

Abstract
Paroxysmal nocturnal hemoglobinuria (PNH) is widely regarded as an archetypal complement-mediated disorder that has propelled complement drug discovery in recent decades. Its pathology is driven by chronic complement dysregulation resulting from the lack of the GPI-linked regulators DAF and CD59 on susceptible erythrocytes. This complement imbalance fuels persistent C3 activation on affected erythrocytes, which culminates in chronic complement-mediated intravascular hemolysis. The clinical application of eculizumab, a humanized anti-C5 antibody that blocks terminal pathway activation, has led to drastic improvement of therapeutic outcomes but has also unveiled hitherto elusive pathogenic mechanisms that are now known to contribute to the clinical burden of a significant proportion of PNH patients. These emerging clinical needs have sparked a true resurgence of complement therapeutics that offer the promise of even more effective, disease-tailored therapies for PNH. Here we review the current state of complement therapeutics with a focus on the clinical development of C3-targeted and alternative pathway-directed drug candidates for the treatment of PNH. We also discuss the relative advantages and benefits offered by each complement-targeting approach, including translational considerations that might leverage a more comprehensive clinical intervention for PNH.
Introduction
Pathophysiology and clinical landscape of paroxysmal nocturnal hemoglobinuria (PNH)
PNH is a chronic, debilitating hematological disorder characterized by the clonal expansion of hematopoietic stem cells (HSCs) and their progeny, mature blood cells, which carry an acquired somatic mutation in the phosphatidyl-inositol glycan class A (PIG-A) gene [1]. PIG-A codes for an enzyme that is essential for the biosynthesis of the glycosyl phosphatidyl inositol (GPI) anchor, a protein modification allowing the attachment of proteins to the cell membrane [2]. The preferential expansion of these PIG-A mutated HSCs leads to the release of red blood cells into the circulation that lack, among other GPI-anchored proteins, the two key complement regulators CD55 and CD59 [3,4]. As a result of this deficiency, PNH erythrocytes are incapable of withstanding physiologic complement activation (i.e., due to spontaneous C3 tick-over or bystander activation) and undergo persistent C3 opsonization and terminal pathway activation that culminate in membrane attack complex (MAC)-mediated intravascular hemolysis. In fact, complement-mediated hemolytic anemia is one of the three cardinal features of PNH, along with bone marrow failure and thrombophilia [4–7].
Several studies have indicated that PIG-A inactivation is likely insufficient to trigger the disease process, indicating that additional mechanisms are causally involved in driving PNH pathogenesis [8–10]. Both preclinical data and clinical observations have provided a concrete framework for adopting the “dual pathophysiology” hypothesis for PNH [8,11,12]. According to this hypothesis, an (auto)-immune attack against normal hematopoiesis, similar to that seen in aplastic anemia, eventually results in the relative expansion of PIG-A mutated HSCs within the bone marrow [12]. An autoimmune basis for PNH pathophysiology is supported by both clinical and experimental observations, including the well-known clinical overlap between PNH and aplastic anemia [13] and the presence of GPI-specific autoreactive T cells that selectively target normal HSCs for immune destruction. This aberrant T-cell repertoire apparently spares the PIG-A mutated hematopoietic progenitors that ultimately expand as a result of this selective immune pressure [14–16]. Whereas mounting evidence lends further credence to this hypothesis the precise molecular mechanisms underlying this “immune escape” of PNH cells still remain ill-defined.
Intravascular hemolysis is the most typical manifestation of the disease, affecting to a variable extent all patients with clinical PNH [4,5,17,18]. The second defining clinical feature is cytopenia that is mostly secondary to the underlying bone marrow disorder, which is embedded in the dual pathophysiology of PNH [8]. The third typical manifestation of PNH is thrombophilia, with thromboembolic complications being the main cause of mortality among PNH patients [19–21].
Current treatment paradigm: Targeting C5
The treatment of PNH has dramatically changed since the introduction of the first clinically approved complement C5 inhibitor, eculizumab (trade name Soliris®, Alexion). Eculizumab is a recombinant humanized monoclonal antibody that selectively targets the terminal complement component C5, preventing its cleavage into C5a and C5b and the assembly of the pore-forming MAC [22]. Notably, the discovery of eculizumab was spearheaded by studies dating back to the late 1980s, when the first monoclonal antibody (mAb) against murine C5 (BB5.1) was generated [23]. Following proof-of-concept studies of therapeutic C5 inhibition in rodent models [24], Alexion developed a primate and human C5-specific mAb and, finally, the humanized antibody eculizumab [25,26].
The efficacy of eculizumab in controlling intravascular hemolysis in PNH patients was consolidated in two large multi-center trials [27,28], which recorded improved clinical responses (i.e., reduced transfusion needs, hemoglobin stabilization, and the resolution of all hemolysis-related symptoms). Sustained inhibition of the terminal complement pathway and abrogation of subsequent intravascular hemolysis may result in transfusion-independence in about half of the treated patients, with some patients also achieving a substantial increase in their hemoglobin levels. Furthermore, eculizumab seems to reduce the risk of thromboembolic events [29], possibly resulting in improved long-term survival [30]. Despite its profound clinical gains in PNH, anti-C5 therapy entails high annual costs for treatment and requires bi-monthly intravenous infusions in a hospital setting in most countries, thereby limiting patient compliance and interfering with the patient’s social activity and productivity at work.
Emerging clinical needs in the era of anti-complement therapy
The advent of anti-C5 therapy has brought clinical benefit to ~70% of PNH patients [31]; however, a significant proportion of patients receiving anti-C5 are either suboptimal responders or unresponsive to therapy and still require blood transfusions to treat residual anemia [32–35].
An insufficient response to anti-C5 therapy may be attributed to several factors. Bone marrow failure is the most obvious reason behind a poor hematological response; however, this condition cannot be treated with complement inhibition and may require alternative measures (i.e., either bone marrow transplantation or immunosuppression) [6]. “Breakthrough” intravascular hemolysis has been described in about 10–15% of PNH patients, potentially requiring an increased dosage of eculizumab (pharmacokinetic breakthrough); in addition, some in vitro observations suggest that, regardless of its levels, eculizumab may fail to completely prevent intravascular hemolysis in circumstances that evoke massive complement activation (“pharmacodynamic breakthrough”) [36]. A pharmacodynamic breakthrough appears to correspond clinically to hemolytic paroxysms observed at the time of infections in PNH patients under eculizumab treatment. This insufficient response to eculizumab may be attributed to a forceful AP-driven amplification of C3b deposition at high surface densities that curtails the full inhibitory effect of anti-C5 agents [37].
More importantly, anti-C5 treatment has unmasked a novel pathogenic mechanism that partly explains the limited hematologic benefit afforded to some PNH patients by C5 blockade. Persistent C3 opsonization of surviving PNH RBCs coupled to uncontrollable alternative pathway (AP) amplification, resulting from the genetic absence of GPI-linked CD55, may lead to C3-mediated extravascular hemolysis and consequent anemia, which exacerbate the patients’ long-term clinical course [32,33]. Complement regulation remains impaired on PNH erythrocytes as a result of the lack of CD55; more importantly, deregulated C3 fragment deposition on PNH cells promotes the phagocytic engulfment of C3-tagged PNH erythrocytes by hepatosplenic macrophages. It is now well established that extravascular hemolysis fueled by persistent AP dysregulation on PNH cells is the most important cause of the insufficient response to eculizumab [35,38]. Remarkably, this mechanism does not apply only to the one-third of PNH patients who remain transfusion-dependent on eculizumab; indeed, C3 opsonization and subsequent extravascular hemolysis can be documented with variable clinical signs in all PNH patients on eculizumab [34,35]. Recent studies have provided mechanistic insight into the basis for C3-mediated extravascular hemolysis, showing that C3dg-opsonized RBCs from eculizumab-treated PNH patients are recognized and efficiently phagocytozed by macrophages/monocytes in vitro [39].
A genetic basis for the refractory phenotype of certain PNH patients receiving anti-C5 treatment has been identified, with complement gene polymorphic variants (i.e., C5, CR1) implicated as genetic modifiers that skew therapy responses, e.g. by modifying recognition of eculizumab’s epitope on C5 or altering the inherent capacity of PNH cells to withstand C3 activation [40,41]. Overall, even though the clinical experience gained so far with eculizumab has consolidated its clinical efficacy for PNH patients, it has also revealed previously elusive pathogenic mechanisms that cannot be addressed by the standard treatment. These clinical observations have accentuated the need for developing anti-complement agents that can afford broader therapeutic efficacy by intercepting multiple disease-exacerbating processes. This effort may yield significant clinical gains with profound socioeconomic impact, if coupled with drug optimization strategies that can enhance patient compliance (i.e., via alternative dosing routes), and offer wider access to a more affordable therapeutic option.
Novel therapeutic avenues for tackling complement dysregulation in PNH
The need for broadly inhibitory complement therapeutics that can further improve hematological responses in PNH has materialized into a creative toolbox of diversified complement-targeting approaches and drug candidates (Figure 1) [38,42,43]. Being the obvious target for tackling intravascular hemolysis, C5 inhibition has retained the lion’s share in the pipelines of pharmaceutical companies with at least six anti-C5 mAbs in preclinical/clinical development, a small-interfering RNA, and two small molecule-based approaches also being evaluated (Table 1) [38,42]. A second and highly promising approach for clinical intervention aims at blocking the central hub of the cascade, C3, with broadly-inhibiting peptidic drugs that protect the native protein from convertase-mediated activation, thereby abolishing all downstream effector functions. A third promising approach targets the AP, the pathway fueling chronic pathology and residual extravascular hemolysis, with inhibitors that either dismantle the AP C3 convertase (C3bBb) or block the function of individual AP components (factors B and D, or properdin) [38,42,44]. This class of therapeutics includes mAbs, small-molecule inhibitors, as well as engineered fusion proteins encompassing the regulatory activity of endogenous AP regulators, such as Factor H (Table 1) [45].
Figure 1. Schematic overview of the complement activation cascade illustrating targets for therapeutic modulation in PNH.
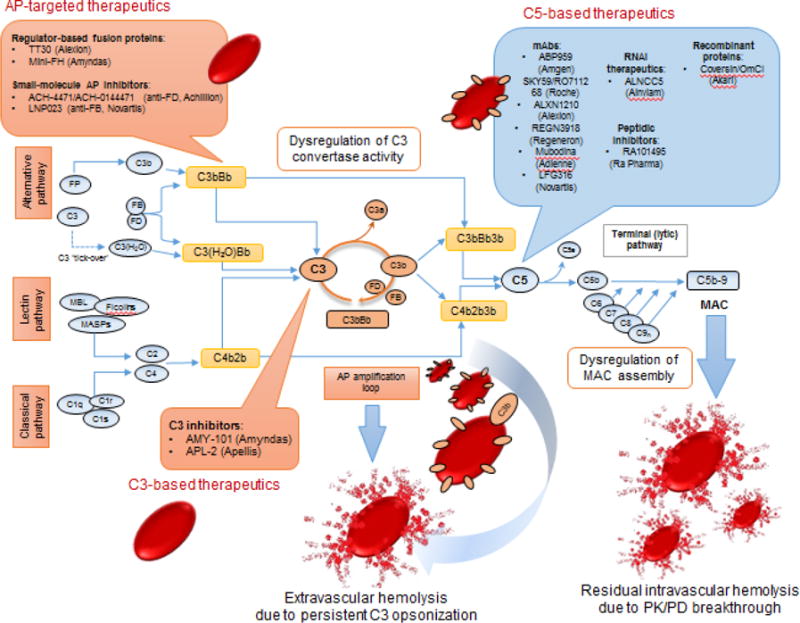
Complement activation via any of the three pathways (Classical, CP, alternative, AP or lectin pathway, LP) triggers a cascade of proteolytic reactions that converge at the cleavage of the central component, C3, by short-lived enzymatic complexes, termed C3 convertases of the CP (C4b3b) or AP (C3bBb). In the absence of the GPI-linked AP regulator DAF/CD55, PNH erythrocytes undergo uncontrollable C3 opsonization that leads to C3b deposition at high surface densities. This AP-amplified opsonic turnover triggers C5 convertase activity that leads to MAC assembly on target cells. In the absence of CD59, regulation of MAC formation on PNH erythrocytes is impaired and consequently this persistent C3 opsonization culminates in chronic, MAC-mediated intravascular hemolysis, one of the cardinal clinical manifestations of PNH. In the era of eculizumab treatment, C3-mediated extravascular hemolysis of C3-opsonized PNH cells in the hepatosplenic compartment has emerged as a significant unmet clinical need for PNH patients who are suboptimal responders to anti-C5 therapy. Furthermore, a fraction of PNH patients may show incomplete response to eculizumab due to residual intravascular hemolysis which is attributed to pharmacodynamic or pharmacologic breakthrough during inhibitor dosing. These unmet needs have sparked the development of next-generation complement therapeutics that can modulate C3 or C5 activation and also target AP convertase activity (shown in callout boxes). In principle, C5 blockage results in complete abrogation of intravascular hemolysis without however affecting upstream AP dysregulation. Conceivably, the application of different anti-C5 agents (or their combination) may offer deeper terminal pathway inhibition and/or improved patient compliance through alternative dosing routes. Upstream complement intervention at the level of C3 is anticipated to afford broader inhibition of complement responses that drive PNH pathology. C3 inhibition can abrogate both intravascular and extravascular C3-mediated hemolysis while AP-targeted inhibitors can also afford broader coverage by controlling AP amplification and downstream effector generation.
Table 1.
Complement therapeutics in various stages of development for PNH and other hematological disorders.
| Complement therapeutic | Entity | Target | Mechanism of action | Mode of administration | Pharmaceutical sponsor | Stage of development |
|---|---|---|---|---|---|---|
| Eculizumab (Soliris) | mAb | C5 | Inhibition of C5 activation/blockag e of C5 convertase activity | IV infusions | Alexion Pharmaceuticals | In the clinic |
| ALXN1210/ravulizumab | mAb | C5 | C5 inhibition/same epitope as ecu | bimonthly IV infusions | Alexion Pharmaceuticals | Phase II/III |
| ABP959 | mAb | C5 | Inhibition of C5 activation/biosimil ar of ecu | IV infusions | Amgen | Phase III |
| SKY59/RO7112689 | mAb | C5 | Inhibition of C5 activation/differen t epitope from ecu | IV and SC injections | Hoffmann-La Roche | Phase I/II |
| LFG-316/tesidolumab | mAb | C5 | Inhibition of C5 activation/differen t epitope from ecu | IV infusions | Novartis | Phase II |
| REGN3918 | mAb | C5 | n.a. | n.a. | Regeneron | Phase I |
| Mubodina | minibody (Fab-based) | C5 | C5 inhibition/different epitope from ecu | n.a. | Adienne | preclinical stage |
| Coversin (OmCI) | recomb. protein | C5 | Inhibition of C5 activation | SC injections | Akari Therapeutics | Phase II |
| RA101495 | peptide | C5 | Allosteric inhibition of C5 activation | daily SC injections | Ra Pharmaceuticals | Phase II |
| Cemdisiran (ALN-CC5) | siRNA | C5 | Silencing of hepatic C5 production | SC injections | Alnylam | Phase II |
| AMY-101 | peptide | C3 | C3 inhibition/blockage of C3 convertase activity | daily SC injections | Amyndas Pharmaceuticals | Phase I completed/Phase II announced |
| APL-2 | PEGylate d peptide | C3 | C3 inhibition/blockage of C3 convertase activity | daily SC injections | Apellis Pharmaceuticals | Phase Ib/II, (Phase III announced) |
| TT30 (CR2/FH) | recomb protein | AP C3 convertase | Surface directed inhibition of AP | SC and IV infusions | Alexion Pharmaceuticals | Phase I (terminated) |
| mini-FH/AMY-201 | recomb protein | AP C3 convertase | Surface directed inhibition of AP | n.a. | Amyndas Pharmaceuticals | preclinical stage |
| LNP023 | small molecule | Factor B | Inhibition of AP C3 convertase formation | orally | Novartis | Phase II |
| ACH-4471/ACH-0144471 | small molecule | Factor D | Inhibition of AP C3 convertase formation | orally | Achillion Pharmaceuticals | Phase II |
| TNT009/BIVV009 | mAb | C1s | CP inhibition/inhibition of C1s protease activity | IV infusions | True North Therapeutics/Bioverat iv | Phase Ib (CAD patients) |
Abbreviations: IV, intravenous; SC, subcutaneous; CP, classical pathway; AP, alternative pathway; siRNA, small interfering RNA; CAD, cold agglutinin disease; n.a., not available
Alternative anti-C5 agents
The ongoing clinical experience with eculizumab has bolstered confidence in complement therapeutics that target C5 [42]. However, the identification of patient sub-groups that fail to optimally respond to eculizumab because of inherited genetic polymorphisms, and the partial hematologic responses observed in certain eculizumab-treated patients as a result of breakthrough hemolysis have accentuated the need for alternative anti-C5 therapeutics that can circumvent such obstacles [35,40]. In this respect, a fully human anti-C5 antibody that binds to a different epitope on C5 than eculizumab (tesidolumab, LFG-316, Novartis) is currently in clinical evaluation as a treatment option for PNH patients refractory to eculizumab therapy [46]. In an effort to improve the bioavailability and plasma residence of anti-C5 agents, Roche, in collaboration with Chugai, has developed a humanized anti-C5 recycling antibody (SKY59/RO7112689) with improved circulatory residence, thanks to a combination of pH-dependent C5 binding and enhanced recycling via the neonatal Fc receptor [47]. Extending the toolbox of antibodies targeting C5 in sites distant from the eculizumab epitope, Adienne has developed a fully human (Fab-based) minibody against C5 (Mubodina) that is still listed as a product in the preclinical discovery phase [48]. On the other hand, Regeneron has recently registered an anti-C5 antibody (REGN3918) into phase 1 trials for PNH without disclosing any information on the exact nature or properties of this agent [49]. Anticipating the expiration of the patent on eculizumab, other companies such as Amgen have focused on developing biosimilars of eculizumab. The anti-C5 Mab ABP959 has been evaluated in Phase I trials [50], and a phase III trial in Europe is currently underway to assess its safety and efficacy in comparison to eculizumab [51]. In addition to antibody-based therapeutics, the toolbox of complement C5 inhibitors has embraced diverse and mechanistically subtle approaches exemplified by peptidic inhibitors, aptamers, recombinant proteins, and RNAi therapeutics [42]. These alternative agents might help circumvent the genetically driven resistance to eculizumab therapy and might also offer advantages in terms of lower production costs and enhanced pharmacokinetic profiles, including the potential for oral administration.
Coversin (OmCI, Akari Therapeutics) is a tick-derived 16-kDa protein that blocks C5 activation and also inhibits leukotriene B4 activity [52]. Coversin has shown preclinical efficacy in in vitro PNH models [53] and has reached Phase II clinical trials, delivered as a subcutaneous injection in untreated PNH patients or poor responders to eculizumab [54]. While the potential immunogenicity of this protein may raise concerns in chronic intervention protocols, no neutralizing antibodies have been reported so far in ongoing trials, with coversin showing biological efficacy and promise as an alternative PNH treatment that could also enable self-administration.
RA101495 (RA Pharma) is a synthetic macrocyclic peptide that binds C5 with high affinity and allosterically inhibits its convertase-mediated cleavage [55]. Daily subcutaneous administration of RA101495 in healthy individuals (Phase I trials) achieved sustained suppression of complement activity [56], and this peptide-based anti-C5 agent is currently being evaluated in two Phase II studies in naive and eculizumab-treated PNH patients, respectively [57,58]. These ongoing studies will discern whether RA101495 can provide therapeutic benefit as a monotherapy or as an add-on therapy in PNH patients with insufficient response to eculizumab. Expanding the C5-targeted therapeutic arsenal, Alnylam has developed a silencing approach for shutting down hepatic C5 production. Cemdisiran (ALN-CC5) is a small-interfering RNA (RNAi) therapeutic that can achieve almost complete inhibition of C5 synthesis by hepatocytes in animal models [59]. Phase 1/2 trials of cemdisiran as a monotherapy in PNH did not appear to reach full efficacy, indicating a potential contribution of locally produced C5 to disease pathogenesis [60] and the need of complete (>99%) C5 inhibition to achieve therapeutic effect. In view of these results, cemdisiran is likely envisioned as a potential add-on to eculizumab that could improve hematological responses in certain poor responders [61].
Capitalizing on the clinical success of eculizumab and in an effort to extend its inhibitory potency across less frequent dosing intervals, Alexion has developed a next-generation, long-acting mAb (ravulizumab, ALXN1210) that shares the same epitope on C5 with eculizumab [62]. Targeted mutagenesis has extended the antibody’s plasma residence through pH-dependent lysosomal release and increased FcRn-mediated recycling [62]. This antibody has shown efficacy in two Phase I/II trials, delivering sustained C5 inhibition that resulted in normalization of key hematological markers of disease [63]. ALXN1210 is being further evaluated in two large Phase III trials that will ascertain its safety and biological efficacy as a therapy for naive PNH patients or as a switch therapy for patients already on eculizumab [64,65]. This new anti-C5 antibody is anticipated to provide a more patient-compliant treatment option for PNH, extending the dosing window of eculizumab from 2 to 8 weeks.
Targeting the core of the cascade: C3 inhibitors
C3 serves as the nodal point of all complement activation pathways [66]. Its activation can trigger a plethora of inflammatory and immunostimulatory processes, with detrimental consequences for the host when regulatory control in the circulation or on host cell surfaces is compromised [66,67]. While C5 blockage abrogates MAC formation thereby preventing intravascular hemolysis, it does not interfere with upstream AP amplification, which is self-perpetuated on PNH cells because of the absence of DAF [68,69]. This process fuels persistent C3 opsonization on PNH cells and culminates in the extravascular phagocytic clearance of C3-osponized PNH cells in the hepatosplenic compartment [39]. In view of this pathogenic mechanism, C3 interception has emerged as a promising and perhaps superior approach for therapeutic intervention in PNH. In principle, agents targeting C3 offer the unique benefit of concomitantly abrogating both intravascular hemolysis and extravascular C3-mediated clearance of PNH RBCs [68]. Hence, this approach may afford greater hematologic benefit to PNH patients than the currently approved treatment.
Compstatin analogues
Since the discovery of the parental compound, compstatin, in the mid 90’s [70], this family of small-sized, peptidic complement inhibitors has grown to include a series of highly potent and selective C3 inhibitors that have propelled the clinical advancement of C3-based therapeutics for PNH and other complement-mediated diseases [71,72]. The discovery, molecular characterization, structure-guided optimization, and most of the preclinical development of these inhibitors were achieved through purely academic endeavours coordinated by the senior author of this paper at the University of Pennsylvania [71,73]. All next-generation compstatin analogs have been built on the scaffold of the original compstatin, a cyclic 13-aminoacid peptide with strong affinity and selectivity for human and non-human primate (NHP) C3 [71]. The most recent analogs have incorporated unnatural aminoacids to increase the inhibitory potency, and possess an extended N–terminus that can afford additional contacts with C3, thus further improving their binding affinity [73]. Compstatin analogs bind to a shallow pocket formed between the MG4 and MG5 domains of the β chain of C3 and impair the binding of the native protein to assembled C3 convertases, regardless of the initiating complement pathway [74]. In this way, compstatin-based drugs prevent propagation and amplification of complement responses and blunt effector generation via any of the three activation pathways (CP, LP, or AP) (Figure 1).
The third-generation compstatin analog Cp40 (clinicaly developed as AMY-101, Amyndas Pharmaceuticals) has demonstrated sustained C3 inhibition and consistent therapeutic efficacy in a number of in vitro, ex vivo, and in vivo (NHP) models of complement-mediated activation [71]. The pharmacological studies conducted with Cp40/AMY-101 include: i) an ex vivo model of PNH (discussed below); ii) an in vitro model of C3 glomerulopathy in which Cp40 effectively restored complement regulation and ameliorated key pathological drivers that promote kidney injury and inflammation [75]; iii) an in vivo NHP model of hemodialysis-induced inflammation in which two discrete Cp40 treatment regimens have led to complete inhibition of hemodialysis-induced complement activation [76]; iv) inhibition of heme-induced complement activation and malarial inflammation [77]; v) early organ protection and improved clinical outcome in an NHP model of trauma-induced hemorrhagic shock [78] and vi) inducible and natural NHP models of periodontitis, in which local application of AMY-101 inhibited complement activation, attenuated clinical indices of periodontal inflammation and, more importantly, decreased inflammatory bone loss [79].
C3-based inhibition has shown clinical promise as a new treatment option for PNH. The Cp40 analog abrogated MAC-mediated hemolysis of PNH RBCs and completely prevented C3 deposition on surviving RBCs from PNH patients receiving anti-C5 therapy (eculizumab) [80]. In this ex vivo PNH model, fresh RBCs obtained from PNH patients were exposed to complement activation, paralleling -or even exceeding- physiological levels of complement activation (i.e., the continuous low-level activation due to spontaneous hydrolysis of C3, also known as C3 “tick-over”) occurring in humans. AMY-101 completely abrogated hemolysis with an efficacy better than that observed with eculizumab under similar conditions; moreover, and in contrast to eculizumab, C3 opsonization on RBCs was completely abolished [80]. PK/PD studies of Cp40 in non-human primates revealed favorable drug elimination profiles, safety, and inhibitory efficacy after repeated subcutaneous administration, making this compound (AMY-101/Cp40) amenable to chronic application [68,73]. With a target affinity in the subnanomolar range, almost 6,000-fold greater that the first-generation compstatin, and an extended plasma half-life of more than 50 h, Cp40 is well suited for prolonged therapeutic application [42,71]. Notably, this pharmacokinetic profile obviates the need for further peptide modifications of Cp40, such as PEGylation, that could entail the risk of eliciting adverse events related to PEG-specific immune responses over extended dosing periods. Furthermore, the pharmacokinetic behaviour of Cp40 potentially enables better control in a clinical setting (i.e, interruption of therapy and swift recovery of complement activity in the case of adverse events) and may have important implications for tissue distribution, dosing, and administration in a disease-tailored context. Given that the production costs of unmodified Cp40 are expected to be lower than those of PEGylated peptides, this drug candidate is well-poised for paving the way to a broadly efficacious and affordable C3-targeted therapy for PNH.
After receiving orphan drug designation for C3G and PNH [81,82], the Cp40-based drug candidate AMY-101 (Amyndas) entered clinical development in 2017. An open-label first–inhuman clinical trial assesed the safety, tolerability, and PK/PD profile of AMY-101 after a single ascending dose (SAD) and multiple doses (MD) administered systemically in healthy volunteers, using both I.V. and S.C. dosing routes [83]. According to preliminary results released by Amyndas, AMY-101 dosing displayed safety and a PK profile that can support further clinical development with sustained C3 inhibition through s.c. dosing every 48 hours [84]. Amyndas has announced plans for Phase II studies of AMY-101 in both untreated PNH patients and patients with poor clinical response to eculizumab, as well as in ABO-incompatible kidney transplantation, periodontitis, and C3 glomeruolopathy [84]. In addition, fourth-generation compstatin analogs are in preclinical development for various indications.
Further credence to the clinical translatability of peptidic C3 inhibitors has been provided by the clinical development of the second-generation compstatin analog 4(1MeW)7W/POT-4, which was licensed to Apellis by the University of Pennsylvania. This drug candidate has been developed as a long-acting, PEGylated derivative for several indications, including PNH and age-related macular degeneration (AMD) (APL-2, Apellis Pharmaceuticals). APL-2 is currently being evaluated in two Phase Ib trials, both as a monotherapy and “add-on” therapy in PNH patients who are poor responders to eculizumab [85,86]. According to preliminary results released by Apellis, daily s.c. dosing of APL-2 has resulted in improvement of key markers of hematologic response (e.g., LDL and Hb levels), both in naive PNH patients and in poor responders to eculizumab [87]. While more concrete data in terms of patient follow-up and optimum choice of therapy (add-on versus monotherapy) have yet to be provided, Apellis has announced plans for advancing this inhibitor to Phase III trials for PNH [87]. Notably, C3 inhibitors have also shown promise as a treatment option for geographic atrophy (GA), an advanced form of AMD [71]. APL-2 met the primary endpoint of lesion reduction in a large multicenter Phase II study following a monthly intravitreal dosing scheme in patients with GA [88].Taken together, these studies attest to the initial safety and feasibility of clinical C3-based intervention, paving the way to more comprehensive, affordable, and patient-compliant complement therapies.
Targeted AP inhibitors for PNH
Surface-directed engineered regulators
The cardinal role of chronic AP dysregulation in PNH pathogenesis has galvanized efforts to develop targeted AP inhibitors that can intercept convertase activity in close proximity to complement-opsonized surfaces [45]. In this respect, several groups have designed chimeric regulator of complement activation (RCA)-type inhibitors exploiting the capacity of endogenous regulators (i.e., Factor H), to destabilize C3 convertases and degrade C3b [45]. Indeed, the opsonin-targeted FH derivative TT30 (Taligen/Alexion) was one of the first engineered AP regulators to reach clinical trials for PNH in the post-eculizumab era [89]. TT30 consists of the complement regulatory domains of FH (CCPs 1-5) fused to the iC3b/C3dg binding region of complement receptor 2 (CR2) (CCPs 1-4) [90]. Although TT30 demonstrated complete abrogation of intravascular hemolysis and C3 opsonization of PNH cells in vitro and showed pharmacological activity in a Phase I trial in untreated PNH patients, its clinical program was terminated for undisclosed reasons, possibly including the very short half-life of this chimeric protein in circulation [91]
Embracing the same concept of surface-directed AP modulation, research efforts have yielded a miniaturized form of human FH (mini-FH/AMY-201, Amyndas) engineered to exert its inhibitory activity on opsonized cells after systemic administration [92]. Mini-FH consists of the regulatory and surface-recognizing segments of FH connected together by a peptide linker. Despite a significant reduction in size, this engineered regulator retains a high or even superior binding affinity for C3-derived opsonic fragments and it has shown therapeutic efficacy abrogating both intravascular hemolysis and C3 opsonization in PNH models [93].
Factor D/Factor B inhibitors
Instead of modulating convertase activity at the C3 breakdown level, one can effectively block convertase assembly by targeting any of its assembling elements. In this respect, small-molecule drug discovery platforms that target serine proteases involved in convertase formation (i.e., Factors B and D) have attracted considerable attention by pharmaceutical companies [42]. FD is considered an attractive drug target due to its relatively low plasma concentration, high specificity, and bottleneck role in AP C3 convertase assembly [94]. Novartis has developed potent, orally available, and highly selective small-molecule FD and factor B inhibitors that have shown efficacy in preventing AP dysregulation on PNH cells in vitro [95,96]. A Phase II study to assess the safety, PK/PD profile, and efficacy of the factor B inhibitor LNP023 as add-on therapy for patients with insufficient response to eculizumab has recently been announced by Novartis (EudraCT Number 2017-000888-33). It remains to be seen whether upstream AP modulation will synergize with anti-C5 therapy in abrogating the residual intravascular hemolysis observed in these patients.
Achillion has adopted a similar approach introducing orally bioavailable small FD inhibitors that have shown efficacy in modulating AP activity in preclinical PNH and aHUS models [97]. One of these FD inhibitors, ACH-4471/ACH-0144471 (Achillion) has shown efficacy in abrogating intravascular hemolysis and attenuating C3 opsonization in PNH models [97] and has entered clinical development for PNH as an orally administered therapeutic. Following a Phase I study that established its safety and tolerability in healthy volunteers, ACH-4471 is being evaluated in two ongoing Phase II trials as a single agent for PNH treatment [98]. Achillion has also announced a Phase II trial to evaluate ACH-4471 in combination with eculizumab (EudraCTNumber 2016-003526-16), based on in vitro results suggesting a potential synergistic effect with eculizumab in improving clinical responses [99]. In view of the recently described FD bypass pathway that enables kallikrein-mediated AP activation in the absence of FD [100] and the failure of the FD-blocking antibody lampalizumab to meet the primary endpoint in one large Phase III trial in patients with GA [101], particular caution should be exercised when projecting the therapeutic outcome of such inhibitors in pathologies involving aberrant AP activation. In principle, factor D/B inhibitors are anticipated to provide broad coverage against both intravascular hemolysis and extravascular clearance of C3-opsonized PNH erythrocytes in patients with PNH. Actual clinical data from ongoing clinical trials evaluating targeted AP inhibitors in PNH patients are therefore highly anticipated.
While inhibitors targeting upstream complement components, such as C3 or FD/FB, offer the promise of a broader control of complement-mediated anaemia and may therefore prove to be efficacious as a monotherapy, there is still a possibility that sub-total inhibition of complement activity at the level of two discrete components (e.g. combined anti-C3 and anti-C5 therapy) would also result in a similar clinical benefit for PNH patients, maintaining also residual complement activity for pathogen immune surveillance. This plausible scenario is worth investigating in future clinical trials.
Translational considerations
Anti-C5 therapy has drastically improved the clinical landscape of PNH affording significant clinical gains as the first complement-specific treatment approved for this debilitating disorder [19,29]. However, the unmasking of previously elusive pathogenic mechanisms that limit the hematologic response of patients, the necessity for more patient-compliant therapies and emerging genetic obstacles that limit patients’ responsiveness to eculizumab have all set forth important scientific questions regarding the optimal choice of therapeutic agents, dosing routes, and treatment algorithms and whether solid scientific rationale lies behind these emerging therapeutic concepts and targets [35,38].
New anti-C5 agents are being evaluated with the goal of achieving a comparable clinical response to that of eculizumab, likely improving issues related to dosing frequency and patient compliance through self-administration of the drug. For example, the development of long-acting anti-C5 antibodies, such as ALXN1210, or other anti-C5 agents delivered via alternative systemic routes (e.g. S.C.) (e.g. coversin, ALNCC5) might signify a major leap forward in terms of increasing patient compliance. The possible combination of anti-C5 drugs might also offer a more effective means of controlling residual terminal pathway activity in case of PK or PD breakthrough. To this end, many of these agents are being clinically evaluated as add-on regimens to eculizumab therapy. Recent studies have shown that in settings of pronounced complement activation, high C3b densities on the target surface can lead to forceful C5 convertase generation in a way that overrides the full inhibitory effect of anti-C5 agents such as eculizumab [37]. Complete abrogation of residual hemolysis can be achieved in these cases by simultaneous use of C5 inhibitors. This perspective might be worth investigating in PNH, given the availability of multiple anti-C5 agents in the drug development pipeline [38]. Genetic resistance due to C5 polymorphisms located within the eculizumab binding epitope may further limit clinical responses [40]. The availability of new anti-C5 agents targeting sites on C5 distal to the epitope of the drug might offer an important alternative for improving clinical responses. Undeniably, the main pathogenic mechanism that remains unaddressed by the standard treatment is C3-mediated extravascular hemolysis, which leads to partial hematologic responses in ~30% of PNH patients under eculizumab treatment [33]. In this context, the clinical advancement of C3-based or AP-targeted therapeutics may constitute a major milestone for PNH therapy, since this strategy will likely afford broader clinical gains in terms of abrogating intravascular hemolysis and intercepting residual anemia resulting from extravascular clearance of C3-opsonized PNH cells [68]. Of note, peptide-based therapeutics (i.e., compstatins) may likely develop into a relatively affordable and broadly accessible treatment option, given the projected lower costs for large-scale peptide manufacturing nowadays [71]. The availability of orally active factor B/factor D-targeting agents provides an option for increasing patient compliance. Furthermore, selective targeting of the AP may allow for maintenance of antimicrobial surveillance in PNH patients through an intact CP or LP. Conversely, the use of such AP-specific agents might come with the risk of fueling hemolytic paroxysms through opportunistic, forceful CP or LP activation.
Longstanding, yet largely hypothetical, discussions concerning the safety of C3 therapeutics need to be placed into a clinically validated context. While clinical observations from rare cases of primary complement deficiencies have indicated an increased susceptibility to certain bacterial infections, especially during early age, this phenotype appears to subside during adulthood, likely due to compensatory mechanisms of pathogen immune surveillance [102,103]. It should be stressed that complement-based pharmacologic intervention cannot recapitulate the phenotype of an inherited deficiency. Importantly, the clinical experience gained with eculizumab should similarly guide antimicrobial prophylaxis in the case of C3 intervention. A comprehensive vaccination program directed against highly virulent encapsulated bacteria such as meningococci, streptococci, and Haemophilus influenza should provide ample antimicrobial coverage in prolonged C3-targeted intervention protocols. A cautionary note, however, is necessary regarding the variable bactericidal activity elicited against meningococci in immunized patients, depending on the mode of complement inhibition [104]. All in all, one should avoid extrapolating clinical phenotypes from theoretical discussions about safety before obtaining actual clinical data from trials evaluating C3-based inhibitors. Of note, preliminary results from small ongoing trials have not revealed any treatment-related adverse events, supporting a favorable safety profile for anti-C3 agents. Overall, long-term monitoring of PNH patients treated with anti-C3 agents will ultimately decide the safety and clinical efficacy of C3 intervention.
Concluding remarks and outlook
Undeniably, the clinical approval of eculizumab has drastically changed the clinical management of PNH, offering an effective therapy that tackles the main clinical hallmark of the disease, intravascular hemolysis. Following this clinical breakthrough, a series of next-generation complement therapeutics have been thrust into the limelight of clinical research with the goal of improving PNH management. More than 10 drug candidates are currently in clinical development for PNH, marking a remarkable resurgence of complement therapeutics. Upstream complement intervention, either at the level of C3 or through targeted AP modulation, offers the promise of broader therapeutic coverage against established and emerging pathogenic mechanisms and may therefore represent a tangible new milestone for further improving the management of PNH patients. It is foreseeable that once new complement therapeutics become approved for PNH, they will likely translate into clinical benefit for more complement-mediated indications that are currently in need of an etiologic therapy or lack effective treatment. These indications include, among others, hematological disorders fueled by autoimmune factors (e.g. cold agglutinin disease, CAD) a spectrum of rare renal pathologies driven by AP dysregulation (i.e., C3 glomerulopathy) and transplant or biomaterial-triggered thromboinflammatory complications (e.g. AMR-induced allograft rejection). Further raising optimism about this approach, drug leads specifically targeting the CP are now in clinical development as another viable option for tackling complement-driven pathology in hematological diseases. Indeed, an anti-C1s antibody (TNT009/BIVV009, True North Therapeutics/Bioverativ) is currently in Phase Ib trials as a treatment option for patients with CAD, a rare autoimmune hemolytic anemia that lacks approved therapy [105].
The integration of solid clinical data from ongoing trials with refined diagnostic algorithms for monitoring complement activity will enable a more comprehensive and unbiased evaluation of the clinical efficacy of these inhibitors. The increasing appreciation of the impact of patient-specific genetic variance on clinical responses to anti-complement agents will enable a more rational stratification strategy for enrolling those patients in clinical trials with the greatest chance to benefit from each anti-complement agent. Hopefully the next decade will witness a long-awaited expansion of the arsenal of clinically approved complement therapeutics for the treatment of PNH and other complement-driven diseases.
Acknowledgments
We thank Dr. Deborah McClellan for her excellent editorial assistance. The authors’ work was supported by grants from the US National Institutes of Health (AI068730, AI030040), the US National Science Foundation (grant No. 1423304), as well as from the Aplastic Anemia & Myelodysplastic Syndrome (AAMDS) International Foundation and the Italian PNH Association (AIEPN).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure statement
J.D.L. and A.M.R. are inventors of patents or patent applications that describe the use of complement inhibitors for therapeutic purposes, some of which are developed by Amyndas Pharmaceuticals. J.D.L. is the founder of Amyndas Pharmaceuticals, which is developing complement inhibitors, including third-generation compstatin analogues such as AMY-101, for the treatment of complement-mediated diseases. J.D.L. is also the inventor of the compstatin technology licensed to Apellis Pharmaceuticals [4(1MeW)7W, also known as POT-4 and APL-1 and PEGylated derivatives such as APL-2]. A.M.R. has received research support from Alexion, Alnylam, Rapharma and Novartis; A.M.R. has received lecture fees and serves as member of an investigator board for Alexion, Novartis and Roche. AMR is involved as an investigator in clinical trials evaluating the following agents: TT30, ALXN1210, SKY59, ACH-4471, LNP023 and AMY-101. The rest of the authors have no competing interests.
References
- 1.Takeda J, Miyata T, Kawagoe K, Iida Y, Endo Y, Fujita T, et al. Deficiency of the GPI anchor caused by a somatic mutation of the PIG-A gene in paroxysmal nocturnal hemoglobinuria. Cell. 1993 May 21;73(4):703–11. doi: 10.1016/0092-8674(93)90250-t. [DOI] [PubMed] [Google Scholar]
- 2.Miyata T, Takeda J, Iida Y, Yamada N, Inoue N, Takahashi M, et al. The cloning of PIG-A, a component in the early step of GPI-anchor biosynthesis. Science. 1993 Feb 26;259(5099):1318–20. doi: 10.1126/science.7680492. [DOI] [PubMed] [Google Scholar]
- 3.Medof ME, Gottlieb A, Kinoshita T, Hall S, Silber R, Nussenzweig V, et al. Relationship between decay accelerating factor deficiency, diminished acetylcholinesterase activity, and defective terminal complement pathway restriction in paroxysmal nocturnal hemoglobinuria erythrocytes. J Clin Invest. 1987 Jul;80(1):165–74. doi: 10.1172/JCI113043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Risitano AM. Paroxysmal nocturnal hemoglobinuria and other complement-mediated hematological disorders. Immunobiology. 2012 Nov;217(11):1080–7. doi: 10.1016/j.imbio.2012.07.014. [DOI] [PubMed] [Google Scholar]
- 5.Parker C, Omine M, Richards S, Nishimura J, Bessler M, Ware R, et al. Diagnosis and management of paroxysmal nocturnal hemoglobinuria. Blood. 2005 Dec 1;106(12):3699–709. doi: 10.1182/blood-2005-04-1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Luzzatto L, Gianfaldoni G, Notaro R. Management of paroxysmal nocturnal haemoglobinuria: a personal view. Br J Haematol. 2011 Jun;153(6):709–20. doi: 10.1111/j.1365-2141.2011.08690.x. [DOI] [PubMed] [Google Scholar]
- 7.Hill A, Richards SJ, Hillmen P. Recent developments in the understanding and management of paroxysmal nocturnal haemoglobinuria. Br J Haematol. 2007 May;137(3):181–92. doi: 10.1111/j.1365-2141.2007.06554.x. [DOI] [PubMed] [Google Scholar]
- 8.Rotoli B, Luzzatto L. Paroxysmal nocturnal haemoglobinuria. Baillieres Clin Haematol. 1989 Jan;2(1):113–38. doi: 10.1016/s0950-3536(89)80010-1. [DOI] [PubMed] [Google Scholar]
- 9.Araten DJ, Nafa K, Pakdeesuwan K, Luzzatto L. Clonal populations of hematopoietic cells with paroxysmal nocturnal hemoglobinuria genotype and phenotype are present in normal individuals. Proc Natl Acad Sci U S A. 1999 Apr 27;96(9):5209–14. doi: 10.1073/pnas.96.9.5209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Araten DJ, Bessler M, McKenzie S, Castro-Malaspina H, Childs BH, Boulad F, et al. Dynamics of hematopoiesis in paroxysmal nocturnal hemoglobinuria (PNH): no evidence for intrinsic growth advantage of PNH clones. Leukemia. 2002 Nov;16(11):2243–8. doi: 10.1038/sj.leu.2402694. [DOI] [PubMed] [Google Scholar]
- 11.Luzzatto L, Bessler M, Rotoli B. Somatic mutations in paroxysmal nocturnal hemoglobinuria: a blessing in disguise? Cell. 1997 Jan 10;88(1):1–4. doi: 10.1016/s0092-8674(00)81850-4. [DOI] [PubMed] [Google Scholar]
- 12.Young NS, Maciejewski JP. Genetic and environmental effects in paroxysmal nocturnal hemoglobinuria: this little PIG-A goes “Why? Why? Why?”. J Clin Invest. 2000 Sep;106(5):637–41. doi: 10.1172/JCI11002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lewis SM, Dacie JV. The aplastic anaemia–paroxysmal nocturnal haemoglobinuria syndrome. Br J Haematol. 1967 Mar;13(2):236–51. doi: 10.1111/j.1365-2141.1967.tb08736.x. [DOI] [PubMed] [Google Scholar]
- 14.Karadimitris A, Manavalan JS, Thaler HT, Notaro R, Araten DJ, Nafa K, et al. Abnormal T-cell repertoire is consistent with immune process underlying the pathogenesis of paroxysmal nocturnal hemoglobinuria. Blood. 2000 Oct 1;96(7):2613–20. [PubMed] [Google Scholar]
- 15.Gargiulo L, Lastraioli S, Cerruti G, Serra M, Loiacono F, Zupo S, et al. Highly homologous T-cell receptor beta sequences support a common target for autoreactive T cells in most patients with paroxysmal nocturnal hemoglobinuria. Blood. 2007 Jun 1;109(11):5036–42. doi: 10.1182/blood-2006-10-052381. [DOI] [PubMed] [Google Scholar]
- 16.Gargiulo L, Papaioannou M, Sica M, Talini G, Chaidos A, Richichi B, et al. Glycosylphosphatidylinositol-specific, CD1d-restricted T cells in paroxysmal nocturnal hemoglobinuria. Blood. 2013 Apr 4;121(14):2753–61. doi: 10.1182/blood-2012-11-469353. [DOI] [PubMed] [Google Scholar]
- 17.Risitano AM. Paroxysmal nocturnal hemoglobinuria and the complement system: recent insights and novel anticomplement strategies. Adv Exp Med Biol. 2013;735:155–72. doi: 10.1007/978-1-4614-4118-2_10. [DOI] [PubMed] [Google Scholar]
- 18.Risitano AM, Rotoli B. Paroxysmal nocturnal hemoglobinuria: pathophysiology, natural history and treatment options in the era of biological agents. Biologics. 2008 Jun;2(2):205–22. doi: 10.2147/btt.s1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hillmen P, Lewis SM, Bessler M, Luzzatto L, Dacie JV. Natural history of paroxysmal nocturnal hemoglobinuria. N Engl J Med. 1995 Nov 9;333(19):1253–8. doi: 10.1056/NEJM199511093331904. [DOI] [PubMed] [Google Scholar]
- 20.Socie G, Mary JY, de GA, Rio B, Leporrier M, Rose C, et al. Paroxysmal nocturnal haemoglobinuria: long-term follow-up and prognostic factors. French Society of Haematology. Lancet. 1996 Aug 31;348(9027):573–7. doi: 10.1016/s0140-6736(95)12360-1. [DOI] [PubMed] [Google Scholar]
- 21.Hill A, Kelly RJ, Hillmen P. Thrombosis in paroxysmal nocturnal hemoglobinuria. Blood. 2013 Jun 20;121(25):4985–96. doi: 10.1182/blood-2012-09-311381. [DOI] [PubMed] [Google Scholar]
- 22.Rother RP, Rollins SA, Mojcik CF, Brodsky RA, Bell L. Discovery and development of the complement inhibitor eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria. Nat Biotechnol. 2007 Nov;25(11):1256–64. doi: 10.1038/nbt1344. [DOI] [PubMed] [Google Scholar]
- 23.Frei Y, Lambris JD, Stockinger B. Generation of a monoclonal antibody to mouse C5 application in an ELISA assay for detection of anti-C5 antibodies. Mol Cell Probes. 1987 Jun;1(2):141–9. doi: 10.1016/0890-8508(87)90022-3. [DOI] [PubMed] [Google Scholar]
- 24.Wang Y, Rollins SA, Madri JA, Matis LA. Anti-C5 monoclonal antibody therapy prevents collagen-induced arthritis and ameliorates established disease. Proc Natl Acad Sci U S A. 1995 Sep 12;92(19):8955–9. doi: 10.1073/pnas.92.19.8955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang H, Arp J, Liu W, Faas SJ, Jiang J, Gies DR, et al. Inhibition of terminal complement components in presensitized transplant recipients prevents antibody-mediated rejection leading to long-term graft survival and accommodation. J Immunol. 2007 Oct 1;179(7):4451–63. doi: 10.4049/jimmunol.179.7.4451. [DOI] [PubMed] [Google Scholar]
- 26.Rinder CS, Rinder HM, Smith BR, Fitch JC, Smith MJ, Tracey JB, et al. Blockade of C5a and C5b-9 generation inhibits leukocyte and platelet activation during extracorporeal circulation. J Clin Invest. 1995 Sep;96(3):1564–72. doi: 10.1172/JCI118195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hillmen P, Young NS, Schubert J, Brodsky RA, Socie G, Muus P, et al. The complement inhibitor eculizumab in paroxysmal nocturnal hemoglobinuria. N Engl J Med. 2006 Sep 21;355(12):1233–43. doi: 10.1056/NEJMoa061648. [DOI] [PubMed] [Google Scholar]
- 28.Brodsky RA, Young Antonioli E, Risitano AM, Schrezenmeier H, Schubert J, et al. Multicenter phase 3 study of the complement inhibitor eculizumab for the treatment of patients with paroxysmal nocturnal hemoglobinuria. Blood. 2008 Feb 15;111(4):1840–7. doi: 10.1182/blood-2007-06-094136. [DOI] [PubMed] [Google Scholar]
- 29.Hillmen P, Muus P, Duhrsen U, Risitano AM, Schubert J, Luzzatto L, et al. Effect of the complement inhibitor eculizumab on thromboembolism in patients with paroxysmal nocturnal hemoglobinuria. Blood. 2007 Dec 1;110(12):4123–8. doi: 10.1182/blood-2007-06-095646. [DOI] [PubMed] [Google Scholar]
- 30.Loschi M, Porcher R, Barraco F, Terriou L, Mohty M, de GS, et al. Impact of eculizumab treatment on paroxysmal nocturnal hemoglobinuria: a treatment versus no-treatment study. Am J Hematol. 2016 Jun;91(4):366–70. doi: 10.1002/ajh.24278. [DOI] [PubMed] [Google Scholar]
- 31.Brodsky RA. How I treat paroxysmal nocturnal hemoglobinuria. Blood. 2009 Jun 25;113(26):6522–7. doi: 10.1182/blood-2009-03-195966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hill A, Rother RP, Arnold L, Kelly R, Cullen MJ, Richards SJ, et al. Eculizumab prevents intravascular hemolysis in patients with paroxysmal nocturnal hemoglobinuria and unmasks low-level extravascular hemolysis occurring through C3 opsonization. Haematologica. 2010 Apr;95(4):567–73. doi: 10.3324/haematol.2009.007229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Risitano AM, Notaro R, Marando L, Serio B, Ranaldi D, Seneca E, et al. Complement fraction 3 binding on erythrocytes as additional mechanism of disease in paroxysmal nocturnal hemoglobinuria patients treated by eculizumab. Blood. 2009 Apr 23;113(17):4094–100. doi: 10.1182/blood-2008-11-189944. [DOI] [PubMed] [Google Scholar]
- 34.Luzzatto L, Risitano AM, Notaro R. Paroxysmal nocturnal hemoglobinuria and eculizumab. Haematologica. 2010 Apr;95(4):523–6. doi: 10.3324/haematol.2009.017848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Risitano AM, Notaro R, Luzzatto L, Hill A, Kelly R, Hillmen P. Paroxysmal nocturnal hemoglobinuria–hemolysis before and after eculizumab. N Engl J Med. 2010 Dec 2;363(23):2270–2. doi: 10.1056/NEJMc1010351. [DOI] [PubMed] [Google Scholar]
- 36.Seregina EA, Tsvetaeva NV, Nikulina OF, Zapariy AP, Erasov AV, Gribkova IV, et al. Eculizumab effect on the hemostatic state in patients with paroxysmal nocturnal hemoglobinuria. Blood Cells Mol Dis. 2015 Feb;54(2):144–50. doi: 10.1016/j.bcmd.2014.11.021. [DOI] [PubMed] [Google Scholar]
- 37.Harder MJ, Kuhn N, Schrezenmeier H, Hochsmann B, von Z I, Weinstock C, et al. Incomplete inhibition by eculizumab: mechanistic evidence for residual C5 activity during strong complement activation. Blood. 2017 Feb 23;129(8):970–80. doi: 10.1182/blood-2016-08-732800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Risitano AM, Marotta S. Toward complement inhibition 2.0: Next generation anticomplement agents for paroxysmal nocturnal hemoglobinuria. Am J Hematol. 2018 Jan 4; doi: 10.1002/ajh.25016. [DOI] [PubMed] [Google Scholar]
- 39.Lin Z, Schmidt CQ, Koutsogiannaki S, Ricci P, Risitano AM, Lambris JD, et al. Complement C3dg-mediated erythrophagocytosis: implications for paroxysmal nocturnal hemoglobinuria. Blood. 2015 Jun 16; doi: 10.1182/blood-2015-02-625871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nishimura J, Yamamoto M, Hayashi S, Ohyashiki K, Ando K, Brodsky AL, et al. Genetic variants in C5 and poor response to eculizumab. N Engl J Med. 2014 Feb 13;370(7):632–9. doi: 10.1056/NEJMoa1311084. [DOI] [PubMed] [Google Scholar]
- 41.Rondelli T, Risitano AM, Peffault de LR, Sica M, Peruzzi B, Ricci P, et al. Polymorphism of the complement receptor 1 gene correlates with the hematologic response to eculizumab in patients with paroxysmal nocturnal hemoglobinuria. Haematologica. 2014 Feb;99(2):262–6. doi: 10.3324/haematol.2013.090001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ricklin D, Mastellos DC, Reis ES, Lambris JD. The renaissance of complement therapeutics. Nat Rev Nephrol. 2018 Jan;14(1):26–47. doi: 10.1038/nrneph.2017.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Morgan BP, Harris CL. Complement, a target for therapy in inflammatory and degenerative diseases. Nat Rev Drug Discov. 2015 Dec;14(12):857–77. doi: 10.1038/nrd4657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mastellos DC, Reis ES, Yancopoulou D, Hajishengallis G, Ricklin D, Lambris JD. From orphan drugs to adopted therapies: Advancing C3-targeted intervention to the clinical stage. Immunobiology. 2016 Oct;221(10):1046–57. doi: 10.1016/j.imbio.2016.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schmidt CQ, Lambris JD, Ricklin D. Protection of host cells by complement regulators. Immunol Rev. 2016 Nov;274(1):152–71. doi: 10.1111/imr.12475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Novartis Pharmaceuticals. Proof of Concept Study to Assess the Efficacy, Safety and Pharmacokinetics of LFG316 in Patients With Paroxysmal Nocturnal Hemoglobinuria. 2018 https://clinicaltrials.gov/ct2/show/NCT02534909.
- 47.Fukuzawa T, Sampei Z, Haraya K, Ruike Y, Shida-Kawazoe M, Shimizu Y, et al. Long lasting neutralization of C5 by SKY59, a novel recycling antibody, is a potential therapy for complement-mediated diseases. Sci Rep. 2017 Apr 24;7(1):1080. doi: 10.1038/s41598-017-01087-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Adienne Pharma and Biotech. 2018 http://www.adienne.com/rnd/molecules-in-development/mubodina.
- 49.Regeneron Pharmaceuticals. 2018 http://adisinsight.springer.com/drugs/800049599.
- 50.Amgen. A Randomized, Double-Blind, Single-Dose, 3-Arm, Parallel Group Study to Determine the Pharmacokinetic Similarity of ABP 959 and Eculizumab (Soliris Registered Trademark) in Healthy Male Subjects. 2018 doi: 10.1111/ejh.13411. https://www.anzctr.org.au/Trial/Registration/TrialReview.aspx?id=369851. [DOI] [PMC free article] [PubMed]
- 51.Amgen. A RANDOMIZED, DOUBLE-BLIND, ACTIVE-CONTROLLED PHASE 3 STUDY EVALUATING THE EFFICACY AND SAFETY OF ABP 959 COMPARED WITH ECULIZUMAB IN ADULT SUBJECTS WITH PAROXYSMAL NOCTURNAL HEMOGLOBINURIA (PNH) 2018 https://www.clinicaltrialsregister.eu/ctr-search/trial/2017-001418-27/ES.
- 52.Nunn MA, Sharma A, Paesen GC, Adamson S, Lissina O, Willis AC, et al. Complement inhibitor of C5 activation from the soft tick Ornithodoros moubata. J Immunol. 2005 Feb 15;174(4):2084–91. doi: 10.4049/jimmunol.174.4.2084. [DOI] [PubMed] [Google Scholar]
- 53.Weston-Davies WH, Nunn MA, Pinto FO, et al. Clinical and Immunological Characterisation of Coversin, a Novel Small Protein Inhibitor of Complement C5 with Potential As a Therapeutic Agent in PNH and Other Complement Mediated Disorders. Blood. 2014;124:4280. [Google Scholar]
- 54.Hill A, Weston-Davies WH, Nunn M, et al. Coversin, a Novel C5 Complement Inhibitor, Is Safe and Effective in the Treatment of PNH: Results of a Phase II Clinical Trial. Blood. 2017;130:4747. [Google Scholar]
- 55.Ricardo A, Arata M, DeMarco S, et al. Preclinical Evaluation of RA101495, a Potent Cyclic Peptide Inhibitor of C5 for the Treatment of Paroxysmal Nocturnal Hemoglobinuria. Blood. 2015;126:939. [Google Scholar]
- 56.Johnston JM, Ricardo A, Arata M, et al. A Phase 1 Multiple-dose Clinical Study of RA101495, a Subcutaneously Administered Synthetic Macrocyclic Peptide Inhibitor of Complement C5 for Treatment of Paroxysmal Nocturnal Hemoglobinuria. Haematologica. 2016;101(s1):415. [Google Scholar]
- 57.Ra Pharmaceuticals. Phase 2 Safety and Efficacy Study of RA101495 to Treat PNH Patients. 2017 https://clinicaltrials.gov/ct2/show/NCT03078582?term=RA101495&rank=3.
- 58.Ra Pharmaceuticals. Phase 2 Safety and Efficacy Study of RA101495 to Treat PNH Patients Who Have an Inadequate Response to Eculizumab. 2017 https://clinicaltrials.gov/ct2/show/NCT03030183?term=RA101495&rank=1.
- 59.Borodovsky A, Yucius K, Sprague A, et al. Development Of RNAi Therapeutics Targeting The Complement Pathway. Blood. 2013;122:2471. [Google Scholar]
- 60.Hill A, Valls AG, Griffin M, et al. A Subcutaneously Administered Investigational RNAi Therapeutic (ALN-CC5) Targeting Complement C5 for Treatment of PNH and Complement-Mediated Diseases: Preliminary Phase 1/2 Study Results in Patients with PNH. Blood. 2016;128:3891. [Google Scholar]
- 61.Alnylam Pharmaceuticals. A Phase 2, Open-label, Single Dose, Study of Subcutaneously Administered ALN-CC5 in Patients with Paroxysmal Nocturnal Hemoglobinuria who are Inadequate Responders to Eculizumab. 2017 https://www.clinicaltrialsregister.eu/ctr-search/trial/2016-002943-40/ES.
- 62.Sheridan D, Yu ZX, Zhang Y, et al. Design and preclinical characterization of ALXN1210: A next generation anti-C5 monoclonal antibody with improved pharmacokinetics and duration of action. Immunobiology. 2016;221:1158. [Google Scholar]
- 63.Roeth A, Rottinghaus ST, Hill A, et al. Optimization of Dose Regimen for ALXN1210, a Novel Complement C5 Inhibitor, in Patients with Paroxysmal Nocturnal Hemoglobinuria (PNH): Results of 2 Phase 1/2 Studies. Blood. 2017;130:3482. [Google Scholar]
- 64.Alexion Pharmaceuticals. ALXN1210 Versus Eculizumab in Complement Inhibitor Treatment-Naïve Adult Patients With Paroxysmal Nocturnal Hemoglobinuria (PNH) 2018 https://clinicaltrials.gov/ct2/show/NCT02946463?term=ALXN1210&rank=4.
- 65.Alexion Pharmaceuticals. ALXN1210 Versus Eculizumab in Adult Patients With Paroxysmal Nocturnal Hemoglobinuria (PNH) Currently Treated With Eculizumab. 2018 https://clinicaltrials.gov/ct2/show/NCT03056040?term=ALXN1210&rank=5.
- 66.Ricklin D, Reis ES, Mastellos DC, Gros P, Lambris JD. Complement component C3 - The “Swiss Army Knife” of innate immunity and host defense. Immunol Rev. 2016 Nov;274(1):33–58. doi: 10.1111/imr.12500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ricklin D, Reis ES, Lambris JD. Complement in disease: a defence system turning offensive. Nat Rev Nephrol. 2016 Jul;12(7):383–401. doi: 10.1038/nrneph.2016.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mastellos DC, Ricklin D, Yancopoulou D, Risitano A, Lambris JD. Complement in paroxysmal nocturnal hemoglobinuria: exploiting our current knowledge to improve the treatment landscape. Expert Rev Hematol. 2014 Oct;7(5):583–98. doi: 10.1586/17474086.2014.953926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Risitano AM. Anti-Complement Treatment in Paroxysmal Nocturnal Hemoglobinuria: Where we Stand and Where we are Going. Transl Med UniSa. 2014 Jan;8:43–52. [PMC free article] [PubMed] [Google Scholar]
- 70.Sahu A, Kay BK, Lambris JD. Inhibition of human complement by a C3-binding peptide isolated from a phage-displayed random peptide library. J Immunol. 1996 Jul 15;157(2):884–91. [PubMed] [Google Scholar]
- 71.Mastellos DC, Yancopoulou D, Kokkinos P, Huber-Lang M, Hajishengallis G, Biglarnia AR, et al. Compstatin: a C3-targeted complement inhibitor reaching its prime for bedside intervention. Eur J Clin Invest. 2015 Apr;45(4):423–40. doi: 10.1111/eci.12419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Reis ES, Mastellos DC, Yancopoulou D, Risitano AM, Ricklin D, Lambris JD. Applying complement therapeutics to rare diseases. Clin Immunol. 2015 Dec;161(2):225–40. doi: 10.1016/j.clim.2015.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Qu H, Ricklin D, Bai H, Chen H, Reis ES, Maciejewski M, et al. New analogs of the clinical complement inhibitor compstatin with subnanomolar affinity and enhanced pharmacokinetic properties. Immunobiology. 2013 Apr;218(4):496–505. doi: 10.1016/j.imbio.2012.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Janssen BJ, Halff EF, Lambris JD, Gros P. Structure of compstatin in complex with complement component C3c reveals a new mechanism of complement inhibition. J Biol Chem. 2007 Oct 5;282(40):29241–7. doi: 10.1074/jbc.M704587200. [DOI] [PubMed] [Google Scholar]
- 75.Zhang Y, Shao D, Ricklin D, Hilkin BM, Nester CM, Lambris JD, et al. Compstatin analog Cp40 inhibits complement dysregulation in vitro in C3 glomerulopathy. Immunobiology. 2015 Aug;220(8):993–8. doi: 10.1016/j.imbio.2015.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Reis ES, Deangelis RA, Chen H, Resuello RR, Ricklin D, Lambris JD. Therapeutic C3 inhibitor Cp40 abrogates complement activation induced by modern hemodialysis filters. Immunobiology. 2015 Apr;220(4):476–82. doi: 10.1016/j.imbio.2014.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lindorfer MA, Cook EM, Reis ES, Ricklin D, Risitano AM, Lambris JD, et al. Compstatin Cp40 blocks hematin-mediated deposition of C3b fragments on erythrocytes: Implications for treatment of malarial anemia. Clin Immunol. 2016 Oct;171:32–5. doi: 10.1016/j.clim.2016.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.van Griensven M, Ricklin D, Denk S, Halbgebauer R, Braun CK, Schultze A, et al. Protective Effects of the Complement Inhibitor Compstatin Cp40 in Hemorrhagic Shock. Shock. 2018 doi: 10.1097/SHK.0000000000001127. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hajishengallis G, Hajishengallis E, Kajikawa T, Wang B, Yancopoulou D, Ricklin D, et al. Complement inhibition in pre-clinical models of periodontitis and prospects for clinical application. Semin Immunol. 2016 Mar 24; doi: 10.1016/j.smim.2016.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Risitano AM, Ricklin D, Huang Y, Reis ES, Chen H, Ricci P, et al. Peptide inhibitors of C3 activation as a novel strategy of complement inhibition for the treatment of paroxysmal nocturnal hemoglobinuria. Blood. 2014 Feb 4; doi: 10.1182/blood-2013-11-536573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.AMYNDAS Pharmaceuticals. Amyndas’ lead candidate AMY-101 receives orphan drug status from the FDA and the EMA for the treatment of C3 glomerulopathy. http://www.fiercepharma.com/pharma/amyndas%E2%80%99-lead-candidate-amy-101-receives-orphan-drug-status-from-fda-and-ema-for-treatment. 4-14-2016.
- 82.AMYNDAS Pharmaceuticals. EMA grants orphan drug designation for PNH to Amyndas Pharmaceuticals’ novel complement inhibitor. 2014 [Google Scholar]
- 83.AMYNDAS Pharmaceuticals. First-In-Human Clinical Study of the C3 Complement Inhibitor AMY-101 in Healthy Male Volunteers. 2018 https://clinicaltrials.gov/ct2/show/NCT03316521?term=AMY-101&rank=1.
- 84.AMYNDAS Pharmaceuticals. Press Release: Amyndas Pharmaceuticals Announces Positive Results from a Phase I Trial of its complement C3 inhibitor AMY-101. 2017 http://amyndas.com/press-release-amyndas-pharmaceuticals-announces-positive-results-from-a-phase-i-trial-of-its-complement-c3-inhibitor-amy-101/
- 85.Apellis Pharmaceuticals. Pilot Study to Assess Safety, Preliminary Efficacy and Pharmacokinetics of S.C. APL-2 in PNH Subjects (PADDOCK) 2018 https://clinicaltrials.gov/ct2/show/NCT02588833?term=APL-2&rank=2.
- 86.Apellis Pharmaceuticals. A Phase I Study to Assess the Safety APL-2 as an Add-On to Standard of Care in Subjects With PNH. 2017 https://clinicaltrials.gov/ct2/show/NCT02264639?term=APL-2&rank=5.
- 87.Apellis Pharmaceuticals. Apellis Pharmaceuticals Announces Positive Data from APL-2 Studies Showing Rapid and Durable Improvements in LDH and Hemoglobin Levels in PNH. 2017 http://investors.apellis.com/news-releases/news-release-details/apellis-pharmaceuticals-announces-positive-data-apl-2-studies.
- 88.Apellis Pharmaceuticals. Apellis Pharmaceuticals Announces that APL-2 Met its Primary Endpoint in a Phase 2 Study in Patients with Geographic Atrophy, an Advanced Form of Age-Related Macular Degeneration. 2017 http://investors.apellis.com/news-releases/news-release-details/apellis-pharmaceuticals-announces-apl-2-met-its-primary-endpoint.
- 89.Alexion Pharmaceuticals. Safety and Pharmacokinetics of TT30 in Subjects With Paroxysmal Nocturnal Hemoglobinuria (PNH) 2014 https://clinicaltrials.gov/ct2/show/NCT01335165?term=TT30&rank=1.
- 90.Fridkis-Hareli M, Storek M, Mazsaroff I, Risitano AM, Lundberg AS, Horvath CJ, et al. Design and development of TT30, a novel C3d-targeted C3/C5 convertase inhibitor for treatment of human complement alternative pathway-mediated diseases. Blood. 2011 Oct 27;118(17):4705–13. doi: 10.1182/blood-2011-06-359646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Risitano AM, Storek M, Sahelijo L, Doyle M, Dai Y, Weitz IC, et al. Safety and Pharmacokinetics of the Complement Inhibitor TT30 in a Phase I Trial for Untreated PNH Patients. Orlando, FL: 2015. 2015. [Google Scholar]
- 92.Schmidt CQ, Bai H, Lin Z, Risitano AM, Barlow PN, Ricklin D, et al. Rational engineering of a minimized immune inhibitor with unique triple-targeting properties. J Immunol. 2013 Jun 1;190(11):5712–21. doi: 10.4049/jimmunol.1203548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Schmidt CQ, Harder MJ, Nichols EM, Hebecker M, Anliker M, Hochsmann B, et al. Selectivity of C3-opsonin targeted complement inhibitors: A distinct advantage in the protection of erythrocytes from paroxysmal nocturnal hemoglobinuria patients. Immunobiology. 2016 Apr;221(4):503–11. doi: 10.1016/j.imbio.2015.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ricklin D, Lambris JD. Complement therapeutics. Semin Immunol. 2016 Jun;28(3):205–7. doi: 10.1016/j.smim.2016.07.001. [DOI] [PubMed] [Google Scholar]
- 95.Schubart A, Maibaum J, Anderson K, et al. Small-molecule Factor B inhibitors for oral treatment of alternative pathway-driven diseases. 2016 [Google Scholar]
- 96.Maibaum J, Liao SM, Vulpetti A, Ostermann N, Randl S, Rudisser S, et al. Small-molecule factor D inhibitors targeting the alternative complement pathway. Nat Chem Biol. 2016 Dec;12(12):1105–10. doi: 10.1038/nchembio.2208. [DOI] [PubMed] [Google Scholar]
- 97.Yuan X, Gavriilaki E, Thanassi JA, Yang G, Baines AC, Podos SD, et al. Small-molecule factor D inhibitors selectively block the alternative pathway of complement in paroxysmal nocturnal hemoglobinuria and atypical hemolytic uremic syndrome. Haematologica. 2017 Mar;102(3):466–75. doi: 10.3324/haematol.2016.153312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Achillion Pharmaceuticals. A Treatment Study of ACH-0144471 in Patients With Paroxysmal Nocturnal Hemoglobinuria (PNH) 2017 https://clinicaltrials.gov/ct2/show/NCT03053102?term=ACH-0144471&rank=2.
- 99.Patel D, Thanassi JA, Yang G, et al. In Vitro Combination Studies of ACH-4471 with Eculizumab to Assess a Potential “Switch” Treatment Approach for Paroxysmal Nocturnal Hemoglobinuria. Blood. 2017;130:2198. [Google Scholar]
- 100.Irmscher S, Doring N, Halder LD, Jo EAH, Kopka I, Dunker C, et al. Kallikrein Cleaves C3 and Activates Complement. J Innate Immun. 2017 Dec 14; doi: 10.1159/000484257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hoffmann-La Roche. A Study Investigating the Safety and Efficacy of Lampalizumab Intravitreal Injections in Participants With Geographic Atrophy Secondary to Age-Related Macular Degeneration (SPECTRI) 2017 https://clinicaltrials.gov/show/NCT02247531.
- 102.Botto M, Fong KY, So AK, Barlow R, Routier R, Morley BJ, et al. Homozygous hereditary C3 deficiency due to a partial gene deletion. Proc Natl Acad Sci U S A. 1992 Jun 1;89(11):4957–61. doi: 10.1073/pnas.89.11.4957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Reis S, Falcao DA, Isaac L. Clinical aspects and molecular basis of primary deficiencies of complement component C3 and its regulatory proteins factor I and factor H. Scand J Immunol. 2006 Mar;63(3):155–68. doi: 10.1111/j.1365-3083.2006.01729.x. [DOI] [PubMed] [Google Scholar]
- 104.Konar M, Granoff DM. Eculizumab treatment and impaired opsonophagocytic killing of meningococci by whole blood from immunized adults. Blood. 2017 Aug 17;130(7):891–9. doi: 10.1182/blood-2017-05-781450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.True North Therapeutics/Bioverativ. Safety, Tolerability and Activity of TNT009 in Healthy Volunteers and Patients With Complement Mediated Disorders (TNT009-01) 2017 https://clinicaltrials.gov/show/NCT02502903.