

Abstract
Mitochondria are sensitive to numerous environmental stresses, which can lead to activation of mitochondrial stress responses (MSRs). Of particular recent interest has been the mitochondrial unfolded protein response (UPRmt), activated to restore protein homeostasis (proteostasis) upon mitochondrial protein misfolding. Several axes of the UPRmt have been described, creating some confusion as to the nature of the different responses. While distinct molecularly, these different axes are likely mutually beneficial and activated in parallel. This review aims at describing and distinguishing the different mammalian MSR/UPRmt axes to define key processes and members and to examine the involvement of protein misfolding.
Mitochondrial protein folding
Mitochondria are highly regulated cellular organelles that fulfill numerous metabolic functions, including the production of ATP by respiration. Function and quality of mitochondria need to be tightly controlled to ensure the supply of metabolic building blocks and to prevent the production of harmful agents such as reactive oxygen species (ROS), produced at increased rates upon malfunctioning respiration [1, 2]. Mitochondrial aging, environmental changes such as fever and medication, and numerous pathologies including cancer, Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis involve mitochondrial dysfunction [3–7]. It is crucial to understand the mitochondrial responses elicited upon these conditions to recognize underlying mechanisms. Indeed, for several mitochondrial stresses, such as hypoxia and oxidative stress, the resulting stress responses are well understood, including the pathways by which they potentially trigger cell death [8, 9]. However, how cells react to perturbation in mitochondrial proteostasis caused by accumulation of misfolded proteins is still unclear, despite the significant impact of protein aggregation on mitochondrial function and cellular health.
Mitochondria are cellular organelles separated from the extra-mitochondrial environment by two membranes—the outer mitochondrial membrane (OMM) and the inner mitochondrial membrane (IMM). The compartment enclosed by the IMM is called the matrix, and the space between the OMM and IMM defines the intermembrane space (IMS). Due to the archetypic origin of mitochondria and the resulting physical separation from the cytosol, the mitochondrial matrix forms a largely independent protein compartment providing its own translation and protein quality control machinery including chaperones and proteases [1, 10–12]. Mitochondria are composed of well over 1000 proteins, the majority located in the matrix [13]. Most of these proteins are encoded in the nuclear genome and imported into mitochondria [14]. Thirteen transmembrane proteins of the respiratory chain are encoded in the mitochondrial genome (mtDNA), together with a set of 22 tRNAs and two rRNAs, required for the assembly of a translation machinery in the matrix [15, 16]. Inside the matrix, both imported and mitochondrially translated proteins are folded and need to be quality controlled to maintain mitochondrial proteostasis [11, 15, 17]. Therefore, mitochondria contain their own set of matrix localized heat shock proteins (HSP) 70 and 90, chaperonins, and proteases.
The proper function of proteins and maintenance of proteostasis entails the tight control of protein folding, including co-translational and post-translational folding, maturation, and degradation of proteins [18–22]. These processes must be maintained in all distinct cellular compartments to function correctly [23]. Upon proteostasis failure, stress responses are rapidly activated—typically in a time-course of several hours—in an attempt to alleviate proteostasis defects by modulating the folding environment through modification of protein synthesis and the availability of folding helpers—chaperones (Fig. 1). A hallmark of these responses is that they are highly acute, pro-survival responses that aim to alleviate transient stresses to restore homeostasis and support cell survival. However, upon chronic activation, they typically shift towards pro-death responses [24]. Stress responses like the heat shock response in the cytosol and the unfolded protein response in the endoplasmic reticulum (UPRER) have been extensively studied and reviewed [25, 26]. However, knowledge about the role, function, and regulation of a mitochondrial stress response to unfolded proteins is lagging behind and details are much more uncertain. Similar to the UPRER that elicits a multi-axis response mediated by several receptors and leading to different effects such as induction of pro-folding factors and inhibition of translation [26], the UPRmt also appears to contain several axes with distinct molecular outcomes (Fig. 2). However, their underlying molecular mechanisms and components remain largely unknown. This review will provide insight into these different axes of the mammalian UPRmt.
Fig. 1.
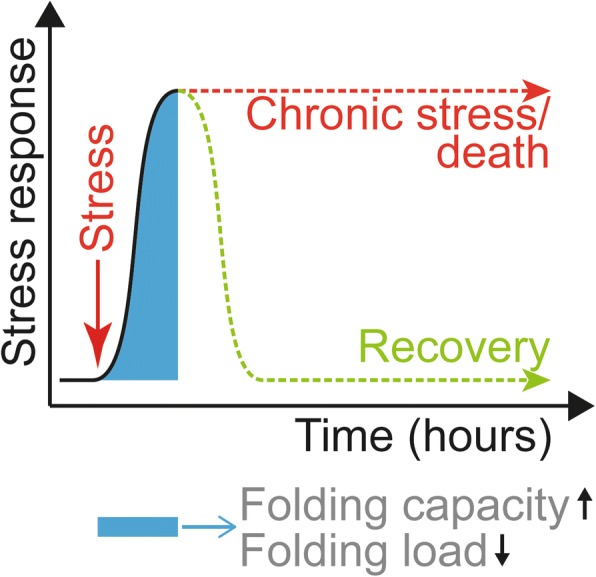
Folding stress responses. Protein misfolding activates transient, pro-survival stress responses that increase the folding capacity (i.e., modulation of chaperone and protease levels) and decrease the folding load (i.e., decrease in translation) to restore proteostasis. Responses typically last several hours. Prolonged stress activation that cannot alleviate the stress causes alternative outcomes, including cell death. Pharmacological induction of protein misfolding allows the study of the acute response to protein misfolding. Chronic activation of the stress, as observed upon genomic modulation or in disease, leads to the activation of alternative pathways and potentially cell death
Fig. 2.
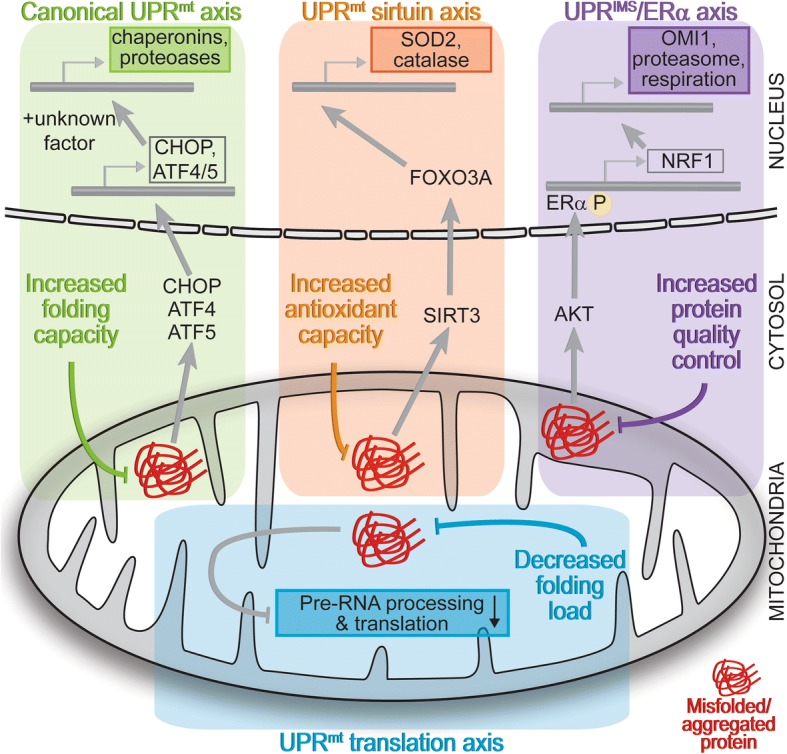
The different mammalian UPRmt axes. Depiction of the different UPRmt axes that are activated upon mitochondrial protein misfolding/aggregation: (1) The canonical UPRmt leads to altered localization and levels of CHOP, ATF4, and ATF5. These, together with other unknown transcription factors, lead to the induction of the chaperonins, chaperones, and proteases to increase the folding capacity inside mitochondria. (2) SIRT3 becomes activated as part of the UPRmt sirtuin axis leading to the deacetylation and relocalization of FOXO3A to the nucleus, where it induces SOD2 and catalase as part of an antioxidant response. (3) Protein misfolding in the intermembrane space activates the UPRIMS–ERα axis, which acts via AKT and ROS-dependent phosphorylation of ERα, causing induction of NRF1. This in turn leads to increased protease levels, modulation of respiration levels, and enhanced proteasome activity to increase the protein quality control capacity. (4) The UPRmt translation axis is a local response, largely independent of transcriptional effects in the nucleus. Protein unfolding in the matrix causes the rapid degradation of components of the pre-RNA processing machinery and a shutdown of mitochondrial translation to decrease the mitochondrial folding load
Discovery of the UPRmt
In 1996, the Hoogenraad laboratory discovered a stress response that is specific to mitochondrial protein misfolding and that was later named the UPRmt. They described the gene locus of the nuclear-encoded, mitochondria-localized chaperonins HSPD1 and HSPE1 (also known as HSP60 and HSP10) [27], to be controlled by a bi-directional promoter, which shows significantly increased activity upon loss of the mtDNA and heat shock [28, 29]. This was proof of a specific mitochondrial response to folding stress within the organelle and it was shown to depend on mitochondrial–nuclear communication to elicit a specific feedback to improve folding conditions in mitochondria, independent of general heat-shock responses.
Extensive studies into the UPRmt in Caenorhabditis elegans have uncovered molecular mechanisms involved in signaling the mitochondrial stress to the nucleus, causing induction of a transcriptional response [4]. This response is largely driven by the release of peptides from mitochondria by the transporter HAF-1 and detection of these peptides in the cytosol, and by a dual-localized transcription factor—activating transcription factor associated with stress–1 (ATFS-1)—whose import into mitochondria is inhibited upon UPRmt, leading to its accumulation in the nucleus and activation of the transcriptional UPRmt [30, 31]. The UPRmt pathways in C. elegans have been comprehensively reviewed [4, 32], but to what extent these mechanisms are conserved in mammalian cells remains unclear.
The canonical UPRmt response
The transcriptional response to mitochondrial protein misfolding described above remains the best understood mammalian UPRmt axis and forms the canonical UPRmt. Its outcome is the induction of genes increasing the folding capacity in mitochondria. Employing misfolding-prone deletion mutants of the mitochondrial protein ornithine transcarbamylase (OTCΔ) aided in defining the UPRmt in mammalian cells and determined its role in response to protein misfolding [33]. Exogenously expressed OTCΔ misfolds and accumulates in the matrix, triggering the induction of the chaperonin promoter via c-Jun N-terminal kinase 2 [33, 34]. Chaperonin induction by OTCΔ is transient and reversible, thus showing hallmarks typical for misfolding stress responses [24, 33]. Analysis of the mitochondrial chaperonin promoter uncovered a transcription factor C/EBP homologous protein (CHOP) binding element essential for its activation during OTCΔ-induced UPRmt [33]. CHOP is known to be part of the integrated stress response (ISR), which is activated by any of four different kinases to integrate various cellular stresses (amino acid deprivation, heme deficiency, ER protein misfolding, or viral infection, mediated by the eukaryotic initiation factor 2 alpha (EIF2A) kinases GCN2, HRI, PERK, or PKR, respectively). Kinase phosphorylation of EIF2A causes alternative initiation and an increase in the translation of activating transcription factor 4 (ATF4), which activates numerous genes including CHOP [35].
Despite the dependence on ISR factors, i.e., CHOP, UPRmt signaling is highly specific, as documented by the fact that the OTCΔ-triggered activation of CHOP does not increase BiP transcript levels, an ER chaperone induced by UPRER in a process also mediated by the ISR [23, 33]. While activation of the chaperonin promoter by CHOP occurs in association with C/EBPβ [33, 36], increasing CHOP and C/EBPβ levels are not sufficient to induce the chaperonins, demonstrating the need for further activating factors [36]. Recently, ATF5, which is induced by CHOP and ATF4 [37, 38], was found to be involved in retrograde signaling of the UPRmt to the nucleus in a function similar to ATFS-1 in C. elegans [39]. Additionally, further analysis of the chaperonin promoter revealed two additional promoter elements—mitochondrial unfolded protein response element (MURE) 1 and 2—in close proximity to the CHOP elements that likely play a role in the specificity of UPRmt signaling [36]. Which transcription factors bind these sites and whether they are essential for UPRmt signaling in cells have not been determined. Luciferase reporter assay testing of potentially UPRmt-regulated promoters pointed towards further genes possibly being activated by the UPRmt [36], some of which could, however, not be confirmed by analysis of endogenous transcripts [40]. Recent analysis of changes in the transcriptome upon acute induction of the UPRmt revealed that induction of the canonical UPRmt leads to specific and extensive transcriptional rearrangements affecting a wide range of biological pathways, mainly involved in protein folding and cellular homeostasis [41]. Combined, these findings described the canonical UPRmt response as an extensive transcriptional response in the nucleus, triggered by mitochondrial unfolded proteins inducing chaperonin transcription via a mechanism involving a CHOP element in the promoter region.
The central role of CHOP in UPRmt signaling is surprising as CHOP is also a key member of the ISR, which is induced by various other stresses and also forms part of the UPRER [23]. Overexpression of OTCΔ in murine intestinal epithelial cells causes induction of protein kinase double-stranded RNA-dependent (PKR), as seen in virus-related ISR, suggesting a possible role of this ISR type in the UPRmt [42]. However, knockdown of any of the four EIF2A kinases capable of eliciting an ISR has no effect on the induction of CHOP upon acute UPRmt induction mediated by mitochondrial HSP90 inhibition [41]. This implies that EIF2A kinases, at least individually, are not required for a UPRmt-mediated CHOP induction. The distinct regulation of CHOP hints towards other factors playing important roles in shaping the transcriptional outcome of the UPRmt, possibly by binding to MURE1 and MURE2 sites. Due to the lack of understanding of co-regulating factors and the inherent non-specificity of CHOP, the induction of CHOP must not be used as a readout for the induction of the UPRmt. Indeed, numerous mitochondrial stresses that are not related to protein misfolding, but instead inhibit central functions like respiration, mitochondrial membrane potential, import, and translation, can quickly induce CHOP (via ATF4) without leading to chaperonin activation [41, 43]. Thus, although ATF4 and CHOP play important roles in the UPRmt, likely regulating hitherto unknown aspects of the modulation of cellular processes by the UPRmt, they alone are not sufficient to elicit the canonical UPRmt transcriptional response, which likely depends on additional signaling factors.
The following conditions causing mitochondrial protein misfolding have been shown to subsequently elicit the canonical UPRmt defined by chaperonin induction: (1) overexpression of misfolding-prone deletion mutants of OTCΔ; (2) inhibition of mitochondrial HSP90 [41, 44, 45]; (3) inhibition of lon peptidase 1 (LONP1) [41, 46], which is crucial for the digestion of misfolded matrix proteins [12]; and (4) expression of another misfolding mitochondrial protein (EndoG, see below) [47, 48]. Due to the high chaperonin protein levels under basal conditions [49], the analysis of changes in their levels has proven difficult as a readout for the UPRmt in mammalian cells, unless the UPRmt is induced chronically, or highly quantitative methods such as mass spectrometry are used [41]. However, this issue is overcome by analyzing chaperonin transcript levels, which provide a robust increase, despite their high abundance in cells [41], and are now widely accepted as the gold standard marker for activation of the canonical UPRmt axis, as documented by numerous publications [39, 41, 48, 50]. Induction of chaperonins exemplifies the role of the canonical UPRmt to increase the mitochondrial folding capacity in response to protein misfolding; however, this is not the only mitochondrial response to protein misfolding.
The UPRmt translation axis
In addition to increasing folding capacity through induction of chaperones—the canonical UPRmt transcriptional response—cells employ a second mechanism of decreasing the unfolded protein load, e.g., achieved by a reduced uptake of proteins into the organelle (for protein-importing compartments) or by reduced translation (for compartments containing a translation machinery). While work in C. elegans has shown a decrease in mitochondrial protein import and translation [31, 51], the effects of mitochondrial protein misfolding on import and translation in mammalian cells, and thus a role of the second principle, are not clear. Recently, J. Wade Harper and myself have provided the first evidence to support the existence of a mammalian UPRmt that reduces the folding load: taking advantage of two inhibitors targeting the mitochondrial HSP90 and LONP1 [45, 46] to acutely induce the UPRmt, we discovered a translational UPRmt axis that controls the folding load within mitochondria upon UPRmt activation [41]. Whole strands of mtDNA are transcribed into long, polycistronic pre-RNAs that are processed by the RNase P complex, consisting of MRPP1–3 [15, 52, 53]. Upon acute induction of the UPRmt, we observed a rapid decrease of MRPP3 transcript and protein levels, causing a markedly lower level of mitochondrial pre-RNA processing and ultimately a reversible reduction in mitochondrial translation [41].
This translational UPRmt axis, which limits the protein folding load by regulating mitochondrial translation, constitutes an interesting new aspect to the UPRmt: due to its post-translational regulation within a single mitochondrion, there is no requirement to pass a cellular signaling threshold for activating the transcriptional UPRmt. Instead, it acts locally in single, damaged mitochondria and could thus form a first line of defense against mitochondrial damage that is most likely independent of extra-mitochondrial stimuli (Fig. 3): under cellular conditions with few stressed mitochondria, only the locally acting UPRmt translation axis becomes activated to rapidly improve proteostasis without cell-wide effects. However, once a certain, larger number of mitochondria show perturbed proteostasis, reflective of possible harmful environmental conditions, the other UPRmt axes are initiated to modulate the cellular proteome via global transcriptional rearrangements. Defects in pre-RNA processing are the cause of several human diseases [54–56]. Also, work in yeast and mouse hepatocytes has shown cellular programs to control the mito-nuclear protein balance, particularly with respect to subunits of the respiratory chain [57, 58], indicating that mitochondrial protein translation may have impacts on the cell. Future work will be required to understand the relationship between these diseases, pre-RNA processing, and the UPRmt. The translational UPRmt axis described here is highly complementary to the transcriptional canonical UPRmt axis described above to decrease the folding load and increase the folding capacity, respectively, in an attempt to overcome mitochondrial protein misfolding. In addition, mitochondria can activate a third and additional UPRmt axis.
Fig. 3.

Integration of mitochondrial misfolding stress and different UPRmt axes. Cells contain numerous mitochondria with a certain, low percentage stressed upon basal conditions, due to aging and metabolic damage. Dealing with these refined incidents of mitochondrial proteostasis defects, which are not due to significant environmental perturbation, requires spatially defined responses. The UPRmt translational response acts via local, posttranslational regulation of MRPP3 levels and can decrease translation, and thus folding load, in an individual mitochondrion. Thus, it acts locally as a first response to mitochondrial protein misfolding in few damaged mitochondria without causing global effects. The transcriptional UPRmt effects occur cell-wide and likely require passing a certain threshold of mitochondrial proteostasis defects. This suggests a model in which mitochondrion-specific UPRmt effects (i.e., the UPRmt translation axis) are activated upon cellular conditions with a certain low percentage of mitochondria suffering from protein misfolding and activation of the cell-wide UPRmt axes upon proteostasis defects in a large percentage of mitochondria
The UPRmt sirtuin axis
Mitochondrial dysfunction often causes a proteotoxic oxidative environment. To counteract this potential source of misfolded proteins, cells can activate the UPRmt sirtuin axis, exerting antioxidant activity. Sirtuins are lysine deacetylases and ADP-ribosyltransferases controlling a wide range of cellular processes, many affecting mitochondrial function [59, 60]. Regulation of metabolism by sirtuins has been associated with longevity and aging [59, 61]. In mammalian cells, there are seven sirtuins (SIRT1–7) with distinct cellular localization and function: (1) SIRT1, 6, and 7 predominantly localize to the nucleus and control the acetylation state of proteins such as histones, PGC1α (a mitochondrial biogenesis factor) and forkhead box O (FOXO) transcription factors [59, 60, 62, 63]; (2) SIRT2 localizes to the cytosol and controls tubulin and PGC1α acetylation [60, 64]; and (3) SIRT3–5 localize to mitochondria and mainly control metabolic processes such as the Krebs cycle and fatty acid oxidation [59, 65]. Strikingly, sirtuins, particularly SIRT1 and SIRT3, have also been shown to be involved in the UPRmt.
SIRT1/SIRT 3 had been known to exert an antioxidant effect by controlling the activity and localization of the transcription factor FOXO3A [66, 67]. Deacetylation of FOXO3A by SIRT1/SIRT3 drives FOXO3A localization to the nucleus [68], where it stimulates the transcription of antioxidant enzymes such as the mitochondrial superoxide dismutase 2 (SOD2) and catalase [69, 70]. Strikingly, the same mechanism is triggered by proteotoxic folding stress in the mitochondrial matrix, leading to activation and increased levels of SIRT3 and subsequently eliciting an antioxidant response via FOXO3A deacetylation and the induction of SOD2 and catalase [48, 71]. The observed effects are dependent on the production of ROS and also entail the lipidation of LC3B, induction of several autophagy genes, and increased autophagy rates, suggesting a stimulation of autophagy and/or autophagic flux [48]. These effects were also confirmed by direct sirtuin activation via chemically increasing NAD+ levels, thereby causing an elevated mitochondrial antioxidant activity in both C. elegans and mammalian cells [72, 73]. Recently, SIRT3 was shown to bind to ATP synthase and to be stimulated upon mitochondrial depolarization via a pH-dependent dissociation from ATP synthase, linking respiratory stress and SIRT3 activity [65]. Additionally, different mitochondrial stresses not related to protein misfolding and directly causing ROS production are also capable of SIRT3 induction, further emphasizing the role of the sirtuin axis as an antioxidant response, but also indicating SIRT3 levels alone cannot serve as a marker for the UPRmt [48]. Importantly, the SIRT3–FOXO3A axis is independent of CHOP, as seen by RNAi-mediated knockdown of CHOP and inhibition of SIRT3 having no effect on the canonical UPRmt transcriptional response [48]. With production of ROS as an ample byproduct of mitochondrial dysfunction, the antioxidant activity of the UPRmt sirtuin axis is likely highly complementary to the canonical UPRmt transcriptional response in securing mitochondrial health.
The UPRIMS–ERα axis
The IMS is separated from the matrix by a membrane forming a distinct compartment in which protein misfolding can occur, leading to a distinct mitochondrial UPR—the UPRIMS. Its underlying features have been largely described by the use of endonuclease G (EndoG), an IMS endonuclease released from mitochondria to fragment DNA, causing caspase-independent apoptosis upon conditions such as heat and oxidative stress [74–77]. Expression of mutant EndoG leads to accumulation of misfolded EndoG in the IMS and clustering of mitochondria [78, 79]. This process elicits an IMS UPR (UPRIMS) that appears to be independent of the matrix UPRmt and does not cause induction of CHOP or HSP60 [79]. Thus, it does not signal through the ISR or the canonical UPRmt transcriptional response. Instead, its signaling is dependent on estrogen receptor alpha (ERα) and mediated by ROS-dependent phosphorylation of ERα by AKT [79]. Activated ERα then leads to (1) increased nuclear respiratory factor 1 (NRF1) transcript and protein levels, a factor known to regulate proteasome levels [80], the mitochondrial transcription machinery, and thus respiration [81], (2) elevated transcript and protein levels of the IMS protease OMI [79, 82], and (3) an increase in proteasome activity [79]. Together, these effects increase the protein quality control (PQC) system to prevent import and accumulation of (defective) IMS proteins in the IMS [78, 79]. Strikingly, in cells not expressing ERα, misfolding within the IMS leads to induction of CHOP and HSP60 similarly to the effects observed upon inducing matrix protein misfolding [47], suggesting that, upon loss of the IMS PQC machinery and accumulation of misfolded proteins in the IMS, either the canonical UPRmt transcriptional response becomes activated directly by unknown mechanisms, or that the severe accumulation of misfolded IMS proteins causes matrix protein misfolding that activates the canonical UPRmt transcriptional response as an indirect response to perturbed IMS proteostasis. The UPRIMS–ERα axis defines a distinct response from the UPRmt axes, attempting to specifically modulate IMS proteostasis to improve folding. Depending on the environment causing protein misfolding, it may act in parallel to the UPRmt axes. UPRmt induction upon UPRIMS failure shows the important role IMS proteostasis exerts on folding in the matrix and suggests possible links between these responses.
Mitochondrial stress responses
Importantly, several mitochondrial stress responses (MSRs) are not apparently induced by mitochondrial protein misfolding, but still show a certain degree of similarity to the UPRmt by relying on overlapping pathways (Fig. 4). Of particular importance are the distinct stresses that lead to activation of the ISR and result in activation of specific transcriptional profiles, as mentioned above. In addition to these stresses that all involve induction of ATF4, several other stresses have been studied in detail, which have not been directly associated with protein unfolding, but trigger a MSR and the ISR: (1) mutations in twinkle, a mtDNA helicase, lead to mtDNA deficiencies resulting in respiratory chain deficiency and mitochondrial myopathy [83]. Skeletal muscle of mutant twinkle mice, carrying a dominant duplication of 13 amino acids in twinkle, accumulate mtDNA deletions and show an induction of Atf4 and Atf5, mediated by mTOR activity [84]. This response also activates markers of the canonical UPRmt transcriptional response, suggesting crosstalk with this pathway [84]. However, whether this response is mediated by protein misfolding in the matrix remains unclear. (2) Knockout of the mitochondrial tRNA synthetase Dars2 in mice causes mitochondrial translation defects and respiratory deficiency without any apparent effects on mitochondrial protein misfolding [85]. DARS2-deficient hearts show upregulation of Chop, Atf4, and Atf5, thereby demonstrating activation of the ISR [85]. Strikingly, the lethality caused by Dars2 knock-out is alleviated by an additional loss of CLPP without modulating the transcriptional response observed upon Dars2 knock-out [86]. (3) Overexpression of CLPX, the AAA+ ATPase unfoldase lid of the mitochondrial CLPP protease [87], induces a retrograde transcriptional pathway mediated by CHOP through an unknown mechanism [50]. Increasing ClpX levels stimulate the degradation capacity of ClpXP [88]. Thus, an accumulation of non-degraded CLPXP substrates is unlikely, suggesting a signaling pathway not defined by a lack of degradation of CLPP substrates. (4) Knockout of Surf1 in the skeletal muscle of mice causes induction of CHOP, HSP60, and LONP [89, 90]. Surf1 is a complex IV assembly factor and knock-out leads to decreased respiration without apparent effects on mitochondrial protein folding [91]. Thus, the observed effects of Surf1−/− are likely mediated by similar mechanisms as observed upon pharmacological ablation of respiration. Strikingly, Surf1−/− mice exhibit an increase in mitochondrial number and longevity [89, 91], pointing towards a mechanism of increased robustness due to the defects in respiration. (5) Inhibition of mitochondrial translation leads to GCN2-dependent ISR induction activating CHOP, without stimulating chaperonins [92].
Fig. 4.
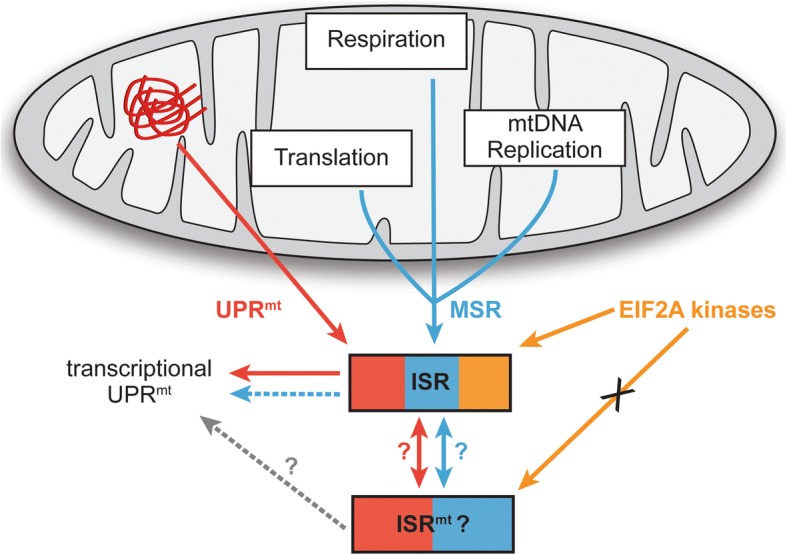
Mitochondrial stress responses. Various mitochondrial stresses, not directly linked to mitochondrial protein misfolding, elicit stress responses that are similar to the UPRmt retrograde signaling and involve the integrated stress response (ISR). These stresses affect important aspects of mitochondria, such as respiration, translation, and mtDNA replication and are distinct from the UPRmt transcriptional responses. Recent findings suggest a specific mitochondrial ISR that shares common factors with the ISR but driven by different signaling pathways and eliciting alternative outputs
Together, these described MSRs are able to induce adaptation mechanisms, largely mediated by retrograde signaling (via the ISR) and transcriptional modulation, and some potentially utilize signaling through the canonical UPRmt transcriptional response (Fig. 4). There appear to be common signaling pathways broadly activated upon mitochondrial stress. To what extent these overlap molecularly with UPRmt signaling and whether some of the described MSRs also involve mitochondrial protein misfolding to activate the UPRmt is an important and challenging question for future research. Due to the current lack of insight and to avoid confusion as to the numerous mitochondrial stresses and responses observed, it is important to distinguish between (1) MSRs and UPRmt, depending on the significant/primary involvement of mitochondrial protein misfolding as causative agent, and (2) the UPRmt axes studied by clearly determining and describing the UPRmt axes monitored and activated.
Many of the MSRs signal, at least in part, via the ISR. One of the future challenges is to determine to what extent MSR and UPRmt signaling through the ISR is overlapping with the activation of the ISR by EIF2A kinases. Some significant differences between classic activation of the ISR by EIF2A kinases and activation by mitochondrial stress have already been described, leading to the proposal of a specific mitochondrial ISR (ISRmt), distinct from the canonical ISR [84]. Further research will be required to determine the role of such a ISRmt in the UPRmt and to determine the temporal control and cross-activation of the ISRmt and the different UPRmt axes in response to mitochondrial misfolding stress.
Summary and future outlook
Various conditions, where mitochondrial protein misfolding is the primary cause of mitochondrial stress, have been shown to activate axes of the UPRmt. Strikingly, inhibition of mitochondrial HSP90 to induce protein misfolding and the UPRmt has been recently explored as a potent therapeutic strategy to target cancer [93, 94]. The different effects elicited by the different UPRmt axes demonstrate that the UPRmt is a multi-pronged response modulating several aspects of mitochondrial proteostasis in an attempt to alleviate folding stress (Fig. 2). The UPRmt axes are distinct with specific sets of defining factors, allowing and encouraging researchers to clearly define, describe, and validate activation of the proposed axis studied. The different UPRmt axes are pro-survival and attempt to maintain mitochondria. However, there must also be destructive pathways activated upon failure to restore proteostasis. Strikingly, there have now been numerous reports of mitochondrial protein misfolding triggering LC3B lipidation, induction of autophagy genes, and mitophagy [41, 44, 48, 94, 95], suggesting that autophagy in general and the selective degradation of damaged mitochondria via this route might play a significant role in the UPRmt. However, substantiated evidence proving a causative relation between mitochondrial protein misfolding and autophagy is still lacking, and the same applies to insight into the mechanisms involved. Thus, the relationship between non-selective autophagy, mitophagy, and the UPRmt will require further investigation. It is tempting to speculate that these studies will reveal autophagy pathways forming an additional UPRmt axis that might initiate upon more severe or prolonged mitochondrial protein misfolding, when the described UPRmt axes fail to restore proteostasis, or are overburdened.
Acknowledgements
I would like to thank the members of the Münch laboratory, Stefan Müller, and Kerstin Koch for helpful comments on the manuscript. This work was supported by the Deutsche Forschungsgemeinschaft Emmy Noether program and the LOEWE Center for Cell and Gene Therapy Frankfurt.
Author’s contributions
CM wrote, read, and approved the final manuscript.
Competing interests
The author declares he has no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Baker MJ, Tatsuta T, Langer T. Quality control of mitochondrial proteostasis. Cold Spring Harb Perspect Biol. 2011;3:1–20. doi: 10.1101/cshperspect.a007559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wolff S, Weissman JS, Dillin A. Differential scales of protein quality control. Cell. 2014;157:52–64. doi: 10.1016/j.cell.2014.03.007. [DOI] [PubMed] [Google Scholar]
- 4.Pellegrino MW, Nargund AM, Haynes CM. Signaling the mitochondrial unfolded protein response. Biochim Biophys Acta Mol Cell Res. 2013;1833:410–416. doi: 10.1016/j.bbamcr.2012.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Selfridge JE, E L, Lu J, Swerdlow RH. Role of mitochondrial homeostasis and dynamics in Alzheimer’s disease. Neurobiol Dis. 2013;51:3–12. doi: 10.1016/j.nbd.2011.12.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abramov AY, Berezhnov AV, Fedotova EI, Zinchenko VP, Dolgacheva LP. Interaction of misfolded proteins and mitochondria in neurodegenerative disorders. Biochem Soc Trans. 2017; 10.1042/BST20170024. [DOI] [PubMed]
- 7.Vuda M, Kamath A. Drug induced mitochondrial dysfunction: mechanisms and adverse clinical consequences. Mitochondrion. 2016;31:63–74. doi: 10.1016/j.mito.2016.10.005. [DOI] [PubMed] [Google Scholar]
- 8.Cenini G, Voos W. Role of mitochondrial protein quality control in oxidative stress-induced neurodegenerative diseases. Curr Alzheimer Res. 2016;13:164–173. doi: 10.2174/1567205012666150921103213. [DOI] [PubMed] [Google Scholar]
- 9.Choudhry H, Harris AL. Advances in hypoxia-inducible factor biology. Cell Metab. 2018;27:281–298. doi: 10.1016/j.cmet.2017.10.005. [DOI] [PubMed] [Google Scholar]
- 10.Pickles S, Vigié P, Youle RJ. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr Biol. 2018;28:R170–R185. doi: 10.1016/j.cub.2018.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Voos W, Jaworek W, Wilkening A, Bruderek M. Protein quality control at the mitochondrion. Essays Biochem. 2016;60:213–225. doi: 10.1042/EBC20160009. [DOI] [PubMed] [Google Scholar]
- 12.Voos W. Chaperone-protease networks in mitochondrial protein homeostasis. Biochim Biophys Acta. 2013;1833:388–399. doi: 10.1016/j.bbamcr.2012.06.005. [DOI] [PubMed] [Google Scholar]
- 13.Rhee H-W, Zou P, Udeshi ND, Martell JD, Mootha VK, Carr SA, et al. Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science. 2013;339:1328–1331. doi: 10.1126/science.1230593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schmidt O, Pfanner N, Meisinger C. Mitochondrial protein import: from proteomics to functional mechanisms. Nat Rev Mol Cell Biol. 2010;11:655–667. doi: 10.1038/nrm2959. [DOI] [PubMed] [Google Scholar]
- 15.Suzuki T, Nagao A, Suzuki T. Human mitochondrial tRNAs: biogenesis, function, structural aspects, and diseases. Annu Rev Genet. 2011;45:299–329. doi: 10.1146/annurev-genet-110410-132531. [DOI] [PubMed] [Google Scholar]
- 16.Ott M, Amunts A, Brown A. Organization and regulation of mitochondrial protein synthesis. Annu Rev Biochem. 2016;85:77–101. doi: 10.1146/annurev-biochem-060815-014334. [DOI] [PubMed] [Google Scholar]
- 17.Patron M, Sprenger H-G, Langer T. m-AAA proteases, mitochondrial calcium homeostasis and neurodegeneration. Cell Res. 2018;28:296–306. doi: 10.1038/cr.2018.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim YE, Hipp MS, Bracher A, Hayer-Hartl M, Hartl FU. Molecular chaperone functions in protein folding and proteostasis. Annu Rev Biochem. 2013;82:323–355. doi: 10.1146/annurev-biochem-060208-092442. [DOI] [PubMed] [Google Scholar]
- 19.Gloge F, Becker AH, Kramer G, Bukau B. Co-translational mechanisms of protein maturation. Curr Opin Struct Biol. 2014;24:24–33. doi: 10.1016/j.sbi.2013.11.004. [DOI] [PubMed] [Google Scholar]
- 20.Holtkamp W, Kokic G, Jager M, Mittelstaet J, Komar AA, Rodnina MV. Cotranslational protein folding on the ribosome monitored in real time. Science. 2015;350:1104–1107. doi: 10.1126/science.aad0344. [DOI] [PubMed] [Google Scholar]
- 21.Dikic I. Proteasomal and autophagic degradation systems. Annu Rev Biochem. 2017;86:193–224. doi: 10.1146/annurev-biochem-061516-044908. [DOI] [PubMed] [Google Scholar]
- 22.Hartl FU. Cellular homeostasis and aging. Annu Rev Biochem. 2016;85:1–4. doi: 10.1146/annurev-biochem-011116-110806. [DOI] [PubMed] [Google Scholar]
- 23.Pilla E, Schneider K, Bertolotti A. Coping with protein quality control failure. Annu Rev Cell Dev Biol. 2017;33:439–465. doi: 10.1146/annurev-cellbio-111315-125334. [DOI] [PubMed] [Google Scholar]
- 24.Lamech LT, Haynes CM. The unpredictability of prolonged activation of stress response pathways. J Cell Biol. 2015;209:781–787. doi: 10.1083/jcb.201503107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Richter K, Haslbeck M, Buchner J. The heat shock response: life on the verge of death. Mol Cell. 2010;40:253–266. doi: 10.1016/j.molcel.2010.10.006. [DOI] [PubMed] [Google Scholar]
- 26.Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 27.Horwich AL, Fenton WA, Chapman E, Farr GW. Two families of chaperonin: physiology and mechanism. Annu Rev Cell Dev Biol. 2007;23:115–145. doi: 10.1146/annurev.cellbio.23.090506.123555. [DOI] [PubMed] [Google Scholar]
- 28.Ryan MT, Herd SM, Sberna G, Samuel MM, Hoogenraad NJ, Høj PB. The genes encoding mammalian chaperonin 60 and chaperonin 10 are linked head-to-head and share a bidirectional promoter. Gene. 1997;196:9–17. doi: 10.1016/S0378-1119(97)00111-X. [DOI] [PubMed] [Google Scholar]
- 29.Martinus RD, Garth GP, Webster TL, Cartwright P, Naylor DJ, Høj PB, et al. Selective induction of mitochondrial chaperones in response to loss of the mitochondrial genome. Eur J Biochem. 1996;240:98–103. doi: 10.1111/j.1432-1033.1996.0098h.x. [DOI] [PubMed] [Google Scholar]
- 30.Haynes CM, Yang Y, Blais SP, Neubert TA, Ron D. The matrix peptide exporter HAF-1 signals a mitochondrial UPR by activating the transcription factor ZC376.7 in C. elegans. Mol Cell. 2010;37:529–540. doi: 10.1016/j.molcel.2010.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nargund AM, Pellegrino MW, Fiorese CJ, Baker BM, Haynes CM. Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science. 2012;337:587–590. doi: 10.1126/science.1223560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shpilka T, Haynes CM. The mitochondrial UPR: mechanisms, physiological functions and implications in ageing. Nat Rev Mol Cell Biol. 2017;19:109–120. doi: 10.1038/nrm.2017.110. [DOI] [PubMed] [Google Scholar]
- 33.Zhao Q, Wang J, Levichkin IV, Stasinopoulos S, Ryan MT, Hoogenraad NJ. A mitochondrial specific stress response in mammalian cells. EMBO J. 2002;21:4411–4419. doi: 10.1093/emboj/cdf445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Horibe T, Hoogenraad NJ. The chop gene contains an element for the positive regulation of the mitochondrial unfolded protein response. PLoS One. 2007;2:e835. doi: 10.1371/journal.pone.0000835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pakos-Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, Gorman AM. The integrated stress response. EMBO Rep. 2016;17:1374–1395. doi: 10.15252/embr.201642195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aldridge JE, Horibe T, Hoogenraad NJ. Discovery of genes activated by the mitochondrial unfolded protein response (mtUPR) and cognate promoter elements. PLoS One. 2007;2:e874. doi: 10.1371/journal.pone.0000874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Teske BF, Fusakio ME, Zhou D, Shan J, McClintick JN, Kilberg MS, et al. CHOP induces activating transcription factor 5 (ATF5) to trigger apoptosis in response to perturbations in protein homeostasis. Mol Biol Cell. 2013;24:2477–2490. doi: 10.1091/mbc.e13-01-0067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fusakio ME, Willy JA, Wang Y, Mirek ET, Al Baghdadi RJT, Adams CM, et al. Transcription factor ATF4 directs basal and stress-induced gene expression in the unfolded protein response and cholesterol metabolism in the liver. Mol Biol Cell. 2016;27:1536–1551. doi: 10.1091/mbc.e16-01-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fiorese CJ, Schulz AM, Lin Y-FF, Rosin N, Pellegrino MW, Haynes CM. The transcription factor ATF5 mediates a mammalian mitochondrial UPR. Curr Biol. 2016;26:2037–2043. doi: 10.1016/j.cub.2016.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Arnould T, Michel S, Renard P. Mitochondria retrograde signaling and the UPRmt: where are we in mammals? Int J Mol Sci. 2015;16:18224–18251. doi: 10.3390/ijms160818224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Münch C, Harper JW. Mitochondrial unfolded protein response controls matrix pre-RNA processing and translation. Nature. 2016;534:710–713. doi: 10.1038/nature18302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rath E, Berger E, Messlik A, Nunes T, Liu B, Kim SC, et al. Induction of dsRNA-activated protein kinase links mitochondrial unfolded protein response to the pathogenesis of intestinal inflammation. Gut. 2012;61:1269–1278. doi: 10.1136/gutjnl-2011-300767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Quirós PM, Prado MA, Zamboni N, D’Amico D, Williams RW, Finley D, et al. Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J Cell Biol. 2017;216:2027–2045. doi: 10.1083/jcb.201702058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fiesel FC, James ED, Hudec R, Springer W, Fiesel FC, James ED, et al. Mitochondrial targeted HSP90 inhibitor Gamitrinib-TPP (G-TPP) induces PINK1/Parkin-dependent mitophagy. Oncotarget. 2017;8:106233–106248. doi: 10.18632/oncotarget.22287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kang BH, Plescia J, Song HY, Meli M, Colombo G, Beebe K, et al. Combinatorial drug design targeting multiple cancer signaling networks controlled by mitochondrial Hsp90. J Clin Invest. 2009;119:454–464. doi: 10.1172/JCI37613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bernstein SH, Venkatesh S, Li M, Lee J, Lu B, Hilchey SP, et al. The mitochondrial ATP-dependent Lon protease: a novel target in lymphoma death mediated by the synthetic triterpenoid CDDO and its derivatives. Blood. 2012;119:3321–3329. doi: 10.1182/blood-2011-02-340075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kenny TC, Germain D. From discovery of the CHOP axis and targeting ClpP to the identification of additional axes of the UPRmt driven by the estrogen receptor and SIRT3. J Bioenerg Biomembr. 2017;49:1–9. [DOI] [PubMed]
- 48.Papa L, Germain D. SirT3 regulates the mitochondrial unfolded protein response. Mol Cell Biol. 2014;34:699–710. doi: 10.1128/MCB.01337-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Beck M, Schmidt A, Malmstroem J, Claassen M, Ori A, Szymborska A, et al. The quantitative proteome of a human cell line. Mol Syst Biol. 2011;7:1–8. doi: 10.1038/msb.2011.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Al-Furoukh N, Ianni A, Nolte H, Hölper S, Krüger M, Wanrooij S, et al. ClpX stimulates the mitochondrial unfolded protein response (UPRmt) in mammalian cells. Biochim Biophys Acta Mol Cell Res. 2015;1853:2580–2591. doi: 10.1016/j.bbamcr.2015.06.016. [DOI] [PubMed] [Google Scholar]
- 51.Nargund AM, Fiorese CJ, Pellegrino MW, Deng P, Haynes CM. Mitochondrial and nuclear accumulation of the transcription factor ATFS-1 promotes OXPHOS recovery during the UPRmt. Mol Cell. 2015;58:123–133. doi: 10.1016/j.molcel.2015.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brzezniak LK, Bijata M, Szczesny RJ, Stepien PP. Involvement of human ELAC2 gene product in 3′ end processing of mitochondrial tRNAs. RNA Biol. 2011;8:616–626. doi: 10.4161/rna.8.4.15393. [DOI] [PubMed] [Google Scholar]
- 53.Holzmann J, Frank P, Löffler E, Bennett KL, Gerner C, Rossmanith W. RNase P without RNA: identification and functional reconstitution of the human mitochondrial tRNA processing enzyme. Cell. 2008;135:462–474. doi: 10.1016/j.cell.2008.09.013. [DOI] [PubMed] [Google Scholar]
- 54.Haack TB, Kopajtich R, Freisinger P, Wieland T, Rorbach J, Nicholls TJ, et al. ELAC2 mutations cause a mitochondrial RNA processing defect associated with hypertrophic cardiomyopathy. Am J Hum Genet. 2013;93:211–223. doi: 10.1016/j.ajhg.2013.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vilardo E, Rossmanith W. Molecular insights into HSD10 disease: impact of SDR5C1 mutations on the human mitochondrial RNase P complex. Nucleic Acids Res. 2015;43:5112–5119. doi: 10.1093/nar/gkv408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Metodiev MD, Thompson K, Alston CL, Morris AAM, He L, Assouline Z, et al. Recessive mutations in TRMT10C cause defects in mitochondrial RNA processing and multiple respiratory chain deficiencies. Am J Hum Genet. 2016;98:993–1000. doi: 10.1016/j.ajhg.2016.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Couvillion MT, Soto IC, Shipkovenska G, Churchman LS. Synchronized mitochondrial and cytosolic translation programs. Nature. 2016;533:1–17. doi: 10.1038/nature18015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Houtkooper RH, Mouchiroud L, Ryu D, Moullan N, Katsyuba E, Knott G, et al. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature. 2013;497:451–457. doi: 10.1038/nature12188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Houtkooper RH, Pirinen E, Auwerx J. Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol Cell Biol. 2012;13:225–238. doi: 10.1038/nrm3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dang W. The controversial world of sirtuins. Drug Discov Today Technol. 2014;12:e9–17. doi: 10.1016/j.ddtec.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Houtkooper RH, Williams RW, Auwerx J. Metabolic networks of longevity. Cell. 2010;142:9–14. doi: 10.1016/j.cell.2010.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1α and SIRT1. Nature. 2005;434:113–118. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- 63.Vaquero A, Scher M, Lee D, Erdjument-Bromage H, Tempst P, Reinberg D. Human SirT1 interacts with histone H1 and promotes formation of facultative heterochromatin. Mol Cell. 2004;16:93–105. doi: 10.1016/j.molcel.2004.08.031. [DOI] [PubMed] [Google Scholar]
- 64.Krishnan J, Danzer C, Simka T, Ukropec J, Walter KM, Kumpf S, et al. Dietary obesity-associated Hif1 activation in adipocytes restricts fatty acid oxidation and energy expenditure via suppression of the Sirt2-NAD+ system. Genes Dev. 2012;26:259–270. doi: 10.1101/gad.180406.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yang W, Nagasawa K, Münch C, Xu Y, Satterstrom K, Jeong S, et al. Mitochondrial sirtuin network reveals dynamic SIRT3-dependent deacetylation in response to membrane depolarization. Cell. 2016;167:985–1000.e21. doi: 10.1016/j.cell.2016.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tao R, Coleman MC, Pennington JD, Ozden O, Park SH, Jiang H, et al. Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol Cell. 2010;40:893–904. doi: 10.1016/j.molcel.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- 68.Sundaresan NR, Gupta M, Kim G, Rajamohan SB, Isbatan A, Gupta MP. Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J Clin Invest. 2009;119:2758–2771. doi: 10.1172/JCI39162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Salminen A, Kaarniranta K, Kauppinen A. Crosstalk between oxidative stress and SIRT1: impact on the aging process. Int J Mol Sci. 2013;14:3834–3859. doi: 10.3390/ijms14023834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tao R, Vassilopoulos A, Parisiadou L, Yan Y, Gius D. Regulation of MnSOD enzymatic activity by Sirt3 connects the mitochondrial acetylome signaling networks to aging and carcinogenesis. Antioxid Redox Signal. 2014;20:1646–1654. doi: 10.1089/ars.2013.5482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kim H, Yang J, Kim MJ, Choi S, Chung J-R, Kim J-M, et al. Tumor necrosis factor receptor-associated protein 1 (TRAP1) mutation and TRAP1 inhibitor gamitrinib-triphenylphosphonium (G-TPP) induce a forkhead box O (FOXO)-dependent cell protective signal from mitochondria. J Biol Chem. 2016;291:1841–1853. doi: 10.1074/jbc.M115.656934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mouchiroud L, Houtkooper RH, Moullan N, Katsyuba E, Ryu D, Cantó C, et al. The NAD+/Sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling. Cell. 2013;154:430–441. doi: 10.1016/j.cell.2013.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cantó C, Houtkooper RH, Pirinen E, Youn DY, Oosterveer MH, Cen Y, et al. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 2012;15:838–847. doi: 10.1016/j.cmet.2012.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li LY, Luo X, Wang X. Endonuclease G is an apoptotic DNase when released from mitochondria. Nature. 2001;412:95–99. doi: 10.1038/35083620. [DOI] [PubMed] [Google Scholar]
- 75.Lee JS, Seo TW, Yi JH, Shin KS, Yoo SJ. CHIP has a protective role against oxidative stress-induced cell death through specific regulation of endonuclease G. Cell Death Dis. 2013;4:e666. doi: 10.1038/cddis.2013.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chinnathambi S, Tomanek-Chalkley A, Bickenbach JR. HSP70 and EndoG modulate cell death by heat in human skin keratinocytes in vitro. Cells Tissues Organs. 2008;187:131–140. doi: 10.1159/000109941. [DOI] [PubMed] [Google Scholar]
- 77.Ishihara Y, Shimamoto N. Involvement of endonuclease G in nucleosomal DNA fragmentation under sustained endogenous oxidative stress. J Biol Chem. 2006;281:6726–6733. doi: 10.1074/jbc.M510382200. [DOI] [PubMed] [Google Scholar]
- 78.Radke S, Chander H, Schäfer P, Meiss G, Krüger R, Schulz JB, et al. Mitochondrial protein quality control by the proteasome involves ubiquitination and the protease Omi. J Biol Chem. 2008;283:12681–12685. doi: 10.1074/jbc.C800036200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Papa L, Germain D. Estrogen receptor mediates a distinct mitochondrial unfolded protein response. J Cell Sci. 2011;124:1396–1402. doi: 10.1242/jcs.078220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhang Y, Nicholatos J, Dreier JR, Ricoult SJH, Widenmaier SB, Hotamisligil GS, et al. Coordinated regulation of protein synthesis and degradation by mTORC1. Nature. 2014;513:440–443. doi: 10.1038/nature13492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Scarpulla RC. Nuclear control of respiratory gene expression in mammalian cells. J Cell Biochem. 2006;97:673–683. doi: 10.1002/jcb.20743. [DOI] [PubMed] [Google Scholar]
- 82.Clausen T, Kaiser M, Huber R, Ehrmann M. HTRA proteases: regulated proteolysis in protein quality control. Nat Rev Mol Cell Biol. 2011;12:152–162. doi: 10.1038/nrm3065. [DOI] [PubMed] [Google Scholar]
- 83.Tyynismaa H, Mjosund KP, Wanrooij S, Lappalainen I, Ylikallio E, Jalanko A, et al. Mutant mitochondrial helicase twinkle causes multiple mtDNA deletions and a late-onset mitochondrial disease in mice. Proc Natl Acad Sci U S A. 2005;102:17687–17692. doi: 10.1073/pnas.0505551102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Khan NA, Nikkanen J, Yatsuga S, Jackson C, Wang L, Pradhan S, et al. mTORC1 regulates mitochondrial integrated stress response and mitochondrial myopathy progression. Cell Metab. 2017;26:419–428.e5. doi: 10.1016/j.cmet.2017.07.007. [DOI] [PubMed] [Google Scholar]
- 85.Dogan SA, Pujol C, Maiti P, Kukat A, Wang S, Hermans S, et al. Tissue-specific loss of DARS2 activates stress responses independently of respiratory chain deficiency in the heart. Cell Metab. 2014;19:458–469. doi: 10.1016/j.cmet.2014.02.004. [DOI] [PubMed] [Google Scholar]
- 86.Seiferling D, Szczepanowska K, Becker C, Senft K, Hermans S, Maiti P, et al. Loss of CLPP alleviates mitochondrial cardiomyopathy without affecting the mammalian UPRmt. EMBO Rep. 2016;17:1–12. doi: 10.15252/embr.201642077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Baker TA, Sauer RT. ClpXP, an ATP-powered unfolding and protein-degradation machine. Biochim Biophys Acta Mol Cell Res. 2012;1823:15–28. doi: 10.1016/j.bbamcr.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Al-Furoukh N, Kardon JR, Krüger M, Szibor M, Baker TA, Braun T. NOA1, a novel ClpXP substrate, takes an unexpected nuclear detour prior to mitochondrial import. PLoS One. 2014;9:e103141. doi: 10.1371/journal.pone.0103141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pulliam DA, Deepa SS, Liu Y, Hill S, Lin A-L, Bhattacharya A, et al. Complex IV-deficient Surf1−/− mice initiate mitochondrial stress responses. Biochem J. 2014;462:359–371. doi: 10.1042/BJ20140291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Pharaoh G, Pulliam D, Hill S, Sataranatarajan K, Van Remmen H. Ablation of the mitochondrial complex IV assembly protein Surf1 leads to increased expression of the UPRMT and increased resistance to oxidative stress in primary cultures of fibroblasts. Redox Biol. 2016;8:430–438. doi: 10.1016/j.redox.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dell’agnello C, Leo S, Agostino A, Szabadkai G, Tiveron C, Zulian A, et al. Increased longevity and refractoriness to Ca(2+)-dependent neurodegeneration in Surf1 knockout mice. Hum Mol Genet. 2007;16:431–444. doi: 10.1093/hmg/ddl477. [DOI] [PubMed] [Google Scholar]
- 92.Michel S, Canonne M, Arnould T, Renard P. Inhibition of mitochondrial genome expression triggers the activation of CHOP-10 by a cell signaling dependent on the integrated stress response but not the mitochondrial unfolded protein response. Mitochondrion. 2015;21:58–68. doi: 10.1016/j.mito.2015.01.005. [DOI] [PubMed] [Google Scholar]
- 93.Siegelin MD, Dohi T, Raskett CM, Orlowski GM, Powers CM, Gilbert CA, et al. Exploiting the mitochondrial unfolded protein response for cancer therapy in mice and human cells. J Clin Invest. 2011;121:1349–1360. doi: 10.1172/JCI44855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chae YC, Caino MC, Lisanti S, Ghosh JC, Dohi T, Danial NN, et al. Control of tumor bioenergetics and survival stress signaling by mitochondrial HSP90s. Cancer Cell. 2012;22:331–344. doi: 10.1016/j.ccr.2012.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jin SM, Youle RJ. The accumulation of misfolded proteins in the mitochondrial matrix is sensed by PINK1 to induce PARK2/Parkin-mediated mitophagy of polarized mitochondria. Autophagy. 2013;9:1750–1757. doi: 10.4161/auto.26122. [DOI] [PMC free article] [PubMed] [Google Scholar]