of the essence of 3D culture in maintaining cell functions and signaling[62], the current ES matrix-based 2D biomolecule printing technology would need to be integrated with other technologies for creating complex 3D structures, for instance, the layer-by-layer stacking strategy to assemble multiple layers of biomolecule-printed ES matrices with or without seeded cells[63]. Thus, the established methodology has also foreseeable applications in tissue engineering by spatially patterning cells and biomolecules.
Materials and Methods
Materials and Methods are available online as Supporting Information
Supplementary Material
Acknowledgments
The authors are grateful for scientific comments from Dr. Philip L. Leopold from Weill Cornell Medical College and editorial assistance from Thomas Cattabiani. This work was made possible by financial support from NIAMS 1R01AR067859 (CJ), NSF-DMR 1508511 (HW), Chinese National High Technology Research and Development Program (863 program) 2015AA020313(YB).
Footnotes
Supporting Information is available from the Wiley Online Library or from the authors.
References
- 1.Rosso F, Giordano A, Barbarisi M, Barbarisi A. Journal of cellular physiology. 2004;199:174. doi: 10.1002/jcp.10471. [DOI] [PubMed] [Google Scholar]
- 2.Visse R, Nagase H. Circulation research. 2003;92:827. doi: 10.1161/01.RES.0000070112.80711.3D. [DOI] [PubMed] [Google Scholar]
- 3.Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Nature reviews. Molecular cell biology. 2002;3:349. doi: 10.1038/nrm809. [DOI] [PubMed] [Google Scholar]
- 4.Shin H, Jo S, Mikos AG. Biomaterials. 2003;24:4353. doi: 10.1016/s0142-9612(03)00339-9. [DOI] [PubMed] [Google Scholar]
- 5.Lutolf MP, Gilbert PM, Blau HM. Nature. 2009;462:433. doi: 10.1038/nature08602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cranford SW, de Boer J, van Blitterswijk C, Buehler MJ. Advanced materials. 2013;25:802. doi: 10.1002/adma.201202553. [DOI] [PubMed] [Google Scholar]
- 7.Place ES, Evans ND, Stevens MM. Nature materials. 2009;8:457. doi: 10.1038/nmat2441. [DOI] [PubMed] [Google Scholar]
- 8.Xu T, Jin J, Gregory C, Hickman JJ, Boland T. Biomaterials. 2005;26:93. doi: 10.1016/j.biomaterials.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 9.Campbell PG, Miller ED, Fisher GW, Walker LM, Weiss LE. Biomaterials. 2005;26:6762. doi: 10.1016/j.biomaterials.2005.04.032. [DOI] [PubMed] [Google Scholar]
- 10.Ilkhanizadeh S, Teixeira AI, Hermanson O. Biomaterials. 2007;28:3936. doi: 10.1016/j.biomaterials.2007.05.018. [DOI] [PubMed] [Google Scholar]
- 11.Piner RD, Zhu J, Xu F, Hong SH, Mirkin CA. Science. 1999;283:661. doi: 10.1126/science.283.5402.661. [DOI] [PubMed] [Google Scholar]
- 12.Wilbur JL, Kumar A, Kim E, Whitesides GM. Advanced materials. 1994;6:600. [Google Scholar]
- 13.Li HW, Muir BVO, Fichet G, Huck WTS. Langmuir. 2003;19:1963. [Google Scholar]
- 14.Derby B. Science. 2012;338:921. doi: 10.1126/science.1226340. [DOI] [PubMed] [Google Scholar]
- 15.Li X, Liang H, Sun J, Zhuang Y, Xu B, Dai J. Advanced healthcare materials. 2015;4:1869. doi: 10.1002/adhm.201500271. [DOI] [PubMed] [Google Scholar]
- 16.Kim DH, Lee H, Lee YK, Nam JM, Levchenko A. Advanced materials. 2010;22:4551. doi: 10.1002/adma.201000468. [DOI] [PubMed] [Google Scholar]
- 17.Tasoglu S, Demirci U. Trends Biotechnol. 2013;31:10. doi: 10.1016/j.tibtech.2012.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Allain LR, Askari M, Stokes DL, Vo-Dinh T. Fresen J Anal Chem. 2001;371:146. doi: 10.1007/s002160100962. [DOI] [PubMed] [Google Scholar]
- 19.Newman JD, Turner APF, Marrazza G. Anal Chim Acta. 1992;262:13. [Google Scholar]
- 20.Blanchard AP, Kaiser RJ, Hood LE. Biosens Bioelectron. 1996;11:687. [Google Scholar]
- 21.Roda A, Guardigli M, Russo C, Pasini P, Baraldini M. Biotechniques. 2000;28:492. doi: 10.2144/00283st07. [DOI] [PubMed] [Google Scholar]
- 22.Roth EA, Xu T, Das M, Gregory C, Hickman JJ, Boland T. Biomaterials. 2004;25:3707. doi: 10.1016/j.biomaterials.2003.10.052. [DOI] [PubMed] [Google Scholar]
- 23.Bucella SG, Luzio A, Gann E, Thomsen L, McNeill CR, Pace G, Perinot A, Chen ZH, Facchetti A, Caironi M. Nature communications. 2015;6 doi: 10.1038/ncomms9394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tao H, Marelli B, Yang MM, An B, Onses MS, Rogers JA, Kaplan DL, Omenetto FG. Advanced materials. 2015;27:4273. doi: 10.1002/adma.201501425. [DOI] [PubMed] [Google Scholar]
- 25.Xu T, Zhao W, Zhu JM, Albanna MZ, Yoo JJ, Atala A. Biomaterials. 2013;34:130. doi: 10.1016/j.biomaterials.2012.09.035. [DOI] [PubMed] [Google Scholar]
- 26.Matsusaki M, Sakaue K, Kadowaki K, Akashi M. Advanced healthcare materials. 2013;2:534. doi: 10.1002/adhm.201200299. [DOI] [PubMed] [Google Scholar]
- 27.Tavana H, Mosadegh B, Takayama S. Advanced materials. 2010;22:2628. doi: 10.1002/adma.200904271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cui X, Dean D, Ruggeri ZM, Boland T. Biotechnology and bioengineering. 2010;106:963. doi: 10.1002/bit.22762. [DOI] [PubMed] [Google Scholar]
- 29.Saunders RE, Gough JE, Derby B. Biomaterials. 2008;29:193. doi: 10.1016/j.biomaterials.2007.09.032. [DOI] [PubMed] [Google Scholar]
- 30.Ker ED, Nain AS, Weiss LE, Wang J, Suhan J, Amon CH, Campbell PG. Biomaterials. 2011;32:8097. doi: 10.1016/j.biomaterials.2011.07.025. [DOI] [PubMed] [Google Scholar]
- 31.Campbell PG, Weiss LE. Expert opinion on biological therapy. 2007;7:1123. doi: 10.1517/14712598.7.8.1123. [DOI] [PubMed] [Google Scholar]
- 32.Sanjana NE, Fuller SB. Journal of neuroscience methods. 2004;136:151. doi: 10.1016/j.jneumeth.2004.01.011. [DOI] [PubMed] [Google Scholar]
- 33.Dawson E, Mapili G, Erickson K, Taqvi S, Roy K. Advanced drug delivery reviews. 2008;60:215. doi: 10.1016/j.addr.2007.08.037. [DOI] [PubMed] [Google Scholar]
- 34.Bognitzki M, Czado W, Frese T, Schaper A, Hellwig M, Steinhart M, Greiner A, Wendorff JH. Advanced materials. 2001;13:70. [Google Scholar]
- 35.Greiner A, Wendorff JH. Angew Chem Int Edit. 2007;46:5670. doi: 10.1002/anie.200604646. [DOI] [PubMed] [Google Scholar]
- 36.Li D, Xia YN. Advanced materials. 2004;16:1151. [Google Scholar]
- 37.Lembach AN, Tan HB, Roisman IV, Gambaryan-Roisman T, Zhang YY, Tropea C, Yarin AL. Langmuir. 2010;26:9516. doi: 10.1021/la100031d. [DOI] [PubMed] [Google Scholar]
- 38.Yang L, Fitie CF, van der Werf KO, Bennink ML, Dijkstra PJ, Feijen J. Biomaterials. 2008;29:955. doi: 10.1016/j.biomaterials.2007.10.058. [DOI] [PubMed] [Google Scholar]
- 39.Moiseeva EP. Cardiovasc Res. 2001;52:372. doi: 10.1016/s0008-6363(01)00399-6. [DOI] [PubMed] [Google Scholar]
- 40.Falconnet D, Csucs G, Grandin HM, Textor M. Biomaterials. 2006;27:3044. doi: 10.1016/j.biomaterials.2005.12.024. [DOI] [PubMed] [Google Scholar]
- 41.Miller ED, Fisher GW, Weiss LE, Walker LM, Campbell PG. Biomaterials. 2006;27:2213. doi: 10.1016/j.biomaterials.2005.10.021. [DOI] [PubMed] [Google Scholar]
- 42.Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Nat Rev Mol Cell Bio. 2002;3:349. doi: 10.1038/nrm809. [DOI] [PubMed] [Google Scholar]
- 43.Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat ML, Gabbiani G. Am J Pathol. 2007;170:1807. doi: 10.2353/ajpath.2007.070112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Midgley AC, Rogers M, Hallett MB, Clayton A, Bowen T, Phillips AO, Steadman R. The Journal of biological chemistry. 2013;288:14824. doi: 10.1074/jbc.M113.451336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Discher DE, Mooney DJ, Zandstra PW. Science. 2009;324:1673. doi: 10.1126/science.1171643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bissell MJ, Radisky D. Nature reviews. Cancer. 2001;1:46. doi: 10.1038/35094059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zuk PA, Zhu M, Mizuno H, Huang J, Futrell JW, Katz AJ, Benhaim P, Lorenz HP, Hedrick MH. Tissue engineering. 2001;7:211. doi: 10.1089/107632701300062859. [DOI] [PubMed] [Google Scholar]
- 48.Lu PF, Weaver VM, Werb Z. J Cell Biol. 2012;196:395. doi: 10.1083/jcb.201102147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shen K, Luk S, Hicks DF, Elman JS, Bohr S, Iwamoto Y, Murray R, Pena K, Wang F, Seker E, Weissleder R, Yarmush ML, Toner M, Sgroi D, Parekkadan B. Nature communications. 2014;5:5662. doi: 10.1038/ncomms6662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dvorak HF, Flier J, Frank H. New Engl J Med. 1986;315:1650. doi: 10.1056/NEJM198612253152606. [DOI] [PubMed] [Google Scholar]
- 51.Hu M, Peluffo G, Chen H, Gelman R, Schnitt S, Polyak K. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:3372. doi: 10.1073/pnas.0813306106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kellermann MG, Sobral LM, da Silva SD, Zecchin KG, Graner E, Lopes MA, Kowalski LP, Coletta RD. Oral oncology. 2008;44:509. doi: 10.1016/j.oraloncology.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 53.Sullivan NJ, Sasser AK, Axel AE, Vesuna F, Raman V, Ramirez N, Oberyszyn TM, Hall BM. Oncogene. 2009;28:2940. doi: 10.1038/onc.2009.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Guo X, Oshima H, Kitmura T, Taketo MM, Oshima M. The Journal of biological chemistry. 2008;283:19864. doi: 10.1074/jbc.M800798200. [DOI] [PubMed] [Google Scholar]
- 55.Noma K, Smalley KS, Lioni M, Naomoto Y, Tanaka N, El-Deiry W, King AJ, Nakagawa H, Herlyn M. Gastroenterology. 2008;134:1981. doi: 10.1053/j.gastro.2008.02.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Weis SM, Cheresh DA. Nature medicine. 2011;17:1359. doi: 10.1038/nm.2537. [DOI] [PubMed] [Google Scholar]
- 57.Soker S, Takashima S, Miao HQ, Neufeld G, Klagsbrun M. Cell. 1998;92:735. doi: 10.1016/s0092-8674(00)81402-6. [DOI] [PubMed] [Google Scholar]
- 58.Jain RK. Nature medicine. 2001;7:987. doi: 10.1038/nm0901-987. [DOI] [PubMed] [Google Scholar]
- 59.Mao Y, Keller ET, Garfield DH, Shen K, Wang J. Cancer Metastasis Rev. 2013;32:303. doi: 10.1007/s10555-012-9415-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kitadai Y. Cancer Microenviron. 2010;3:109. doi: 10.1007/s12307-009-0032-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dvir T, Timko BP, Kohane DS, Langer R. Nat Nanotechnol. 2011;6:13. doi: 10.1038/nnano.2010.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fischbach C, Chen R, Matsumoto T, Schmelzle T, Brugge JS, Polverini PJ, Mooney DJ. Nature methods. 2007;4:855. doi: 10.1038/nmeth1085. [DOI] [PubMed] [Google Scholar]
- 63.Yang X, Shah JD, Wang H. Tissue Eng Part A. 2009;15:945. doi: 10.1089/ten.tea.2007.0280. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.