

Abstract
Puberty is a critical period for changes in genetic effects for binge eating in girls. Previous twin studies show increases in genetic influences on binge eating from pre-puberty (~0%) to mid-puberty and beyond (~50%). However, little is known about the factors that drive these shifts in genetic effects. A small pilot study showed that pubertal activation of estrogen may contribute to increases in genetic influences, possibly via hormonally induced changes in gene expression. However, large-scale studies investigating hormone effects on genetic risk are lacking. Thus, the purpose of the present study was to examine the effects of estrogen on genetic influences for binge eating in 964 female twins (ages 8-16 years) from the Michigan State University Twin Registry. Binge eating was assessed with the Minnesota Eating Behaviors Survey, while afternoon saliva samples were assayed for estradiol levels using standard enzyme immunoassays. Twin moderation models showed substantial differences in genetic influences on binge eating across estradiol levels. Stronger genetic effects were observed at lower (rather than higher) estradiol levels, even when controlling for the effects of age, body mass index, the physical changes of puberty, and the onset of menses. Overall, findings suggest that comparatively lower levels of estradiol during this critical period may disrupt normative developmental processes and enhance genetic influences on binge eating.
Keywords: Binge eating, estrogen, progesterone, genetic effects, twins
Binge eating is defined as the consumption of a large amount of food, in a short period of time, with a loss of control over the eating episode (American Psychiatric Association [APA], 2013). It is a core symptom of bulimia nervosa (BN) and binge eating disorder (BED) and is a common feature of many eating disorders (e.g., anorexia nervosa binge/purge type, other specified eating and feeding disorders (OSFED; APA, 2013). Given the transdiagnostic nature of binge eating, understanding its etiology is likely to contribute to knowledge of risk factors for a range of eating pathology.
Puberty is a key phenotypic and genetic risk period for the development of binge eating (Hildebrandt, Alfano, Tricamo, & Pfaff, 2010; Klump, Culbert, O’Connor, Fowler, & Burt, 2017) and eating disorders (Harden, Kretsch, Moore, & Mendle, 2014; Young, 2010) in girls. Rates of binge eating are low in pre-puberty but increase significantly in late-to-post puberty and into young adulthood in girls (Klump, 2013 for a review) and female animals (Klump, Culbert, & Sisk, 2017; Klump, Suisman, Culbert, Kashy, & Sisk, 2011). This pattern of phenotypic effects mimics changes in genetic influences on binge eating across puberty as well. The heritability of binge eating is essentially 0% in pre-early puberty, but increases linearly across puberty to ~50% in late-post puberty and adulthood (Klump, Culbert, O’Connor, et al., 2017). An interesting aspect of these pubertal shifts is that they appear to be specific to girls. Rates of binge eating do not increase during puberty in boys (Klump, 2013), and the heritability of more general disordered eating symptoms remains constant across puberty in boys as well (Klump et al., 2012). Although recent data suggests that shifts in genetic risk for boys may occur during earlier developmental stages (e.g., during adrenarche; Culbert, Burt, & Klump, 2017), there appear to be sex-specific factors that substantially increase the risk for binge eating in girls during puberty.
One sex-specific factor that may contribute is estrogen. Increases in estrogen drive pubertal development in girls but not boys (Wilson, Foster, Kronenberg, & Larsen, 1998) and may contribute to the phenotypic and genetic differences described above. Estrogen increases linearly throughout puberty and maps on to the linear increases in genetic effects on binge eating observed previously. Although progesterone is also involved in pubertal development in girls, progesterone becomes activated much later during the pubertal period (i.e., after first ovulation; Wilson et al., 1998); an activation that is likely too late to be able to account for increases in heritability that are commonly observed around mid-puberty in girls (Culbert, Burt, McGue, Iacono, & Klump, 2009; Klump, Culbert, O’Connor, et al., 2017; Klump, McGue, & Iacono, 2003; Klump, Perkins, Burt, McGue, & Iacono, 2007). Thus, estrogen has been the primary focus of pubertal theories of genetic risk (Hildebrandt et al., 2010; Klump, 2013; Klump, Culbert, O’Connor, et al., 2017; Klump, Keel, Sisk, & Burt, 2010; Klump et al., 2003; Young, 2010).
An additional reason for the focus on estrogen is its known genomic effects within the central nervous system (CNS). A critical function of estradiol during development is the regulation of gene transcription (Ostlund, Keller, & Hurd, 2003). When activated by hormones, intracellular receptors act as transcription factors to regulate mRNA expression in neurobiological systems via activating or inhibiting target genes. Increased genetic effects on binge eating during puberty may therefore result from the activation of estradiol and the genomic effects of the hormone on the structure and/or function of the CNS (e.g., the production of neurotransmitters, their receptors, or their signal transduction mechanisms) or other biological systems (e.g., appetitive signals from the periphery).
Extant data indicate that estrogen regulates gene transcription within several neural systems (e.g., serotonin, dopamine, opioids; Becker, 2009; Craft, 2008; Ostlund et al., 2003) that are disrupted in binge eating and that could contribute to increases in genetic effects during puberty. Given the timing of pubertal increases in genetic effects (from early to mid-puberty) versus increases in phenotypic rates of binge eating (late-to-post puberty), theories (Klump, 2013; Klump, Culbert, & Sisk, 2017) have proposed that estrogen acts organizationally to impact binge eating, with estrogen mediated genomic changes leading to (permanent) neural changes in brain structure/function during early-mid puberty that later lead to increased phenotypic risk for binge eating in late adolescence and adulthood.
Unfortunately, it is difficult to test organizational hormone effects in humans, and the specific genes involved in binge eating remain unknown. Thus, as a first step, twin studies should examine differences/changes in heritability across puberty and hormonal milieus. Differences in heritability would reflect changes in the influence of genetic factors/genes. This approach would be a straightforward (and cost effective) way to investigate whether estrogen may contribute to differences in genetic effects across puberty in girls.
Only one small pilot twin study has examined estrogen effects during puberty (Klump, Keel, et al., 2010). In this preliminary study, the degree of genetic influence on binge eating was significantly greater in twins with higher versus lower estradiol levels during puberty (Klump, Keel, et al., 2010). Although sample sizes were small (N = 98 pairs), the effects of estrogen were found to be independent of age, body mass index (BMI), and the physical changes of puberty, suggesting specific effects of estrogen on genetic influences for binge eating.
To date, there have been no large-scale studies of estrogen effects during puberty. Thus, the aim of the current study was to examine the effects of estrogen on genetic influences on binge eating during puberty in a sample of 964 pre-adolescent and adolescent female twins (ages 8-16). We took advantage of our larger sample size and conducted contemporary twin moderation models that allowed us to examine linear and non-linear effects of estrogen on genetic influences while controlling for many confounding variables (i.e. age, BMI, physical changes of puberty, onset of menses) that could account for any estrogen moderation effects. We used a continuous measure of binge eating that has shown increases in genetic effects across pubertal development in this sample (Klump, Culbert, O’Connor, et al., 2017) and that is appropriate for use in girls across our broad age range. Finally, we directly ruled out the effects of progesterone (and estrogen × progesterone interactions) on genetic influences by exploring progesterone moderation in preliminary analyses.
Methods
Participants
The sample consisted of 964 (MZ = 464 (48%); DZ = 500 (52%)) female twins ages 8 to 16 (M = 11.75, SD = 2.03) who participated in the Twin Study of Mood, Behavior, and Hormones during Puberty (TSMBH; O’Connor, Burt, VanHuysse, & Klump, 2016) from within the population-based, Michigan State University Twin Registry (MSUTR; for MSUTR and recruitment details, Burt & Klump (2013) and Klump & Burt (2006)). The MSUTR recruits twins using birth records in collaboration with the Michigan Department of Health and Human Services (see Klump & Burt, 2006 and Burt & Klump, 2012 for recruitment details), and then the TSMBH (and other follow-up studies – Klump et al., 2013; Burt, Klump, Gorman-Smith, & Neiderhiser, 2016) recruit twins directly from the MSUTR pool. Response rates for the MSUTR (Burt & Klump, 2013) and the TSMBH (~65%) are quite good and are on par or better than response rates for other large-scale twin registries (Baker, Barton, & Raine, 2002; Hay, McStephen, Levy, & Pearsall-Jones, 2002). MSUTR twins are representative of the recruitment region in terms of race, ethnicity, and socioeconomic status (Burt & Klump, 2013).
Because the TSMBH was focused on examining hormone regulation during puberty, twins were required to meet the following inclusion/exclusion criteria to ensure the validity/reliability of the hormone samples: 1) no hormonal contraceptive use within the past 3 months; 4) no psychotropic or steroid medications within the past 4 weeks; 5) no pregnancy or lactation within the past 6 months; and 6) no history of genetic or medical conditions known to influence hormone functioning or appetite/weight. Despite these criteria, the TSMBH twins are representative of the recruitment region in terms of race/ethnicity (81.4% White, 7.9% Black, 0.6% Asian/Pacific Islander, 0.2% Native American, 7.7% Multiracial, 2.1% missing data; 94.4% Non-Hispanic, 4.0% Hispanic, 1.7% missing data), and they do not differ from other MSUTR twins on measures of binge eating (Cohen’s d = .12). This study was approved by the Michigan State University Institutional Review Board (protocol #01-052M).
Measures
Zygosity Determination
Zygosity was determined using a well-validated physical similarity questionnaire that has been shown to be over 95% accurate when compared to genotyping (Lykken, Bouchard, McGue, & Tellegen, 1990; Peeters, Van Gestel, Vlietinck, Derom, & Derom, 1998). The twins’ mother and trained research assistants independently completed the questionnaire for all twins. Reports were compared amongst raters, and discrepancies were resolved using questionnaire responses, review of photographs of the twins by the principal investigator (KLK), and twin concordance across several nucleotide polymorphisms.
Binge Eating
Binge eating was assessed using the 7-item binge eating scale (assessing the tendency to engage in binge eating and to think about binge eating) from the Minnesota Eating Behavior Survey1 (von Ranson, Klump, Iacono, & McGue, 2005). The MEBS binge eating scale is appropriate for use in pre-pubertal children (Luo, Donnellan, Burt, & Klump, 2016) and shows adequate internal consistency in past studies (alpha = .65-.69; von Ranson et al., 2005) and our current sample (alpha = .69). Importantly, the internal consistency of the MEBS binge eating scale was very similar across hormone levels in the current study (i.e., using median splits, alpha at lower estradiol levels = .68, alpha at higher levels = .70) (see Supplemental Table S1), and deleting items from the scale decreased (i.e., alphas = .62-.66) rather than increased the internal consistency. Moreover, the item-total correlations (r’s = .38-.48; Mean r = .40) were within the recommended range (i.e., .40-.50) (Clark and Watson, 1995) and were indicative of very good item discrimination. Finally, the binge eating scale exhibits good criterion-related validity, as women with bulimia nervosa (BN) score significantly higher on the scale than controls (von Ranson et al., 2005) and, in the current sample, twins who self-reported one or more episodes of objective binge eating (on the Eating Disorders Examination Questionnaire (EDEQ); Fairburn & Beglin, 1994) scored significantly higher on the MEBS binge eating scale than controls (t(992) = 15.67, p < .0001; d = 1.02). Pearson correlations with related phenotypes (e.g., body mass index (BMI), levels of body dissatisfaction/weight preoccupation, etc.) were significant and nearly identical across hormone levels as well (see Supplemental Table S1).
As noted above, the MEBS has been employed in numerous developmental twin studies of eating disorders (Culbert et al., 2009; Klump, Burt, McGue, & Iacono, 2007; Klump, Culbert, O’Connor, et al., 2017; Klump, Keel, et al., 2010; Klump et al., 2003; Klump, McGue, & Iacono, 2000; Klump, Perkins, et al., 2007), including a recent study of pubertal differences in genetic influences in the current TSMBH sample (Klump, Culbert, O’Connor, et al., 2017). Our use of the same, developmentally appropriate measure allowed us to explore whether estrogen accounts for previously observed pubertal differences in genetic effects.
Hormone Levels
Salivary samples were used to obtain estradiol and progesterone levels. Saliva samples are preferred over other methods (e.g., blood spots) because they represent a less invasive collection method, particularly for pre-adolescent and adolescent participants who, in our lab studies, are less likely to agree to blood draws. Moreover, previous research has shown that saliva samples are associated with higher compliance and more robust hormone-behavior associations than blood-spot sampling (Edler, Lipson, & Keel, 2007). Finally, estradiol values from matched serum and saliva samples show a strong linear relationship for females (r = .80; Shirtcliff et al., 2000).
Following previously reported procedures (Klump, Keel, et al., 2010), twins were asked to refrain from eating or drinking for four hours prior to providing a saliva sample and to refrain from smoking, brushing their teeth, or chewing gum for 30 minutes prior to the saliva collection. Twins were then instructed to passively drool down a straw into a cryovial until ≥ 4 ml was produced. All saliva collections occurred between 2:00 pm and 5:00 pm. We chose to collect during these times because afternoon-early evening diurnal variations in ovarian hormones during puberty tend to be minimal (Angold, Costello, Erkanli, & Worthman, 1999; Grumbach & Styne, 1998), and most families were available for assessments during this time. Saliva samples were frozen immediately and stored until transport.
We collected saliva samples on three consecutive days: two samples were collected by the twins in their home, while the third was collected in our MSU laboratory during the primary, in-person lab assessment. Unfortunately, hormone values were less correlated (r’s = .53-.58) across the assessment days than would be expected. Reasons for this are unclear, although it seems likely that twin difficulties adhering to our saliva collection procedures (e.g., collection between 2 and 5 pm, fasting for 4 hours, immediately freezing samples and keeping them frozen during transport) likely contributed. Thus, in the current report, we focus on the in-person assessment hormone values only as they were collected on the same day as the binge eating scores, and our in-lab collection procedures were carefully standardized and minimized error. However, our results were similar when using the average of the three assessment days (see Figure S1 in the Supplemental Material), suggesting that our findings are robust to saliva collection procedures and sampling frequency.
All saliva samples were processed in duplicate by Salimetrics, LLC (State College, PA) using enzyme immunoassay kits designed specifically for analyzing saliva. These assays show excellent intra- and inter-assay coefficients of variation (estradiol = 7.1% and 7.5%; progesterone = 6.2% and 7.6%), as well as assay sensitivity (measured by interpolating the mean optical density minus 2 SDs of 10-20 replicates at the 0-pg/ml level; estradiol = 0.10 pg/ml; progesterone = 5 pg/ml) and method accuracy (determined by spike recovery and linearity; estradiol = 104.2% and 99.4%; progesterone = 99.6% and 91.8%). Notably, all of the twins in the current study had estradiol and progesterone levels that fell above the minimum detection limits for the assays.
Physical Changes of Puberty
The physical changes of puberty were measured with twin self-report on the Pubertal Development Scale (Petersen, Crockett, Richards, & Boxer, 1988). The PDS assesses growth spurts, skin changes, body hair growth, and breast development on a 4-point scale: 1) development has not yet begun, (2) development has barely started, (3) development is definitely underway, or (4) development seems complete. Menses was rated as either present (coded 1) or absent (coded 4). Maternal reports on the PDS were used for a subset of twins (n = 16; 1% of sample) who were missing self-reported PDS scores. Consistent with past studies (Culbert et al., 2009; Klump et al., 2003), PDS items were summed and averaged for all analyses. Previous studies of the PDS psychometric properties have supported its reliability (median alpha = .77; Petersen et al., 1988) and validity, including showing high correlations with clinician ratings of these same physical characteristics (r’s = .61-.67; Petersen et al., 1988).
BMI
BMI was calculated (weight in kg/height in m2) using height and weight measured with a wall-mounted ruler and digital scale, respectively. We used raw (instead of age- and sex-adjusted) BMIs in analyses to be consistent with our recent examining differences in genetic effects across pubertal development in binge eating (Klump, Culbert, O’Connor, et al., 2017) and prior developmental twin studies (Culbert et al., 2009; Culbert, Burt, Sisk, & Klump, 2013; Klump et al., 2012, 2003; Klump, Burt, et al., 2007; Klump, Burt, McGue, Iacono, & Wade, 2010; Klump, Holly, Iacono, McGue, & Willson, 2000; Klump, Keel, et al., 2010). Raw and age-adjusted BMI scores were nearly identical (r > .99; M difference = 0.04, SD = 0.03).
Statistical Analyses
Data Preparation
Missing scores on the binge eating scale were prorated when ≤10% of the items were missing, and coded missing if > 10% of items were missing. These scores were then log transformed prior to analyses to account for positive skew. We conducted all analyses with the covariates age, BMI, and PDS scores regressed out of each twin’s binge eating score to ensure that differences in etiologic effects were specific to hormone levels rather than these potentially confounding variables. Notably, however, results were similar when using raw binge eating scores that did not partial out these variables (see Figure S2 in the Supplemental Online Material), suggesting that they had minimal impact on differences in genetic effects across hormone levels during puberty.
Twin Correlations
Twin intra-class correlations were used as initial indicators of differences in genetic and environmental influences on binge eating across estradiol levels. Because MZ twins share approximately 100% of their segregating genes while DZ twins share, on average, 50%, significantly greater MZ relative to DZ twin correlations indicate the presence of additive genetic effects (i.e., effects that add or sum across genes). By contrast, MZ and DZ twin correlations that are similar in magnitude and are significantly greater than 0 signify a lack of genetic effects, but significant shared environmental influences (i.e., environmental factors that are common to siblings growing up in the same family and contribute to their behavioral similarity). Finally, MZ twin correlations that are less than 1.00 indicate the presence of nonshared environmental factors (i.e., factors that are unique to siblings growing up in the same family and contribute to behavioral differences) and measurement error.
Although continuous estradiol levels were used in model-fitting (see below), a dichotomous estrogen group (i.e., low/high) was developed for the twin correlations based on median splits for estradiol levels (cut-off = 1.26 pg/ml). Only pairs concordant on estrogen group (e.g., both co-twins had low estradiol levels) could be included in correlations (N = 534 twins from 267 pairs; 55% of the total sample), although concordant and discordant co-twins were included in model-fitting analyses (see below).
Twin Moderation Models
We used extended, univariate twin moderation models (see Figure 1; van der Sluis, Posthuma, & Dolan, 2012) to examine differences in additive genetic (A), shared environmental (C), and nonshared environmental (E) influences on binge eating across estradiol levels. These models estimate: 1) path coefficients (i.e., a, c, e) assessing genetic/environmental influences at the lowest level of estradiol; 2) linear moderators (βXM, βYM, βzM) assessing linear increases/decreases in etiologic influences across estradiol levels; and 3) quadratic moderators (βX2M, βY2M, βZ2M) assessing non-linear increases/decreases in genetic/environmental influences across estradiol levels.
Figure 1. Partial Path Diagram for the Full Moderation Model for One Twin Only.
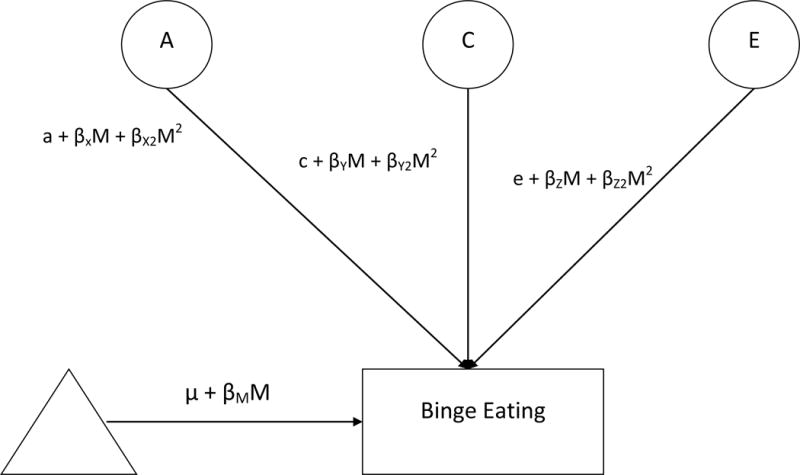
Abbreviations include: A = additive genetic effects; C = shared environmental effects; E = nonshared environmental effects; M = Moderator (i.e., estradiol levels); triangle = mean for binge eating; βM = phenotypic regression coefficient; a, c, and e = paths or intercepts; βX, βY, βZ = linear moderators; βX2M2, βY2M2, βz2M2 = quadratic moderators.
We first fit the “full” model that included all parameters. We then compared the fit of this model to nested submodels that differentially constrained the linear and quadratic genetic, shared environmental, and nonshared environmental moderators to 0. We initially focused on submodels that are commonly tested in twin moderation analyses (i.e., models that constrained all moderators to 0, models that constrained the quadratic moderators only to 0, etc.) as well as those that directly tested our theory of genetic moderation (i.e., models that constrained the genetic moderators to 0). Because of the large number of submodels that could be fit, however, we used parameter estimates from the full model to identify additional submodels to fit. This approach allowed us to test relevant submodels without unduly increasing the number of tests.
We used the raw estradiol values in all analyses, although results were nearly identical when “binning” the estradiol values into two groups (see Figure S3 in Supplemental Material). Prior to model-fitting, binge eating scores were standardized and minimum estradiol levels were “floored” to 0. Models were then fit to the raw data using the maximum likelihood option in Mx (Neale, Boker, Xie, & Maes, 2003). This option treats missing data as missing-at-random and is expected to produce less biased and more consistent estimates than other techniques (e.g., listwise deletion). Model fit comparisons were made by taking the difference in minus twice the log-likelihood (−2lnL) between the full and nested models, which is chi-squared distributed under the null hypothesis implied by the reduced model. Large (statistically significant) differences led to a rejection of the nested model. Akaike’s information criterion (AIC), Bayesian information criterion (BIC), sample-size adjusted BIC, and deviance information criterion (DIC) were also used to select the best-fitting model, where models that minimized the largest number of these scores were preferred. Unstandardized parameter estimates from the full and best-fitting models are reported in figures, as they more accurately depict absolute differences in etiologic effects than standardized estimates that represent differences as proportions of the total variance. However, we report standardized estimates in the text, as these estimates allow for a direct comparison of our findings to previous studies.
Notably, the twin moderation models described above rest on one important assumption – that the moderator (i.e., estradiol levels) is genetically independent of the dependent variable (i.e., binge eating). If there is not independence, then genetic mediation (i.e., termed gene-environment correlations or rGE) may be present for estradiol levels and binge eating, such that the same genes that influence hormone levels also influence binge eating. These types of rGE are troublesome in the context of twin moderation models, as rGE could conceivably “masquerade” as hormone moderation effects. For example, if binge eating stems, in part, from genes common to hormone levels, then increases in genetic influences at high levels of hormones could be a reflection of shared genes and/or environments, rather than an independent effect of the hormone levels on genetic influences on binge eating. To avoid this interpretive difficulty, twin researchers are advised to use the “gene × environment (GxE) in the presence of rGE” model (Purcell, 2002) that tests the significance of genetic and/or environmental mediation by comparing the fit of models that allow for genetic/environmental covariance between the moderator and the dependent variable to models that constrain the covariance to be zero. Consequently, before fitting the models described above, we fit “GxE in the presence of rGE” models and found that the moderation of the covariance between estrogen and binge eating was non-significant (i.e., the parameter could be constrained to 0 without a worsening of model fit - comparison with full model: χ2Δ (3) = 3.53, p = .32). Moreover, the best fitting model retained the parameter that tests for the unique/independent moderating effects of estrogen on binge eating (i.e., the unique estrogen moderator could not be constrained to 0 without a worsening of model fit - comparison with full model: χ2Δ (3) = 18.45, p = .0004), outside of any rGE processes. These findings provided the first indication that our twin models would show significant effects of estrogen on genetic and/or environmental risk for binge eating that reflect independent hormone effects rather than rGE.
Finally, as noted in the Introduction, we also conducted preliminary model-fitting analyses to confirm that progesterone did not significantly moderate etiologic effects on binge eating during puberty. We examined these effects using two-moderator models that allowed us to examine whether estrogen alone, progesterone alone, or estrogen × progesterone interactions moderated etiologic effects on binge eating. Importantly, the best-fitting model constrained all of the progesterone moderators (including the estrogen × progesterone interaction) to 0 (i.e., comparison with full model: χ2Δ (6) = 5.79, p = .45), but retained the estrogen moderators (see a description of these models in Figure S3 in Supplemental Material). These preliminary results supported our theories of estrogen (rather than progesterone) moderation (Klump, 2013; Klump, Culbert, & Sisk, 2017) and our focus on estrogen in the primary analyses.
Results
Descriptive Statistics and Phenotypic Correlations
Descriptive statistics and intercorrelations among our study variables are presented in Table 1. A range of binge eating was present in our sample, as scores varied across the full spectrum of possible scores (i.e., 0-7), and 3.2% of twins scored above the clinical cut-off for binge eating (i.e., clinical cut-off = the mean of individuals with bulimia nervosa score ≥ 4.36; von Ranson et al., 2005). Intercorrelations among the study variables were as expected with binge eating showing significant (albeit modest) correlations with PDS scores and BMI, and estradiol levels showing significant and positive correlations with age, PDS scores, and BMI as well. Binge eating scores did not correlate significantly with estradiol levels, a finding that has been reported previously in pre-adolescent/adolescent girls (Klump, Keel, et al., 2010). Importantly, however, the lack of significant phenotypic associations does not negate a moderating effect of the hormone on genetic effects, as the presence of genetic moderation would be expected to attenuate phenotypic associations. This pattern has been observed previously in twin studies, where pubertal development was a significant moderator of genetic effects despite modest phenotypic associations with binge eating or other types of disordered eating (Culbert et al., 2009; Klump, Culbert, O’Connor, et al., 2017; Klump, Keel, et al., 2010; Klump et al., 2003; Klump, Perkins, et al., 2007).
Table 1.
Descriptive Statistics and Intercorrelations for Study Variables (N = 964 twins).
| Variable | Range | Mean (SD) | Pearson Correlations
|
||||
|---|---|---|---|---|---|---|---|
| Binge Eating | Estradiol | Age | PDS | BMI | |||
| Binge Eating | 0-7 | 0.90 (1.34) | 1.00 | −.01 | .04 | .11** | .13*** |
| Estradiol (pg/mL) | 0.11-4.69 | 1.43 (.82) | – | 1.00 | .24*** | .23*** | .22** |
| Age | 8-16 | 11.75 (2.03) | – | – | 1.00 | .80*** | .41*** |
| PDS Score | 1-4 | 2.25 (.90) | – | – | – | 1.00 | .49*** |
| Body Mass Index (BMI) | 10.70-46.55 | 19.53 (4.47 | – | – | – | – | 1.00 |
Note. Binge eating scores were log transformed prior to analysis, but raw values are presented in the table for descriptive purposes.
p < .01,
p < .001
Twin Correlations
Twin correlations suggested significant moderation of genetic effects by estradiol levels (see Table 2), but the pattern of effects was opposite to that found previously in pilot work (Klump, Keel, et al., 2010). Lower levels of estradiol were associated with stronger genetic influences, as the MZ twin correlation was significantly greater than the DZ twin correlation, and the MZ/DZ difference was of large effect size (see q estimates in Table 2). By contrast, no significant MZ/DZ twin differences were observed at higher estradiol levels (and effect sizes were small – see Table 2), suggesting minimal genetic influences but possible shared environmental effects. Nonshared environmental influences were present across all estradiol levels, as the MZ twin correlation was less than 1.00, but seemingly increased at higher estradiol levels where the MZ twin correlation was quite small. Notably, in the higher estradiol group, the DZ twin correlation was larger than the MZ twin correlation, but the MZ/DZ correlation difference was non-significant and of small effect size. Overall then, findings from the twin correlations seemed to show stronger genetic effects on binge eating at lower versus higher estradiol levels, and stronger nonshared environmental influences as estradiol levels increased.
Table 2.
Twin Correlations for Binge Eating by Estradiol Levels.
| Estrogen Groups | MZ Pairs | DZ Pairs | Z Test of Independence | p | q |
|---|---|---|---|---|---|
| Full Sample (N =267 pairs (534 twins)) | |||||
| Lower Estradiol (MZ n = 67 pairs; DZ n = 62 pairs) |
.70*** (.55, .80) |
.10 (.15, .34) |
4.25 | <.001 | .77 |
| Higher Estradiol (MZ n = 69 pairs; DZ n = 69 pairs) |
.11 (-.13, .34) |
.27* (.04, .48) |
0.96 | .34 | .16 |
| Pre-Menarcheal Twins Only (N = 141 pairs (282 twins)) | |||||
| Lower Estradiol (MZ n = 39 pairs; DZ n = 31 pairs) |
.74*** (.55, .86) |
.50** (.18, .73) |
1.59 | .11 | .40 |
| Higher Estradiol (MZ n = 36 pairs; DZ n = 35 pairs) |
.001 (-.33, .33) |
.22 (-.12, .52) |
0.90 | .37 | .22 |
Note. MZ = monozygotic; DZ = dizygotic; Z Test of Independence = a z test of significant differences between the MZ and DZ twin correlations; q = effect size for the difference between the two twin correlations (no effect q < .10, small effect q = .10-.30, medium effect q = .30-.50, large effect q > .50. The 95% confidence intervals are presented in parentheses. Binge eating scores were log transformed prior to analysis.
p < .05,
p < .001. The twin correlation is significantly different from zero.
Twin Moderation Models
Twin model-fitting analyses confirmed these impressions. Examination of the parameter estimates from the full model (see Figure 2a) suggested significant (and linear) decreases in genetic influences across estradiol levels, quadratic/non-linear changes in nonshared environmental effects, but minimal differences in shared environmental effects. Model fit comparisons (see Table 3) indicated that the model constraining all of the shared environmental moderators (linear and quadratic) to 0 provided a good fit to the data (i.e., a non-significant chi-square change and lower AIC, BIC, sample-adjusted BIC, and DIC values than most models – see Table 3), whereas models constraining all of the genetic, shared environmental, and nonshared environmental moderators to 0 provided a poorer fit (i.e., significant chi-square change tests and higher values for some fit indices - see Table 3). Importantly, models constraining the genetic moderators (linear and quadratic) only, and the nonshared environmental moderators (linear and quadratic only) only, also resulted in a significant worsening of model fit (significant chi-square change tests and higher values for fit indices - see Table 3). Overall, these initial model fit comparisons confirmed that there is significant moderation of genetic effects (and nonshared environmental influences) but no significant shared environmental moderation by estradiol levels during puberty.
Figure 2. Unstandardized Estimates of Additive Genetic (a), Shared Environmental (c), and Nonshared Environmental (e) Effects from the Full and Best-Fitting Models in All Twins (N = 964) and Premenarcheal Twins Only (N = 530).
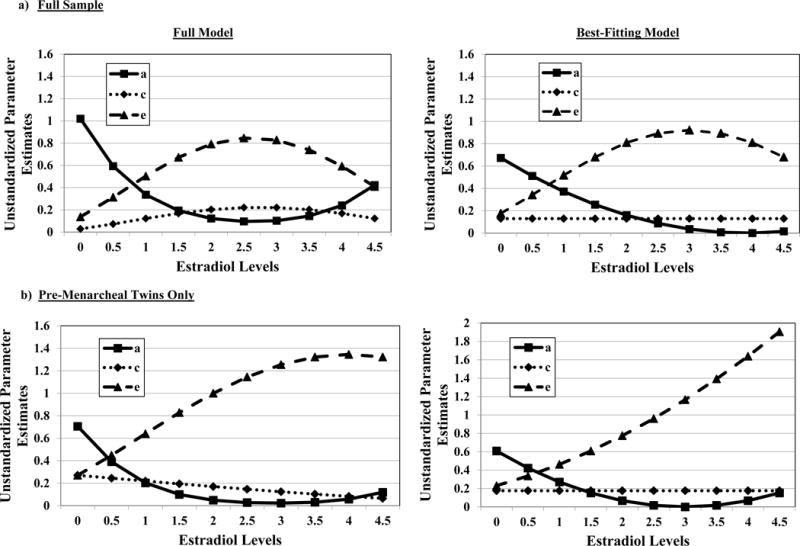
For the full sample, the best-fitting model was the model that constrained all of the shared environmental moderators to 0 as well as the genetic quadratic moderator. The best fitting model for pre-menarcheal twins was the model that constrained all of the shared environmental moderators as well as the genetic and nonshared environmental quadratic moderators to 0. It is important to note that the estradiol values in the figures were floored to 0 for the twin models (see Methods), and that the raw estradiol values range from 0.11-4.69 in the full sample, and 0.11-4.56 in the sample of pre-menarcheal twins only.
Table 3.
Model Fit Comparisons for the Twin Moderation Models.
| Model | −2lnL (df) | χ2Δ (df) | p | AIC | BIC | Adj. BIC |
DIC |
|---|---|---|---|---|---|---|---|
| Full Sample (N = 964 Twins) | |||||||
| Full Model | 2227.64 (780) | – | – | 667.64 | −1226.73 | 10.78 | −509.96 |
| Nested Submodels: | |||||||
| Constrain A, C, E quad mods | 2232.75 (783) | 5.11 (3) | .16 | 666.75 | −1233.18 | 9.09 | −513.65 |
| Constrain linear and quad A mods | 2255.69 (782) | 28.05 (2) | <.001 | 691.69 | −1218.71 | 21.98 | −500.10 |
| Constrain linear and quad C mods | 2228.25 (782) | 0.61 (2) | .74 | 664.25 | −1232.43 | 8.26 | −513.82 |
|
| |||||||
| Constrain all C mods and A quad only | 2229.97 (783) | 2.33 (3) | .51 | 663.97 | −1234.57 | 7.71 | −515.04 |
|
| |||||||
| Constrain all C mods and E quad only | 2232.68 (783) | 5.04 (3) | .17 | 666.68 | −1233.21 | 9.06 | −513.68 |
| Constrain all C mods, A, E quad | 2232.82 (784) | 5.18 (4) | .27 | 664.82 | −1236.14 | 7.72 | −515.70 |
| Constrain linear and quad E mods | 2241.62 (782) | 13.98 (2) | <.001 | 677.62 | −1225.74 | 14.95 | −507.13 |
| Constrain all linear and quad moderators for A, C, and E | 2242.05 (786) | 14.41 (6) | .03 | 670.05 | −1237.53 | 9.50 | −515.25 |
| Pre-Menarcheal Twins Only (N = 530 Twins) | |||||||
| Full Model | 1420.77 (510) | – | – | 400.77 | −713.50 | 95.09 | −244.75 |
| Nested Submodels: | |||||||
| Constrain A, C, E quad mods | 1421.80 (513) | 1.03 (3) | .79 | 395.80 | −721.27 | 91.98 | −249.85 |
| Constrain linear and quad A mods | 1423.33 (512) | 2.56 (2) | .28 | 399.33 | −717.71 | 93.95 | −247.22 |
| Constrain linear and quad C mods | 1420.97 (515) | 0.20 (2) | .90 | 396.97 | −718.89 | 92.77 | −248.40 |
| Constrain all C mods, A quad only | 1421.62 (513) | 0.85 (3) | .84 | 395.62 | −721.36 | 91.89 | −249.94 |
| Constrain all C mods, E quad only | 1421.63 (513) | 0.96 (3) | .81 | 395.73 | −721.30 | 91.95 | −249.89 |
|
| |||||||
| Constrain all C mods, A, E quad | 1422.05 (514) | 1.28 (4) | .86 | 394.05 | −723.93 | 90.90 | −251.69 |
|
| |||||||
| Constrain linear and quad E mods | 1431.66 (512) | 10.89 (2) | .004 | 407.66 | −713.55 | 98.12 | −243.05 |
| Constrain A, C, Moderators | 1433.49 (516) | 12.72 (6) | .047 | 401.49 | −723.80 | 94.21 | −249.63 |
Note. −2lnL = minus twice the log-likelihood; χ2Δ = chi-square change; AIC = Akaike information criterion; BIC = Bayesian information criterion; Adj. BIC = sample-size adjusted BIC; DIC = deviance information criterion; Full Model = model with paths and all moderators; mods = moderators; quad = quadratic or non-linear moderators; A = additive genetic effects; C = shared environmental effects; E = nonshared environmental effects. Each nested model is compared to the full model when calculating the χ2Δ and degrees of freedom. The best-fitting models are noted by bolded text and borders. Unstandardized estimates (with 95% confidence intervals in parentheses) from the best-fitting models were as follows: Full Sample: a = −0.81 (−1.04, −0.49), c = 0.36 (−0.59, .59), e = −0.42 (−0.64, −0.26), βXM = 0.21 (.02, 0.43), βYM = 0, βZM = −0.36 (−0.58, −0.11), βX2M2 = 0; βY2M2 = 0; βZ2M2 = 0.05 (−0.01, .12); Premenarcheal Twins Only: a = 0.78 (0.34, 1.06), c = −0.41 (−0.62, 0.62), e = −0.48 (−0.66, −0.33), βXM = −0.26 (−0.48, −0.04), βYM = 0, βZM = −0.20 (−0.31, −0.09), βX2M2 = 0; βY2M2 = 0; βZ2M2 = 0.
In terms of additional submodels to fit, parameter estimates from the full model seemed to show linear decreases in genetic, but non-linear differences in nonshared environmental effects, across estradiol levels. We therefore fit three additional submodels that constrained all of the shared environmental moderators to 0, but then differentially constrained the quadratic genetic moderator, quadratic nonshared environmental moderator, or both to 0 (see Table 3). Importantly, the benefit of these models is that they directly tested whether the significant differences in genetic and nonshared environmental influences across estradiol levels were linear (i.e., the quadratic moderator could be constrained to 0) or non-linear in nature (i.e., the quadratic moderator could not be constrained without a worsening of model fit). Two of these models (i.e., the model that constrained the genetic quadratic moderator only and the model that constrained both the genetic and nonshared environmental quadratic moderators) showed essentially equivocal evidence for being the best-fitting model. Both models exhibited non-significant chi-square change values, but two out of the four fit indices favored one model over the other (i.e., the AIC and sample-adjusted BIC favored the model constraining the genetic quadratic moderator only, while the BIC and DIC favored the model constraining both the genetic and nonshared environmental quadratic moderators - see Table 3). We took the more conservative approach and chose the model that retained the nonshared environmental quadratic moderator (i.e., the model that constrained the genetic quadratic model only) as best fitting, as the nonshared environmental quadratic moderator was statistically significant (i.e., 95% confidence interval did not include 0 – see the Note in Table 3) in this model. Unstandardized (see Figure 2a) and standardized (see parentheses below) estimates from this final model confirmed the presence of substantial differences in genetic and nonshared environmental influences across estradiol levels, with significantly greater genetic effects at lower (69%) versus higher (2%) estradiol levels, and significantly greater nonshared environmental influences at higher (68%) versus lower (18%) estradiol levels. Shared environmental influences remained stable (13%) and non-significant (i.e., 95% confidence intervals included 0 – see Table 3 Note) across all estradiol levels.
Because findings for genetic effects were opposite to those of our small pilot study (Klump, Keel, et al., 2010), we conducted post hoc analyses examining only those twins who were pre-menarcheal to determine if our inclusion of pre- and post-menarcheal twins may have influenced our results. Our pilot study also included pre/post-menarcheal twins, but the age range of the current sample was much broader and included a larger proportion of post-menarcheal twins (Klump, Keel, et al., 2010). Hormone values would be expected to be more variable in post-menarcheal twins and dependent upon menstrual cycle phase. In the current study, we attempted to standardize the day of the lab assessment for our menstruating twins (i.e., day 15 of the menstrual cycle) to ensure more stable hormone levels, but this was not always possible given twin scheduling constraints and the error associated with relying on twin self-reports for estimating cycle days.
Twin correlations in the pre-menarcheal only group (see Table 2) showed a very similar pattern of results to the full sample (i.e., stronger genetic influences at lower as compared to higher estradiol levels). The only exception was that the MZ/DZ twin correlation differences for high estradiol levels was no longer significant (but still represented a medium effect size (q) – see Table 2). Model-fitting results in pre-menarcheal twins were also very similar to the full sample, with stronger genetic influences at lower estradiol levels, stronger nonshared environmental effects at higher estradiol levels (see Figure 2b), and minimal differences in shared environmental influences. In this smaller sample, the best-fitting model constrained all shared environmental moderator and the quadratic genetic moderator to 0 (like in the full sample) and also constrained the quadratic nonshared environmental moderator to 0. Unstandardized (see Figure 2) and standardized (see parentheses) estimates from the best-fitting model once again showed stronger genetic effects at lower (60%) versus higher (7%) estradiol levels, significantly greater nonshared environmental influences at higher (85%) versus lower (23%) estradiol levels, and minimal/non-significant shared environmental influences (18%).2
Discussion
This was the first large-scale study of the effects of estrogen on genetic influences on binge eating during puberty in girls. Estradiol levels significantly moderated genetic and environmental influences on binge eating, and these effects were independent of age, BMI, the physical changes of puberty, and rGE processes. Findings were consistent across pre-menarcheal and post-menarcheal girls, and preliminary analyses confirmed that effects were due to estrogen, not progesterone or estrogen × progesterone interactions, during the pubertal period. Overall, these results suggest that estrogen activation at puberty is associated with substantial etiologic shifts for binge eating that may have longer lasting effects on binge eating risk into adulthood.
The direction of effects for estrogen was opposite to what we observed in a small pilot study (Klump, Keel, et al., 2010). In the current sample, genetic influences were significantly stronger in twins with lower estradiol levels, whereas in the pilot study, genetic effects were stronger in twins with higher estradiol levels. This pattern of findings replicated across several different ways of analyzing the data (i.e., in the full sample of twins, in premenarcheal twins only, with and without covariates, using saliva samples on the assessment day only versus all three assessment days, etc.), suggesting that the results were robust to sample composition, analytic approach, and study design decisions. Taken together, it seems that differences in results could be due to the much smaller sample sizes in the pilot study and that study’s inability to include discordant twin pairs in analyses. The current study took full advantage of a larger sample of twins and modeled hormone effects in concordant and discordant twins using continuous twin moderation models that simultaneously examined linear and non-linear differences in etiologic effects in all twins. Using this much larger sample and more powerful analytic techniques, we found evidence that lower estradiol levels are associated with stronger genetic effects (rather than the reverse) in the full sample of twins as well as in pre-menarcheal twins only.
Clearly, more replication studies are needed to definitively confirm our findings. If results replicate, it will be important to understand how/why genetic estimates for binge eating increase across the pubertal period when estradiol levels are increasing. It is possible that estrogen does not play any role in pubertal increases in genetic influences, and that the estrogen effects we observed reflect completely independent processes. In that case, it may be that other factors that come on-line and/or increase during puberty (e.g., leptin, kisspeptins, growth hormone; Saenger, 2003; Sanchez-Garrido & Tena-Sempere, 2013) are driving pubertal effects on genetic risk for binge eating rather than estrogen. Although these other factors should be examined in future research, we think that estrogen’s primary and central role in pubertal development and regulation of gene transcription within the CNS make this possibility less likely. Perhaps more importantly, increases in genetic effects during puberty/gonadarche occur in girls but not in boys (Klump et al., 2012), further suggesting that female-specific pubertal factors (like estrogen) rather than processes present in girls and boys (e.g., leptin, kisspeptins, and growth hormone) likely contribute to changes in genetic effects during puberty in girls.
If estrogen is involved in these pubertal effects, then our findings point to a potentially more complicated picture for estrogen than originally proposed (Klump, 2013; Klump, Culbert, & Sisk, 2017; Klump, Keel, et al., 2010). Our results suggest that increasing genetic effects on binge eating during puberty may be driven by girls who with lower levels of estradiol relative to other girls at similar ages and levels of development. Prior to the activation of the reproductive axis during puberty, all girls have relatively low levels of estradiol; however, once the reproductive axis is activated during puberty, girls begin to vary in their estradiol levels, and these individual differences appear to translate into stronger genetic influences on binge eating in girls with relatively lower (rather than higher) hormone levels. From an evolutionary perspective, these findings make intuitive sense, as all girls experience increases in estrogen during puberty that drive normative development and reproductive success. What our current findings suggest are that lower levels of estradiol during puberty may disrupt this developmental process and increase genetic effects on binge eating that (potentially) lead to the later development of binge eating in adulthood. Interestingly, although genetic effects were not examined, a recent animal study showed that complete removal of estrogen (via ovariectomy) before puberty in female rats resulted in substantially increased rates of binge eating in adulthood (Klump et al., 2016). These findings provide additional support for our theory that lower levels of estradiol during puberty may disrupt normative developmental processes and increase risk for binge eating.
Importantly, these data also imply that pubertal estrogen may be protective against binge eating. Estrogen is protective against binge eating in adulthood. Higher levels of estradiol are associated with low phenotypic rates of binge eating (Klump et al., 2013, 2014) and lower heritability (i.e., ~12%; Klump et al., 2015). By contrast, lower levels of estradiol are associated with higher phenotypic rates (Edler et al., 2007; Klump et al., 2013, 2014) and higher heritability (~40-50%; Klump et al., 2015, 2016) of the same binge-eating phenotypes. Studies in adulthood have used strong cross-sectional (Klump et al., 2016) and longitudinal (i.e., across the menstrual cycle; Klump et al., 2013, 2014, 2015) designs to establish these effects, and they directly replicate 40+ years of animal work showing that estradiol is protective against excessive food intake in adulthood (Asarian & Geary, 2013; Klump, Culbert, & Sisk, 2017). Taken together, data across puberty, adulthood, and animal and human studies suggest that estrogen may be protective against binge eating, and that lower levels of estradiol are associated with increased genetic effects and increased phenotypic rates of binge eating in adulthood.
Critically, there is one difference in the effects of estrogen during puberty versus adulthood – significant phenotypic estrogen/binge eating associations exist in adulthood, but no significant phenotypic associations have been observed before/during puberty in our current analyses (see Table 1) or past human (Klump, Keel, et al., 2010) and animal work (Klump, Culbert, & Sisk, 2017). These differential phenotypic effects imply the presence of organizational and activational effects of estrogen on binge eating risk across development. The organizational/activational hypothesis of gonadal hormone action predicts that: 1) gonadal hormones organize risk for binge eating during puberty through changes in gene transcription and resulting changes in brain structure/function; and 2) these changes organize the brain to respond to circulating levels of hormones in adulthood which activate and/or influence the expression of behavior (Sisk & Zehr, 2005). Importantly, when hormone organizational/activational effects are present, modest phenotypic associations between gonadal hormones and behavior are observed during puberty when organizational changes are taking place, whereas significant phenotypic associations are present in adulthood (Sisk & Zehr, 2005). This pattern of phenotypic associations completely mimics the pattern observed for estrogen and binge eating in puberty/adulthood and suggests that estrogen’s actions during puberty might be organizational in nature, while its effects in adulthood are activational.
Indeed, based on current and prior data, it seems likely that relatively lower levels of estradiol may organize risk for binge eating during puberty via genomic changes within the CNS that lead to differential organization of neural circuitry and significant binge eating/estrogen associations in adulthood. These genomic changes may involve decreased expression/transcription of genes involved in normative development within key brain regions (e.g., brain reward pathways) or other, yet-to-be identified processes. Given the polygenic nature of binge eating, and the range of genetic/neural processes involved in pubertal brain development, the genomic changes likely involve a wide array of genes and neural networks that may code for mood/negative affect (e.g., serotonin genes; Barth, Villringer, & Sacher, 2015; Hildebrandt et al., 2010; Ostlund et al., 2003), ingestive behavior/obesity (e.g., FTO, TAS1R2, TAS1R3; Grimm & Steinle, 2011; Micali, Field, Treasure, & Evans, 2015; Tanofsky-Kraff et al., 2009), neuropeptides/hormones involved in satiety and food intake (e.g., cholecystokinin (CCK), ghrelin, leptin; Grimm & Steinle, 2011; de Krom et al., 2007; Monteleone, Tortorella, Castaldo, Di Filippo, & Maj, 2007), and/or reward system neurocircuitry (e.g., dopamine, opioids; Barth et al., 2015; Becker, 2009; Craft, 2008). To account for our findings, these systems must be regulated by estrogen and show pubertal changes in gene expression. Although hormonal regulation is known for some of these genes/systems (e.g., serotonin, dopamine, leptin, CCK; Barth et al., 2015; Becker, 2009; Brown & Clegg, 2012; Geary, 2001; Hildebrandt et al., 2010; Ostlund et al., 2003), more is needed in terms of identifying estrogen modulation during puberty and/or other developmental milestones. Nonetheless, the presence of hormone-mediated, pubertal sculpting of the CNS in animals (Ahmed et al., 2008; Barth et al., 2015; Nunez, Sodhi, & Juraska, 2002; Primus & Kellogg, 1991; Spear, 2000; Zehr, Todd, Schulz, McCarthy, & Sisk, 2006) and genetically-mediated associations between estradiol levels and changes in neural structures (i.e., less estradiol associated with more neural changes) in girls (Herting et al., 2014; Lenroot et al., 2009; Peper, Brouwer, et al., 2009; Peper, Schnack, et al., 2009) provides indirect support for key aspects of our organizational/activational theory and the candidate systems that may be involved.
Before ending, we should comment on findings regarding estrogen moderation of environmental effects. Similar to previous work, we found evidence for increasing nonshared environmental influences across estradiol levels (Klump, Keel, et al., 2010). The specific factors contributing to these environmental differences remain unclear, but they could reflect non-genomic, membrane-mediated actions of estrogen receptors (Santollo, Yao, Neal-Perry, & Etgen, 2012) that become more prominent as levels of estradiol increase. Importantly, increases in nonshared environmental factors are unlikely to reflect differential environmental experiences for the twins based on age, BMI, or pubertal physical changes, as our analyses controlled for all of these factors. We found minimal differences in shared environmental effects across estradiol levels in the full twin sample or pre-menarcheal twins. This was somewhat surprising given twin studies showing significant differences in shared environmental factors across pubertal development (Klump, Culbert, O’Connor, et al., 2017) and estradiol levels (Klump et al., 2015, 2016; Klump, Keel, et al., 2010). It is possible that we were underpowered to detect differences in these effects, given some evidence for shared environmental influences in the pre-menarcheal twin sample (i.e., more modest MZ/DZ twin correlation differences - see Table 2) and known difficulties of identifying genetic and shared environmental influences in classical twin models (Martin, Eaves, Kearsey, & Davies, 1978). Additional studies with larger sample sizes are needed to clarify whether estrogen moderates shared environmental influences on binge eating during puberty.
We should note a few additional limitations of our work. First, we used a continuous, self-report measure of binge eating rather than interview-based diagnoses and/or symptoms. Although self-reports might over-estimate binge eating (Berg, Peterson, Frazier, & Crow, 2011), the percentage of our twins (3.1%) scoring above the binge eating scale cut-off for individuals with BN (von Ranson et al., 2005) is on par with what would be expected. Moreover, our measure performs well across the age range of pubertal development (Luo et al., 2016), and range of hormone values (see Supplemental Table S1) examined, and it reliably distinguishes between girls who binge eat (see Methods) and/or have BN versus controls (von Ranson et al., 2005). Nonetheless, given discrepant results between current analyses and past pilot study results (Klump et al., 2010), additional replication studies are needed.
Second, we focused on binge eating rather than binge-related disorders. Sample sizes for disorders were too small for analysis, but binge eating appears to be dimensional rather than categorical in nature (Luo et al., 2016), and heritabilities of binge eating and binge-related disorders are similar (e.g., Bulik, Sullivan, & Kendler, 1998). Clearly, more studies of clinical disorders with interview-based measures are needed, but initial data suggest that results might extend to these disorders and measures as well.
Third, we used salivary hormone measures instead of serum. We focused on saliva in order to increase participation rates, ease data collection, and ensure that we were measuring active/unbound hormones (rather than the bound and unbound hormones in serum). Nonetheless, there is less published literature on the reliability/validity of salivary hormone values, and normative data for girls in our age range and during puberty are lacking. Normative data would be helpful for determining if our findings are driven by twins with non-normative/abnormally low levels of estradiol or are present across the full spectrum of estradiol values. Given our findings of linear (rather than non-linear) differences in genetic effects, it seems unlikely that our results are accounted for by girls with abnormally low estradiol levels, but normative data could help confirm these impressions. In addition, more data on reliability/validity of salivary measures could confirm that our findings are not unduly influenced by measurement error, particularly at the lower levels of estradiol. It is important to note, however, that measurement error loads on the nonshared environment in twin models, and thus would result in increased nonshared environmental influences at lower estradiol levels (where presumably, error of measurement would be greater). This pattern of results is opposite to what we observed, where genetic effects were stronger, and nonshared environmental influences were weaker, at lower estradiol levels. This pattern of results was observed when modeling estradiol levels continuously as well as categorically in the full sample of twins and premenarcheal twins only (see Tables 2 and 3, and Figures 2 and S3). Given these results, it seems unlikely that measurement error unduly influenced our results, but additional replications using salivary and serum measures of estradiol are needed to confirm our findings.
Finally, our data were cross-sectional rather than longitudinal in nature and focused on relatively discreet hormone measurements (i.e., 1-3 days of hormone values). It is therefore unclear whether our findings extend to within-subject/sample changes in genetic effects across multiple pubertal hormone assessments. We controlled for many variables that could account for differences in etiologic effects across our hormone levels (i.e., age, BMI, physical changes of puberty), and longitudinal and cross-sectional studies in adulthood (Klump et al., 2015, 2016) have confirmed that estrogen effects exist both within- and between-groups. Nonetheless, longitudinal studies that collect multiple hormone and behavioral measures across pubertal development are needed to confirm that findings reflect changes in estrogen across puberty. Ideally, these studies would capitalize on the existence of multiple animal models of binge eating (Klump, Culbert, & Sisk, 2017) and conduct longitudinal assessments in both girls and female animals to directly test our theories of estrogen’s organizational/activational effects and substantially advance our understanding of ovarian hormone influences on binge eating and binge-related disorders in females.
Supplementary Material
General Scientific Summary.
This study suggests that the activation of estrogen during pubertal development in girls may impact genetic risk for binge eating. Specifically, genetic influences on binge eating become more prominent in girls who have comparatively lower levels of estradiol during this critical period. These lower levels of estradiol may disrupt normative development and lead to increased risk for binge eating in later adolescence and adulthood.
Acknowledgments
This work was supported by grants awarded to KLK, CLS, and SAB by the National Institute of Mental Health (NIMH) (R01 MH092377). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIMH.
Footnotes
The Minnesota Eating Behavior Survey (MEBS; previously known as the Minnesota Eating Disorder Inventory [M-EDI]) was adapted and reproduced by special permission of Psychological Assessment Resources, Inc., 16204 North Florida Avenue, Lutz, FL 33549, from the Eating Disorder Inventory (collectively, EDI and EDI-2) by Garner, Olmstead, and Polivy. (1983) by the Psychological Assessment Resources, Inc. Further reproduction of the MEBS is prohibited without prior permission from Psychological Assessment Resources, Inc.
As an additional check, we also fit a series of two moderator models (i.e., estrogen × menses) examining whether etiologic effects differed in pre- versus post-menarcheal twins. The menses moderator could be constrained to 0 without a significant worsening of model fit (χ2Δ (6) = 8.55, p = .20; see Figure S4 in the Supplemental Material), suggesting that there were no statistically significant differences in etiologic effects between pre-menarcheal and post-menarcheal twins.
References
- Ahmed EI, Zehr JL, Schulz KM, Lorenz BH, Doncarlos LL, Sisk CL. Pubertal hormones modulate the addition of new cells to sexually dimorphic brain regions. Nature Neuroscience. 2008;11:995–997. doi: 10.1038/nn.2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders: DSM-5. Washingon, DC: American Psychiatric Association; 2013. [Google Scholar]
- Angold A, Costello EJ, Erkanli A, Worthman CA. Pubertal changes in hormone levels and depression in girls. Psychological Medicine. 1999;29(5):1043–1053. doi: 10.1017/s0033291799008946. [DOI] [PubMed] [Google Scholar]
- Asarian L, Geary N. Sex differences in the physiology of eating. American Journal of Physiology: Regulatory, Integrative, and Comparative Physiology. 2013;305(11):R1250–R1267. doi: 10.1152/ajpregu.00446.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker LA, Barton M, Raine A. The southern california twin register at the university of southern california. Twin Research and Human Genetics. 2002;5(5):456–459. doi: 10.1375/136905202320906273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth C, Villringer A, Sacher J. Sex hormones affect neurotransmitters and shape the adult female brain during hormonal transition periods. Frontiers in Neuroscience. 2015;9(37):1–20. doi: 10.3389/fnins.2015.00037. http://doi/10.3389/fnins.2015.00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker JB. Sexual differentiation of motivation: A novel mechanism? Hormones and Behavior. 2009;55(5):646–654. doi: 10.1016/j.yhbeh.2009.03.014. https://doi.org/S0018-506X(09)00063-4 [pii] 10.1016/j.yhbeh.2009.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg KC, Peterson CB, Frazier P, Crow SJ. Convergence of scores on the interview and questionnaire versions of the eating disorder examination: A meta-analytic review. Psychological Assessment. 2011;23(3):714–724. doi: 10.1037/a0023246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown LM, Clegg DJ. Estrogen and leptin regulation of endocrinological features of anorexia nervosa. Neuropsychopharmacology. 2012;38(1):237. doi: 10.1038/npp.2012.176. http://doi/10.1038/npp.2012.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulik CM, Sullivan PF, Kendler KS. Heritability of binge-eating and broadly defined bulimia nervosa. Biological Psychiatry. 1998;44(12):1210–1218. doi: 10.1016/s0006-3223(98)00280-7. [DOI] [PubMed] [Google Scholar]
- Burt SA, Klump KL. The Michigan State University Twin Registry (MSUTR): An Update. Twin Research and Human Genetics. 2012;16(1):344–350. doi: 10.1017/thg.2012.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burt SA, Klump KL. Delinquent peer affiliation as an etiological moderator of childhood delinquency. Psychological Medicine. 2013;43(6):1269–78. doi: 10.1017/S0033291712000013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burt SA, Klump KL, Gorman-Smith D, Neiderhiser JM. Neighborhood disadvantage alters the origins of childrens nonaggressive conduct problems. Clinical Psychological Science. 2016;4(3):511–526. doi: 10.1177/2167702615618164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craft RM. Sex differences in analgesic, reinforcing, discriminative, and motoric effects of opioids. Experimental and Clinical Psychopharmacology. 2008;16(5):376–385. doi: 10.1037/a0012931. [DOI] [PubMed] [Google Scholar]
- Clark LA, Watson D. Constructing validity: Basic issues in objective scale development. Psychological Assessment. 1995;7(3):309–319. doi: 10.1037/pas0000626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culbert KM, Burt SA, Klump KL. Expanding the developmental boundaries of etiologic effects: The role of adrenarche in genetic influences on disordered eating in males. Journal of Abnormal Psychology. 2017;126(5):593–606. doi: 10.1037/abn0000226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culbert KM, Burt SA, McGue M, Iacono WG, Klump KL. Puberty and the genetic diathesis of disordered eating attitudes and behaviors. Journal of Abnormal Psychology. 2009;118(4):788–796. doi: 10.1037/a0017207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culbert KM, Burt SA, Sisk CL, Klump KL. The emergence of sex differences in disordered eating attitudes and behaviors during puberty: A role for prenatal testosterone exposure. Journal of Abnormal Psychology. 2013;122(2):420–432. doi: 10.1037/a0031791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culbert KM, Burt SA, Sisk CL, Nigg JT, Klump KL. The effects of circulating testosterone and pubertal maturation on risk for disordered eating symptoms in adolescent males. Psychological Medicine. 2014;44(11):2271–2286. doi: 10.1017/S0033291713003073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Krom M, van der Schouw YT, Hendriks J, Ophoff RA, van Gils CH, Stolk RP, Grobbee DE, Adan R. Common genetic variations in CCK, leptin, and leptin receptor genes are assocaited with specific human eating patterns. Diabetes. 2007;56(1):276–80. doi: 10.2337/db06-0473. [DOI] [PubMed] [Google Scholar]
- Edler C, Lipson SF, Keel PK. Ovarian hormones and binge eating in bulimia nervosa. Psychological Medicine. 2007;37(1):131–141. doi: 10.1017/S0033291706008956. [DOI] [PubMed] [Google Scholar]
- Fairburn CG, Beglin SJ. Assessment of eating disorders: Interview or self-report questionnaire? International Journal of Eating Disorders. 1994;16(4):363–70. [PubMed] [Google Scholar]
- Geary N. Estradiol, CCK, and satiation. Peptides. 2001;22(8):1251–63. doi: 10.1016/s0196-9781(01)00449-1. [DOI] [PubMed] [Google Scholar]
- Grimm ER, Steinle NI. Genetics of eating behavior: Established and emerging concepts. Nutrition Reviews. 2011;69(1):52–60. doi: 10.1111/j.1753-4887.2010.00361.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grumbach MM, Styne DM. Puberty: Ontogeny, neuroendocrinology, physiology, and disorders. In: Wilson JD, Foster DW, Kronenberg HM, Larsen PR, editors. Williams Textbook of Endocrinology. 9th. Philadelphia, PA: W.B. Saunders Company; 1998. pp. 1509–1625. [Google Scholar]
- Harden KP, Kretsch N, Moore SR, Mendle J. Descriptive review: Hormonal influences on risk for eating disorder symptoms during puberty and adolescence. International Journal of Eating Disorders. 2014;47(7):718–726. doi: 10.1002/eat.22317. [DOI] [PubMed] [Google Scholar]
- Hay DA, McStephen M, Levy F, Pearsall-Jones J. Recruitment and attrition in twin register studies of childhood behavior: The example of the australian twin ADHD Project. Twin Research and Human Genetics. 2002;5(5):324–328. doi: 10.1375/136905202320906039. [DOI] [PubMed] [Google Scholar]
- Herting M, Gautam P, Spielberg JM, Kan E, Dahl RE, Sowell ER. The role of testosterone and estradiol in brain volume changes across adolescence: A longitudinal structural MRI study. Human Brain Mapping. 2014;35(11):5633–5645. doi: 10.1002/hbm.22575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrandt T, Alfano L, Tricamo M, Pfaff DW. Conceptualizing the role of estrogens and serotonin in the development and maintenance of bulimia nervosa. Clinical Psychology Review. 2010;30(6):655–668. doi: 10.1016/j.cpr.2010.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klump KL. Puberty as a critical risk period for eating disorders: A review of human and animal studies. Hormones and Behavior. 2013;64(2):399–410. doi: 10.1016/j.yhbeh.2013.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klump KL, Burt SA. The Michigan State University Twin Registry (MSUTR): Genetic, environmental and neurobiological influences on behavior across development. Twin Research and Human Genetics. 2006;9(6):971–977. doi: 10.1375/183242706779462868. [DOI] [PubMed] [Google Scholar]
- Klump KL, Burt SA, McGue M, Iacono WG. Changes in genetic and environmental influences on disordered eating across adolescence: A longitudinal twin study. Archives of General Psychiatry. 2007;64(12):1409–1415. doi: 10.1001/archpsyc.64.12.1409. [DOI] [PubMed] [Google Scholar]
- Klump KL, Burt SA, McGue M, Iacono WG, Wade TM. Age differences in genetic and environmental influences on weight and shape concerns. International Journal of Eating Disorders. 2010;43(8):679–688. doi: 10.1002/eat.20772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klump KL, Culbert KM, O’Connor SM, Fowler N, Burt SA. The significant effects of puberty on the genetic diathesis of binge eating in girls. International Journal of Eating Disorders. 2017;50(8):984–989. doi: 10.1002/eat.22727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klump KL, Culbert KM, Sisk CL. Sex differences in binge eating: Gonadal hormone effects across development. Annual Review of Clinical Psychology. 2017;13:183–207. doi: 10.1146/annurev-clinpsy-032816-045309. [DOI] [PubMed] [Google Scholar]
- Klump KL, Culbert KM, Slane JD, Burt SA, Sisk CL, Nigg JT. The effects of puberty on genetic risk for disordered eating: Evidence for sex difference. Psychological Medicine. 2012;42(3):627–638. doi: 10.1017/S0033291711001541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klump KL, Hildebrandt BA, O’Connor SA, Keel PK, Neale MC, Sisk CL, Burt SA. Changes in genetic risk for emotional eating across the menstrual cycle: A longitudinal study. Psychological Medicine. 2015;45(15):3227–3237. doi: 10.1017/S0033291715001221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klump KL, Holly A, Iacono WG, McGue M, Willson L. Physical similarity and twin resemblance for eating attitudes and behaviors: A test of the equal environments assumption. Behavior Genetics. 2000;30(1):51–58. doi: 10.1023/a:1002038610763. [DOI] [PubMed] [Google Scholar]
- Klump KL, Keel PK, Racine SE, Burt SA, Neale M, Sisk CL, Hu JY. The interactive effects of estrogen and progesterone on changes in emotional eating across the menstrual cycle. Journal of Abnormal Psychology. 2013;122(1):131–137. doi: 10.1037/a0029524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klump KL, Keel PK, Sisk CL, Burt SA. Preliminary evidence that estradiol moderates genetic influences on disordered eating attitudes and behaviors during puberty. Psychological Medicine. 2010;40(10):1745–1753. doi: 10.1017/S0033291709992236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klump KL, McGue M, Iacono WG. Age differences in genetic and environmental influences on eating attitudes and behaviors in female adolescent twins. Journal of Abnormal Psychology. 2000;109(2):239–251. [PubMed] [Google Scholar]
- Klump KL, McGue M, Iacono WG. Differential heritability of eating attitudes and behaviors in prepubertal versus pubertal twins. International Journal of Eating Disorders. 2003;33(3):287–292. doi: 10.1002/eat.10151. [DOI] [PubMed] [Google Scholar]
- Klump KL, O’Connor SAM, Hildebrandt BA, Keel PK, Neale M, Sisk CL, Burt SA. Differential effects of estrogen and progesterone on genetic and environmental risk for emotional eating in women. Clinical Psychological Science. 2016;4(5):895–908. doi: 10.1177/2167702616641637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klump KL, Perkins P, Burt SA, McGue M, Iacono WG. Puberty moderates genetic influences on disordered eating. Psychological Medicine. 2007;37(5):627–634. doi: 10.1017/S0033291707000189. [DOI] [PubMed] [Google Scholar]
- Klump KL, Racine SE, Hildebrandt B, Burt SA, Neale M, Sisk CL, Keel PK. Ovarian hormone influences on dysregulated eating: A comparison of associations in women with versus without binge episodes. Clinical Psychological Science. 2014;2(5):545–559. doi: 10.1177/2167702614521794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klump KL, Sinclair EB, Hildebrandt BA, Serfinski R, Sisk CL. A critical role for puberty and ovarian hormones in the development of binge eating in female rats: A longitudinal study. Paper presented at the Eating Disorder Research Society Meeting; New York, New York. 2016. [Google Scholar]
- Klump KL, Suisman JL, Culbert KM, Kashy DA, Sisk CL. Binge eating proneness emerges during puberty in female rats: A longitudinal study. Journal of Abnormal Psychology. 2011;120(4):948–955. doi: 10.1037/a0023600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenroot RK, Schmitt JE, Ordaz SJ, Wallace GL, Neale MC, Lerch JP, Giedd JN. Differences in genetic and environmental influences on the human cerebral cortex associated with development during childhood and adolescence. Human Brain Mapping. 2009;30(1):163–174. doi: 10.1002/hbm.20494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo X, Donnellan MB, Burt SA, Klump KL. The dimensional nature of eating pathology: Evidence from a direct comparison of categorical, dimensional, and hybrid models. Journal of Abnormal Psychology. 2016;125(5):715–726. doi: 10.1037/abn0000174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lykken DT, Bouchard TJ, McGue M, Tellegen A. The Minnesota Twin Family Registry: Some initial findings. Acta Geneticae Medicae et Gemellologiae. 1990;39(1):35–70. doi: 10.1017/s0001566000005572. [DOI] [PubMed] [Google Scholar]
- Martin MG, Eaves LJ, Kearsey MJ, Davies P. The power of the classical twin study. Heredity. 1978;40(1):97–116. doi: 10.1038/hdy.1978.10. [DOI] [PubMed] [Google Scholar]
- Micali N, Field AE, Treasure JL, Evans DM. Are obesity risk genes associated with binge eating in adolescence? Obesity. 2015;23(8):1729–1736. doi: 10.1002/oby.21147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteleone P, Tortorella A, Castaldo E, Di Filippo C, Maj M. The Leu72Met polymorphism of the ghrelin gene is significantly associated with binge eating disorder. Psychiatric Genetics. 2007;17(1):13–16. doi: 10.1097/YPG.0b013e328010e2c3. [DOI] [PubMed] [Google Scholar]
- Neale MC, Boker SM, Xie G, Maes HM. Mx: Statistical modeling. Richmond, VA: 2003. [Google Scholar]
- Nunez JL, Sodhi J, Juraska JM. Ovarian hormones after postnatal day 20 reduce neuron number in the rat primary visual cortex. Journal of Neurobiology. 2002;52(4):312–321. doi: 10.1002/neu.10092. [DOI] [PubMed] [Google Scholar]
- O’Connor SM, Burt SA, VanHuysse JL, Klump KL. What drives the association between weight-conscious peer groups and disordered eating? Disentangling genetic and environmental selection from pure socialization effects. Journal of Abnormal Psychology. 2016;125(3):356–368. doi: 10.1037/abn0000132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostlund H, Keller E, Hurd YL. Estrogen receptor gene expression in relation to neuropsychiatric disorders. Annals of the New York Academy of Sciences. 2003;1007:54–63. doi: 10.1196/annals.1286.006. [DOI] [PubMed] [Google Scholar]
- Peeters H, Van Gestel S, Vlietinck R, Derom C, Derom R. Validation of telephone zygosity questionnaire in twins of known zygosity. Behavior Genetics. 1998;28(3):159–161. doi: 10.1023/a:1021416112215. [DOI] [PubMed] [Google Scholar]
- Peper JS, Brouwer RM, Schnack HG, van Baal GC, van Leeuwen M, van den Berg SM, Hulshoff Pol HE. Sex steroids and brain structure in pubertal boys and girls. Psychoneuroendocrinology. 2009;34(3):332–342. doi: 10.1016/j.psyneuen.2008.09.012. [DOI] [PubMed] [Google Scholar]
- Peper JS, Schnack HG, Brouwer RM, Van Baal GC, Pjetri E, Szekely E, Hulshoff Pol HE. Heritability of regional and global brain structure at the onset of puberty: A magnetic resonance imaging study in 9-year-old twin pairs. Human Brain Mapping. 2009;30(7):2184–2196. doi: 10.1002/hbm.20660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen AC, Crockett L, Richards M, Boxer A. A self-report measure of pubertal status: Reliability, validity, and initial norms. Journal of Youth and Adolescence. 1988;17(2):117–133. doi: 10.1007/BF01537962. [DOI] [PubMed] [Google Scholar]
- Primus RJ, Kellogg CK. Gonadal status and pubertal age influence the responsiveness of the benzodiazepine/GABA receptor complex to environmental challenge in male rats. Brain Research. 1991;561(2):299–306. doi: 10.1016/0006-8993(91)91608-4. [DOI] [PubMed] [Google Scholar]
- Purcell S. Variance components models for gene-environment interaction in twin analysis. Twin Research. 2002;5(6):554–571. doi: 10.1375/136905202762342026. [DOI] [PubMed] [Google Scholar]
- Saenger P. Does effects of growth hormone during puberty. Hormone Research. 2003;60(Suppl 1):52–57. doi: 10.1159/000071226. [DOI] [PubMed] [Google Scholar]
- Sanchez-Garrido M, Tena-Sempere M. Metabolic control of puberty: Roles of leptins and kisspeptins. Hormones and Behavior. 2013;64(2):187–194. doi: 10.1016/j.yhbeh.2013.01.014. [DOI] [PubMed] [Google Scholar]
- Santollo J, Yao D, Neal-Perry G, Etgen AM. Middle-aged female rats retain sensitivity to the anorexigenic effect of exogenous estradiol. Behavioural Brain Research. 2012;232(1):159–164. doi: 10.1016/j.bbr.2012.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirtcliff EA, Granger DA, Schwartz EB, Curran MJ, Booth A, Overman WH. Assessing estradiol in biobehavioral studies using saliva and blood spots: Simple radioimmunoassay protocols, reliability, and comparative validity. Hormones and Behavior. 2000;38(2):137–147. doi: 10.1006/hbeh.2000.1614. [DOI] [PubMed] [Google Scholar]
- Sisk CL, Zehr JL. Puberty hormones organize the adolescent brain and behavior. Frontiers in Neuroendocrinology. 2005;26(3-4):163–174. doi: 10.1016/j.yfrne.2005.10.003. [DOI] [PubMed] [Google Scholar]
- Spear LP. The adolescent brain and age-related behavioral manifestations. Neuroscience and Biobehavioral Reviews. 2000;24(2):417–63. doi: 10.1016/S0149-7634(00. [DOI] [PubMed] [Google Scholar]
- Tanofsky-Kraff M, Han JC, Anandalingam K, Shomaker LB, Columbo KM, Wolkoff LE, Yanovski JA. The FTO gene rs9939609 obesity-risk allele and loss of control over eating. American Journal of Clinical Nutrition. 2009;90(6):1483–8. doi: 10.3945/ajcn.2009.28439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Sluis S, Posthuma D, Dolan CV. A note on false positives and power in G × E modelling of twin data. Behavior Genetics. 2012;42(1):170–186. doi: 10.1007/s10519-011-9480-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Ranson KM, Klump KL, Iacono WG, McGue M. The Minnesota Eating Behavior Survey: A brief measure of disordered eating attitudes and behaviors. Eating Behaviors. 2005;6(4):373–392. doi: 10.1016/j.eatbeh.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Wilson JD, Foster DW, Kronenberg HM, Larsen PR. Williams Textbook of Endocrinology. 9th. Philadelphia, PA: W.B. Saunders Company; 1998. [Google Scholar]
- Young JK. Anorexia nervosa and estrogen: Current status of the hypothesis. Neuroscience and Biobehavioral Reviews. 2010;34(8):1195–1200. doi: 10.1016/j.neubiorev.2010.01.015. [DOI] [PubMed] [Google Scholar]
- Zehr JL, Todd BJ, Schulz KM, McCarthy MM, Sisk CL. Dendritic pruning of the medial amygdala during pubertal development of the male Syrian hamster. Journal of Neurobiology. 2006;66(6):578–590. doi: 10.1002/neu.20251. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.