

The table of contents entry
A dimeric drug polymeric micelle with acid-active tumor targeting and FRET-traceable drug release is successfully fabricated. It has such unique features as a long blood circulation through shielding of the cationic charges of TAT, an enhanced cellular internalization by regenerating the original TAT in acidic tumor tissue and a self-reported drug release via FRET signal triggered by the intracellular GSH.
Keywords: tumor targeting, drug delivery, acid-active, reduction sensitive, FRET
Cancer is the leading cause of death in the world today, with over 1,600,000 new cancer cases diagnosed and over 580,000 cancer deaths in the U.S. in 2015, according to the American Cancer Society.[1] Although traditional chemotherapy remains to be a prior method against cancer, chemotherapeutics usually fail due to the poor water solubility, high toxicity and low bioavailability. To solve these problems, nanotechnology-mediated drug delivery system (NDDS) has emerged. Nanoparticles (NPs), especially those that are made from biodegradable and biocompatible polymers, have been widely utilized as NDDS for cancer therapy.[2–4] Using nanotechnology, it is possible to achieve improved delivery of poorly water-soluble drugs, targeted delivery of drugs in a cell- or tissue-specific manner, codelivery of two or more drugs for combination therapy, etc.[5] Hence with all these advantages, NPs can be an outstanding candidate for loading chemotherapeutics.
A key goal of NPs is to quickly deliver their payloads into desired cells. Cell-penetrating peptides (CPPs), highly cationic peptides usually rich in arginine and lysine amino acids,[6] have been extensively explored for decorating NPs to promote their cellular uptake. This is ascribed to the high affinity of positively charged CPPs for negatively charged cell membranes. Some sequences, such as the protein transduction domain from the HIV transactivating transcriptional activator (TAT) peptide (CYGRKKRRQRRR),[7] can be recognized by the nuclear pore complexes (NPCs),[8] and thus actively transport cargos from the cytosol into the cell nucleus. Therefore, TAT peptide is one of the most thoroughly studied and extensively used CPPs for intracellular and nuclear delivery.
However, TAT-modified NPs with a positively charged surface undergo substantial phagocytosis by the reticular endothelin system (RES) and interact strongly with serum components, causing severe aggregation and a short blood circulation half-life.[9] Moreover, TAT peptides are internalized by both normal cells and tumor cells due to their lack of tumor targeting property. In order to effectively use TAT-modified NPs for cancer treatment, a new method that can prevent aggregation and increase both the half-life time in the blood circulation and the tumor targeting capability needs to be developed. Previous studies have proven that the NPs containing amides with β-carboxylic acid groups are acid labile; as a result, they are negatively charged under a neutral condition while becoming positively charged in an acidic environment, followed by a selective internalization by tumor cells instead of normal cells.[10,11] Therefore, the TAT’s positive charge can be shielded by converting their amines to β-carboxylic amides when triggered by an environmental pH change, making the modified TAT exhibit pH-dependent hydrolysis and achieving a negative-to-positive charge reversal.
Apart from high stability and tumor targeting, an ideal drug delivery vehicle ought to precisely control the release of cargo as well. In order to achieve site-specific drug delivery, strategies have been developed by introducing stimuli responsiveness into NPs, making them respond to modest environmental changes, which in turn leads to alterations in their structures or chemistry subsequently inducing the release of drug.[12-14] Owing to the differential glutathione tripeptide (γ-glutamyl-cysteinyl-glycine, GSH) level between the intracellular compartment (2~10 mM) and the extracellular microenvironment (2~20 μΜ).[15] the significant redox potential difference can be used as a unique trigger for designing NDDS with programmed release of chemotherapeutics intracellularly.[16,17] Compared to the disulfide bond that has been widely applied for constructing reduction-responsive systems, another bioreducible bond, dithiomaleimides, produced by reaction of 2,3-dibromomaleimide and thiols,[18,19] possess better thermal- and photo-stability than disulfide bonds.[20] Furthermore, the strong green fluorescence of dithiomaleimides can be taken advantage of for tracking the ndds.[21,22]
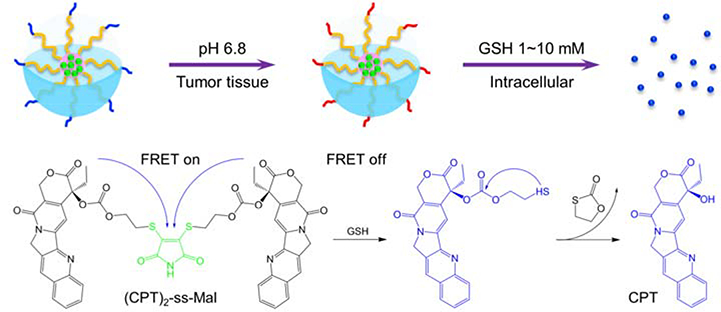
Here, we develop an acid-triggered tumor targeting polymeric micelle system loaded with a reduction-sensitive camptothecin (CPT) dimer, which is capable of self-reporting drug release via FRET. Polymeric micelles that possess a unique core/shell architecture and nanoscale size are widely used as drug carriers for loading anticancer drugs. In this nanoplatform, polyethylene (PEG) and poly(ε-caprolactone) (PCL) were used as the hydrophilic segment and hydrophobic segment, respectively. To allow the micelles to be internalized by cancer cells, TAT peptide was conjugated to the PEG segment. Further, the lysine and glutamine residues’ amines of TAT were amidized by 2,3-dimethylmaleic anhydride (DA) to prevent nonspecific interactions in vivo resulting from the cationic nature of the amines (Figure 1).
Figure 1.
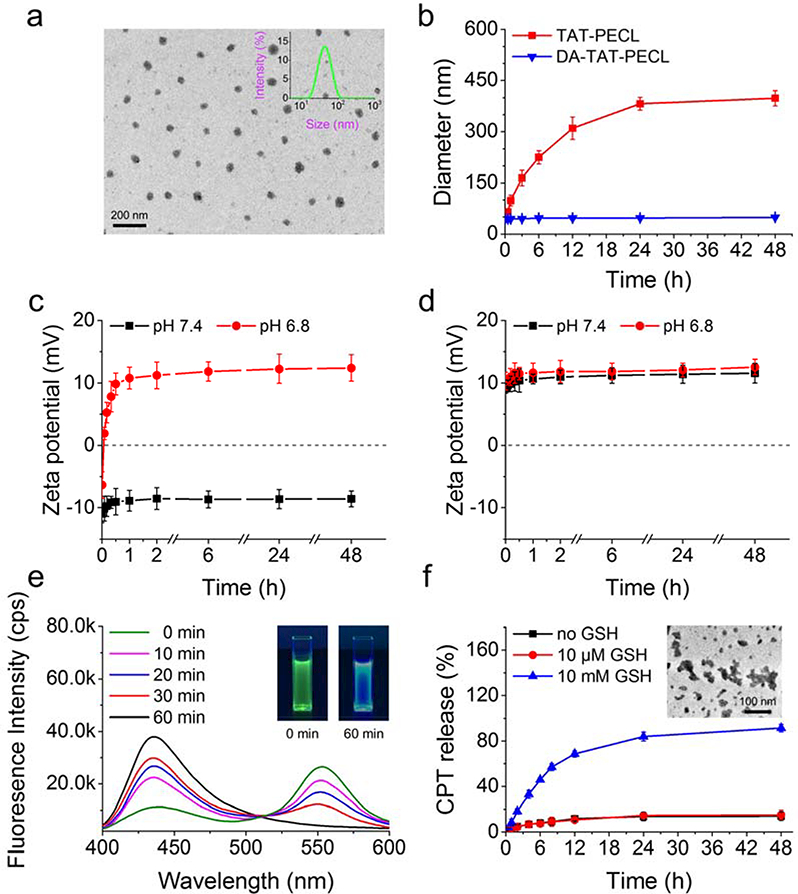
a) Size distribution detected by DLS and TEM image of DA-TAT-PECL micelles. b) Size change in TAT-PECL and DA-TAT-PECL micelles after incubation with 0.5 mg ML−1 bovine serum albumin (BSA). Zeta-potential change in DA-TAT-PECL c) and TAT-PECL d) micelles at pH 7.4 and pH 6.8. e) Fluorescence spectra of (CPT)2-ss-Mal in the presence of 10 mM GSH at different time points. Excitation wavelength was set at 370 nm. Inset: pictures of (CPT)2-ss-Mal in the presence of 10 mM GSH under UV irradiation (365 nm) for 0 min (left) and 60 min (right). f) Percent CPT released from the (CPT)2-ss-Mal-loaded DA-TAT-PECL micelles in the absence or presence of 10 μΜ and 10 Mm GSH. Inset: TEM image of the (CPT)2-ss-Mal-loaded DA-TAT-PECL micelles after treatment of 10 mM GSH for 48 h.
Next, the anticancer drug, CPT was modified into a dimer (CPT)2-ss-Mal, whereby two CPT molecules were connected by a reduction-labile maleimide thioether bond. The dimer featured an emission peak at 550 nm (dithiomaleimide emission wavelength, green fluorescence) rather than at 438 nm (CPT emission wavelength, blue fluorescence) when excited at 370 nm (CPT excitation wavelength), indicating the existence of the FRET phenomenon between CPT (donor) and maleimide thioether bond (acceptor). However, in the presence of a reducing environment, the maleimide thioether bond can be cleaved, subsequently releasing CPT via cyclization reaction. As a result, the FRET effect was abolished accordingly with the transition of emission peak to 438 nm and the recovery of blue fluorescence when excited at 370 nm. By monitoring the change of the FRET signal on the CPT dimer-loaded micelles in real time, we can visualize and quantify the drug release process (Scheme 1).
Scheme 1.
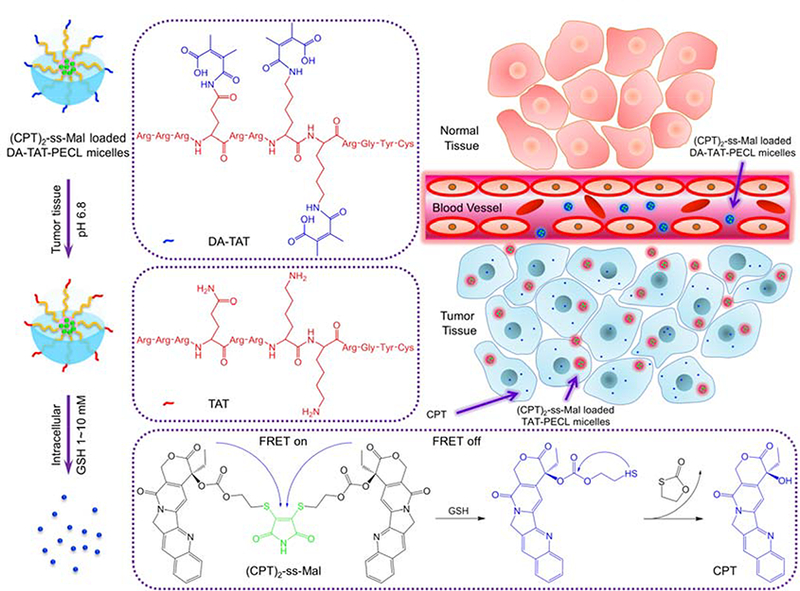
Schematic illustration of acid-triggered tumor targeting DA-TAT-PECL nanoplatform loading with reduction-sensitive (CPT)2-ss-Mal. Shielding of the cationic charges of TAT with DA suppresses its nonspecific interactions in blood circulation, while regenerating the original TAT in weakly acidic tumor tissue (pH 6.8) for cellular internalization or in more acidic endo/lysosomes (pH 4~5) for endo/lysosomal escape. Release of CPT from (CPT)2-ss-Mal is selectively triggered by the high concentration of intracellular GSH (2~10 mM), and the concomitant change of FRET signal can be used to report the drug release process.
The (CPT)2-ss-Mal-loaded DA-TAT-PECL micelles with a negatively charged surface possessed high stability in blood circulation due to the electrostatic effect. Once they accumulated at the acidic tumor tissues through highly permeable blood vessels via the enhanced permeability and retention (EPR) effect.[23] the amides in the DA-TAT quickly hydrolyzed, fully restoring TAT’s functions and facilitating the cellular entry. Afterwards, CPT were released from (CPT)2-ss-Mal triggered by the intracellular GSH with FRET-indicated signal change. This development of combined acid-active targeting with reducible drug dimer and FRET-based self-reporting drug release provides a novel approach for efficient cancer therapy.
DA-TAT-PECL was synthesized by amidization of the TAT lysine and glutamine residues’ amines using 2,3-dimethylmaleic anhydride (DA), followed by cojugating DA-TAT to Mal- PECL which was synthesized through ring-opening polymerization (ROP) of ε-caprolactone initiated by Mal-PEG-OH in the presence of stannous octoate (Sn(Oct)2) (Figure S1a, Supporting Information). (CPT)2-ss-Mal was synthesized by activation of CPT with triphosgene and then reaction with 2-hydroxyethyl disulfide to get disulfide linked-(CPT)2-ss, which was further conjugated to 2,3-dibromomaleimide in the presence of tris(2-carboxyethyl)phosphine (TCEP) to yield (CPT)2-ss-Mal (Figure S1b, Supporting Information).
According to the ratio of integral areas of different peaks in 1Η NMR, the average degree of polymerization of PCL segment can be calculated as 59 (Figure S2a, Supporting Information). Correspondingly, the molecular weight of Mal-PECL is 11700. As for the TAT-PECL, the characteristic resonance of maleimide at 6.68 ppm disappeared, indicating the successful conjugation of TAT to Mal-PECL (Figure S2b, Supporting Information).[24] Besides, the appearance of the peak at 1.84 ppm in 1H NMR of DA-TAT-PECL indicated the complete amidation of TAT amines by DA (Figure S2c, Supporting Information).[10] The signals of hydrogen protons adjacent to difsulfide bond in (CPT)2-ss appeared at 2.83 ppm and 4.02 ppm, while transferred to 3.08 ppm and 4.41 ppm after reaction with 2,3-dibromomaleimide (Figure S2d,e, Supporting Information). This change in chemical shift is caused by the electron-withdrawing inductive effect of maleimide.[25]
The average size of DA-TAT-PECL micelles was 43.9 nm with low polydispersity index (PDI) of 0.135, indicating a narrow distribution of diameters (Figure 1a). The TEM image showed that the DA-TAT-PECL micelles exhibited a spherical shape and a nearly uniform distribution. The diameter of TEM image was consistent with that detected by DLS (Figure 1a).
Shielding of the cationic charges of TAT with DA was supposed to inhibit the nonspecific interaction in blood circulation, as the negatively charged components in the surface of blood vessels and the surface of cells would repel negatively charged nanoparticles.[26] To validate this, the average size of TAT-PECL and DA-TAT-PECL micelles in the presence of 0.5 mg mL-1 bovine serum albumin (BSA) was measured by DLS at different time points. There’s no significant size change in cationic charge-shielded DA-TAT-PECL micelles after 48 h’ BSA incubation (Figure 1b). In contrast, the cationic charge-unshielded TAT-PECL micelles increased their size rapidly in a short time. The result indicated that the DA-decorated micelles could effectively resist protein adsorption as expected.
Amides with β-carboxylic acid groups are acid labile,[27] in which DA-TAT-PECL micelles ought to be negatively charged in neutral condition while reversing to be positively charged triggered by acid due to the recovery of amines upon amides hydrolysis.[11] As shown in Figure 1c, DA-TAT-PECL micelles were negatively charged at the beginning regardless of the environmental pH value. However, the ζ-potential of these micelles quickly increased and became positive within 5 min at pH 6.8, while it remained negative within 48 h at pH 7.4, implying the high stability. Considering the negative charge of cell membranes, the charge-reversal behavior of DA-TAT-PECL micelles at tumor extracellular pH would enhance their internalization by tumor cells.[28] Besides, the exposure of TAT peptide with the cleavage of amides in DA-TAT-PECL micelles would further facilitate the cellular uptake, followed by endo/lysosomal escape in more acidic endo/lysosomal compartment.[29] In contrast to DA- TAT-PECL micelles, TAT-PECL micelles always stayed positively charged with ζ-potentials around 10 mV at any environmental pH value (Figure 1d).
To confirm the reduction-responsive FRET inactivation, the fluorescence properties of (CPT)2-ss-Mal in the presence of GSH were evaluted by fluorescence spectrometer and digital photos. When excited at 370 nm (CPT excitation wavelength), the fluorescence spectrum of the solution showed an emission peak at 550 nm (dithiomaleimide emission wavelength) instead of 438 nm (CPT emission wavelength), indicating the existence of the FRET phenomenon between CPT and maleimide thioether bond (Figure 1e). However, a decrease in dithiomaleimide fluorescence and an increase in CPT fluorescence were observed with GSH incubation, while the peak at 550 nm even disappeared 60 min later, indicating a complete cleavage of maleimide thioether bond. This was also reflected in a significant color change of the solution from green to blue under UV irradiation (365 nm) (Figure 1e, inset). According to these results, it can be inferred that (CPT)2-ss-Mal rapidly released CPT in a reducing environment accompanied by change in the FRET signal.
Owing to the π-π interaction of the planar pentacyclic aromatic ring structure, CPT molecules self-aggregated to micrometer-size large particles when encapsulated into micelles, leading to a very low drug loading (usually less than 5%).[30] By covalently linking two CPT molecules into a dimer, formation of large aggregates was prevented due to the inhibition of long-range-ordered drug molecule packing. Consequently, high drug-loading content (LC) and encapsulation efficiency (EE) would be expected by loading the CPT dimers into micelles. To confirm this, the size and drug content of (CPT)2-ss-Mal-loaded DA-TAT-PECL micelles were measured by DLS and UV-vis, respectively. The results demonstrated that these particles were a little bigger than the blank micelles with a diameter of 51.6 nm. Besides, the LC and EE were as high as 42% and 84%, respectively. Next, we detected the in vitro CPT release from the (CPT)2-ss-Mal-loaded DA-TAT-PECL micelles in the absence of GSH and in the presence of 10 μΜ or 10 mM GSH. Negligible CPT release was observed over 48 h without GSH treatment or treated with 10 μM GSH (Figure 1f). In comparison, a rapid release of CPT was immediately turned on in the presecne of 10 mM GSH, in which over 90% of CPT was released from the micelles within the experimental time. This trigger-responsive property is due to the cleavable maleimide thioether bond in (CPT)2-ss-Mal, which was ruptured upon reducing environment, followed by a preferential release of CPT instead of releasing in physiological condition. Therefore, the ideal drug release behavior of this system was confirmed. Considering the elevated level of thiol species in a variety of cells,[31] this drug delivery system is shown to be stable in the physiological condition (~10 μM GSH), while resopnding to intracellular reducing environment (~10 mM GSH) and subsequently releasing the cargo. Furthermore, the disassembly of micelles and partial aggregation were observed in the TEM image after 48 h of GSH treatment (Figure 1f, inset), mainly due to the instability of micelles after releasing the cargo.
To study the cytocompatibility of these micelles as potential drug delivery systems, we performed alamarBlue assay and live/dead staining. MDA-MB-231 cell viability was over 90% with incubation of different concentrations of PECL, TAT-PECL and DA-TAT-PECL micelles for 48 h (Figure S3a, Supporting Information). The fluorescence images showed that there were no dead cells unless treated with a high concentration of micelles at 500 μg mL−1 (Figure S3b, Supporting Information). Taken together, we believe these vehicles possess excellent cytocompatibility and potential application as drug carreris.
The reducing intracellular compartment is expected to trigger the cleavage of maleimide thioether bond in (CPT)2-ss-Mal once it is internalized by cells, followed by the release of CPT. Interestingly, this prcoess can be self-reporting via FRET signal. To demonstrate this, we treated MDA-MB-231 cells with 1 mM (CPT)2-ss-Mal and observed the real-time fluorescence images using 365 nm as excitation wavelength. As shown in Figure 2a, the cells overall showed green fluorescence within 30 min, indicating a cellular uptake process without CPT leakage. After 45 min of incubation, the blue fluorescence appeared in the cells and increased in intensity as time passed by. Finally, all the cells showed blue fluorescence with the disappearance of green fluorescence at 90 min. The change in FRET signal indicated the in situ release of CPT from (CPT)2-ss-Mal between 45 min to 90 min. It is also worth mentioning that the intracellular GSH-induced FRET inactivation process took as long as that measured by the fluorescence spectrometer (Figure 1e). On the basis of these images, we were able to confirm that the drug release occurred in response to the intracellular GSH.
Figure 2.
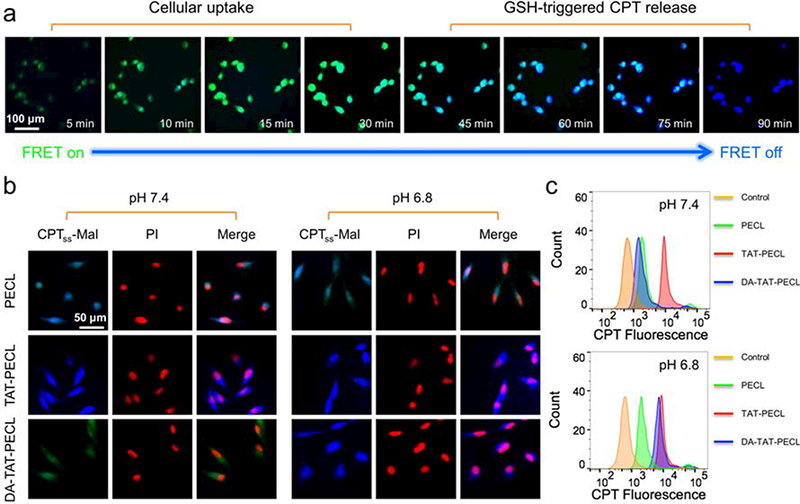
a) Real-time fluorescence images in MDA-MB-231 cells showing the GSH-triggered FRET inactivation after treating with (CPT)2-ss-Mal (CPT-equivalent dose: 1 mM). Green fluorescence (FRET on) indicated the uptake of (CPT)2-ss-Mal by cells while blue fluorescence (FRET off) indicated the cleavage of maleimide thioether bond in (CPT)2-ss-Mal and release of CPT from the drug dimer. b) Fluorescence images and c) flow cytometry analysis in MDA-MB-231 cells showing the pH-dependent cellular uptake. Cells were treated with (CPT)2-ss-Mal-loaded PECL, TAT-PECL and DA-TAT-PECL micelles at pH 7.4 and pH 6.8 for 2 h (CPT-equivalent dose: 1 mM). Nuclei were stained with PI (red).
Compared to the negatively charged NPs, vehicles with a positively charged surface tend to possess a higher rate of cellular uptake.[32] The charge reversal of DA-TAT-PECL micelles in weak acidic condition was confirmed (Figure 1c), consequently, they ought to be more efficiently internalized by cells once reversing to positive charge and exposure of TAT peptide. To observe the pH-dependent cellular uptake, MDA-MB-231 cells were treated with (CPT)2-ss-Mal-loaded PECL, TAT-PECL and DA-TAT-PECL micelles at pH 7.4 and pH 6.8 for 2 h, and imaged by the fluorescence microscope. As shown in Figure 2b, weak green fluorescence was observed in DA-TAT-PECL group at pH 7.4, indicating a slow cellular uptake of these micelles. However, they were remarkably internalized at pH 6.8 and completely released CPT within 2 h, in which blue fluorescence as strong as the TAT-PECL group was displayed. The cellular uptake of the neutral PECL micelles without TAT decoration was irrelevant to environmental pH, where similar fluorescence intensity was observed in the cells at pH 7.4 and 6.8. The flow cytometry analysis further proved that the endocytosis of DA-TAT-PECL micelles by MDA-MB-231 cells was pH dependent. After 2 h of incubation, the CPT fluorescence in the DA-TAT-PECL group was significantly enhanced at pH 6.8 compared to that at pH 7.4 (Figure 2c). There was no obvious change of the CPT fluorescence in cells treated with either PECL micelles or TAT-PECL micelles at pH 7.4 and pH 6.8.
We have shown that DA-TAT-PECL micelles could quickly deliver (CPT)2-ss-Mal into MDA-MB-231 cells at pH 6.8, which were able to release CPT in response to intracellular GSH (Figure 2). In this case, cell cytotoxicity should also be pH-dependent for DA-TAT-PECL micelles. To demonstrate this, MDA-MB-231 cells were treated with various (CPT)2-ss-Mal formulations at different pH values. Cell viability by alamarBlue assay indicated that (CPT)2-ss-Mal-loaded DA-TAT-PECL micelles were more toxic at pH 6.8 relative to that at pH 7.4 (Figure 3a). This difference is attributed to the higher uptake of these micelles by MDA-MB-231 cells in acidic condition. There is no significant difference in the IC50 values of (CPT)2-ss-Mal at pH 7.4 and pH 6.8, and the same is true for the (CPT)2-ss-Mal-loaded PECL and TAT-PECL micelles. Live/dead staining was performed to further evaluate the pH-dependent cell cytotoxicity, the result was consistent with the alamarBlue assay as more cells were dead if treated with (CPT)2-ss-Mal-loaded DA-TAT-PECL micelles at pH 6.8 (Figure 3b).
Figure 3.
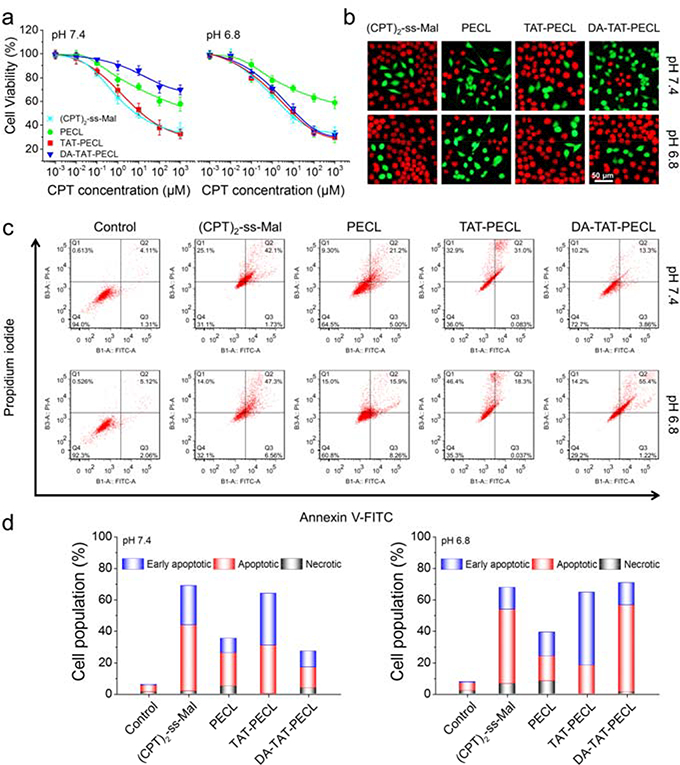
pH-dependent cell cytotoxicity and cell apoptosis. a) Viability of MDA-MB-231 cells showing the pH-dependent cytotoxicity by alamarBlue assay. Cells were treated with different concentrations of (CPT)2-ss-Mal and (CPT)2-ss-Mal-loaded PECL, TAT-PECL and DA-TAT-PECL micelles at pH 7.4 and pH 6.8 for 48 h (n = 5). b) Fluorescence images showing the pH-dependent viability of MDA-MB-231 cells by calcein AM and propidium iodide double staining. Cells were treated with (CPT)2-ss-Mal and (CPT)2-ss-Mal-loaded PECL, TAT-PECL and DA-TAT-PECL micelles at pH 7.4 and pH 6.8 for 48 h. The live cells were stained green while the dead cells were stained red. c) Flow cytometry analysis in MDA-MB-231 cells showing the pH-dependent cell apoptosis by Annexin V-FITC/PI double-staining assay. Cells were treated with (CPT)2-ss-Mal or (CPT)2-ss-Mal-loaded PECL, TAT-PECL and DA-TAT-PECL micelles at pH 7.4 and 6.8 for 48 h. X-axis represented FITC-labeled Annexin V positive cells and Y-axis represented PI positive cells. d) Population of early apoptotic, apoptotic and necrotic MDA-MB-231 cells summarized from flow cytometry analysis. CPT-equivalent dose for each cytotoxicity and apoptosis test was 1 mM.
Annexin V-FITC/PI double-staining assay was used to evaluate the apoptosis in MDA-MB-231 cells induced by various (CPT)2-ss-Mal formulations at different pH values. Compared to the treatment at pH 7.4, the percentage of MDA-MB-231 cells undergoing apoptosis after incubation with (CPT)2-ss-Mal-loaded DA-TAT-PECL micelles at pH 6.8 was significantly increased (Figure 3c,d). Similar to the cell cytotoxicity study, cells exposed to (CPT)2-ss-Mal, (CPT)2-ss-Mal-loaded PECL and TAT-PECL micelles showed pH-independent apoptosis characteristics. Therefore, it can be concluded that the DA-TAT-PECL system was able to induce more significant cell death and apoptosis upon acid stimuli.
To investigate the delivery efficiency of micelles in vivo, the biodistribution of IR-780- loaded PECL, TAT-PECL and DA-TAT-PECL was tested on 4T1 tumor bearing nude mice through intravenous administration. In vivo imaging system (IVIS) was utilized to monitor the IR-780 fluorescence in mice. As shown in Figure 4a, all mice displayed significant fluorescence in tumors at 1 h post-injection, mainly due to the enhanced permeation and retention (EPR) effect of nanoparticles.[33] In comparison with PECL group, mice injected with TAT-PECL micelles showed much higher tumor accumulation, however, a high localization was also found in liver. This confirms the RES uptake and nonspecific internalization of TAT peptide. As we expected, DA decoration efficiently decreased the distribution of micelles in normal tissues. And the strong signal at tumor site demonstrated that DA group could be shedable in tumor microenvironment and the restored TAT peptide facilitated the uptake of nanoparticles by tumor cells. As time extended, TAT-PECL group displayed much weaker fluorescence intensity in tumor than DA-TAT-PECL group after administration for 6 h, implying the fast elimination and short blood circulation of positively charged TAT peptide. At 24 h post-injection, the tumor of DA-TAT-PECL group remained strong fluorescence signal, indicating the prolonged accumulation in tumor. Then mice were sacrificed to separate the organs and tumors for ex vivo imaging. There’s no significant difference in biodistribution between PECL and TAT-PECL (Figure 4b). Notably, the IR-780 fluorescence obtained from region-of-interest (ROI) analysis at the tumor site in DA-TAT-PECL group was 2.6-fold and 2.1-fold that of PECL and TAT-PECL, respectively (Figure 4c). These results proved the efficient tumor targeting ability of DA-TAT-PECL, which took advantage of EPR effect-driven accumulation in tumor tissue and the restored TAT peptide-induced internalization by tumor cells.
Figure 4.
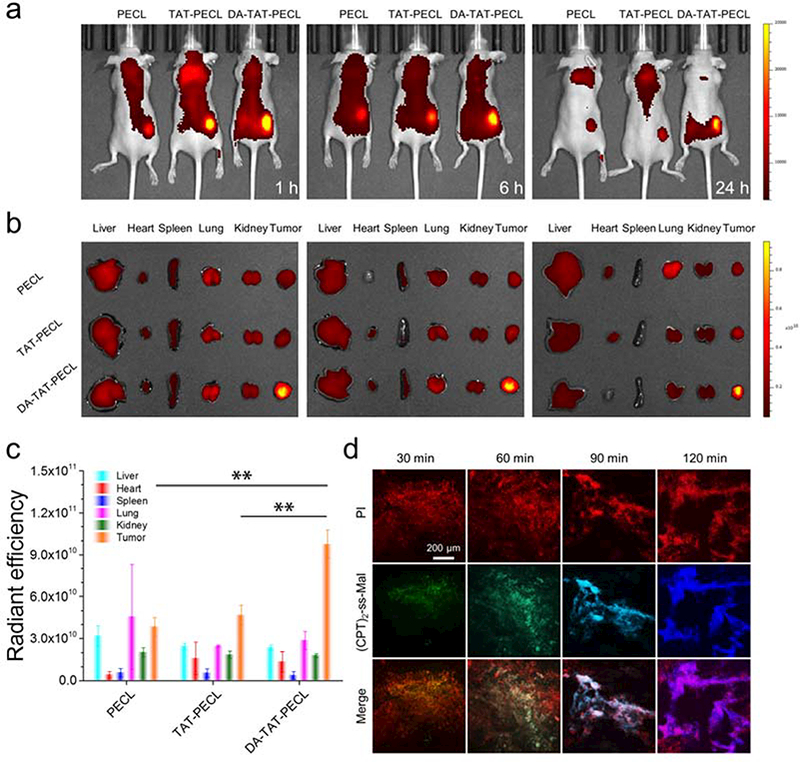
a) In vivo fluorescence images of 4T1 tumor bearing nude mice after intravenous injection of IR780-loaded PECL, TAT-PECL and DA-TAT-PECL micelles. b) Ex vivo fluorescence images of isolated tissues at 24 h post-injection. c) Quantitative analysis of fluorescence intensity based on ex vivo images. An unpaired t-test was used to discriminate the significant differences between two groups (two-tailed, **, p < 0.01). d) Fluorescence images of tumor tissue sections isolating from 4T1 tumor-bearing mice at indicated time points after intratumoral injection of (CPT)2-ss-Mal. Red, PI-stained cell nuclei; green (FRET on), (CPT)2-ss-Mal; blue (FRET off), CPT.
It has been confirmed that the intracellular cleavage of maleimide thioether bond in (CPT)2-ss-Mal could be self-reported via FRET signal (Figure 2a). We expected to see whether this property can be achievable in the tumor tissues. To demonstrate the intratumoral FRET inactivation, 4T1 tumor bearing BALB/c mice were administrated with (CPT)2-ss-Mal through intratumoral injection. Then mice were sacrificed at different time periods to separate the tumors for making slices. The sections were stained with propidium iodide (PI) and observed using a fluorescence microscopy. As shown in Figure 4d, a significant alteration in fluorescence from green (FRET on) to blue (FRET off) was observed in tumor tissues during 120 min. This process is similar to the intracellular FRET inactivation (Figure 2a), indicating the efficient cleavage of maleimide thioether bond in (CPT)2-ss-Mal and subsequent release of CPT upon tumor microenvironment.
The in vivo antitumor study was carried out in the 4T1 tumor-bearing BALB/c mice. As expected, tumors grew rapidly in the saline group, which reached up to ~1200 mm3 on day 21 (Figure 5a,b). Treating mice with (CPT)2-ss-Mal-loaded DA-TAT-PECL micelles resulted in remarkable suppression of tumors, with an inhibition rate of ~97%, while that of the TAT-PECL group and PECL was ~81% and ~67%, respectively. Single use of (CPT)2-ss-Mal was less effective for tumor suppression in comparison with drug-loaded micelles, and the inhibition rate was only ~17%. As an indicator of systemic toxicity, the body weight of mice was monitored during 21 days. The results demonstrated that no obvious weight loss was observed in mice treated with drug-loaded micelles, especially the DA-TAT-PECL group showing an increase of weight (Figure 5c). In contrast, mice treated with (CPT)2-ss-Mal exhibited obvious weight loss, implying the serious toxicity of free CPT. Consistent with the tumor inhibition and body weight change, the survival time of mice was significantly prolonged when treated with drug-loaded micelles (Figure 5d). It’s worth mentioning that no mice were dead in DA-TAT-PECL group during 42 days, indicating the efficient treatment and high safety of this acid-active tumor targeting nanoplatform in combination with the reduction-sensitive drug dimer.
Figure 5.
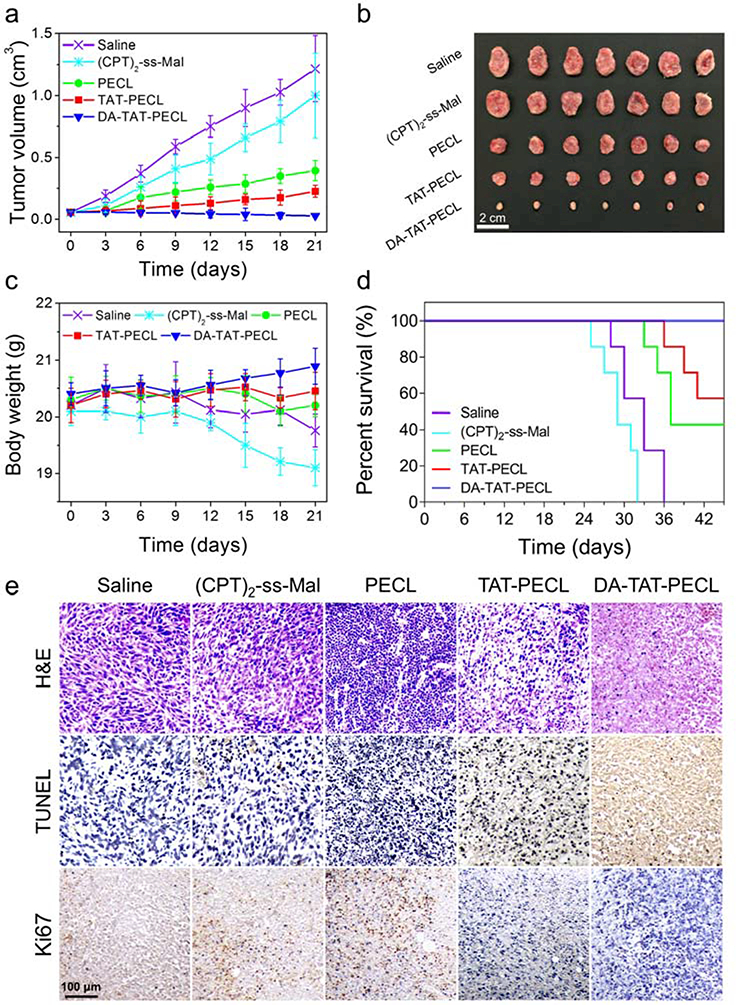
In vivo tumor inhibition and systemic toxicity in 4T1 tumor-bearing Balb/c mice after intravenous injection of different formulations (n = 7, dose: 5 mg CPT per kg mouse body weight). a) Tumor volume. b) Excised 4T1 solid tumors at the 21st day. c) Body weight. d) Survival curves. e) H&E, TUNEL and Ki67 analyses of tumor tissues after treatment. In H&E staining, nuclei are stained blue, and extracellular matrix and cytoplasm are stained red. In TUNEL staning, normal cells are stained blue and apoptotic cells are stained brown. In Ki67 staining, the proliferating cells are stained brown.
To further investigate the cell apoptosis in tumors of mice after treatment, haematoxylin and eosin (H&E), terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) and Ki-67 staining were performed. The H&E staining showed that saline and (CPT)2-ss-Mal groups appeared to be most hypercellular and possessed significant nuclear polymorphism (Figure 5e). In contrast, micellar groups showed fewer tumor cells and more tumor necrosis, validating that proliferation had been effectively halted. Compared with other formulations, DA-TAT-PECL markedly increased the number of TUNEL-apoptotic cells and reduced the percentage of Ki67-positive proliferating cells, implying the enhanced therapeutic efficiency in inducing the apoptosis and inhibiting the proliferation of tumor cells.
In summary, we developed an acid-active tumor targeting DA-TAT-PECL nanoplatform loaded with reduction-sensitive (CPT)2-ss-Mal. Shielding of the cationic charges of TAT with DA effectively inhibited its nonspecific interactions with proteins in blood circulation. Meanwhile, the original TAT is regenerated in the acidic tumor microenvironment, facilitating the cellular entry of the nanoparticles via cell-penetrating peptide-mediated endocytosis and electrostatic interaction. Once the nanoparticles were internalized by tumor cells and escaped from the endo/lysosomes, CPT would be released from (CPT)2-ss-Mal in response to the high concentration of intracellular GSH. The change in FRET signals between CPT and maleimide thioether bond was able to report the drug release process. The in vivo studies evidenced the efficient tumor targeting and high tumor inhibition capacities of this multifunctional nanoplatform. We believe this drug delivery system will provide a promising potential to be further developed as a real therapeutic for effective cancer therapy.
Supplementary Material
Acknowledgements
This work was partially supported by the Society in Science Branco Weiss Fellowship, administered by ETH Zürich. The research reported in this paper was in part supported by the National Cancer Institute of the National Institutes of Health under Award (No. U01CA202123) and the National Natural Science Foundation of China (Nos. 51603172, 21574105). The authors thank Prof. Jiangbing Zhou, Dr. Zeming Chen and Dr. Pan Zou in Yale University for assistance in animal experiments.
Footnotes
EXPERIMENTAL SECTION
See the Supporting Information for experimental details
The authors declare no competing financial interest.
References
- [1].Lee A, Ng V, Gao S, Hedrick J, Yang Y, Adv. Funct. Mater 2014, 24, 1538. [Google Scholar]
- [2].Wang AZ, Langer R, Farokhzad OC, Annu. Rev. Med 2012, 63, 185. [DOI] [PubMed] [Google Scholar]
- [3].Petros RA, DeSimone JM, Nat. Rev. Drug Discov. 2010, 9, 615. [DOI] [PubMed] [Google Scholar]
- [4].Yu XJ, Trase I, Ren MQ, Duval K, Guo X, Chen Z, J. Nanomater. 2016, 2016, 1087250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Farokhzad OC, Langer R, ACS Nano 2009, 3, 16. [DOI] [PubMed] [Google Scholar]
- [6].Lindgren M, Hällbrink M, Prochiantz A, Langel Ü, Trends Pharmacol. Sci 2000, 21, 99. [DOI] [PubMed] [Google Scholar]
- [7].Vives E, Brodin P, Lebleu B, J. Biol. Chem 1997, 272, 16010. [DOI] [PubMed] [Google Scholar]
- [8].Lange A, Mills RE, Lange CJ, Stewart M, Devine SE, Corbett AH, J. Biol. Chem 2007, 282, 5101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Oupicky D, Ogris M, Howard KA, Dash PR, Ulbrich K, Seymour LW, Mol. Ther 2002, 5, 463. [DOI] [PubMed] [Google Scholar]
- [10].Guo X, Shi C, Yang G, Wang J, Cai Z, Zhou S, Chem. Mater 2014, 26, 4405. [Google Scholar]
- [11].Guo X, Wei X, Jing Y, Zhou S, Adv. Mater 2015, 27, 6450. [DOI] [PubMed] [Google Scholar]
- [12].Mura S, Nicolas J, Couvreur P, Nat. Mater 2013, 12, 991. [DOI] [PubMed] [Google Scholar]
- [13].Ge Z, Liu S, Chem. Soc. Rev 2013, 42, 7289. [DOI] [PubMed] [Google Scholar]
- [14].Torchilin VP, Nat. Rev. Drug Discov 2014, 13, 813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Schafer FQ, Buettner GR, Free Radical Bio. Med 2001, 30, 1191. [DOI] [PubMed] [Google Scholar]
- [16].Huo M, Yuan J, Tao L, Wei Y, Polym. Chem 2014, 5, 1519. [Google Scholar]
- [17].Deng B, Ma P, Xie Y, Nanoscale 2015, 7, 12773. [DOI] [PubMed] [Google Scholar]
- [18].Jones MW, Strickland RA, Schumacher FF, Caddick S, Baker JR, Gibson MI, Haddleton DM, J. Am. Chem. Soc 2012, 134, 1847. [DOI] [PubMed] [Google Scholar]
- [19].Robin MP, Jones MW, Haddleton DM, O’Reilly RK, ACS Macro Lett 2011, 1, 222. [DOI] [PubMed] [Google Scholar]
- [20].Wang H, Xu M, Xiong M, Cheng J, Chem. Commun 2015, 51, 4807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Robin MP, O’Reilly RK, Chem. Sci 2014, 5, 2717. [Google Scholar]
- [22].Robin MP, Wilson P, Mabire AB, Kiviaho JK, Raymond JE, Haddleton DM, O’Reilly RK, J. Am. Chem. Soc 2013, 135, 2875. [DOI] [PubMed] [Google Scholar]
- [23].Maeda H, Wu J, Sawa T, Matsumura Y, Hori K, J. Control. Release 2000, 65, 271. [DOI] [PubMed] [Google Scholar]
- [24].Li Y, Li P, Jin M, Jiang C, Gao Z, Int. J. Mol. Sci 2014, 15, 23571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Gobbo P, Workentin MS, Synlett 2016, 27, 1919. [Google Scholar]
- [26].Davis ME, Shin DM, Nat. Rev. Drug Disco 2008, 7, 771. [DOI] [PubMed] [Google Scholar]
- [27].Kluger R, Lam CH, J. Am. Chem. Soc 1978, 100, 2191 [Google Scholar]
- [28].Gratton SE, Ropp PA, Pohlhaus PD, Luft JC, Madden VJ, Napier ME, DeSimone JM, Proc. Natl. Acad. Sci. U.S.A 2008, 105, 11613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Jin E, Zhang B, Sun X, Zhou Z, Ma X, Sun Q, Tang J, Shen Y, Van Kirk E, Murdoch WJ, J. Am. Chem. Soc 2013, 135, 933. [DOI] [PubMed] [Google Scholar]
- [30].Cai K, He X, Song Z, Yin Q, Zhang Y, Uckun FM, Jiang C, Cheng J, J. Am. Chem. Soc 2015, 137, 3458. [DOI] [PubMed] [Google Scholar]
- [31].Lee MH, Yang Z, Lim CW, Lee YH, Dongbang S, Kang C, Kim JS, Chem. Rev 2013, 113, 5071. [DOI] [PubMed] [Google Scholar]
- [32].Alexis F, Pridgen E, Molnar LK, Farokhzad OC, Mol. Pharm 2008, 5, 505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Maeda H, Wu J, Sawa T, Matsumura Y, Hori KJ, J Control. Release 2000, 65,271. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.