

Abstract
Human pluripotent stem cells (hPSCs) represent a formidable tool for disease modeling, drug discovery, and regenerative medicine using human cells and tissues in vitro. Evolving techniques of targeted genome editing, specifically the CRISPR/Cas9 system, allow for the generation of cell lines bearing gene-specific knock-outs, knock-in reporters, and precise mutations. However, there are increasing concerns related to the transfection efficiency, cell viability, and maintenance of pluripotency in genome editing techniques. The presented protocol employs transient antibiotic selection that overcomes reduced transfection efficiency, avoids cytotoxic flow sorting for increased viability, and generates multiple genome-edited pluripotent hPSC lines expanded from a single parent cell. Avoidance of xenogeneic contamination from feeder cells and reduced operator workload, owing to single cell passaging rather than clump passaging, are additional benefits. The outlined methods may enable researchers of limited means and technical experience to create human stem cell lines containing desired gene-specific mutations.
Keywords: Stem cell, CRISPR/Cas9, genome editing, disease modeling
Introduction
By virtue of two intrinsic properties, pluripotency and self-renewal, human pluripotent stem cells (hPSCs) theoretically have the ability to generate unlimited quantities of organ-specific human tissue for use in developmental studies, toxicity testing, disease modeling, and regenerative medicine. To date, hPSCs have proven useful in the generation of human heart, liver, kidney, pancreas, stomach, lung, retina, intestines, and brain tissues (Witty et al. 2014; Takebe et al., 2013; Morizane et al., 2015; Yamaguchi et al., 2016; Morizane et al. 2017; Rezania et al., 2014; Dye et al.; 2015; Zhong et al., 2014; Watson et al., 2014; Lancaster et al., 2013). The two cell types considered hPSCs are embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs). While ESCs are derived from isolation of cells from the inner cell mass of the blastocyst (Thomson et al., 1998), iPSCs allowed for the generation of pluripotent cells through transcriptional reprogramming of terminal cells from adult subjects (Takahashi and Yamanaka, 2006). The newfound ability to create patient-specific human tissue in vitro has enabled genetic disease models where no faithful model previously existed.
Coupling the advances in stem cell biology with genome-editing techniques has enabled researchers to generate knock-out, knock-in, and reporter hPSC lines. Such hPSC lines that bear targeted genomic manipulations serve as useful tools in the fabrication of organ-specific human tissue, as well as understanding the inheritable and acquired pathophysiology in such tissue. Given the variable penetrance of many monogenetic diseases (MacArthur et al., 2014), it follows that disease-harboring iPSCs have demonstrated a greater utility for human disease modelling than genetically engineered mutant hPSCs (Sterneckert et al., 2014). Correction of the naturally occurring mutation in patient-derived iPSC lines can serve as an isogenic control, thus correlating the genetic defect with any observed phenotypic variation.
The basis of genome-editing techniques involves the generation of site-specific double-strand breaks (DSBs) by nucleases, such as zinc finger nuclease (ZFN), transcription activator-like effector nuclease (TALENs), and Cas9 nuclease (Urnov et al., 2010; Miller et al., 2011; Ran et al., 2013). DSBs activate cellular DNA repair pathways to fix the defect via non-homologous end-joining (NHEJ) or homologous recombination (HR) (Johnson et al., 1999). The process of NHEJ involves blunt end ligation of DSB ends in an error-prone fashion, often generating small insertions or deletions (indels) (Lieber, 2010). Indels have been associated with frameshift mutations and premature stop codons (Perez et al., 2008), thereby generating gene-specific knock-outs. Meanwhile, HR faithfully maintains genome integrity through the presence of a DNA template homologous to the region surrounding the DSB and can be used to introduce point mutations or additional DNA fragments (e.g. GFP) using constructs that include surrounding sequence homology. NHEJ predominates in the G1 phase of the cell cycle whereas HR predominates in the G2/M phases (Chapman et al., 2012), suggesting the utility of the sister chromatid to serve as template for HR.
Although each nuclease has been implemented in genome-editing of hPSCs, Cas9 has gained traction due to its ease of use (Gaj et al., 2013). Adapting a humoral immunity method of prokaryotes, the clustered regularly-interspaced short palindromic repeat (CRISPR)/Cas9 system can generate site-specific DNA breaks. A CRISPR synthetic guide RNA (sgRNA) consists of a CRISPR RNA (crRNA) fused to a transactivating RNA (tracrRNA). The crRNA contains a variable 20 base pair protospacer, which determines DNA-binding specificity, linked to additional nucleotides complementary to the constant tracrRNA. The tracrRNA facilitates the association of Cas9 nuclease with the crRNA/tracrRNA complex. When the protospacer binds a complementary DNA sequence that is followed by a 3 nucleotide downstream protospacer adjacent motif (PAM), Cas9 cleaves the DNA three base pairs upstream of the PAM sequence. The most commonly used Cas9 is from Streptococcus Pyogenes and has a PAM sequence of 5′-NGG-3′.
The facility of the CRISPR/Cas9 system stems from the ease of sgRNA design and the efficiency of site-specific DSB creation. The variable 20 base pair protospacer can be designed complementary to any unique sequence in the targeted gene, provided it is immediately upstream of a PAM sequence. Manipulations of the PAM sequence required by Cas9 has expanded the possible target sites for DSB creation (Kleinstiver et al., 2015).
Concerns exist regarding off-target cleavage using the CRISPR/Cas9 system due to conflicting reports of occurrence (Veres et al., 2014; Wang et al., 2015). Certain methodologies of the CRISPR/Cas9 system serve to limit such off-target DSBs. One such method includes the development of Cas9 nickase (Ran et al., 2013), which introduces single stranded breaks (SSBs) when the protospacer binds a complementary DNA sequence. The combination of two distinct sgRNAs, one for each opposing DNA strand, generates a targeted DSB. As SSBs are repaired in a genome preserving fashion, off-target genome modifications may be reduced. Interestingly, reducing the length of the protospacer to 17 base pairs serves as an alternative method to increase site-specific genome-editing (Fu et al., 2014). Regardless of the approach, it is important to reduce the chance of off-target indels and, where possible, determine the impact. This may be done using Next Gen Sequencing (NGS) of the modified cell genome and comparing to the parental line, though this may be cost prohibitive. We have adopted the method of comparing several resulting modified clones (3+). Since off-target events occur at a lower efficiency and inconsistently between clones, we expect the likelihood that an off-target mutation would identically impact our experiments in all clones to be minimal.
The transfection efficiency of different Cas9 and sgRNA plasmids is variable, necessitating methods for the selection of successfully transfected cells (Modeno-Mateos et al., 2015). Many previously published protocols involving CRISPR/Cas9 genome-editing in hPSCs have relied on flow sorting to select for transfected cells (Hendriks et al., 2015; Santos et al., 2016). The commercial availability of plasmids, which may incorporate variable protospacers, that contain both Cas9 and an antibiotic resistance gene allow for antibiotic selection of transfected cells (Ran et al., 2013). Here we present a basic protocol that employs an optimized antibiotic selection method for the generation of multiple clones of genome-edited cell lines. Each hPSC line is derived from a single parent cell, with theoretically isogenic progeny, made possible using a feeder-free culture system that allows for single cell plating (Nakagawa et al., 2014). The outlined methods may reduce the off-target variability of previous protocols that have employed FACS or colony passaging. Deep sequencing of individual clones is used to identify hPSC lines that contain the desired targeted mutation and the status at both allelic locations.
Basic Protocol 1: CRISPR/Cas9-based targeted genome editing in human pluripotent stem cells (hPSCs)
Subprotocol #1: sgRNA design
Introductory Paragraph
The most critical step in CRISPR/Cas9 mediated genome editing is guide RNA design. The site specificity for the generation of DSBs is determined by the number of sites, adjacent to a PAM sequence, that the protospacer binds through complementary base pairing. However, off-target effects may occur at sites without significant sequence homology (i.e. tolerated mismatches). Additionally, the efficiency of site specific DNA breaks may vary considerably between nearby gRNA target sites within the same gene. For these reasons, we recommend attempting gene-editing with three distinct gRNAs for each gene of interest. An alternative approach to increase site specificity, while reducing efficiency, would be to utilize Cas9n (“nickase”). The instructions include modifications that enable the ligation of the designed gRNA into available plasmids from repositories that contain either spCas9 or spCas9n. Following identical steps, gRNAs can be designed for incorporation into the PX462 plasmid that contains spCas9n and the puromycin resistance gene. Two gRNAs would have to be designed in nearby locations on complementary DNA strands, thereby generating a DSB from two opposing SSBs.
Various gRNA design tools allow users to input genomic DNA sequences and report the specificity of all possible target sequences for Cas9 nuclease (see Table 1, Wiki). gRNA design tools have evolved to include multiple species and differing PAM sequences. Some of those tools such as CRISPRdirect also provide the prediction of off-targets sites. We have had success using CRISPRdirect (Naito et al., 2015).
Table 1.
CRISPR/Cas9 sgRNA Design Tools
| Tool Name | Provider | Searches whole genome for targets | Returns all targets of genome | Seed span and location can be defined | Maximum number of mismatches supported | Predicts gRNA activity | Available PAM sequences | gRNA suggestion or scoring |
|---|---|---|---|---|---|---|---|---|
| Benchling CRISPR gRNA Design | Benchling | Yes | Yes | Yes | 4 | Yes | User customizable | Yes |
| Breaking-Cas | Spanish National Center for Biotechnology | Yes | Yes | Yes | 4 | No | User customizable | Yes |
| Cas-OFFinder | Seoul National University | Yes | Yes | No | 0–10 | No | NGG, NRG, NNAGAAW, NNNNGMTT | Yes |
| CCTop | University of Heidelberg | Yes | Yes | Partial | 5 (0–5) | No | NGG, NRG, NNGRRT, NNNNGATT, NNAGAAW, NAAAAC | Yes |
| CHOPCHOP | Harvard University | Yes | Yes | Partial | 0, 2 | No | NGG, NNAGAA, NNNNGANN | Yes |
| CHOPCHOP v2 | University of Bergen | Yes | Yes | Yes | 3 (0–3) | Yes | User customizable | Yes |
| COD | Dayong Guo | No | No | No | 0, 3, 5, 8 | No | NGG and NAG | Yes |
| CRISPR Configurator & Specificity Tool | Dharmacon, Inc. | Yes (over 30 species) | Yes | Yes | 8 (gaps or mismatches) | Internally | NGG and NAG | No |
| CRISPR Design | Zhang Lab, MIT | Yes | No | No | 4 | No | NGG and NAG | Yes |
| CRISPRdirect | Database Center for Life Science (DBCLS) | Yes (over 200 species) | Yes | No | Any number | No | NNN | Yes |
| CRISPR gRNA Design Tool | DNA2.0 | Yes | Yes | No | 0–10 | No | NGG, NAG | Yes |
| CRISPRseek | Bioconductor | Yes | Yes | No | Any number | No | User customizable | Yes |
| DESKGEN | Desktop Genetics | Yes | Yes | Yes | Any number | Yes | Fully user customizable | Yes |
| Off-Spotter | Thomas Jefferson University | Yes | Yes | Yes | 0–5 | Yes | NGG, NAG, NNNNACA, NNGRRT (R is A or G) | User customizable |
| sgRNA Designer | Broad Institute | No | No | No | 0 | Yes | NGG | Yes |
Materials
No material required
Protocol steps
-
Obtain the human genomic DNA sequence of the desired gene to be modified from one of the online repositories (e.g. genome.ucsc.edu) and import into a bioinformatics program of choice (see Subprotocol 8 for an example).
Be certain the DNA sequence used is correct and ideally includes annotations (e.g. each exon and the coding region) provided by the online genome databases. -
Identify the 5′ coding sequence (~200 base pairs) of your gene of interest for generation of knock-out lines.
Targeting the translated region near the N-terminus causes an early frameshift mutation that increases the likelihood of generating a non-functional protein by introducing a premature stop codon that truncates the protein. For generation of reporter lines, we are targeting the reporter to the C-terminus of the endogenous protein. Therefore, we identify the STOP codon and select approximately 200 bp around that site for gRNA searches (see subprotocol 8). -
Copy the identified 5′ coding sequence into CRISPRdirect, maintaining the PAM sequence setting as ‘NGG’ and the species setting as ‘human’, and click ‘design’. Other CRISPR/Cas9 sgRNA design tools may be employed as an alternative to CRISPRdirect (see Table 1).
CRISPRdirect searches for potential target sites on both the entered sequence and the reverse complement of that sequence, corresponding to the complement DNA strand. -
Choose the 20 base pair target sequence with the fewest number of off-target sites, corresponding to the gene of interest’s most unique genomic sequence immediately upstream of a PAM sequence. Target sites containing 40–80% GC content are preferred. (Figure 1)
Higher GC content increases RNA:DNA duplex stability, while destabilizing off-target hybridization. Ideally, there will be a single target site for the ‘12mer + PAM’ and as few as possible target sites for the ‘8mer + PAM’. -
Write out the desired 20 base pair target sequence in 5′ to 3′ format. Transcribe the complementary strand.
Note that the ‘boxed’ PAM sequence is not part of the 20 base pair target sequence. -
Add a ‘CACCG’ to the 5′ end of your target sequence. Add an ‘AAAC’ to the 5′ end of the complementary strand. Lastly, add a ‘C’ on the 3′ end of the complementary strand. The result is your guide RNA sequence. [see Figure 2].
The required modifications allow for target sequence ligation into Bbs1-digested, spCas9-bearing plasmids, including PX459 (Addgene, Catalog #62988). -
Order the guide RNA sequence as custom duplexed DNA oligos. On receipt, briefly centrifuge the tubes to ensure that dried DNA is brought down to the bottom of the tube. Then resuspend duplexed DNA oligos to a stock concentration of 100μM using nuclease-free water.
Reconstituted 100 μM stock DNA oligos are stable at 4°C for at least 6 months and −20°C for at least 24 months.
Figure 1. gRNA selection from search output.
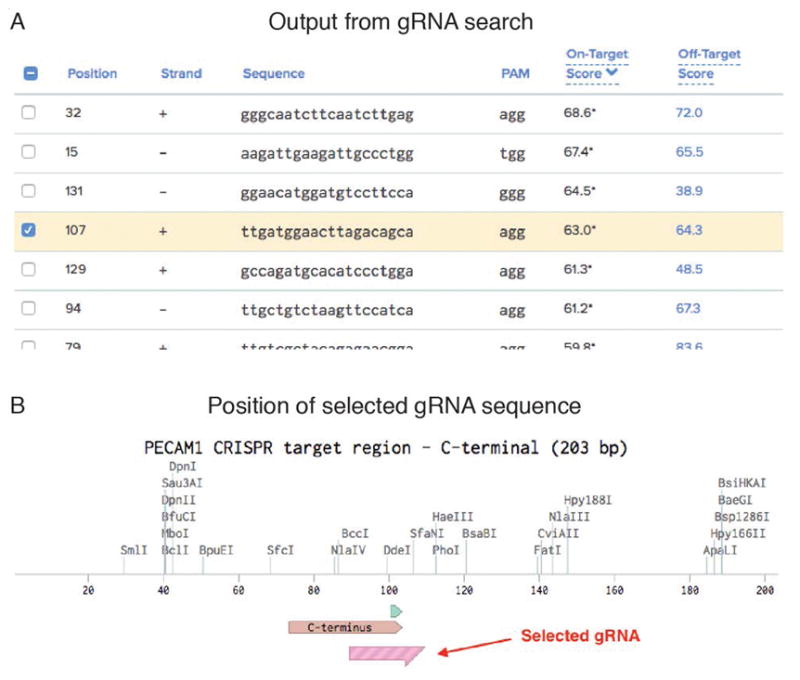
A) The tools listed in Table 1 output potential gRNA sequences ranked by various scoring criteria (see individual tools for details). From a list of 20 potential sites found in the 203 bp input sequence, we selected the fourth highest because it also fits our preferred location near the STOP codon. B) Map showing the relative location of the C-terminus, the STOP codon (green), and the selected gRNA sequence. The PAM site associated with the gRNA is 9 bp downstream of the STOP codon.
Figure 2. sgRNA design.

A target sequence is reported as a 20mer + PAM by CRISPRdirect. The presence of a PAM sequence in genomic DNA is necessary for Cas9 nuclease activity, but must be removed in sgRNA design. The 20 base pair protospacer is the variable region of an sgRNA responsible for complementary base pairing with genomic DNA. The reverse complement of the protospacer is required for incorporation into Cas9-bearing plasmids. Specific end modifications are required for the ligation of the designed sgRNA into Bbs1-digested PX459/462, linking the protospacer element with the constant sgRNA scaffold within the plasmid.
Subprotocol #2: sgRNA expression plasmid generation
Introductory Paragraph
Successful CRISPR/Cas9 genome editing requires the cellular delivery of active Cas9 with an sgRNA targeting sequence linked to an sgRNA scaffold. There are commercially available plasmids that encode Streptococcus Pyogenes Cas9 (spCas9) or Cas9n (spCas9n), contain an sgRNA scaffold, and additionally carry an antibiotic resistance gene. The PX459 plasmid, available from Addgene (Ran et al., 2013), bears spCas9 and allows for integration of a designed protospacer linked to an sgRNA scaffold under Bbs1 digestion. The PX459 plasmid contains the puromycin resistance gene, thus the low transfection efficiency of hPSCs can be overcome using puromycin selection for transfected cells [see Figure 3].
Figure 3. sgRNA expression plasmid generation.

Following digestion with Bbs1 restriction endonuclease, the sticky ends of PX459 plasmid complementary base pair with the designed sgRNA. The target specific protospacer of the sgRNA is ligated directly to an sgRNA scaffold, which complexes with the constitutively expressed spCas9 under a chicken β-actin promoter.
Materials
Synthesized DNA Oligos
PX459 (pSpCas9(BB)-2A-Puro V2.0) Plasmid, Addgene, Catalog #62988
FastDigest Bpil (Bbs1), Thermo Fisher Scientific, Catalog #FD1014
QIAquick gel extraction kit, Qiagen, Catalog #28704
Nuclease-free water, Thermo Fisher Scientific, Catalog #AM9932
Quickligation kit, New England Biolabs, Catalog #M2200S
Plasmid-Safe ATP-dependent DNase, Epicentre, Catalog #E3101K
Protocol steps
-
Digest PX459 plasmid with Bbs1, by combining 1 μg of plasmid, 1 μl of FastDigest Bbs1, 1 μl of FastAP, 2 μl of 10X FastDigest buffer, and bring the total reaction volume up to 20 μl using nuclease free water. Incubate overnight at 37°C to ensure complete digestion.
A number of cited plasmids that contain a form of Cas9 are commercially available from Addgene for use in mammalian cells (https://www.addgene.org/crispr/mammalian/). Briefly, PX458 bears Cas9 + GFP, PX459 bears Cas9 + puromycin resistance, PX460 bears Cas9 nickase, PX461 bears Cas9 nickase + GFP, and PX462 bears Cas9 nickase + puromycin resistance. Notably, Cas9 nickase generates single-stranded DNA breaks, and thus requires two complementary sgRNAs to generate a double-stranded break. Although Cas9 nickase suffers from poor efficiency, it may significantly reduce off-target effects. -
Gel purify Bbs1 digested PX459 plasmid with QIAquick Gel Extraction Kit, eluting in the supplied elution buffer.
Note that BbsI cuts outside its recognition sequence and the design of the cloning site in these plasmids yields incompatible overhangs that prevent self-ligation. Thus, removal of the 5′ phosphate from the plasmid vector is unnecessary, and likewise the phosphorylation of the synthetic duplexed oligos is unnecessary. Carefully consider the needs of each plasmid in this regard when planning other experiments. -
Ligate the Bbs1-digested plasmid with duplexed oligos. Combine 50 ng of Bbs1 digested plasmid with 1 μl of 500nM phosphorylated oligo duplexes, 5 μl of 2X Quickligation buffer, and then take the reaction volume up to 10 μl using nuclease-free water. Finally, add 1 μl of Quick ligase and incubate at room temperature for 10 min.
We resuspend the duplexed oligos at 100μM, then dilute 1:200 in nuclease free water to a concentration of 500nM to approach a 1:1 molar ratio of Bbs1 digested vector and duplex oligos in the ligation reaction. Also include a vector-only ligation control consisting of the Bbs1-digested plasmid without any added duplexed oligos. -
Subject ligation product from step 3 with PlasmidSafe exonuclease. Combine the 11 μl of ligation product with 1.5 μl of 10X Plasmid-Safe ATP-dependent DNase buffer, 1.5 μl of 10mM ATP, and 1 μl of nuclease-free water. Incubate at 37°C for 30 min.
This step removes unwanted unligated DNA from the ligated plasmid. PlasmidSafe DNase has a high selectivity for digesting linear double-stranded DNA, so that the ligated plasmid can be purified for subsequent protocols. Samples can be stored at −20 °C for at least 1 week following PlasmidSafe treatment.
Subprotocol #3: Plasmid amplification and purification
Introductory Paragraph
The generated plasmid is next introduced into competent E. coli. using standard bacterial transformation and plated on agar plates with the appropriate antibiotic for selection. Well separated single colonies are picked for miniprep cultures and screening of correct cloning events. For good screening, it is required to confirm much more colonies can be seen on the ligation product plate than on the vector-only ligation control plate. Amplified plasmid DNA in E. coli. needs to be purified for transfection to stem cells. Since human stem cells are sensitive to endotoxin derived from E. coli. (Lieder et al., 2013), it is important to use a DNA purification kit that removes endotoxin (Stadler et al., 2004).
Materials
ice
competent cells (e.g. Alpha-Select Gold Efficiency Competent Cells, BIOLINE, Catalog #BIO85027)
antibiotic-containing agar plates (e.g. LB-Ampicillin)
antibiotic-containing liquid media
50 mL conical tube
autoclaved 300 ml Erlenmeyer flask
microcentrifuge tube
plasmid DNA miniprep kits (e.g. QIAGEN Plasmid Mini Kit, QIAGEN, Catalog #12123)
endotoxin-free plasmid DNA large-scale purification kits (e.g. QIAGEN EndoFree Plasmid Maxi Kit, Catalog #12362)
isopropanol
70% ethanol
TE buffer (10 mM Tris, 1mM EDTA. pH 8.0)
Protocol steps
The following transformation protocol is a typical method using frozen chemically competent cells from various manufacturers. For a method to make competent cells see the reference (Green and Sambrook, 2012).
Warm the appropriate antibiotic-containing agar plates (e.g. LB-Ampicillin [100 μg/ml] for PX459) at 37°C.
Thaw 50 μl of competent cells on ice
Add 1μl of ligation product (~10 ng vector) for each ligation and the vector-only ligation control.
Mix by gently flicking the tube 3–4 times.
Incubate on ice for 30 min.
Place the tubes in a 42°C water bath for 30 seconds to heat shock the cells.
Place back on ice for 2 min.
Add 950 μl S.O.C. media and grow for 1 hour at 37°C with shaking.
-
Plate the competent cells onto the warmed plates and distribute evenly with a bacterial spreader.
We recommend plating with 10 μl of the cell mixture and 50 μl of cell mixture to ensure well-spaced colonies. Immediately incubate plate at 37°C overnight.
On the next day, observe growth of transformed colonies. If the screening is successful, more than 10 times as many colonies will appear on the ligation product plate compared with the vector-only ligation control plate.
Pick some colonies from the ligation product plate with a clean tip and inoculate it into 15 ml of selection liquid media (e.g. LB-Ampicillin [100 μg/ml] liquid media).
Incubate at 37°C in a shaking incubator at 200–300 rpm overnight.
On the next day, make glycerol stocks from the culture. Move 300 μl of media containing E-coli into a 2 ml cryotube, then add 200 μl of 50 % glycerol and store them at −80C.
Extract plasmid DNA from the culture using standard miniprep method. Various commercial kits are available. Follow the manufacturer’s protocol. Dissolve the obtained plasmid DNA with TE buffer.
Measure the concentration of plasmid DNA. Store at −20°C. Plasmid DNA is stable for many years at −20°C.
Survey Sanger sequences to confirm that these plasmid DNA are carrying the objective gRNA.
After the confirmation, pick the glycerol stock in which the objective gRNA is contained with a clean tip and inoculate it into 100 ml of selection liquid media in an autoclaved 300 ml Erlenmeyer flask.
Incubate at 37°C in a shaking incubator at 200–300 rpm overnight.
On the next day, extract plasmid DNA from the culture using the endotoxin-free large scale plasmid purification method. Various commercial kits are available. Follow the protocols attached with the kit. Dissolve the obtained plasmid DNA with TE buffer.
Measure the concentration of plasmid DNA. Store at −20°C. Plasmid DNA is stable for many years at −20°C.
Support Protocol #1: Coating plates with Geltrex
Introductory Paragraph
Geltrex provides a basement membrane matrix for the culture of hPSCs under feeder free conditions.
Materials
LDEV-free, hESC-qualified Geltrex, Thermo Fisher Scientific, Catalog #A1413302
DMEM/F12, Life Technologies, Catalog #11320033
6-well tissue culture plates, Falcon, Catalog #353046
Protocol Steps
-
Thaw Geltrex stock solution at 2–8°C overnight and place on ice. Mix thoroughly to ensure a homogenous solution. Make 1 ml aliquots from the stock solution. Store the working solution at 2 to 8°C and unused aliquots at −20 to −80°C. Geltrex is stable for 18 months from the date of manufacture at −20°C.
Make aliquots quickly, as Geltrex undergoes liquid to solid transition within 5–10min above 15°C. It is unnecessary to prechill pipette tips. Add LDEV-Free hESC-qualified Geltrex to cold DMEM/F12 at a 1:100 dilution ratio. Add 1 ml total volume per single well of a 6-well plate, or 60 μl per single well of a 96-well plate.
Incubate plates at 37°C for a minimum of 1 hour.
Parafilm the edges of plates and store in a 4°C refrigerator for up to one week prior to use.
Subprotocol #4: hPSC culture technique
Introductory Paragraph
Numerous culture methods have been employed for the culture of hPSCs since their inception more than two decades ago (Thomson et al., 1998). Variations in stem cell media and culture technique have yielded feeder-free culture systems, as opposed to on-feeder systems, and single cell passaging, as opposed to clump passaging (Xu et al., 2001; Nakagawa et al., 2014). The benefits of such advances include a reduction of xenogeneic contamination with mouse embryonic fibroblasts, decreased risk of pathogenicity, increased use of defined factors (Villa-Diaz et al., 2013), a reduction in operator workload, and the ability to reconstitute an unlimited number of isogenic cells from a single parent cell. Although differing hPSC culture methods permit successful genome-editing (Freedman et al., 2015; Morizane et al., 2017; Oldershaw et al,. 2010), common concerns exist between them regarding the potential for karyotypic abnormalities and the maintenance of pluripotency (Taapken et al., 2011). The hPSC media, StemFit Basic02 (SF02), has proven faithful for hPSC maintenance without chromosomal aberration (Nakagawa et al., 2014). Compared with other feeder-free systems, SF02 has added benefits of single cell passaging, twice weekly feeding, and a low passage ratio of 1:200–300 that allows for once weekly passaging. SF02 supplemented with bFGF 10 ng/ml on a 1% Geltrex coating has enabled successful generation of gene-specific, compound heterozygotic knock-out mutants in our hands.
The outlined hPSC culture techniques were designed to allow researchers with limited access to equipment and of limited experience in the CRISPR/Cas9 system to perform targeted genome editing in hPSCs. However, a significant background in basic tissue culture is required and a strict maintenance to proper sterile technique presumed.
Materials
Desired hPSC line
StemFit Basic02 (SF02), Ajinomoto, Catalog #ASB01
Recombinant Human FGF-basic (bFGF), Peprotech, Catalog #100-18B
10mgY27632 (ROCK inhibitor), TOCRIS, Catalog #1254
Accutase, STEMCELL Technologies, Catalog #07920
1X PBS, Thermo Fisher Scientific, Catalog #10010049
15 ml Falcon tubes
Equipment
37°C Waterbath
37°C Incubator
4°C refrigerator
−80°C refrigerator
Liquid nitrogen tank
Centrifuge
Brightfield microscope
Cell counter/hemocytometer
Protocol steps
Pre-warm Geltrex-coated plates for a minimum of 30 min at room temperature within a laminar flow culture hood.
-
Warm desired amount of StemFit Basic02 (SF02) supplemented with bFGF 10 ng/ml to 37°C.
Include 5 ml for diluting the DMSO in the cryovial, plus 1.4 ml per well of a 6-well plate, and 0.2 ml for resuspension of the cell pellet. Thaw cryovial containing 200 thousand hPSCs (see Freezing hPSCs in cryovials to follow) in 37°C water bath until a small portion is melted to liquid (30–60 seconds). Immediately add 1 ml of SF02 supplemented with bFGF 10 ng/ml and transfer to a 15 ml falcon tube containing 4 ml of SF02.
-
Centrifuge the thawed cell suspension for 4 min at 300 × g and resuspend the cell pellet in 200 μl of SF02 supplemented with bFGF 10ng/ml and 10 μM Y27632.
Reconstitute 10 mg of Y27632 at a stock concentration of 10 mM by resuspending in 3.0792 ml of 1X PBS. Dilute stock 10 mM Y27632 by 1:1000 to a working concentration of 10 μM. -
Aspirate Geltrex coating solution and add 1.4 ml of SF02 supplemented with bFGF 10ng/ml and 10 μM Y27632 per well to 2 wells of a 6-well plate.
We usually use bFGF 10 ng/ml; however, some lines require higher concentrations of bFGF up to 100 ng/ml to maintain their pluripotency. If you see a substantial number of differentiated cells, test higher doses of bFGF for maintenance of pluripotency. Plate 100 μl of the cell suspension (i.e. 100 thousand cells) per well and swirl the plate to mix evenly.
Incubate at 37°C and 5% CO2.
-
One day following cell plating, change media with 1.5 ml of SF02 supplemented with bFGF 10ng/ml.
Removal of Y27632. -
Visualize hPSC culture daily, monitoring for confluency and cell/colony morphology
hPSCs may spontaneously differentiate in colonies which grow too large. Differentiating cells take on a fibroblast-like appearance. Four days after plating, change media to feed hPSCs with 1.5 ml SF02 supplemented with bFGF2 10ng/ml.
Six days after plating, change media to feed with 3 ml of similar media.
Prepare the necessary Geltrex-coated plates, as outlined above.
Aspirate the Geltrex coating solution. Add 1.5 ml of SF02 supplemented with bFGF 10 ng/ml and 10 μM Y27632 per Geltrex-coated well of a 6-well plate.
-
Once hPSCs reach ~70% confluency, aspirate media, wash cells with 2 ml of 1X PBS, and add 500 μl of Accutase per well of a 6-well plate.
hPSCs typically reach ~70% confluence 7-8 days after initial thawing. Incubate at 37°C for 10 min.
-
Check for cell dissociation by gently rocking back and forth. Alternatively, confirm cells are non-adherent under brightfield microscopy.
If hPSCs remain adherent, re-incubate for one additional minute and recheck for dissociation. Repeat as necessary. Using a P1000 pipette set to more than 500 μl, manually dissociate the cells to a homogenous mixture of single cells.
Add 500 μl of the cell suspension to 500 μl of pre-warmed SF02 and mix well.
Perform a cell count on 20 μl of the cell suspension and centrifuge the remaining volume for 4 min at 300 × g.
Resuspend the cell pellet to a concentration of 10,000 cells per 1 μl in SF02 supplemented with bFGF 10ng/ml.
-
Plate 12 thousand cells (i.e. 1.2 μl) per Geltrex-coated well from step (18) and incubate at 37°C.
The plating density should be titrated between 10–25 thousand cells to yield ~3–4 million cells with once weekly passaging.
Support Protocol 2: Freezing hPSCs in cryovials
Introductory Paragraph
Freezing hPSCs is essential to establishing frozen stocks of cell lines, both the parental control line and each of the genome-edited lines generated from following the outlined protocol.
Materials
1–2ml cryovials
StemFit Basic02 (SF02), Ajinomoto, Catalog #ASB01
Recombinant Human FGF-basic (bFGF), Peprotech, Catalog #100-18B
DMSO, Sigma-Aldrich, Catalog #2650
Label the desired number of sterile screw-top 1–2 ml cryovials with the cell line, passage number, cell number (200 thousand in our protocol), tube number, date, and initials.
Under sterile conditions, fill each cryovial with 160 μl of cold SF02 supplemented with bFGF 10 ng/ml.
Add 20 μl of DMSO to each cryovial
From Subprotocol #4, hPSC culture technique, step (20), add 20 μl of cell suspension (10,000 cells per 1 μl) to each cryovial.
-
Place cryovials in storage boxes at −80°C for overnight, then transfer to a liquid nitrogen tank for long term storage. Cryovials of frozen stock hPSCs are stable for >5 years when stored in liquid nitrogen.
We usually sandwich cryovials with two styrofoam tube stands of 15 ml Falcon tubes to gradually lower the temperature of cryovials.
Subprotocol #5: Transfection and antibiotic selection
Introductory Paragraph
The amplified plasmids carrying Cas9 and gRNA need to be transfected into hPSCs. Transfection of nucleic acid is one technique to modify gene expression in cells, which is also used to engineer stem cells. Human pluripotent stem cells (hPSCs) may be difficult to transfect, as stem cell transfection depends on many conditions such as cell type, species, and molecule being delivered (Sharifi Tabar et al., 2015). Lipid-mediated delivery, which we describe here, has been the most common method. Lipid-mediated delivery often results in very low transfection efficiency into hPSCs; however, our subsequent protocols of puromycin selection and single cell expansion will allow for sufficient number of mutant lines without using viral vectors or latest electroporation systems such as 4D-nucleofector [see Figure 4].
Figure 4. Schematic representation of transfection, selection, and proliferation of transfected hPSCs.

PX459 plasmids carrying sgRNA are transfected to proliferating hPSCs which are 70–80% confluency. Transfected hPSCs are selected by appropriate concentration of antibiotics for which the plasmids contain antibiotic resistance genes. After nearly 99% of cells are removed by selection, new colonies will be formed, which are composed of successfully transfected hPSCs.
Materials
6-well tissue culture plates, Falcon, Catalog #353046
DMEM/F12, Life Technologies, Catalog #11320033
LDEV-free, hESC-qualified Geltrex, Thermo Fisher Scientific, Catalog #A1413302
10mg Y27632 (ROCK inhibitor), TOCRIS, Catalog #1254
Recombinant Human FGF-basic (bFGF), Peprotech, Catalog #100-18B
StemFit Basic02 (SF02), Ajinomoto, Catalog #ASB01
Opti-MEM, Thermo Fisher Scientific, Catalog #31985062
Lipofectamine 3000 Transfection Reagent, Thermo Fisher Scientific, Catalog #L3000008
puromycin, Santa Cruz, Catalog #sc-108071
Protocol steps
-
Culture hPSCs on 6-well plates coated with hESC-qualified Geltrex in SF02 supplemented with 10 ng/ml FGF2 until they reach 70–80% confluency. Prepare at least two wells of hPSCs because a negative control without plasmid transfection is required to determine whether transient puromycin selection is successful.
Although the timeline is shown as day of the week, it is possible to change for your convenience. We demonstrate the case of H9 hESCs (WiCell Research Institute). We plate 80–100 thousand cells per well of 6-well plates on Friday, and culture them in SF02 supplemented with 10 μM Y27632 and 10 ng/ml FGF2 until Monday without changing media. On Monday, the cells will reach 70–80% confluency. Since the growth rate of cells varies between hPSC lines, the plating number will vary between different hPSCs. However, the plating density is between 70 and 120 thousand per well of 6-well plates for all hESC and hiPSC lines tested in our lab. -
Prior to transfection, replace the old media and pre-treat the hPSCs cultures with ROCK inhibitor (10 μM Y27632) in SF02 supplemented with 10 ng/ml FGF2 for 2–6 hr.
If the cells have already been cultured with ROCK inhibitor, media replacement can be performed just before transfection. Also in this case, use fresh SF02 supplemented with 10 μM Y27632 and 10 ng/ml FGF2. Dilute 7.5 μl of Lipofectamine 3000 Reagent in 125 μl of Opti-MEM Medium, and mix well. Prepare two tubes of the diluted Lipofectamine 3000 Reagent.
Prepare master mix of plasmid DNA by diluting 2.5 μg of DNA in 125 μl of OptiMEM Medium. Add 5 μl of P3000 Reagent, and mix well. For a negative control, prepare a mixture of 125 μl of OptiMEM Medium and P3000 Reagent without plasmid DNA.
Add the master mix of plasmid DNA to the tube of diluted Lipofectamine 3000 Reagent (1:1 ratio). For a negative control, add diluted P3000 reagent without DNA to the tube of diluted Lipofectamine 3000 Reagent.
Incubate for 5 min at room temperature.
Add DNA-lipid complex directly to the well of hPSCs. For a negative control, add the mixture without DNA directly to the well of hPSCs.
-
Incubate at 37°C for 24 hours.
The subsequent selection will be performed by overnight treatment with puromycin; therefore, we usually transfect plasmid DNA into hPSCs in the evening. After 24 hours of transfection with plasmid DNA, transfected cells will be selected by transient antibiotic treatment. For effective selection, cells should be subconfluent, because confluent non-proliferating cells are resistant to the antibiotics (Gibco Education, 2016). -
As a pilot experiment, determine the appropriate concentration of an antibiotic.
The shorter treatment with an antibiotic is better for successful selection of transfected cells, since transfected plasmid DNA will be diluted after cell proliferation. Based on our experiences, overnight treatment with an antibiotic is sufficient to eliminate non-transfected cells. As a preliminary experiment, varied concentrations of an antibiotic will be tested to find the lowest concentration which eliminates all non-transfected cells by overnight treatment. We describe an example case using H9 hESCs and puromycin. Culture H9 cells until they reach 70–80% confluency, then treatment cells with 0, 0.5, 1, 3, 5 and 10 μg/ml of puromycin overnight. On the next day, find the lowest concentration which eliminates all cells. -
On Tuesday evening (the next day from transfection), remove all the media containing transfection reagents and replace with 1.5 ml of fresh Stemfit supplemented with puromycin (at the optimal concentration decided at step 1; e.g. 1 μg/ml for H9), 10 μM Y27632 and10 ng/ml bFGF.
A negative control without plasmid DNA will be used to determine whether the transient antibiotic treatment was sufficient to eliminate non-transfected cells. It is also recommended to assign several different concentrations of an antibiotic for actual selection. Although the timeline is shown as day of the week, it is possible to change the timing for your convenience. On Wednesday morning (the 2nd day from transfection), remove all old media and replace with 1.5 ml of fresh SF02 supplemented with 10 μM Y27632 and 10 ng/ml bFGF.
-
On Friday or Saturday (the 4th or 5th day from transfection), remove all old media and replace with 1.5 ml of fresh Stemfit supplemented with 10 ng/ml FGF2.
When you can recognize small colony formation, you can remove Y27632. This is usually the 4th or 5th day from transfection. On the next Monday, Wednesday and Friday (7th, 9th and 11th day), remove all old media and replace with 1.5 ml of fresh SF02 supplemented with 10 ng/ml FGF2.
Confirm that a few hundred colonies have appeared. Also, confirm that no colonies can be seen in a negative control.
Subprotocol #6: Single cell plating and expansion
Introductory Paragraph
This method is very useful, rapid, and simple, and generates many clones by expansion from a single cell without costly devices such as a flow cytometer. Since 10–15% of seeded cells grow into available colonies, it is possible to determine how many wells of 96-well plates to prepare based on the desired number of clones [see Figure 5].
Figure 5. Single cell plating for the development of distinct transfected hPSC clones.
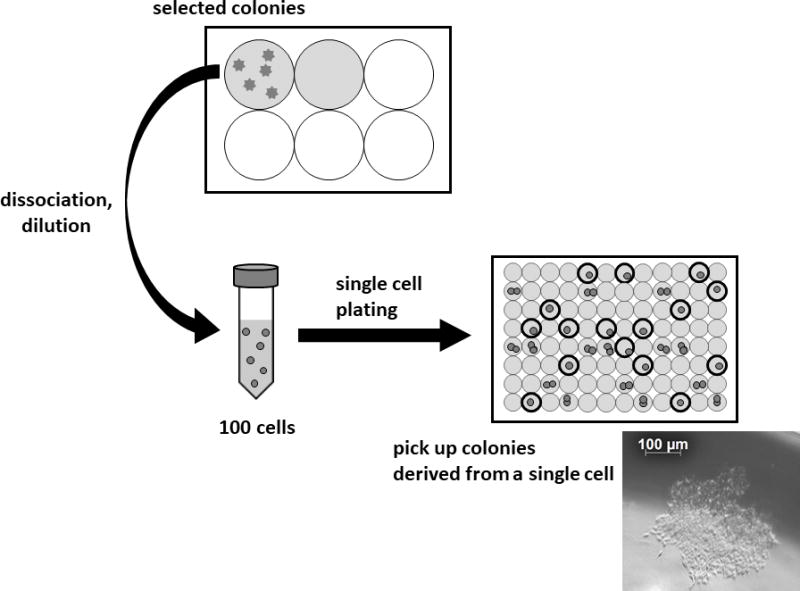
Selected colonies composed of transfected hPSCs are dissociated and diluted to 100 cells / 10 ml of fresh media for single cell cloning. Subsequently 10 ml of media containing 100 cells are divided into 96 wells on a flat-bottomed 96-well plate equally (100 μl / well). If the cells tolerate single cell dilution, colonies derived from single cells will be obtained in 10–15% of wells. An image of a typical colony on the 7th day after the single cell plating is shown.
Materials
6-well tissue culture plates, Falcon, Catalog #353046
96-well tissue culture plate, Flat Bottom, Falcon, Catalog #353072
15 ml conical tube
DMEM/F12, Life Technologies, Catalog #11320033
LDEV-free, hESC-qualified Geltrex, Thermo Fisher Scientific, Catalog #A1413302
10mg Y27632 (ROCK inhibitor), TOCRIS, Catalog #1254
Recombinant Human FGF-basic (bFGF), Peprotech, Catalog #100-18B
StemFit Basic02 (SF02), Ajinomoto, Catalog #ASB01
Accutase, STEMCELL Technologies, Catalog #07920
1X PBS, Thermo Fisher Scientific, Catalog #10010049
Protocol steps
-
After transfection with the Cas9 and gRNA bearing plasmid and subsequent antibiotic selection, expanded colonies need to be replated onto 96-well plates at 1 cell/well to obtain a clonal population. On the 11th day from transfection (Friday), remove all of the media and wash with 2 ml of PBS once.
The day of the week shown in this section is based on the case in which transfection was performed on Monday. It is possible to change for your convenience. Remove PBS and add 500 μl of Accutase. Incubate for 10 min at 37°C.
Dissociate cells with 1 ml pipette, and transfer cells to 10 ml tube filled with 500 μl of SF02.
Count the cell number, and centrifuge at 300× g for 4 min at room temperature.
Aspirate supernatant.
Resuspend cells in SF02 at 10,000 cells/μl. Take 1 μl cell suspension and dilute cells with 99 μl SF02 at 100 cells/μl.
-
Take 1 μl (100 cells) and resuspend cells in 10 ml of fresh SF02 supplemented with 10 μM Y27632 and 100 ng/ml bFGF. Mix gently.
Since 10–15% of seeded cells grow into colonies, 10 to 15 colonies will be obtained from 100 cells. If you desire more colonies, increase the number of cells and wells. To maintain pluripotency, the higher concentration at 100 ng/ml bFGF is recommended. -
To plate one cell into each well, transfer 100 μl of SF02 (1 cell/100 μl) to each well on a 96-well flat bottom plates coated with Geltrex.
Since cell viability of hPSCs is substantially lower than standard cell lines, we plate cells onto 96-well plates at 1 cell/well by a single cell dilution protocol without flow sorting. Culture cells at 37°C.
Observe cells after 2–3 days to confirm that colonies appear. Generally 10–15% of wells will yield a single colony. Mark those wells.
After 3–4 days of single cell plating, remove the old media from the wells in which only one colony has appeared and replace with 200 μl of SF02 supplemented with 100 ng/ml FGF2. Feed cells every 2–3 days.
Select wells with single colonies, and remove all of the media and wash with 100 μl of PBS once.
Remove PBS and add 20 μl of Accutase. Incubate for 10 min at 37°C.
Gently tap the plate, and add 180 μl SF02 supplemented with 10 μM Y27632 and 100 ng/ml FGF2. Dissociate the cells with 200 μl pipette.
-
Take 100 μl and seed cells in each well of two 96-well plates coated with Geltrex.
The first plate is for genomic DNA extraction, and the second plate is for freezing cells. Incubate at 37°C.
The day after passaging, remove media and replace with SF02 supplemented with 100 ng/ml FGF2.
Feed cells with 100 μl SF02 supplemented with 100 ng/ml FGF2 every 2–3 days until the cells reach 70–80% confluency. It usually takes 2–5 days.
After cells reach 70–80% confluency, cells can be harvested for DNA extraction and deep sequencing. Another plate of cells can be frozen in 200 μl SF02 supplemented 10% DMSO after dissociation with 20 μl Accutase.
Subprotocol #7: Genomic DNA Preparation for Deep Sequencing
Introductory Paragraph
Genomic DNA must be extracted to evaluate for targeted genome-editing. DNA samples can be sent to any number of deep sequencing core facilities across the country. These facilities employ a cost-effective, high-throughput method for detection of CRISPR/Cas9-induced mutations via deep sequencing of PCR amplicons.
Materials
Transfected hPSCs in 96-well plate after selection with an antibiotic
NaCl/ethanol solution
20 mg/ml Proteinase K, Thermo Fisher Scientific, Catalog #2546
1X PBS, Thermo Fisher Scientific, Catalog #10010049
70% ethanol
Nuclease-free water, Thermo Fisher Scientific, Catalog #AM9932
-
hPSC lysis buffer:
Solution Stock [M] Final [mM] Volume per 200 ml (ml) Tris (pH 8.0) 1 10 2.0 EDTA (pH 8.0) 0.5 10 4.0 NaCl 4 10 0.5 DNAse-free H20 - - 193.5
Equipment
12 channel pipette
Dry incubator at 65°C
Centrifuge
Protocol steps
Genomic DNA Preparation
-
Prepare NaCl/ethanol solution: Add 937 μl of 4M NaCl per 50 ml of 100% ethanol. Mix well.
There will be some sodium chloride precipitate, but this is of little consequence. -
Grow the transfected hPSCs until achieving ~80% confluence in 96-well plates.
Typically, it takes ~4–7 days after splitting to achieve ~80% confluency. The timing is not critical, rather this confluency reflects sufficient cells to obtain an appropriate amount of genomic DNA. Quickly invert the plate to remove media and blot with paper towels
Wash each well with 150 μl of 1X PBS using a 12-channel pipette, then invert the plate to remove PBS
-
Add 250 μl of fresh 20mg/ml Proteinase K to 4.75 ml of hPSC lysis buffer
Add fresh Proteinase K each time to final concentration 1 mg/ml, store aliquots at −20°C. To prepare the hPSC lysis buffer, please see the table in the Materials section. Add 50 μl of 1 mg/ml Proteinase K in hPSC lysis buffer to each well using a 12-channel pipette
Line the inside of a plastic container with several wet paper towels.
Parafilm the edges of a 96-well plate of transfected hPSCs and place the plate on the wet paper towels.
-
Incubate in a dry incubator at 65°C overnight.
Keep the incubator door closed to avoid condensation. On the following day, add 100 μl of ice cold NaCl/ethanol solution to each well using a 12-channel pipette and mix well.
Incubate at room temperature for 30 min.
Centrifuge the 96-well plate at 3000 rpm for 20 min.
Gently decant the plates to remove supernatant and press on to dry paper towels.
Wash each well with 150 μl of cold 70% ethanol using a 12-channel pipette
Centrifuge the 96-well plate at 3000 rpm for 10 min.
Gently decant the plates and repeat the cold 70% ethanol wash with 150 μl per well.
Centrifuge the 96-well plate at 3000 rpm for 10 min.
-
Air dry for 2–3 hours.
Avoid overdrying genomic DNA after ethanol washes. Vacuum drying should be avoided. -
Resuspend DNA in 30 μl of nuclease-free water. Parafilm the plate and store genomic DNA at 2 to 8 °C and send for deep sequencing within 1 week of isolation.
Storing genomic DNA at −15 to −25 °C can cause shearing of DNA, particularly if the DNA is exposed to repeated freeze-thaw cycles. Pipetting of genomic DNA through small tip openings causes shearing or nicking, therefore we recommend use of wide opening tips for resuspension. Parafilm the 96-well plate
-
Identifying CRISPR/Cas-induced mutations through deep sequencing of PCR products. Send the full 96-well plate to a deep sequencing facility. We are using the Center for Computational and Integrative Biology (CCIB) at Massachusetts General Hospital (MGH, Boston, MA, Contact of Core Director of CCIB - nstange-thomann@ccib.mgh.harvard.edu), however, there are deep sequencing facilities near most major academic centers.
In the Boston area alone, there are facilities affiliated with the MGH, Beth Israel Deaconess Medical Center, Boston Children’s Hospital, Boston Medical Center, Tufts Medical Center, and the University of Massachusetts Medical Center.
Subprotocol #8: Donor DNA construct design and sgRNA design example
Introductory Paragraph
CRISPR/Cas-9 can be used to create mutations to disrupt target proteins, but also for creating sequence-specific genomic modifications through homologous recombination with donor DNA constructs. The example design described below is to create a fluorescent reporter of PECAM1 expression by knocking in a RFP reporter into the PECAM1 locus in hPSCs. Homologous arms are created to align to the correct region of the endogenous genes, and these arms flank the RFP reporter such that a recombination event leaves the RFP in the endogenous target location. By co-transfecting donor DNA construct with a Cas9 / sgRNA expression vector targeting the locus, we increase the efficiency of homology directed repair (HDR) and thus favor donor DNA incorporation.
We will use CRISPR/Cas-9 to target this DNA donor construct to the C-terminal region of the endogenous PECAM1 gene. By supplying a DNA donor template (our construct with homologous arms), we are encouraging HDR as the mechanistic response to Cas9-generated DSBs. When the endogenous DNA repair machinery is supplied a homologous template to initiate repair, a consequence is the incorporation of our engineered reporter. In the absence of a homologous repair template, NHEJ results in indels as described earlier. Our design includes selectable markers in the DNA donor construct to enrich for correctly targeted clones. Note that there are many tools available for examining genomic sequences and planning out cloning projects (free and commercial, see Table 1). For this example, we will be using the free version of an online bioinformatics software called Benchling along with the UCSC and Ensembl genome browsers.
Gene Structure – Where to target?
When choosing how to approach a particular gene, we look for evidence of alternative splicing or a potential phenotype that may dictate where to target the gene. For example, if there are several transcript variants, is there an exon that is shared so we may be inclusive of all (Fig. 6)? Or is there a dominant transcript variant in our target tissue of interest that is best to use? If we use a location at the N-terminal of the endogenous protein, this may knockout function leaving the resulting cell line with less of that target. If this is a crucial gene this may be a disadvantage for the cell, or alter the differentiation ability of that cell for a specific tissue. Thus, we consider known phenotypes in mice for the gene target, especially if there is a phenotype in heterozygous animals that suggests any diminished amount of protein may impact downstream studies.
Figure 6. Spliced transcripts reported in the UCSC genome browser suggest one primary transcript for PECAM1.

By displaying all known spliced transcripts reported we can identify any common alternative transcripts. In this case, the single reported transcript (indicated by the curated GENCODE and RefSeq tracks) is the dominant version of the gene with nearly all the other reported spliced transcripts (“Human mRNAs from GenBank”) matching the curated ones.
After deciding on the transcript to target, we then consider options for where within that gene to place our reporter. For our next steps, we determine the matching PECAM1 transcript annotated in the Ensembl genome browser (Gene ID ENSG00000261371, the longest coding transcript, PECAM1-203, ENST00000563924.5) and import this into Benchling (Fig. 7). This brings the gene annotation into the software as well.
Figure 7. Identify the fully annotated PECAM1 transcript in Ensembl.

The Ensembl genome browser includes well annotated gene data that is useful in downstream design steps. After confirming the single best transcript for a gene from above, we locate the same transcript in Ensembl for download into our software of choice. The long coding transcript is labeled PECAM1-203 with a corresponding transcript ID and the full length coding region.
CRISPR Target Region, Design, and Selection
We select an approximately 200 bp region that includes the PECAM1 stop codon as the target region and use any of the available online tools to determine what potential sgRNA sites exist in this region. The output from the gRNA tools can be ranked by different scoring criteria that considers both on-target and off-target effects. In Figure 1, we have selected not the top scored gRNA, but the highest that fits the location we desire (i.e. near the PECAM1 STOP codon).
DNA construct design
PECAM1 is an endothelial marker and our intended use is to study how endothelial cell interact with other cell types in organoids derived from these modified iPSCs. Our goal was to not alter endogenous PECAM1 protein function and instead place the RFP in-frame at the C-terminus. For this, a P2A sequence is placed in-frame with the end of the endogenous PECAM1 and leading into the RFP protein. The resulting bicistronic transcript will produce both the PECAM1 and RFP proteins due to the ribosome-skipping induced by the P2A sequence (Szymczak et al., 2005; Osborn et al., 2005; Hsiao et al., 2008). In choosing the homologous arms, we include the C-terminus of the PECAM1 protein up to the planned junction with the P2A sequence in the upstream arm (we refer to this as the “Left Arm”, “LA”, Fig. 8).
Figure 8. Select flanking homology arms surrounding the target site.

A) 750 bp left and right homology arms are selected from the endogenous gene sequence. B) Building out the construct in the software allows careful examination and planning to ensure the chimeric protein is in-frame.
Protocol Steps
Select approximately 750 bp upstream and 750 bp downstream of the target integration site for the Left Arm and Right Arm respectively.
-
Combine the flanking homology arms and the reporter, in-frame if necessary, in the software of choice to generate the desired donor DNA construct in silico.
The reporter cassette chosen should include a selectable marker to enrich for transfectants that retain the construct. -
Order the designed construct to be synthesized from a provider.
There are many services that can synthesize large DNA fragments at a reasonable cost (e.g. Genewiz, Integrated DNA Technologies, Twist Bioscience).
Prepare the DNA for transfection using the same technique described above for sgRNA/Cas9 plasmid preparation.
Commentary
Background Information
The advent of human stem cell research began with isolation of human embryonic stem cells (hESCs) from in vitro fertilized human embryos (Thomson et al., 1998). The discovery of reprogramming methods to generate induced pluripotent stem cells (iPSCs) from terminal adult cells enabled human stem cells to be generated from any individual (Takahashi et al., 2006). As iPSCs remain isogenic with the reprogrammed host cell, applications for human stem cell research expanded and included developmental studies, toxicity assays for drug development, congenital and genetic disease modeling, and immunocompatible bioartificial organ engineering. As directed differentiation protocols continue to be elucidated and improved, such applications may now be applied to human organ cells and tissues in vitro.
The past few years have brought considerable advances in stem cell culture methods, including the ability to transition from colony passaging on a bed of irradiated murine embryonic fibroblasts (MEF) feeder culture to single cell passaging in feeder-free conditions (Nakagawa et al., 2014). Benefits of new methods include reduced xenogeneic contamination, reduced operator workload, and the generation of isogenic progeny from a single host cell, important for the techniques involving genomic manipulation. Coupling advances in human stem cell research with evolving genome-editing techniques has enabled researchers to generate gene-specific knock-outs, knock-ins, and reporter lines. Homozygotic knock-outs may facilitate modeling of recessive human monogenic diseases. Likewise, Heterozygotic knock-ins may allow for dominant human monogenic disease modeling. Reporter lines may help to determine directed differentiation protocols to generate organ specific tissue, determine the roles of specific genes in developmental studies, and monitor for toxicity in live human organ-like tissue in vitro. Moreover, genome-editing in hPSCs has obviated the requirement for patient-specific iPSCs to model genetic disease and allowed for the unaltered parent line to serve as an isogenic control.
CRISPR/Cas9 has become a popular technique for genome-editing owing to ease of use, further supported by plasmid repositories and numerous available protocols. Primary concerns regarding CRISPR/Cas9 genome modification include variable transfection efficiency, cell viability, and maintenance of pluripotency. Transient antibiotic selection surmounts potential reduced transfection efficiency in the presented protocol. The puromycin resistance gene was incorporated into the PX459 plasmid for the generation of stable transfectants; however, the sensitivity of hPSCs to antibiotics allows for transient puromycin selection. This antibiotic selection further replaces the need for flow sorting, thereby increasing cellular viability to permit single cell expansion and the generation of multiple hPSC lines derived from a single parent cell. Additionally, the outlined methods reduce xenogeneic contamination from MEF feeder cells and markedly reduce operator workload, due to single cell rather than clump passaging. Researchers of limited means and technical experience may generate human stem cell lines containing desired gene-specific mutations following the presented protocol.
Critical Parameters and Troubleshooting
Subprotocol #1: sgRNA design
It may be difficult to find an appropriately unique target sequence within your gene of interest. However, as the outlined protocol generates multiple clones, each with varying degrees of off-target effects, the best experimental clone can be identified. We advise designing 3 independent sgRNA for each gene of interest due to variable efficiencies, even between nearby target sequences.
Subprotocol #2: sgRNA expression plasmid generation
The outlined subprotocol conforms to use of Addgene’s PX458-462 plasmids; however, subprotocol 5 requires the presence of the puromycin resistance gene found in PX459 (spCas9) and PX462 (spCas9n).
Subprotocol #3: Plasmid amplification and purification
If the amount of DNA for transformation is over 10 ng, the efficiency can be significantly low. When no colonies can be seen after transfection to E. coli. competent cells, confirm whether the amount of DNA is between 1 and 10 ng.
Support Protocol #1: Coating plates with Geltrex
Due to the relatively high viscosity of 1% Geltrex in DMEM/F12, swirl the plate to ensure an even coating. hPSCs will not survive on areas that lack a Geltrex coating.
Subprotocol #4: hPSC culture technique
It is advisable to plate two wells of a 6-well plate, each with 100 thousand cells from the freshly thawed cryovial of hPSCs. When stored at 4°C, bFGF is stable for approximately 1 week. It is best to store weekly aliquots of bFGF at −20°C. Differentiating hPSCs that resemble fibroblasts tend to have stronger adherence to the plate. Reducing the incubation time with Accutase and the aggressiveness of dissociation in the well may reduce the transfer of differentiated cells.
Support Protocol 2: Freezing hPSCs in cryovials
It is best to limit the amount of time that cells are in contact with DMSO prior to placement at −80°C. As each well of a 6-well plate will yield ~3 million cells per week, ~15 aliquots cells can be generated with a single passage.
Subprotocol #5: Transfection and antibiotic selection
Transfection efficiency decreases when cells are not actively proliferating. Avoid using hPSCs which have reached 100% confluent. However, lower cell densities (less than 70%) are also worsen transfection efficiency. Low transfection efficacy are also caused by inappropriate DNA:transfection reagent or too short complexing mixing period. It is advisable to vary DNA (μg) : P3000 reagent from 1:0.5 to 1:5, and to prolong incubation period in step 6 into 20 minutes. Since human stem cells are fragile, cells are sometimes damaged simply because they are exposed to Lipofectamine. When many of cells are detached on the next day of transfection, efficiency of transfection should be too low. In such a case, decrease of Lipofectamine (e.g. half amount) might improve the viability of cells and efficiency of transfection. When no colonies appeared after the selection, there are two possibilities. One is that the exposure to antibiotics is too toxic. The treatment duration or concentration of antibiotics should be reduced. As described in the protocol, before starting selection, we recommend to choose an appropriate selectable marker and determine the selective concentration of antibiotics using cells to which nothing is transfected. The other possibility is that efficiency of transfection is too low. See the troubleshooting of transfection above.
When colonies survived even in negative control cells, it means that concentration of the antibiotic is too low. Similarly, it is advisable to choose appropriate treatment duration and concentration of antibiotics using hPSCs which is not transfected.
Subprotocol #6: Single Cell Plating and amplification
Although stem cells generally tolerates single cell dilution, some cell lines might not be tolerant. When the cell line does not tolerate single cell dilution, alternatively cells can be plated to 6-well plates and individual colonies picked. Put picked colonies into eppendorf tubes separately, then wash with PBS and 20 μl of Accutase. Subsequent steps are same as since step 14.
Subprotocol #7: Genomic DNA Preparation and Deep Sequencing
If the DNA yield is lower than what is required for deep sequencing (generally 30ng/μl), then genomic DNA preparation will have to be repeated. Repeat the subprotocol using fresh Proteinase K, while ensuring the sample remains humid when incubating at 65 °C overnight, and carefully perform 70% ethanol washes.
Subprotocol #8: Donor DNA construct design and sgRNA design example
The design of a specific sgRNA complementary to your target site for both the generation of knock-out lines (indels) and sequence-specific mutagenesis (homology-directed DNA repair) is the single most critical step. Likewise, the design of donor DNA for homology-directed DNA repair is essential for genomic conservation of the regions flanking your target site for sequence-specific mutagenesis.
Major Pitfalls
| Transfection efficiency is low. | Ensure that transfection occurs on actively proliferating cells at high density (~70–80% confluent). The ratio or mixing time of DNA and transfecting reagent may need to be varied, we suggest a DNA (μg) to P3000 (μl) of 1 to 0.5–5.0 and extending the incubation time to 20 minutes. |
| Many of cells are detached on the next day after transfection. | Varying hPSC lines demonstrate differing cytotoxicity to Lipofectamine; we suggest reducing the Lipofectamine volume by 2-fold. |
| No colonies appeared after antibiotic selection. | Either the puromycin concentration overcame the puromycin resistance of transfected cells or the cells were not efficiently transfected. Repeat the puromycin titration to ensure proper puromycin concentration and see troubleshooting for ‘Transfection efficiency is low’ above. |
| Colonies survived in negative control cells. | Repeat the puromycin titration to identify the lowest concentration which eliminates all control cells. |
| No colonies appeared after single cell plating. | Ensure use of StemFit Basic02, which allows for single cell expansion. However, differing cell lines demonstrate differing tolerance of single cell plating. Alternatively, we recommend plating the puromycin selected cells at low concentration in 6-well plates (10–50 cells/well) and picking individual colonies that were generated from a single parental cell. Place picked colonies in separate 1.5ml eppendorf tubes, wash with PBS, add 20ul of Accutase, and proceed with the outlined protocol. |
Anticipated Results
Subprotocol #1: sgRNA design
The design of a highly specific sgRNA to the DNA sequence of interest is anticipated.
Subprotocol #2: sgRNA expression plasmid generation
The expected concentration of the ligation reaction product is ~3ng/μl.
Subprotocol #3: Plasmid amplification and purification
E. coli. competent cells which are successfully transformed will form dozen of colonies on the selection culture plate. Any of colonies are expectable to carry the plasmid. After amplification and purification, 100–2,000 ng/μl of plasmid DNA will be obtained.
Support Protocol #1: Coating plates with Geltrex
Circumferential coating of plates to allow for hPSC proliferation under feeder-free conditions is expected.
Subprotocol #4: hPSC culture technique
We anticipate hPSC lines to proliferate at a rate of 28 per week. However, it is important to visualize each line daily and passage before exceeding 90% confluency, titrating the plating density as necessary.
Support Protocol #2: Freezing hPSCs in cryovials
The generation and long-term maintenance of multiple aliquots of frozen stocks of hPSC lines is anticipated. We expect 5–10% of hPSCs to survive after thawing a cryovial aliquot.
Subprotocol #5: Transfection and antibiotic selection
The efficiency of transfection is generally as low as 1–2%. Even after transfection the exterior of cells will not be changed.
A few hours later from the start of selection, some cells will be getting detached. By 12 hours later all of cells which are not transfected will be detached. It is sometimes difficult to distinguish attaching dead cells from live cells; however, some colonies will be formed from live cells within 5 days from the start of selection. Generally from a few hundred colonies will appear. The number of obtained colonies depends on the cell confluency, cell lines, size of plasmid DNA, and other various conditions.
Subprotocol #6: Single cell plating and amplification
If the cells tolerates single cell dilution, in 10–15% of wells single colony derived from single cell will be seen.
Subprotocol #7: Genomic DNA preparation and deep sequencing
The expected genomic DNA yield from a 100% confluent well of a 96-well plate is 5 μg. Following dilution with 30 μl of nuclease-free water, the expected final concentration of genomic DNA is ~150ng/μl. Deep sequencing results from the University of Washington’s Next Generation Sequencing core facility will return in ~2 weeks. It can be expected that 1 out of ~3 colonies will contain a homozygotic mutant, though the number changes depending on the gRNA design and the target gene.
Subprotocol #8: Donor DNA construct design and sgRNA design example
The generation of a site-specific sgRNA at the preferred site in your target gene is anticipated. For desired homology-directed repair, the design and purchase of flanking homologous arms surrounding your preferred site is anticipated.
Time considerations
sgRNAs can be designed and generated in a matter of days. sgRNA expression plasmids can be generated in 1 to 2 weeks. The estimated period for plasmid amplification and purification is a few weeks, and culture of stem cells for the following procedures can be prepared in parallel. After these components are assembled, it is possible to start transfection and selection. Although the duration required for transfection and selection depends on the speed of cell growth, it will be 2 to 3 weeks. Single cell plating and amplification of transfected cells can be performed in approximately 2 weeks. The following genomic DNA preparation and deep sequencing can be accomplished in 2 weeks (variable by outsourced institutions).
Significance Statement.
Advances in stem cell biology have enabled researchers to develop human organ-like tissue in a dish. By subjecting human pluripotent stem cells (hPSCs) to sequential factors that mimic human organ development, tissue models of human heart, muscle, kidney, liver, lung, brain, and intestines have been generated. The application of 3-dimensional culture techniques has yielded representative organoids for use in human disease modeling, drug screening, and regenerative medicine. Coupling this technology with the emerging technique of CRISPR/Cas9 genome editing has facilitated genome modification in hPSCs to create models of genetic (inheritable) diseases in human organoids, create reporter lines, and study human pathophysiology. The following protocol enables groups with limited experience to make targeted DNA mutations in human pluripotent stem cells.
Acknowledgments
Research reported in this publication was supported by a National Institutes of Health (NIH) T32 fellowship training grant (DK007527, to N.G.), the Program for Advancing Strategic International Networks to Accelerate the Circulation of Talented Researchers (from the Japan Society for the Promotion of Science, to KS), an NIH R37 grant (DK039773, to J.V.B), an NIH R01 grant (DK072381, to J.V.B.), an NIH UG3 grant (TR002155, to J.V.B, T.V., and R.M.), a Rebuilding a Kidney (RBK) Grant (to T.V.), an NIH Subaward (U01DK107350, to T.V.), a Brigham and Women’s Hospital Research Excellence Award (to R.M.), a Brigham and Women’s Hospital Faculty Career Development Award (to R.M.), a Harvard Stem Cell Institute Seed Grant (to R.M.) and a Grant-in-Aid for a Japan Society for the Promotion of Science (JSPS) Postdoctoral Fellowship for Research Abroad (to R.M.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Conflicts of Interests
J.V.B. is a co-founder, consultant to, and owns equity in, Goldfinch Bio.
Literature Cited
- Chapman JR, Taylor Martin RG, Boulton Simon J. Playing the End Game: DNA Double-Strand Break Repair Pathway Choice. Molecular Cell. 2012;47:497–510. doi: 10.1016/j.molcel.2012.07.029. [DOI] [PubMed] [Google Scholar]
- Dye BR, Hill DR, Ferguson MAH, Tsai Y-H, Nagy MS, Dyal R, Wells JM, Mayhew CN, Nattiv R, Klein OD, White ES, Deutsch GH, Spence JR. In vitro generation of human pluripotent stem cell derived lung organoids. eLife. 2015;4:e05098. doi: 10.7554/eLife.05098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freedman BS, Brooks CR, Lam AQ, Fu H, Morizane R, Agrawal V, Saad AF, Li MK, Hughes MR, Werff RV, Peters DT, Lu J, Baccei A, Siedlecki AM, Valerius MT, Musunuru K, McNagny KM, Steinman TI, Zhou J, Lerou PH, Bonventre JV. Modelling kidney disease with CRISPR-mutant kidney organoids derived from human pluripotent epiblast spheroids. Nat Commun. 2015;6:8715. doi: 10.1038/ncomms9715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Sander JD, Reyon D, Cascio VM, Joung JK. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat Biotech. 2014;32:279–84. doi: 10.1038/nbt.2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaj T, Gersbach CA, Barbas CF. ZFN, TALEN and CRISPR/Cas-based methods for genome engineering. Trends in biotechnology. 2013;31:397–405. doi: 10.1016/j.tibtech.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibco Education. Cell Culture Basics Handbook. Thermo Fisher Scientific; Massachusetts: 2016. [Google Scholar]
- Green MR, Sambrook J. Molecular cloning: A laboratory manual. Cold Spring Harbor Laboratory Press; New York: 2012. [Google Scholar]
- Hendriks WT, Jiang X, Daheron L, Cowan CA. TALEN- and CRISPR/Cas9-Mediated Gene Editing in Human Pluripotent Stem Cells Using Lipid-Based Transfection. Curr Protoc Stem Cell Biol. 2015;34:5B.3.1–25. doi: 10.1002/9780470151808.sc05b03s34. [DOI] [PubMed] [Google Scholar]
- Hsiao EC, Yoshinaga Y, Nguyen TD, Musone SL, Kim JE, Swinton P, et al. Marking embryonic stem cells with a 2A self-cleaving peptide: a NKX2-5 emerald GFP BAC reporter. PLoS ONE. 2008;3(7):e2532. doi: 10.1371/journal.pone.0002532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RD, Liu N, Jasin M. Mammalian XRCC2 promotes the repair of DNA double-strand breaks by homologous recombination. Nature. 1999;401:397–9. doi: 10.1038/43932. [DOI] [PubMed] [Google Scholar]
- Kleinstiver BP, Prew MS, Tsai SQ, Topkar VV, Nguyen NT, Zheng Z, Gonzales APW, Li Z, Peterson RT, Yeh J-RJ, Aryee MJ, Joung JK. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature. 2015;523:481–5. doi: 10.1038/nature14592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster MA, Renner M, Martin C-A, Wenzel D, Bicknell LS, Hurles ME, Homfray T, Penninger JM, Jackson AP, Knoblich JA. Cerebral organoids model human brain development and microcephaly. Nature. 2013;501:373–9. doi: 10.1038/nature12517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieber MR. The Mechanism of Double-Strand DNA Break Repair by the Nonhomologous DNA End Joining Pathway. Annual review of biochemistry. 2010;79:181–211. doi: 10.1146/annurev.biochem.052308.093131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieder R, Gaware VS, Thormodsson F, Einarsson JM, Ng CH, Gislason J, Masson M, Petersen PH, Sigurjonsson OE. Endotoxins affect bioactivity of chitosan derivatives in cultures of bone marrow-derived human mesenchymal stem cells. Acta Biomater. 2013;9:4771–8. doi: 10.1016/j.actbio.2012.08.043. [DOI] [PubMed] [Google Scholar]
- MacArthur DG, Manolio TA, Dimmock DP, Rehm HL, Shendure J, Abecasis GR, Adams DR, Altman RB, Antonarakis SE, Ashley EA, Barrett JC, Biesecker LG, Conrad DF, Cooper GM, Cox NJ, Daly MJ, Gerstein MB, Goldstein DB, Hirschhorn JN, Leal SM, Pennacchio LA, Stamatoyannopoulos JA, Sunyaev SR, Valle D, Voight BF, Winckler W, Gunter C. Guidelines for investigating causality of sequence variants in human disease. Nature. 2014;508:469–76. doi: 10.1038/nature13127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JC, Tan S, Qiao G, Barlow KA, Wang J, Xia DF, Meng X, Paschon DE, Leung E, Hinkley SJ, Dulay GP, Hua KL, Ankoudinova I, Cost GJ, Urnov FD, Zhang HS, Holmes MC, Zhang L, Gregory PD, Rebar EJ. A TALE nuclease architecture for efficient genome editing. Nat Biotech. 2011;29:143–8. doi: 10.1038/nbt.1755. [DOI] [PubMed] [Google Scholar]
- Moreno-Mateos MA, Vejnar CE, Beaudoin J-D, Fernandez JP, Mis EK, Khokha MK, Giraldez AJ. CRISPRscan: designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nat Meth. 2015;12:982–8. doi: 10.1038/nmeth.3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morizane R, Lam AQ, Freedman BS, Kishi S, Valerius MT, Bonventre JV. Nephron organoids derived from human pluripotent stem cells model kidney development and injury. Nat Biotech. 2015;33:1193–200. doi: 10.1038/nbt.3392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morizane R, Bonventre JV. Generation of nephron progenitor cells and kidney organoids from human pluripotent stem cells. Nat Protoc. 2017 Jan;12(1):195–207. doi: 10.1038/nprot.2016.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morizane R, Bonventre JV. Kidney Organoids: A Translational Journey. Trends Mol Med. 2017 Feb 7; doi: 10.1016/j.molmed.2017.01.001. pii: S1471-4914(17)30001-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa M, Taniguchi Y, Senda S, Takizawa N, Ichisaka T, Asano K, Morizane A, Doi D, Takahashi J, Nishizawa M, Yoshida Y, Toyoda T, Osafune K, Sekiguchi K, Yamanaka S. A novel efficient feeder-free culture system for the derivation of human induced pluripotent stem cells. Scientific reports. 2014;4:3594. doi: 10.1038/srep03594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldershaw RA, Baxter MA, Lowe ET, Bates N, Grady LM, Soncin F, Brison DR, Hardingham TE, Kimber SJ. Directed differentiation of human embryonic stem cells toward chondrocytes. Nat Biotech. 2010;28(11):1187–1194. doi: 10.1038/nbt.1683. [DOI] [PubMed] [Google Scholar]
- Osborn MJ, Panoskaltsis-Mortari A, McElmurry RT, Bell SK, Vignali DAA, Ryan MD, et al. A picornaviral 2A-like sequence-based tricistronic vector allowing for high-level therapeutic gene expression coupled to a dual-reporter system. Mol Ther. 2005 Sep 1;12(3):569–74. doi: 10.1016/j.ymthe.2005.04.013. [DOI] [PubMed] [Google Scholar]
- Perez EE, Wang J, Miller JC, Jouvenot Y, Kim KA, Liu O, Wang N, Lee G, Bartsevich VV, Lee Y-L, Guschin DY, Rupniewski I, Waite AJ, Carpenito C, Carroll RG, Orange JS, Urnov FD, Rebar EJ, Ando D, Gregory PD, Riley JL, Holmes MC, June CH. Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat Biotech. 2008;26:808–16. doi: 10.1038/nbt1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos DP, Kiskinis E, Eggan K, Merkle FT. Comprehensive Protocols for CRISPR/Cas9-based Gene Editing in Human Pluripotent Stem Cells. Curr Protoc Stem Cell Biol. 2016;38:5b.6.1–5b.6.60. doi: 10.1002/cpsc.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protocols. 2013;8:2281–308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezania A, Bruin JE, Arora P, Rubin A, Batushansky I, Asadi A, O’Dwyer S, Quiskamp N, Mojibian M, Albrecht T, Yang YH, Johnson JD, Kieffer TJ. Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat Biotech. 2014;32:1121–33. doi: 10.1038/nbt.3033. [DOI] [PubMed] [Google Scholar]
- Sharifi Tabar M, Hesaraki M, Esfandiari F, Sahraneshin Samani F, Vakilian H, Baharvand H. Evaluating Electroporation and Lipofectamine Approaches for Transient and Stable Transgene Expressions in Human Fibroblasts and Embryonic Stem Cells. Cell J. 2015;17:438–50. doi: 10.22074/cellj.2015.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadler J, Lemmens R, Nyhammar T. Plasmid DNA purification. J Gene Med. 2004;6(Suppl 1):S54–66. doi: 10.1002/jgm.512. [DOI] [PubMed] [Google Scholar]
- Sterneckert JL, Reinhardt P, Scholer HR. Investigating human disease using stem cell models. Nat Rev Genet. 2014;15:625–39. doi: 10.1038/nrg3764. [DOI] [PubMed] [Google Scholar]
- Szymczak AL, Vignali DAA. Development of 2A peptide-based strategies in the design of multicistronic vectors. Expert opinion on biological therapy. 2005 May 1;5(5):627–38. doi: 10.1517/14712598.5.5.627. [DOI] [PubMed] [Google Scholar]
- Taapken SM, Nisler BS, Newton MA, Sampsell-Barron TL, Leonhard KA, McIntire EM, Montgomery KD. Karyotypic abnormalities in human induced pluripotent stem cells and embryonic stem cells. Nat Biotech. 2011;29(4):313–314. doi: 10.1038/nbt.1835. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell. 2006;126:663–76. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, Jones JM. Embryonic Stem Cell Lines Derived from Human Blastocysts. Science. 1998;282:1145. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- Takebe T, Sekine K, Enomura M, Koike H, Kimura M, Ogaeri T, Zhang R-R, Ueno Y, Zheng Y-W, Koike N, Aoyama S, Adachi Y, Taniguchi H. Vascularized and functional human liver from an iPSC-derived organ bud transplant. Nature. 2013;499:481–4. doi: 10.1038/nature12271. [DOI] [PubMed] [Google Scholar]
- Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, Jones JM. Embryonic Stem Cell Lines Derived from Human Blastocysts. Science. 1998;282:1145. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- Urnov FD, Rebar EJ, Holmes MC, Zhang HS, Gregory PD. Genome editing with engineered zinc finger nucleases. Nat Rev Genet. 2010;11:636–46. doi: 10.1038/nrg2842. [DOI] [PubMed] [Google Scholar]
- Veres A, Gosis Bridget S, Ding Q, Collins R, Ragavendran A, Brand H, Erdin S, Cowan CA, Talkowski Michael E, Musunuru K. Low Incidence of Off-Target Mutations in Individual CRISPR-Cas9 and TALEN Targeted Human Stem Cell Clones Detected by Whole-Genome Sequencing. Cell Stem Cell. 2014;15:27–30. doi: 10.1016/j.stem.2014.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villa-Diaz LG, Ross AM, Lahann J, Krebsbach PH. The evolution of human pluripotent stem cell culture: from feeder cells to synthetic coatings. Stem Cells. 2013;31(1):1–7. doi: 10.1002/stem.1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Wang Y, Wu X, Wang J, Wang Y, Qiu Z, Chang T, Huang H, Lin R-J, Yee J-K. Unbiased detection of off-target cleavage by CRISPR-Cas9 and TALENs using integrase-defective lentiviral vectors. Nat Biotech. 2015;33:175–8. doi: 10.1038/nbt.3127. [DOI] [PubMed] [Google Scholar]
- Watson CL, Mahe MM, Munera J, Howell JC, Sundaram N, Poling HM, Schweitzer JI, Vallance JE, Mayhew CN, Sun Y, Grabowski G, Finkbeiner SR, Spence JR, Shroyer NF, Wells JM, Helmrath MA. An in vivo model of human small intestine using pluripotent stem cells. Nature medicine. 2014;20:1310–4. doi: 10.1038/nm.3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witty AD, Mihic A, Tam RY, Fisher SA, Mikryukov A, Shoichet MS, Li R-K, Kattman SJ, Keller G. Generation of the epicardial lineage from human pluripotent stem cells. Nat Biotech. 2014;32:1026–35. doi: 10.1038/nbt.3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Inokuma MS, Denham J, Golds K, Kundu P, Gold JD, Carpenter MK. Feeder-free growth of undifferentiated human embryonic stem cells. Nat Biotechnol. 2001;19(10):971–974. doi: 10.1038/nbt1001-971. [DOI] [PubMed] [Google Scholar]
- Yamaguchi S, Morizane R, Homma K, Monkawa T, Suzuki S, Fujii S, Koda M, Hiratsuka K, Yamashita M, Yoshida T, Wakino S, Hayashi K, Sasaki J, Hori S, Itoh H. Generation of kidney tubular organoids from human pluripotent stem cells. Sci Rep. 2016 Dec 16;6:38353. doi: 10.1038/srep38353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong X, Gutierrez C, Xue T, Hampton C, Vergara MN, Cao L-H, Peters A, Park TS, Zambidis ET, Meyer JS, Gamm DM, Yau KW, Canto-Soler MV. Generation of three-dimensional retinal tissue with functional photoreceptors from human iPSCs. Nature communications. 2014;5:4047. doi: 10.1038/ncomms5047. [DOI] [PMC free article] [PubMed] [Google Scholar]