

Abstract
A continual dialogue exists between the central nervous and immune system that contributes to neural homeostasis as well as protection from microbes, repair following damage, autoimmune disease, and neurodegeneration. Characterization of resident and peripherally-derived leukocyte populations within the central nervous system can provide valuable information regarding how these cells contribute to steady state and inflammatory conditions. Flow cytometry provides a method to conduct detailed multi-parameter analyses of immune cells isolated from various tissues. This protocol provides a method to isolate leukocytes from brain, spinal cord and meninges for flow cytometric analysis and provides a basic framework for phenotyping these cells.
Keywords: CNS, leukocyte isolation, flow cytometry, FACS, meninges
INTRODUCTION
The central nervous system (CNS) consists of the brain and spinal cord and is enclosed by three membranes collectively referred to as the meninges. The CNS and surrounding meninges are inhabited by resident myeloid cells (e.g., microglia, meningeal macrophages, perivascular macrophages, choroid plexus macrophages) that are highly dynamic and constantly survey their surroundings under steady state conditions (Davalos et al., 2005; Goldmann et al., 2016; Nayak, Roth, & McGavern, 2014; Nayak, Zinselmeyer, Corps, & McGavern, 2012; Nimmerjahn, Kirchhoff, & Helmchen, 2005; Prinz, Erny, & Hagemeyer, 2017). These cells can also respond rapidly to inflammatory challenges and are usually among the first responders to states of infection, injury, neurodegeneration and autoimmune disease (Hanisch & Kettenmann, 2007; Nayak et al., 2014). In general, the meninges are more immunologically active than the CNS parenchyma, and under steady state conditions, it is possible to isolate B cells, T cells, natural killer (NK) cells, dendritic cells (DCs), and myelomonocytic cells from this compartment (Korin et al., 2017). During states of inflammation, additional inflammatory cells can enter CNS barrier structures from the blood. From there, these cells will sometimes enter the brain and/or spinal cord parenchyma where they can either ameliorate or exacerbate neuroinflammation depending on the context (Engelhardt, Vajkoczy, & Weller, 2017; Greenwood et al., 2011; Pachter, de Vries, & Fabry, 2003; Wu et al., 2009). Understanding the complex dialogue between the CNS and immune system during steady state and inflammatory conditions is an area of active investigation that is reliant on many different experimental approaches (Kipnis, 2016; Russo & McGavern, 2016).
First developed in the 1960s, flow cytometry is a technology that allows analysis of cell populations by creating a single cell fluid stream that passes through a flow cell (Fulwyler, 1965). Lasers tuned to specific wavelengths are used to excite the single cell stream, and the emitted light is then collected and amplified for analysis (Chattopadhyay, Hogerkorp, & Roederer, 2008; Herzenberg et al., 2002; Hulett, Bonner, Sweet, & Herzenberg, 1973). Since its inception, flow cytometry has evolved significantly. Development of new lasers, cellular labels, and processing technology has allowed investigators to use polychromatic flow cytometry to assess heterogeneous immune populations (Chattopadhyay et al., 2008; Chattopadhyay et al., 2006; Perfetto, Chattopadhyay, & Roederer, 2004) and determine their proliferation rates (Lyons, 2000; Lyons & Parish, 1994), activation status, signaling events (Krutzik, Clutter, & Nolan, 2005; Krutzik & Nolan, 2003), etc. in an automated fashion (Aghaeepour et al., 2013). Furthermore, recent advances in single cell genomic techniques, such as RNA sequencing, have provided cellular transcriptomes of individual cells, allowing us to identify and profile heterogeneous immune cell populations from various tissues (Buettner et al., 2015; Keren-Shaul et al., 2017; Prinz et al., 2017; Xue et al., 2014).
Flow cytometry and other single cell techniques are reliant on the generation of viable cell suspensions. For example, fluorescence-activated cell sorting (FACS) allows the isolation of viable cells based on fluorescent labels (Julius, Masuda, & Herzenberg, 1972). Techniques such as FACS require efficient extraction of cells from tissues of interest and staining with spectrally distinct fluorophores. The viability and purity of isolated cells often determines the quality of the resultant data. This unit describes the techniques needed to extract the brain, spinal cord parenchyma, and meninges from the cranium and vertebral bodies (see Support Protocol 1) as well as methods for extracting leukocytes from the brain, spinal cord (see Basic Protocol 1), and meninges (see Basic Protocol 2). A guideline for analyzing and immunophenotyping populations (Figure 2 and 3) is also provided.
Figure 2. Gating strategy to phenotype leukocytes isolated from brain tissue.
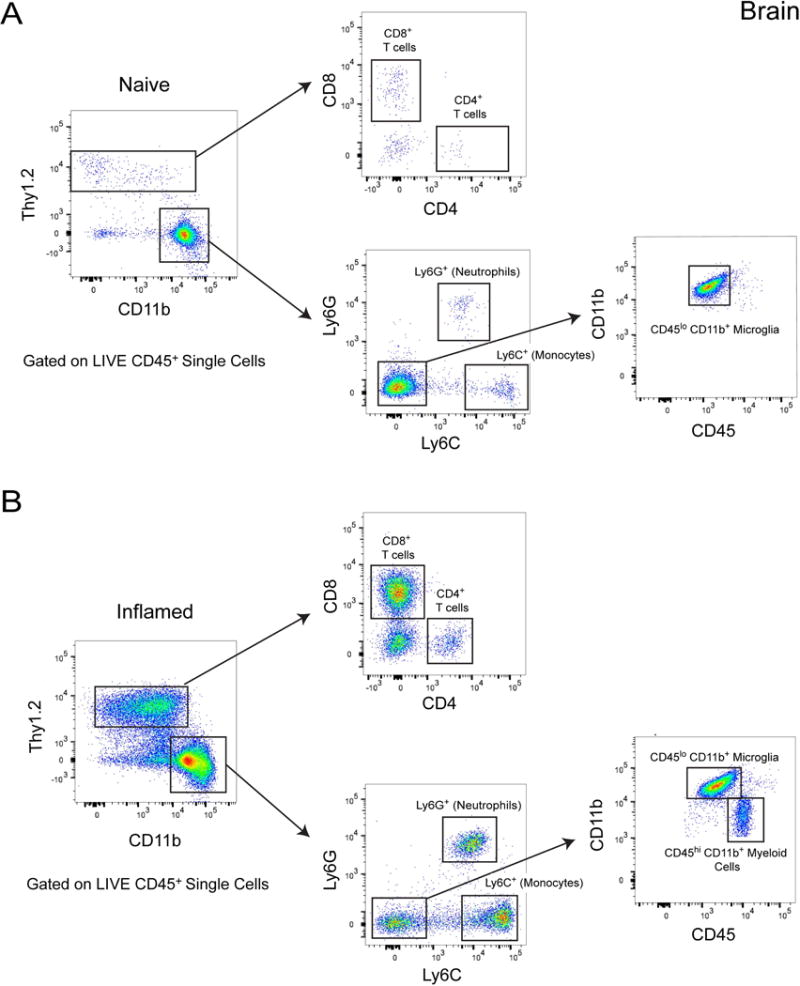
A. Flow cytometric analysis of leukocytes isolated from a naïve mouse brain. Live single cells were pre-gated on CD45.2+ (not shown). Thy1.2+ cells can be subdivided into CD8+ and CD4+ T cells. CD11b+ myeloid cells can be subdivided into neutrophils (Ly6G+), monocytes (Ly6C+) and microglia (Ly6G− Ly6C− CD11b+ CD45lo). B. Flow cytometric analysis of leukocytes isolated from a mouse brain six days following an intracerebral lymphocytic choriomenigitis (LCMV) infection. Live single cells were pre-gated on CD45.2+ (not shown). Thy1.2+ cells are subdivided into CD8+ and CD4+ T cells. CD11b+ cells are split into neutrophils (Ly6G+) and monocytes (Ly6C+). Because peripherally-derived Ly6C− Ly6G− myeloid cells can enter the brain following infection, it is necessary to further analyze this population based on CD45 and CD11b expression. This will result in a cleaner separation of microglia (CD45lo) and peripherally-derived myeloid cell (CD45hi) subsets.
Figure 3. Gating strategy to phenotype leukocytes isolated from the meninges.
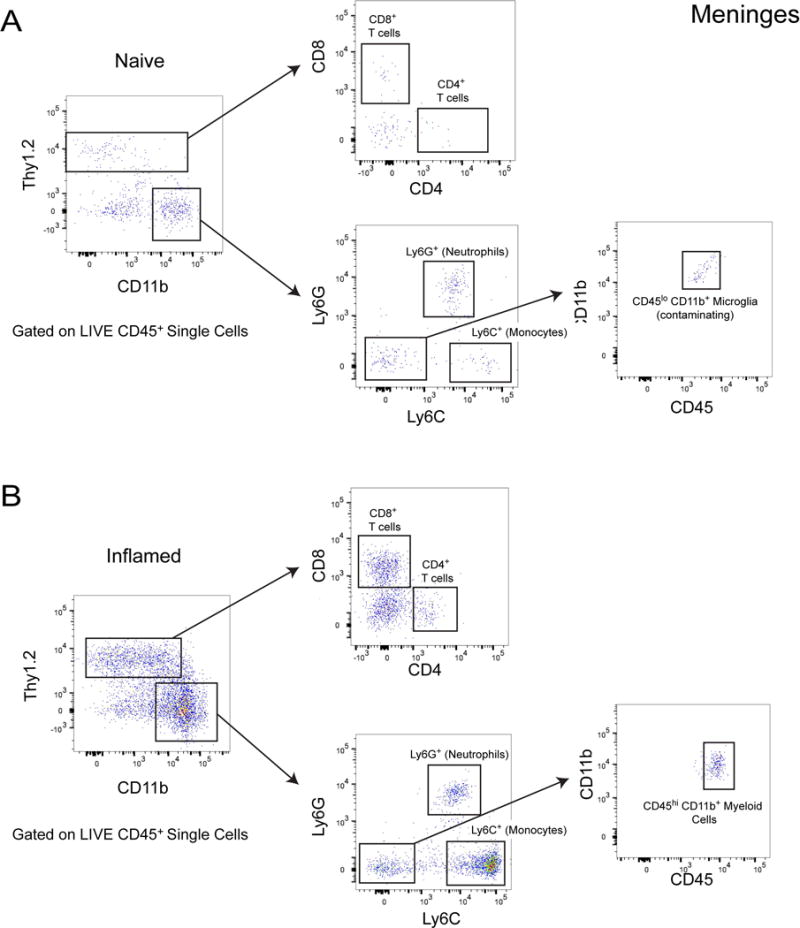
A. Flow cytometric analysis of leukocytes isolated from a naïve mouse meninges. Live single cells were pre-gated on CD45.2+ (not shown). Thy1.2+ cells can be subdivided into CD8+ and CD4+ T cells. CD11b+ myeloid cells be subdivided into neutrophils (Ly6G+) and monocytes (Ly6C+). Some microglia may be inadvertently extracted from the meninges during isolation and will appear as Ly6C− Ly6G− CD45lo in the preparation. B. Flow cytometric analysis of leukocytes isolated a mouse meninges six days following intracerebral inoculation with LCMV. Live single cells were pre-gated on CD45.2+ (not shown). Thy1.2+ cells can be subdivided into CD8+ and CD4+ T cells. CD11b+ cells can be subdivided into neutrophils (Ly6G+) and monocytes (Ly6C+). Following infection, peripherally-derived Ly6C− Ly6G− myeloid cells can enter the meninges parenchyma. These cells will appear as CD45hi on a CD45 vs. CD11b plot.
SUPPORT PROTOCOL 1: PREPARATION OF BRAIN AND SPINAL CORD SAMPLES FOR CELL ISOLATION
This protocol provides guidance on the removal of the brain, spinal cord, and their respective meninges to isolate cells for FACS. For guidance on animal handling techniques, please see Chapter 1.
Materials
Reagents
Normal Saline
RPMI 1640 Medium (Thermo Fisher, Cat No. 11875093)
Bucket of ice
Equipment
Perfusion apparatus
Surgical scissors and forceps
15 ml conical tubes
-
Transcardially perfuse mouse with 20 ml of saline. See Unit 15.1 for detailed instructions on mouse perfusion.
It is imperative that the perfusion blanches both the liver and the lung. Poorly perfused mice could result in inaccurate flow cytometric analysis of immune infiltration into the CNS and/or meninges due to the presence of contaminating blood.
Remove head with scissors. Cut the scalp by creating a midline excision caudal to rostral, from between ears to immediately superior to eyes. Pull the scalp laterally to expose the skull.
-
Use fine surgical scissors to begin cutting the skull. Place the scissor blade within the foramen magnum. Keeping scissors close to the skull, cut around the cortices and anteriorly toward the olfactory bulb. Complete the same cut on the other side. The complete removal of the skull cap is shown in Figure 1.
Incisions should be above the mandibles and eye.
Excisions on the right and left side of the skull should meet immediately superior to the olfactory bulb. Using forceps, remove the skull by pulling upward from the base of the head. Place the skull into a 15ml tube containing ice cold RPMI and keep the tube on ice while extracting other tissues.
Using forceps, remove brain and place it into a tube containing ice cold RPMI.
To dissect the spinal column from remaining tissue, remove the pelt from cervical to sacral vertebrae.
Place two incisions immediately lateral to the spinal column on each side, cutting through the ribs and supporting muscles.
Place another incision at the base of the tail. Remove the spinal column.
Place scissors within the spinal column and cut along the lateral edge of the vertebra. Complete the same on the other side of the vertebra.
This will separate the spinal column from the vertebra. Using fine forceps, lift the spinal cord and place in a vial of ice cold RPMI.
Place both vertebral columns in ice cold RPMI.
Figure 1. Removal of brain and meninges from cranium.
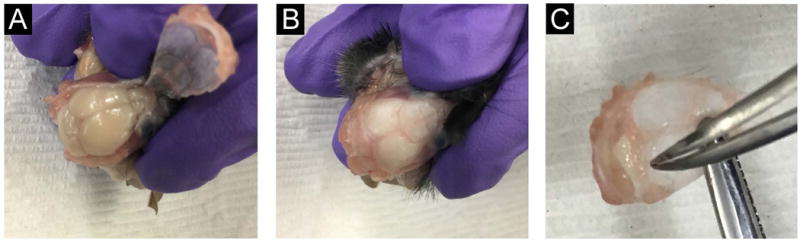
A. This picture shows the head of a perfused mouse. The skull cap has been cut and lifted, exposing the underlying brain. Note that the perfused brain tissue is free of blood and appears white. Blood in the brain tissue is a sign of a poor perfusion. The skull cap with attached meninges is removed from the cranium by using fine scissors to cut from the foramen magnum, laterally to the superior aspect of the olfactory bulb. This is done on both sides. B-C. Once the skull cap is lifted, it can be removed entirely. Immune cells can be isolated from the brain in panel B using Basic Protocol 1 and from the meninges attached to the skull cap in panel C using Basic Protocol 2. The meninges are removed from the surface of the skull cap by gently pulling off the membranous layer using curved forceps.
BASIC PROTOCOL 1: ISOLATION OF MONONUCLEAR CELLS FROM THE BRAIN AND SPINAL CORD
This protocol provides guidance on how to isolate immune cells from the brain and spinal cord for flow cytometry. Cells will be dissociated from CNS tissue, separated on a density gradient, blocked for nonspecific antibody binding, and stained. This protocol should be completed carefully and continuously (without breaks) to limit cell death. Detailed information on flow cytometric settings and acquisition can be found in Chapter 5. We provide a basic framework in Figure 2 for immunophenotyping cell populations extracted from the CNS parenchyma.
Materials
Reagents
RPMI 1640 Medium (Thermo Fisher, Cat No. 11875093)
Collagenase D (Roche, Cat No. 11088858001)
DNase I (Roche, Cat No. 10104159001)
1 X Phosphate Buffered Saline (PBS)
Fetal Bovine Serum (FBS), ultra-low IgG (Thermo Fisher, Cat No. 16250078)
Percoll (Fisher, Cat No. 17-0891-01)
10 X Hanks Balanced Salt Solution (HBSS) (Thermo Fisher, Cat No. 14185052)
Distilled deionized water
TruStain fcX (anti-mouse CD16/32) antibody (Biolegend, Cat No.101320)
Mouse IgG, whole molecule (Jackson Immunoresearch, Cat No. 015-000-003)
Bucket of ice
Equipment
15 ml conical tubes
50 ml conical tubes
Nylon mesh strainer with 100 μm pore size (Corning, Cat No. 352360)
Corning non-treated culture dishes
-
Glass pestle
We utilize custom made glass pestles to homogenize CNS tissue. However, a suitable alterative would be Dounce Tissue Grinder (Corning, Cat No. 772-15).
Falcon™ Round-Bottom Polypropylene Tubes (Corning, Cat No. 14-959-11A)
Falcon 5mL Round Bottom Polystyrene Test Tube with Cell Strainer Snap Cap (Corning, Cat No. 352235)
Hemocytometer
Remove RPMI from brain and spinal cord collection tubes.
Place tissue into petri dish using blunt forceps. Mince tissue finely with surgical scissors.
-
Add 1 ml of digestion mix to petri dish containing the minced tissue and transfer to 50 ml conical tube.
Certain digestion mixes cause cleavage of cell surface proteins. We have found that the use of collagenase D and DNAse I leaves most immune markers intact.
-
Rinse petri dish with 1 ml collagenase/DNAse solution and place in collection tube. Incubate samples for 30 minutes at 37°C.
Incubating samples in a bacterial shaker set to 37°C is ideal. However, if unavailable, place samples in a water bath and shake samples by hand occasionally.
-
Remove samples from incubator and place on ice after 30-minute incubation period. Pipette samples onto a 100-μm nylon mesh strainer within a petri dish. Gently mash the tissue and liquid through the strainer using a pestle.
It is very important to gently press the tissue through mesh. Perform gentle circular motions with the pestle, and avoid applying too much downward pressure, as this can cause significant cell death. If possible, utilize a glass pestle (or, another type of pestle with a smooth surface) to mash tissue. We have found that using smooth glass pestles reduces cell death when compared to plastic and metal pestles.
Place filtered sample into a 15-ml conical tube.
-
Wash strainer and petri dish with 11 ml of ice cold RPMI and add to the 15-ml conical tube with the rest of the filtered sample.
Most of the CNS tissue should pass through filter. If necessary, repeat mashing until most the CNS tissue has passed through the filter.
Centrifuge samples at 1200 RPM for 5 minutes at 4°C.
Aspirate supernatant.
Resuspend pellet in 4 ml of ice-cold 90% Percoll.
-
Overlay 3 ml of 60% Percoll, 4 ml of 40% Percoll and 3 ml of 1x HBSS very carefully.
It is imperative to pipette slowly when adding each layer of the Percoll gradient; otherwise, the gradient will not form properly. Watch for the formation of distinct layers when creating the gradient.
-
Centrifuge the gradients at 1700 RPM for 20 minutes at 4°C with the brake on the centrifuge disengaged.
It is important to disengage the centrifuge brake when spinning. Failure to do so will result in disruption of the mononuclear cell layer in the gradient as the centrifuge slows too abruptly. Following successful centrifugation, you should see a thin white band of cells in the gradient corresponding to the mononuclear cells.
Following centrifugation, carefully aspirate and discard the top 4 ml of the gradient (~0 to 40%). This is a thick white foamy layer containing mostly myelin.
Collect the solution from the tube until there is only 4 ml remaining. The remaining 4 ml will be discarded, and the solution you have collected will contain the mononuclear cells. Place this solution into new tube.
-
In the new tube with the mononuclear cells, add 10 ml of 1x HBSS. Centrifuge at 1400 RPM for 10 minutes at 4°C.
This step is necessary to wash away the Percoll. Failure to mix the HBSS with the extracted mononuclear layer could result in the reformation of the percoll gradient. This can lead to aspiration of cells with supernatant.
-
Remove supernatant and be careful not to suck up the cell pellet at the bottom of the tube. An additional washing step can be performed if needed.
The cell pellet at the bottom of the tube is often barely visible. If a pellet is not visible, you should still proceed to the next step.
-
Resuspend pellet in 3 ml of FACS buffer. Transfer solution into a 5-ml round bottom polystyrene test tube with cell strainer snap cap. Spin at 1200 RPM for 5 minutes at 4°C.
Filtering through filter cap tubes reduces clogging of the flow cytometer.
-
Resuspend cells in 50 μl blocking solution and incubate for 10 minutes at 4°C.
Incubation with the blocking solution reduces non-specific binding when staining with primary antibodies. If you need an absolute cell count, remove a small aliquot of the cell suspension and count using a hemocytometer or automated cell counter.
-
Stain cells with primary antibody cocktail for 30 minutes at 4°C.
Staining solution should be at a 2X concentration since the antibodies will be incubated with blocking solution. After staining, samples should be acquired using a flow cytometer as soon as possible. Keeping living cells on ice for an extended time period can result in cell death. If you cannot acquire your cells the same day, they can be washed and fixed with 1% PFA and run the next day. Refer to Chapter 5 For more information on flow cytometric settings and acquisition. In Figure 2 we provide a basic framework for profiling extracted immune cell populations.
BASIC PROTOCOL 2: ISOLATION OF MONONUCLEAR CELLS FROM THE BRAIN AND SPINAL CORD MENINGES
This protocol provides guidance on how to isolate immune cells from the meninges for flow cytometry. Cells will be dissociated from CNS meninges, separated on a density gradient, blocked for nonspecific antibody binding, and stained. This protocol should be completed carefully and continuously (without breaks) to limit cell death. Detailed information on flow cytometric settings and acquisition can be found in Chapter 5. We provide a basic framework in Figure 3 for immunophenotyping cell populations extracted from the CNS meninges.
Materials
Reagents
RPMI 1640 Medium (Thermo Fisher, Cat No. 11875093)
Collagenase D (Roche, Cat No. 11088858001)
DNase I (Roche, Cat No. 10104159001)
1 X Phosphate Buffered Saline (PBS)
Fetal Bovine Serum (FBS), ultra-low IgG (Thermo Fisher, Cat No. 16250078)
TruStain fcX (anti-mouse CD16/32) antibody (Biolegend, Cat No.101320)
Mouse IgG, whole molecule (Jackson Immunoresearch, Cat No. 015-000-003)
Bucket of ice
Equipment
Falcon™ Round-Bottom Polypropylene Tubes (Corning, Cat No. 14-959-11A)
Nylon mesh strainer with 100 μm pore size (Corning, Cat No. 352360)
Corning non-treated culture dishes
-
Glass pestle
We utilize custom made glass pestles to homogenize CNS tissue. However, a suitable alterative would be Dounce Tissue Grinder (Corning, Cat No. 772-15).
Falcon 5 mL Round Bottom Polystyrene Test Tube with Cell Strainer Snap Cap (Corning, Cat No. 352235)
Hemocytometer
-
Remove skullcap from RPMI. Using curved forceps, peel meninges from edges of the skull cap (see Figure 1) – the side that was facing the brain. Gently scrape the meninges downward to nose bone, and remove all the meninges as a single thin layer from the skull.
The meninges are fragile so it is very important to remove them slowly. If you have difficulty seeing the meninges, a dissecting scope can be used to facilitate this process. We have observed that this procedure results in isolation of cells from the dura and arachnoid mater. The pia remains attached to the brain.
To extract spinal cord meninges, remove the vertebral columns from the RPMI. Using the same technique described in step 1, gently peel the meninges away from the edge of the vertebra until you can completely remove the meninges a single thin layer.
Place meninges in a 5-ml polypropylene tube containing 400 μl of digestion mix.
-
Incubate sample for 30 minutes at 37°C.
Incubating samples in a bacterial shaker set to 37°C is ideal. However, if unavailable, place samples in a water bath and shake samples by hand occasionally.
-
Remove samples from the incubator and place on ice. Pipette samples onto a 100 μm nylon mesh strainer within a petri dish. Gently mash the tissue and liquid through the strainer using a pestle.
It is very important to gently press the tissue through mesh. Perform gentle circular motions with the pestle, and avoid applying too much downward pressure, as this can cause significant cell death. If possible, utilize a glass pestle (or, another type of pestle with a smooth surface) to mash tissue. Some of the meningeal fibers may not completely digest and may remain in the filter
Placed filtered sample into a 5-ml polystyrene tube.
Wash strainer with 4 ml of RPMI and add to the 5-ml polystyrene tube with the rest of the filtered sample.
Centrifuge samples at 1200 RPM for 5 minutes at 4°C.
-
Aspirate supernatant.
Be careful when aspirating the sample not to suck up the cell pellet at the bottom of the tube.
-
Resuspend cells in 3 ml of FACS buffer. Transfer solution into a 5-ml round bottom polystyrene test tube with cell strainer snap cap. Spin at 1200 RPM for 5 minutes at 4°C.
Filtering through filter cap tubes reduces clogging of the flow cytometer.
-
Resuspend in 50 μl blocking solution and incubate for 10 min at 4°C.
If you need an absolute cell count, remove a small aliquot of the cell suspension and count using a hemocytometer or automated cell counter. If you need an absolute cell count, remove a small aliquot of the cell suspension and count using a hemocytometer or automated cell counter.
-
Stain cells with primary antibody cocktail for 30 minutes at 4°C.
Staining solution should be at a 2X concentration since the antibodies will be incubated with blocking solution. Samples should be processed for FACS analysis immediately. Keeping living cells on ice for extended time periods can result in cell death. If you cannot acquire your cells the same day, samples can be washed, fixed with 1% PFA, and run the next day. Refer to Chapter 5 For more information on flow cytometric settings and acquisition. In Figure 3 we provide a basic framework for profiling extracted meningeal immune cell populations.
REAGENTS AND SOLUTIONS
Digestion Mix: 1 mg/ml collagenase D, 50 μg/ml DNAse I in RPMI
FACS buffer: PBS with 2% FBS
Blocking Solution: 1:100 dilution of CD16/CD32 antibody, 1:50 dilution of mouse IgG in FACS buffer
90% Percoll: 90 ml of Percoll, 10 ml of 10x HBSS
60% Percoll: 60 ml of Percoll, 10 ml of 10x HBSS, 30 ml of distilled deionized water
30% Percoll: 30 ml of Percoll, 10 ml of 10x HBSS, 60 ml of distilled deionized water
1x HBSS Solution: 10 ml of 10x HBSS, 90 ml of distilled deionized water
COMMENTARY
Background Information
The advent of flow cytometric methods, including FACs, has greatly enhanced our ability to study immune cells throughout the body (Fulwyler, 1965; Herzenberg et al., 2002; Julius et al., 1972). Use of these and other single cell techniques is reliant on efficient cell extraction methods. Isolation of the immune cells from the CNS and meninges can aid our understanding of neural-immune interactions during steady and inflammatory conditions. Because immune cells have the potential to help and cause great harm to the CNS, the combination of cell isolation techniques and multi-parameter analyses (Chattopadhyay et al., 2008; Perfetto et al., 2004) can help us better understand how these cells contribute to states of health and disease (Kipnis, 2016; Russo & McGavern, 2015). Multi-parameter flow cytometry has become a mainstream technique that comes with many advantages including the ease of use, speed of acquisition, maintenance of cell viability, etc. This technique does not, however, provide information about their position and interactions in the tissue from which they were extracted. In addition, some cell populations including macrophages (Epelman, Lavine, & Randolph, 2014) and tissue resident memory cells (Steinert et al., 2015) are difficult to extract from tissues and can be significantly underrepresented using standard immune cell isolation techniques. It is, therefore, essential to supplement flow cytometry with other approaches such as histo-cytometry (Epelman et al., 2014) and intravital imaging (Epelman et al., 2014). Histo-cytometry combines immunohistochemical and flow cytometric methods to allow multi-parameter analysis of cell populations in tissue sections. Intravital imaging, on the other hand, can provide dynamic information about immune cells operating in living tissues.
Another limitation of flow cytometry is the number of spectrally distinct fluorophores that are readily available (Chattopadhyay et al., 2008; Perfetto et al., 2004). Although some contemporary flow cytometry labs are pushing the envelope with 30 colors, multi-parameter flow cytometry with more than 18-20 colors can be extremely challenging due to spectral overlap, even with compensation matrices and automated analyses (Bendall, Nolan, Roederer, & Chattopadhyay, 2012; Perfetto et al., 2004). A newly developed technique, known as mass cytometry (Unit 5.11), has overcome the spectral limitations of flow cytometry. Using antibodies conjugated to metal isotopes, mass cytometry can identify 45 markers simultaneously (Bendall et al., 2012; Korin et al., 2017). Additionally, since each metal isotope is unique, there is no spectral overlap between the metals (Bendall et al., 2012). The development of mass cytometry has allowed us to gain specific insights into unique cellular proteomics and population heterogeneity (Horowitz et al., 2013; Korin et al., 2017; van Unen et al., 2016). However, compared to flow cytometry, mass spectroscopy processes samples 25 times slower, is currently limited in its ability to assess parameters such as calcium dynamics / cell division and does not result in viable cells that can be sorted similar to FACS (Bendall et al., 2012). Thus, mass cytometry is still a technique that should be used in combination with (rather than replace) flow cytometric methods.
Critical Parameters
Spectral Overlap
Spectral overlap of fluorochromes can occur when using fluorophores with overlapping emission profiles. This issue can be resolved by titering antibodies before experimentation and utilizing single color controls to set appropriate experimental voltages prior to sample collection. More information on selecting appropriate non-overlapping fluorophores can be found in Unit 5.4, Basic Multicolor Flow Cytometry.
Too Few Cells for Analysis
Even the naïve CNS and meninges should provide enough cells for flow cytometric analysis without having to pool samples. Following isolation, there are several potential issues to consider if there are too few cells available for analysis:
Inefficient tissue processing. When isolating meninges from the skull and vertebra, it is easy to tear the tissue and leave parts behind. Be sure to remove all the meninges to maximize cellular yield. When preparing brain and spinal cord tissue, it is important to make sure that nearly all the tissue goes through the 100-μm nylon mesh strainer. Failure to do so will result in inefficient cell extraction.
Disruption of Percoll gradient. When isolating brain and spinal cord leukocytes, it is important to carefully and slowly pipette each layer of Percoll gradient. Failure to do so will result in the inappropriate positioning of the leukocytes in the Percoll solution following centrifugation and ultimately a low cellular yield. It is also necessary to pay close attention to the position of the leukocyte layer in the gradient during the extraction phase. Removing too much of the upper layers of the gradient can result in loss of cells.
Loss of cell pellet. Because the cell pellet following immune cell isolation will often be small and difficult to see (especially in naïve mice), it is easy to inadvertently suck some or all the pellet when removing supernatant following centrifugation.
High Level of Autofluorescence in Isolated Cells
Following flow cytometric acquisition, immune cells isolated from the CNS or meninges will sometimes show a high level of autofluorescence with signal appearing in multiple channels, making it difficult to interpret primary antibody stains. This usually results from aggressive cell preparations, allowing the tissue to sit on ice for several hours without processing, and / or vortexing the cell suspensions. When processing CNS or meningeal tissue, it is best to begin isolating the cells shortly after harvesting, rather than letting the tissue sit on ice for prolonged time periods. It is also important to be gentle when using the pestle to mash the tissue through the 100-μm mesh. Aggressive mashing can result in significant autofluorescence and cell death. Lastly, cell pellet from the CNS and meninges should be resuspended by flicking the tube or pipetting up and down. Use of a cell vortexer can cause undesirable autofluorescence.
Understanding Results
Many surface markers can be used for analyzing immune composition of isolated cells from the CNS. Chapter 5 provides a framework for FACS analysis. In Figure 2 and 3, we provide guidance on immune-phenotyping extracted cells.
Time Considerations
Completion of the protocol from the time of sample acquisition to staining time is approximately 4 hours. It is challenging to complete both the meningeal and brain / spinal cord extraction procedures at the same time without involving multiple people. Isolation of the meninges can be performed during centrifugation of the Percoll gradients for the brain / spinal cord extraction procedure.
Acknowledgments
This work was supported by the Intramural Research Program of the National Institute of Neurological Disease and Stroke (NINDS), NIH.
LITERATURE CITED
- Aghaeepour N, Finak G, Flow, C.A.P.C., Consortium, D. Hoos H, Mosmann TR, Scheuermann RH. Critical assessment of automated flow cytometry data analysis techniques. Nat Methods. 2013;10(3):228–238. doi: 10.1038/nmeth.2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendall SC, Nolan GP, Roederer M, Chattopadhyay PK. A deep profiler’s guide to cytometry. Trends Immunol. 2012;33(7):323–332. doi: 10.1016/j.it.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buettner F, Natarajan KN, Casale FP, Proserpio V, Scialdone A, Theis FJ, Stegle O. Computational analysis of cell-to-cell heterogeneity in single-cell RNA-sequencing data reveals hidden subpopulations of cells. Nat Biotechnol. 2015;33(2):155–160. doi: 10.1038/nbt.3102. [DOI] [PubMed] [Google Scholar]
- Chattopadhyay PK, Hogerkorp CM, Roederer M. A chromatic explosion: the development and future of multiparameter flow cytometry. Immunology. 2008;125(4):441–449. doi: 10.1111/j.1365-2567.2008.02989.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyay PK, Price DA, Harper TF, Betts MR, Yu J, Gostick E, Roederer M. Quantum dot semiconductor nanocrystals for immunophenotyping by polychromatic flow cytometry. Nat Med. 2006;12(8):972–977. doi: 10.1038/nm1371. [DOI] [PubMed] [Google Scholar]
- Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Gan WB. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005;8(6):752–758. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- Engelhardt B, Vajkoczy P, Weller RO. The movers and shapers in immune privilege of the CNS. Nat Immunol. 2017;18(2):123–131. doi: 10.1038/ni.3666. [DOI] [PubMed] [Google Scholar]
- Epelman S, Lavine KJ, Randolph GJ. Origin and functions of tissue macrophages. Immunity. 2014;41(1):21–35. doi: 10.1016/j.immuni.2014.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulwyler MJ. Electronic separation of biological cells by volume. Science. 1965;150(3698):910–911. doi: 10.1126/science.150.3698.910. [DOI] [PubMed] [Google Scholar]
- Goldmann T, Wieghofer P, Jordao MJ, Prutek F, Hagemeyer N, Frenzel K, Prinz M. Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat Immunol. 2016;17(7):797–805. doi: 10.1038/ni.3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwood J, Heasman SJ, Alvarez JI, Prat A, Lyck R, Engelhardt B. Review: leucocyte-endothelial cell crosstalk at the blood-brain barrier: a prerequisite for successful immune cell entry to the brain. Neuropathol Appl Neurobiol. 2011;37(1):24–39. doi: 10.1111/j.1365-2990.2010.01140.x. [DOI] [PubMed] [Google Scholar]
- Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10(11):1387–1394. doi: 10.1038/nn1997. [DOI] [PubMed] [Google Scholar]
- Herzenberg LA, Parks D, Sahaf B, Perez O, Roederer M, Herzenberg LA. The history and future of the fluorescence activated cell sorter and flow cytometry: a view from Stanford. Clin Chem. 2002;48(10):1819–1827. [PubMed] [Google Scholar]
- Horowitz A, Strauss-Albee DM, Leipold M, Kubo J, Nemat-Gorgani N, Dogan OC, Blish CA. Genetic and environmental determinants of human NK cell diversity revealed by mass cytometry. Sci Transl Med. 2013;5(208):208ra145. doi: 10.1126/scitranslmed.3006702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulett HR, Bonner WA, Sweet RG, Herzenberg LA. Development and application of a rapid cell sorter. Clin Chem. 1973;19(8):813–816. [PubMed] [Google Scholar]
- Julius MH, Masuda T, Herzenberg LA. Demonstration that antigen-binding cells are precursors of antibody-producing cells after purification with a fluorescence-activated cell sorter. Proc Natl Acad Sci U S A. 1972;69(7):1934–1938. doi: 10.1073/pnas.69.7.1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, Amit I. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell. 2017;169(7):1276–1290 e1217. doi: 10.1016/j.cell.2017.05.018. [DOI] [PubMed] [Google Scholar]
- Kipnis J. Multifaceted interactions between adaptive immunity and the central nervous system. Science. 2016;353(6301):766–771. doi: 10.1126/science.aag2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korin B, Ben-Shaanan TL, Schiller M, Dubovik T, Azulay-Debby H, Boshnak NT, Rolls A. High-dimensional, single-cell characterization of the brain’s immune compartment. Nat Neurosci. 2017;20(9):1300–1309. doi: 10.1038/nn.4610. [DOI] [PubMed] [Google Scholar]
- Krutzik PO, Clutter MR, Nolan GP. Coordinate analysis of murine immune cell surface markers and intracellular phosphoproteins by flow cytometry. J Immunol. 2005;175(4):2357–2365. doi: 10.4049/jimmunol.175.4.2357. [DOI] [PubMed] [Google Scholar]
- Krutzik PO, Nolan GP. Intracellular phospho-protein staining techniques for flow cytometry: monitoring single cell signaling events. Cytometry A. 2003;55(2):61–70. doi: 10.1002/cyto.a.10072. [DOI] [PubMed] [Google Scholar]
- Lyons AB. Analysing cell division in vivo and in vitro using flow cytometric measurement of CFSE dye dilution. J Immunol Methods. 2000;243(1–2):147–154. doi: 10.1016/s0022-1759(00)00231-3. [DOI] [PubMed] [Google Scholar]
- Lyons AB, Parish CR. Determination of lymphocyte division by flow cytometry. J Immunol Methods. 1994;171(1):131–137. doi: 10.1016/0022-1759(94)90236-4. [DOI] [PubMed] [Google Scholar]
- Nayak D, Roth TL, McGavern DB. Microglia development and function. Annu Rev Immunol. 2014;32:367–402. doi: 10.1146/annurev-immunol-032713-120240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayak D, Zinselmeyer BH, Corps KN, McGavern DB. In vivo dynamics of innate immune sentinels in the CNS. Intravital. 2012;1(2):95–106. doi: 10.4161/intv.22823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308(5726):1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- Pachter JS, de Vries HE, Fabry Z. The blood-brain barrier and its role in immune privilege in the central nervous system. J Neuropathol Exp Neurol. 2003;62(6):593–604. doi: 10.1093/jnen/62.6.593. [DOI] [PubMed] [Google Scholar]
- Perfetto SP, Chattopadhyay PK, Roederer M. Seventeen-colour flow cytometry: unravelling the immune system. Nat Rev Immunol. 2004;4(8):648–655. doi: 10.1038/nri1416. [DOI] [PubMed] [Google Scholar]
- Prinz M, Erny D, Hagemeyer N. Ontogeny and homeostasis of CNS myeloid cells. Nat Immunol. 2017;18(4):385–392. doi: 10.1038/ni.3703. [DOI] [PubMed] [Google Scholar]
- Russo MV, McGavern DB. Immune Surveillance of the CNS following Infection and Injury. Trends Immunol. 2015;36(10):637–650. doi: 10.1016/j.it.2015.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo MV, McGavern DB. Inflammatory neuroprotection following traumatic brain injury. Science. 2016;353(6301):783–785. doi: 10.1126/science.aaf6260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinert EM, Schenkel JM, Fraser KA, Beura LK, Manlove LS, Igyarto BZ, Masopust D. Quantifying Memory CD8 T Cells Reveals Regionalization of Immunosurveillance. Cell. 2015;161(4):737–749. doi: 10.1016/j.cell.2015.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Unen V, Li N, Molendijk I, Temurhan M, Hollt T, van der Meulen-de Jong AE, Koning F. Mass Cytometry of the Human Mucosal Immune System Identifies Tissue- and Disease-Associated Immune Subsets. Immunity. 2016;44(5):1227–1239. doi: 10.1016/j.immuni.2016.04.014. [DOI] [PubMed] [Google Scholar]
- Wu C, Ivars F, Anderson P, Hallmann R, Vestweber D, Nilsson P, Sorokin LM. Endothelial basement membrane laminin alpha5 selectively inhibits T lymphocyte extravasation into the brain. Nat Med. 2009;15(5):519–527. doi: 10.1038/nm.1957. [DOI] [PubMed] [Google Scholar]
- Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I, Schultze JL. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity. 2014;40(2):274–288. doi: 10.1016/j.immuni.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]