

Abstract
A group of heterogeneous muscle diseases are caused by dystrophin gene (DMD) mutations. We hereby present a male patient with a diagnosis of symptomatic dilated cardiomyopathy at 44 years-old who developed, soon after, weakness of distal right upper limb. At the age of 58, neurological examination revealed severe atrophy of right thenar muscles, flexion contractures on the right elbow, wrist and fingers, bilateral calf hypertrophy, myotatic areflexia in the upper limbs and hyporeflexia in the lower limbs. Manual muscle examination showed distal weakness of right upper limb muscles, severe on abductor pollicis brevis and extensor pollicis longus, and milder on interossei, finger extensors and brachioradialis muscles. Further testing revealed CK of 1500 U/L, a myopathic pattern on electromyography, and myopathic changes on right deltoid muscle biopsy, with immunohistochemistry showing focal sub-expression of dystrophin. Cardiac workup revealed a severe reduction in left ventricular ejection fraction, with a left ventricle of increased dimensions and global hypo-contractibility. A next-generation sequencing based panel for muscular diseases was performed and a nonsense mutation (c.C7525T) was identified in exon 51 of DMD gene, present in 70% of the gene readings (consistent with mosaicism).
Key words: dystrophin gene, dilated cardiomyopathy, next generation sequencing
Introduction
The dystrophinopathies comprise a spectrum of different muscular phenotypes, with variable involvement of skeletal and cardiac muscle (1). The mildest end of the spectrum includes the phenotypes of asymptomatic hyper-CK, while the severe end includes Duchenne muscular dystrophy (DMD) and DMD-associated dilated cardiomyopathy. Becker muscular dystrophy (BMD) in the middle spectrum of clinical severity of dystrophinopathies, together with DMD, present cardiac involvement – in DMD it is a late feature in the disease course, whilst in BMD it can be the presenting and most disabling feature (2, 3).
The dystrophinopathies result from mutations in the DMD gene located in the X-chromosome (Xp21.2-p21). Clinical phenotype can be predicted in most patients by the reading-frame rule. Out-of-frame mutations cause absence of dystrophin expression in muscle originating a severe DMD phenotype, whereas in-frame mutations result in a milder BMD phenotype (2, 3). The most frequent mutations are deletions and duplications, while point mutations are responsible for 25% of the genetic changes in the gene (4).
We hereby present a patient with distal asymmetric weakness of the upper limbs and dilated cardiomyopathy, caused by a novel somatic mosaic mutation in the DMD gene.
Clinical findings
The patient was a Portuguese male, aged 58 years-old, born from a non-consanguineous couple. His family history was positive for coronary heart disease in his elder sister, due to proven atherosclerotic heart disease.
He had a personal history of type 2 diabetes mellitus, essential hypertension, dyslipidaemia and paroxysmal atrial fibrillation. At the age of 44, after an event of pre-cordial pain, a symptomatic dilated cardiomyopathy was diagnosed. It was initially managed with medical treatment but, due to its symptomatic progression reaching New York Heart Association class IV, a cardiodisfibrillator was implanted at age 57. He is currently on medication for cardiac insufficiency and comorbidities, including diuretics, anti-arrhythmics, oral anticoagulants, and oral glucose-lowering agents.
He was evaluated in the neuromuscular disease unit because of weakness of the distal right upper limb beginning soon after the diagnosis of dilated cardiomyopathy. He did not complain of muscle cramps or myoglobinuria. Neurological examination revealed flexion contractures of the right elbow, wrist, thumb and 2nd, 3rd and 4th fingers (Fig. 1), and severe atrophy of thenar eminence muscles. Bilateral calf hypertrophy was present. Manual muscle strength examination of the right upper limb (graded according to MRC scale) showed weakness of the abductor pollicis brevis and extensor pollicis longus muscles (grade 0/5), interossei muscles (grade 3–/5), and finger extensors and brachioradialis muscles (grade 4/5 and 4+/5, respectively). In the left upper limb, only finger abduction was mildly impaired (grade 4/5). Tendon reflexes were absent in the upper limbs and diminished in the lower limbs. Cranial nerves, sensory examination and gait were unremarkable.
Figure 1.
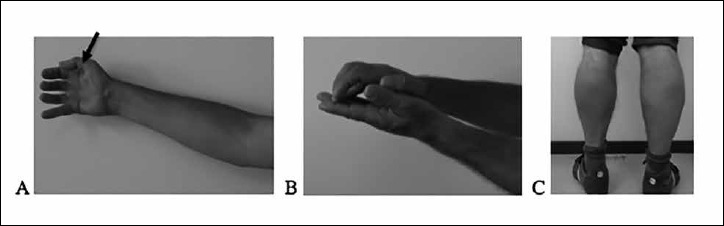
Atrophy of the right thenar eminence (arrow) and flexion contracture of the right elbow (A); Flexion contractures of the right wrist and fingers (B); Calf hypertrophy (C).
Complementary exams
A transthoracic echocardiography revealed a severe global left ventricle (LV) systolic dysfunction, with global hypo-contractility and severely compromised LV ejection fraction, and moderately compromised systolic function of right ventricle; radionuclide cardiac angiogram showed a global LV ejection fraction of 24%, with a LV of increased dimensions and global hypokinesia. Cardiac angiography did not show signs of coronary disease. An evaluation of respiratory function revealed a moderate restrictive syndrome (64.6% of predicted forced vital capacity), with normal alveolocapillary diffusion study.
Laboratory workup revealed an elevated BNP of 948.8 pg/mL (upper reference value 100 pg/mL), and hyper-CK of approximately 1500 U/L (upper reference value 171 U/L).
Electromyography showed a myopathic pattern, with normal motor and sensory nerve conduction studies.
Muscle biopsy
Right deltoid muscle biopsy revealed a myopathic pattern, with fibres of different sizes, internalized nuclei, and rare necrotic fibres (Fig. 2A-B). No fibrosis or fatty replacement was present. Careful re-evaluation of the immunohistochemical staining after molecular studies revealed focal sub-expression of dystrophin with Dys1, Dys2 and Dys3 antibodies (Fig. 2C). In this patchy pattern of dystrophin expression, the majority of fibres still showed normal dystrophin labelling.
Figure 2.
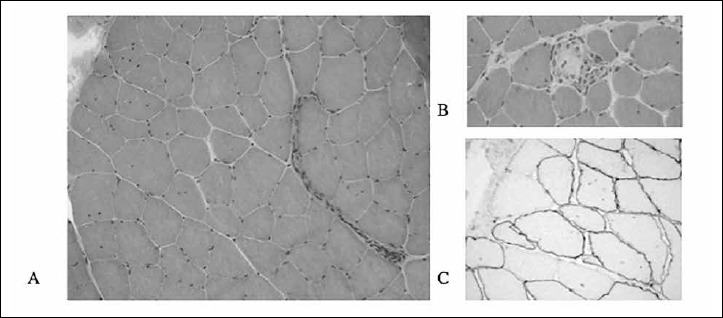
Deltoid muscle biopsy. Increased variability in fibre size and rare necrotic fibres (H&E - 200x) (A, B); Immunohistochemical staining for Dys3 showing focal sub-expression of dystrophin (200x) (C).
Molecular studies
A next generation sequencing panel for muscle diseases was performed through a custom targeted next generation sequencing panel, followed by Sanger sequencing validation of the identified mutations. This study revealed a nonsense novel mutation (c.C7525T) in exon 51 of the DMD gene in somatic mosaicism. Only 70% of X chromosomes from the patient carried the mutation (Fig. 3). This previously undescribed variant predicts a stop codon (p.Gln2509*). This variant was not registered in 1000 genomes, Single Nucleotide Polymorphism (dbSNP) databases, or in the Leiden Open Variation Database (LOVD) associated to disease. It was not detected in 401 chromosomes analysed from the Iberian population. Since this variant was not found in the control population and is a nonsense mutation, we consider it as likely pathogenic.
Figure 3.
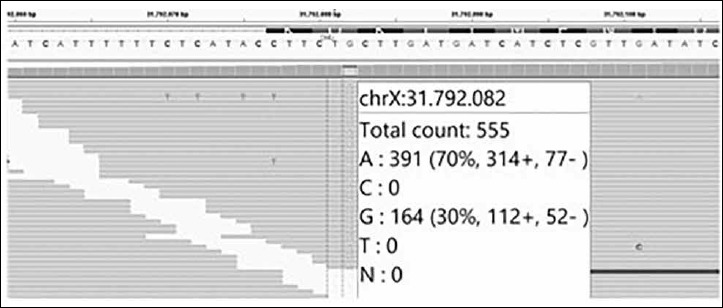
IGV image showing the variant found in the patient. Of the 555 reads reached for this nucleotide, 70% are carriers of the nonsense allele while 30% are carriers of the wild type allele.
The patient had been previously tested (single-gene) for mutations in the MYH7, FKRP and LMNA genes, with a negative result.
Family genetic testing
The proband’s parents were already deceased, and the sister was not available for gene testing. The patient has two daughters, who were tested for DMD gene carrier status, and only the eldest daughter inherited the mutation. On neurological examination, she presented a mild calf hypertrophy without muscular weakness.
Discussion
We present a male patient with a DMD gene mutation in somatic mosaicism, presenting a severe dilated cardiomyopathy and a predominantly distal, asymmetric upper limb muscular weakness.
We believe that both the myocyte and skeletal muscle involvement are related to the novel mutation found – the former confirmed by the absence of coronary disease or other pathology causing cardiac failure, and the latter by the muscle biopsy, revealing a myopathic muscle with sub-expression of dystrophin.
The asymmetric involvement in DMD gene mutations has been previously described in female manifesting carriers in various proportions (5, 6), and in males harbouring somatic mutations in the DMD gene (including a hemi-atrophy pattern, mild muscular limb girdle weakness, and overt cardiomyopathy with minor muscle symptoms) (7-9). The genetic normalization process, where dystrophin-negative muscle is replaced by dystrophin-positive muscle as a function of age, was considered to mitigate muscular symptoms in some of this patients and the predominantly cardiac phenotype (9). We propose that this could account for both the late presentation of the muscular phenotype and its asymmetry in our patient.
However, the predominantly distal presentation of the muscular weakness is uncommon, and, to our knowledge, has never been described in males or female-carriers with DMD gene mutations. Although calf hypertrophy was present, there were no limb-girdle related complaints or weakness. Further studies involving genetic analysis of muscle dystrophin may help in clarifying the real expression and proportion of the normal and the abnormal dystrophin in the patient’s skeletal muscle, especially if a more distal muscle is studied.
The typical transmission pattern for this X-linked disorder would be that all female descendants were carriers for the mutation. The atypical familial transmission presented is probably due to the fact that the patient is also a mosaic for germline cells (10).
This clinical case expands the genotype and phenotypic presentation of DMD gene mutations.
Conclusions
We present a rare and very atypical phenotype of muscular involvement in a male patient with a novel DMD gene mutation in somatic mosaicism – asymmetric distal weakness of the upper limbs – associated with a severe dilated cardiomyopathy.
References
- 1.Brandsema JF, Darras BT. Dystrophinopathies. Semin Neurol 2015. 35:369-84. [DOI] [PubMed] [Google Scholar]
- 2.Muntoni F, Torelli S, Ferlini A. Dystrophin and mutations: one gene, several proteins, multiple phenotypes. Lancet Neurol 2003;2:731-40. [DOI] [PubMed] [Google Scholar]
- 3.Amato AA, Griggs RC. Overview of the muscular dystrophies. Handb Clin Neurol 2011;101:1-9. [DOI] [PubMed] [Google Scholar]
- 4.Ferlini A, Neri M, Gualandi F. The medical genetics of dystrophinopathies: molecular genetic diagnosis and its impact on clinical practice. Neuromuscular Disorders 2013;23:4-14. [DOI] [PubMed] [Google Scholar]
- 5.Hoogerwaard EM, Bakker E, Ippel PF. Signs and symptoms of Duchenne muscular dystrophy and Becker muscular dystrophy among carriers in The Netherlands: a cohort study. Lancet 1999;353:2116-9. [DOI] [PubMed] [Google Scholar]
- 6.Soltanzadeh P, Friez M, Dunn D, et al. Clinical and genetic characterization of manifesting carriers of DMD mutations. Neuromusc Disord 2010;20:499-504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Punetha J, Mansoor S, Bertorini TE, et al. Somatic mosaicism due to a reversion variant causing hemi-atrophy: a novel variant of dystrophinopathy. Eur J Hum Genet 2016;24:1511-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kesari A, Neel R, Wagoner L, et al. Somatic mosaicism for Duchenne dystrophy: evidence for genetic normalization mitigating muscle symptoms. Am J Med Genet A 2009;149:1499-503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Juan-Mateu J, Paradas C, Olivé M, et al. Isolated cardiomyopathy caused by a DMD nonsense mutation in somatic mosaicism: genetic normalization in skeletal muscle. Clin Genet 2012;82:574-8. [DOI] [PubMed] [Google Scholar]
- 10.Freed D, Stevens E, Pevsner J. Somatic mosaicism in the human genome. Genes (Basel) 2014;5:1064-94. [DOI] [PMC free article] [PubMed] [Google Scholar]