

Abstract
Mucor circinelloides is a fungus that belongs to the genus Mucorales. It grows as mold in the environment and can cause mucormycosis, a potentially fatal infection in immunocompromised patients. M. circinelloides is a biodiesel producer, and serves as a model organism for studying several biological processes such as light responses and RNAi mediated silencing. Over the past decade, the increasing number of molecular tools has allowed us to manipulate its genome. This unit outlines the fundamental protocols for the in vitro growth, maintenance, and genetic manipulation of M. circinelloides in the laboratory.
Keywords: Mucor, Mucor circinelloides, RNAi, gene knock out, mucormycosis
INTRODUCTION
Mucor circinelloides is a naturally growing mold that can cause potentially fatal infection in immunocompromised patients. Hence, its predominant growth form is filamentation, but under specific conditions it can also grow as yeast (Bartnicki-Garcia & Nickerson, 1962; Lee et al., 2013; Pasteur, 1876). The current unit focuses on the laboratory growth, maintenance, and genetic manipulation of M. circinelloides.
The commonly used M. circinelloides strains are – CBS277.49, R7B, and MU402. These strains grow as hyphae in the laboratory. The R7B strain is a leucine auxotrophic (leuA−) mutant strain derived from the sequenced CBS277.49 strain by the Joint Genome Institute (https://genome.jgi.doe.gov/Mucci2/Mucci2.home.html) (Roncero et al., 1984). The MU402 strain was derived from R7B, carrying a point mutation in the pyrG gene (Nicolas et al., 2007). Hence, MU402 is both uracil and leucine autotroph. The protocols described in this unit are suitable for the three strains. It is important to note that while using R7B, the recommended growth medium should be supplemented with leucine; and when using MU402, uracil and leucine should be added to the recommended growth medium. The Basic protocols 1–3 outline the methods for growth and maintenance of M. circinelloides on solid and liquid media, which is a pre-requisite for carrying out experiments. Basic Protocol 4 highlights the long-term storage requirements of the spores which is essential for maintaining laboratory stocks. Basic Protocol 5–6 will provide the step-by-step guidance for genetic manipulations by gene knock-out or RNAi mediated gene knock-down. Finally, Basic Protocol 7 provides the directions for measuring steady-state gene expression in M. circinelloides samples.
CAUTION: M. circinelloides is a biosafety level 1 (BSL-1) organism; however, it is recommended to manipulate it as a BSL-2 pathogen. Follow all appropriate guidelines and regulations for the use and handling of pathogenic microorganisms. See UNIT 1A.1 and other pertinent resources (APPENDIX 1B) for more information.
NOTE: Carry out all procedures in this unit using sterile technique.
BASIC PROTOCOL 1: GROWTH AND SPORANGIOSPORE PRODUCTION OF M. CIRCINELLOIDES MYCELIUM ON SOLID PLATE
The growth and sporangiospore production of M. circinelloides on the solid plate is the first step in any experiment. The commonly used medium for generation of mycelium are – YPG and MMC agars (see reagent and solutions). Also, M. circinelloides growth requires the presence of light for the activation of carotenoid biosynthesis and asexual sporangiospore production (Silva et al., 2006). The growth pattern from day 1–4 post inoculation of spores on solid YPG media is shown in Figure 1.
Figure 1. Compares the growth of M. circinelloides from day 1 to day 4.
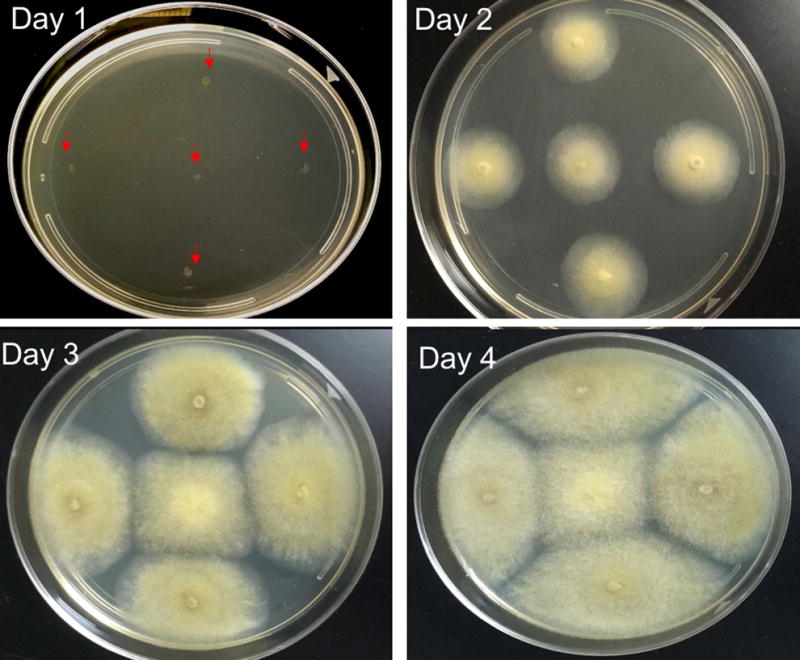
On Day 1, the spores are inoculated as a spot, indicated by arrows. During day 2 to 4, there is expansion in size of the colonies and production of spores. On day 4, the spores can be collected for experiments.
Materials
M. circinelloides frozen stock or sporangiospore (denoted as spore) suspension (see Basic Protocol 4)
YPG or MMC plate (see reagents and solutions)
Sterile wooden toothpick
- 26°C incubator with light or room temperature under the lights
- Bring the plate to room temperature before use.
- Divide the plate roughly into four quadrants and use a sterile toothpick to transfer frozen spores or spores in suspension onto each quadrant of the plate, and on center as shown in Figure 1 (Day 1).
- Place the plates in the incubator (26°C and equipped with light) or grow them at room temperature under light with the lid of the plates facing upwards. It is recommended to tape the plates with masking tapes to avoid the release of spores from the plates in the event of dropping them.
-
Maintain the plates in the incubator for 3 to 4 days’ post inoculation.
- Proceed to the collection of spores (Basic Protocol 2).
Figure 2. Sporangia containing numerous sporangiospores.
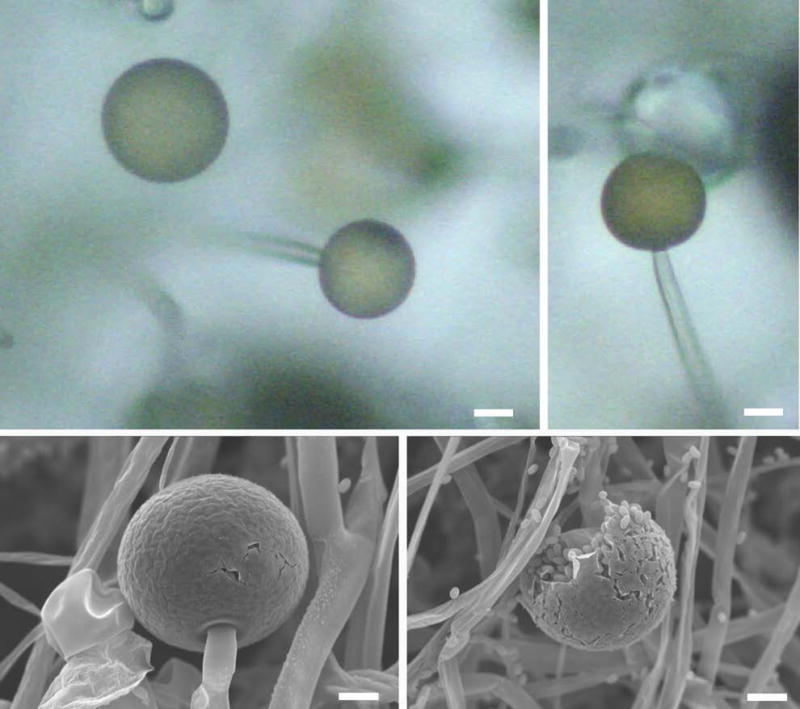
The SEM image shows intact sporangia (top panel). Each sporangium can produce several sporangiospores (bottom panel). Scale = 20 μm (adapted from Li et al., (2011) Sporangiospore size dimorphism is linked to virulence of Mucor circinelloides. PLOS pathogens).
BASIC PROTOCOL 2: COLLECTION OF M. CIRCINELLOIDES SPORES FROM SOLID CULTURE PLATE
The collection of fresh spores is required for subsequent generation of mycelium on a new solid plate (See Basic Protocol 1) or in liquid culture (see Basic Protocol 3), preparation of glycerol stocks (Basic Protocol 4) or for gene manipulation (Basic Protocol 5–6). After collection of spores, the remaining mycelium mat on the plate (as shown in Figure 3A) can be used for genomic DNA isolation. The fungal spores should be collected in a laminar flow hood to prevent the escape of fungal spores. After collection of spores, UV light should be turned on for at least 15 min for complete sterilization followed by appropriate disinfection of the surface of the hood. The spore suspension collected (Basic Protocol 2) can be temporarily stored at 4°C up to 6 months.
Figure 3. Production of mycelial mass by M. circinelloides.
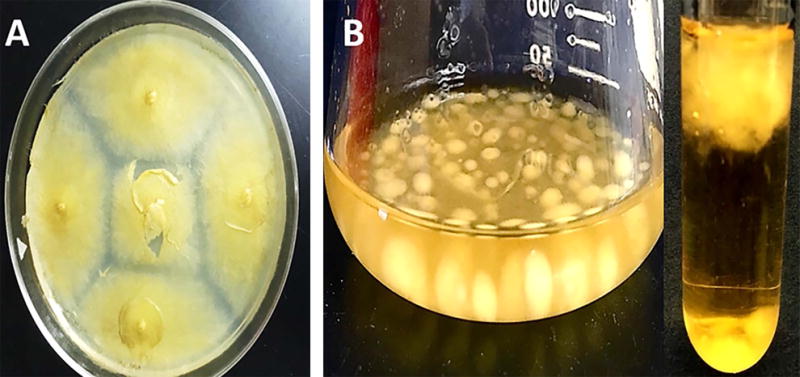
A) Solid culture –The mycelial mass is visible on a solid plate after collection of spores’ post-confluence (Basic Protocol 2), B) Suspension culture - The spores grown overnight in an Erlenmeyer flask or glass culture tube containing YPG produce mycelial mass (Basic Protocol 3).
Materials
M. circinelloides spores/hyphae on solid plate
Autoclaved deionized distilled water
Sterile L- shaped spreader
Microcentrifuge tubes (1.5 or 2ml)
- Hemocytometer (Neubauer) or automated cell counter (Biorad)
- Add 1–3 ml of deionized distilled water onto the plate with spores. If micropipette is used to add water, then change pipette tip every time water is added to avoid cross contaminations.
- Gently scrape the plate with a spreader to release spores.
- Tilt the plates by raising one end of the plate such that all the spores accumulate on the opposite end. Transfer spore suspension into microcentrifuge tubes with a micropipette.
- The number of spores can be determined using a hemocytometer or automated cell counter.
BASIC PROTOCOL 3: SUSPENSION CULTURE OF M. CIRCINELLOIDES
The suspension culture of M. circinelloides is required for applications such as protein precipitation or large scale genomic DNA extraction. Liquid YPG or YPD medium is commonly used for the suspension culture.
Materials
Liquid YPG or YPD medium (see reagent and solutions)
M. circinelloides spore suspension (see Basic Protocol 2)
Culture tube with a cap or Erlenmeyer flask
Aluminum foil
Miracloth (MilliporeSigma).
- Incubator equipped with a shaker
- Add 1–30ml of liquid YPG or YPD medium in a sterile culture tube or Erlenmeyer flask.
- Use a micropipette to add spores to the YPG or YPD medium containing glass culture tube or conical flask. The concentration of spores may vary depending on the experiment. Usually 10 μl spore suspension (1 × 106 spores/ml) per 1 ml of liquid media is enough to generate mycelial mass after overnight culture.
- For culture tubes, do not tighten the cap entirely. If using an Erlenmeyer flask, wrap the top of the flask with a sterile aluminum to prevent contamination.
- Incubate overnight at 26°C or 30°C with shaking at 250 rpm.
Mycelial mass is visible after overnight shaking culture (Figure 3B).
Collect the mycelial mass by filtering the liquid culture with sterile Miracloth. Wash the fungal mass with sterile deionized water and freeze dry for protein extraction or large scale genomic DNA extraction. This step can be done in a non-sterile condition.
BASIC PROTOCOL 4: SHORT TERM AND LONG-TERM STORAGE OF M. CIRCINELLOIDES
The spore suspensions can be stored at 4°C for six months and used as initial inoculum to produce fresh spores for any experiments. For long-term storage and maintenance of laboratory stocks, M. circinelloides should be stored at -80°C. We recommend creating duplicate stocks as a backup for frequently used strains, as the viability of spores may be reduced if the cryo-tubes are thawed to room temperature repeatedly.
Materials
15% glycerol (v/v)
M. circinelloides spore suspension (see Basic Protocol 2)
Cryo-vials
Microcentrifuge
Vortex
- −80°C freezer
- Centrifuge the microcentrifuge tubes containing spores (Basic Protocol 2) for 2 min at 6000–12000 rpm.
- Use a micropipette to carefully discard supernatant.
- Resuspend the spores in 15% (v/v) sterile glycerol and transfer the content to a cryovial.
- Store the glycerol stock in a −80°C freezer.
BASIC PROTOCOL 5: GENE MANIPULATION: DELETION
M. circinelloides spores are maintained on suitable medium for 3–5 days (Basic Protocol 1 or 4) and then the fresh spores are collected (Basic Protocol 2). The spores or germlings are grown under specific conditions to allow it to germinate. The germinated spores are then treated with lysing enzyme and chitosanase to degrade fungal cell walls, and this facilitates the transformation of knock out (KO) construct into the host. By homologous recombination (HR) the target gene is replaced with the generic marker. The construction of KO is explained in our previous papers (Garcia et al., 2017; Lee et al., 2015). The protocol outlines gene deletion protocol in the context of MU402 strain. Depending on strain requirements, the medium should be modified.
Materials
1×107 freshly collected MU402 spores (see Basic Protocol 1–2)
25 mL liquid YPG (see reagent and solutions)
100X UU (see reagent and solutions)
PS Buffer (pH 6.5; see reagent and solutions)
Lysing Enzymes from Trichoderma harzianum (Sigma-Aldrich)
Chitosanase RD (US Biological) suspended in PS buffer (see reagent and solutions)
10-ml syringe
0.22-μm syringe filter
Autoclaved, cold 0.5 M sorbitol in water
Cold YPGS (see reagents and solutions)
MMC (pH 3.2) containing 0.5 M sorbitol plates (see reagents and solutions)
Cold 0.2 cm gap cuvettes
The Gene Pulser Xcell™ electroporation system (Biorad) or equivalent
26°C incubator with a light
MMC Agar plates (pH 4.5)
DNA extraction kit (Epicenter)
Sterile Loop
Sterile water
5 μm filter
5 ml Syringe
5.1 PROTOPLAST PREPARATION FOR TRANSFORMATION OF THE M. CIRCINELLOIDES
-
1)
Collect fresh spores from a culture grown (Basic Protocols 1–2).
-
2)
Resuspend the spores in 20–25 ml of YPG medium containing UU. The final concentration of UU should be 1X.
-
3)
Incubate the spores in the YPG + UU medium overnight at 4°C without shaking.
-
4)Incubate the spores at 26°C while shaking at 300 rpm until the germ tube is about four times the size of the now swollen spore diameter. The emergence of germ tube from a spore is also visible (Figure 4A).
- This will usually take between 3–4 hrs (see critical parameters and troubleshooting).
-
5)
Transfer the spores to a 50-ml tube and centrifuge at 2000 rpm for 5 min. Discard the supernatant.
-
6)
Wash the cells twice by centrifugation at 2000 rpm for 5 min in 10 ml of PS buffer.
-
7)
Resuspend the washed pellet in 4 ml of PS buffer.
-
8)
Transfer the solution to a 50-ml Erlenmeyer flask.
-
9)
Add 5 mg of lysing enzyme to 1 ml of PS buffer in a microcentrifuge tube and mix well until there are no clumps of lysing enzyme left. Filter it using a 10-ml syringe equipped with a 0.22 μm filter (Using 25 mg of lysing enzyme in 5 ml of PS buffer is also an alternative to facilitate the measuring of the lysing enzyme).
-
10)
Take 1 ml of the filtered lysing enzyme solution and add it to the Erlenmeyer flask containing the resuspended spores.
-
11)
Add 100 μl of 0.15 U Chitosanase RD to the Erlenmeyer flask.
-
12)
Incubate at 26°C with gentle shaking for 90 min at 60 rpm or until the majority of fungal germlings become protoplasts. To ensure protoplasting, take 10 μl of mixture and place on a slide glass with a coverslip covered. Add water to the one edge of the coverslip and observe under the microscope, where fungal germlings should undergo lysis by osmosis.
Figure 4. Key observations in M. circinelloides gene deletion protocol.
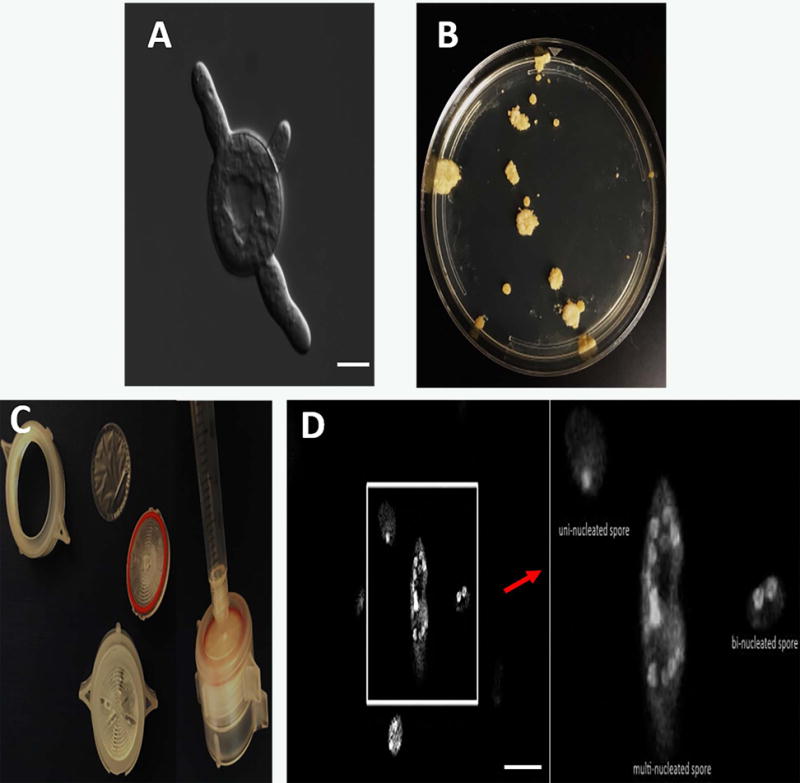
A) Emergence of germ tube from a spore, scale = 10 μm (Basic Protocol 5.1), B) Several isolated colonies are observed on a MMC containing sorbitol media post-transformation (Basic Protocol 5.3), C) Assembling components required for filtering spores to avoid excessive vegetative cycles (spores not shown) (Basic Protocol 5.3), D) Nuclei were stained with DAPI and cell walls were stained with calcofluor. Combined Z-stack images show a difference in the number of nuclei in M. circinelloides spores, scale = 10 μm (Basic Protocol 5.3).
5.2 TRANSFORMATION OF KO CONSTRUCT INTO M. CIRCINELLOIDES
-
13)
Transfer the 5 ml solution with protoplasts (from step 12) into a round bottom centrifuge tube and fill it with 10 ml of cold 0.5 M sorbitol. Centrifuge the tube at 800 rpm for 5 min, and discard the supernatant.
-
14)
Wash by centrifugation in 10 ml of cold 0.5 M sorbitol twice at 800 rpm. Discard the supernatant after the last wash.
-
15)Resuspend the pellet in 800 μl of cold 0.5 M sorbitol.
- Be gentle with resuspending the spores being careful not to pipette too vigorously. This 800 μl solution allows for 8 different transformations.
-
16)Add 100 μl of the 800 μl solution to 8 individual microcentrifuge tubes. Each tube must contain 100 μl of the protoplast solution and about 10 μl of DNA sample.
- We recommend using 1 μg total of DNA for circular plasmids or 3 μg total DNA for linear fragments. DNA must be dissolved in double distilled water. As a control use 10 μl of double distilled water instead of DNA.
-
17)
Mix by gentle tapping or inverting and transfer each solution to individual 0.2-cm gap cuvettes on ice.
-
18)
Electroporation - Apply an electrical pulse with the following conditions: field strength 0.8 KV, capacitance 25 μF, and constant resistance 400 Ω. Immediately after the pulse, add 1 ml of cold YPGS, and keep on ice until all cuvettes have been pulsed.
-
19)
Repeat the electroporation for rest of the cuvettes.
-
20)
Transfer the content of each cuvette to 1.5 ml centrifuge tubes.
-
21)
Incubate at 26°C for 1 hr while shaking at about 150 rpm.
-
22)
Centrifuge the tubes at 1100 rpm for 5 min, and discard the supernatant.
-
23)
Gently resuspend the pellet in a final volume of 400 - 600 μl of 0.5 M sorbitol.
-
24)
Add 200 μl of the resuspended solution containing the transformants onto the MMC + 0.5 M sorbitol plates. Gently spread by using a spreader.
-
25)
Cover with tin foil, and place at 26°C for 3 days (see troubleshooting) or until colonies emerge.
5.3 CONFIRMATION OF THE GENE KNOCKOUT
-
26)
Uncover the plates containing the transformants, and check for colony formation. There should be isolated colonies formed over the media (Figure 4B).
-
27)Dip a sterile loop into the sterile water and gently tap one of the colonies to collect the spores. Touch the center of an MMC agar plate with the loop containing the spores to inoculate the plate.
- Repeat for all the colonies. For each colony use a new MMC agar plate.
-
28)
Grow the plates for 3–4 days at 26°C in the presence of light source (see Basic protocol 1 or 3).
-
29)
Collect the spores from all the plates into separate microcentrifuge tubes, and store them at 4°C (See Basic Protocol 2).
-
30)
Collect the mycelial mat from each plate for genomic DNA extraction.
If the mycelial mat covers the entire surface of the plate, then a quarter of the mat should be sufficient to isolate DNA.
-
31)
Identify a homologous recombinant via junction PCR, as described previously (Lee et al., 2013; Lee et al., 2015).
Upon identification of homologous recombinants, one needs to isolate homokaryotic mutant in the gene of interest. M. circinelloides spores are multinucleated, and the hyphae are co-enocytic, thus one needs to proceed vegetative cycles to eliminate wild-type nuclei from transformants. Spore filtering method can be applied to avoid excessive vegetative cycles. The components of the filter are shown in Figure 4C. Although M. circinelloides produces multi-nucleate larger spores in general, it also produces a subpopulation of uni- or bi-nucleate smaller spores (Li et al., 2011). Figure 4D compares a multi-nucleate, bi-nucleate and a uni-nucleate spore.
-
32)
Filter the spore suspension (from step 29) of transformants positive from junction PCR using a 5 μm pore filter, which only allows smaller uni- or bi-nucleate spores.
-
33)
Streak MMC plate (pH 3.2) with the filtered spore solution.
Low pH suppresses mycelial growth so that M. circinelloides produces compact colonies that are easy to separate.
-
34)
Select several colonies isolated from Step 33) and check the genome of them by PCR to ensure homokaryotypes at the deleted allele.
In addition to junction PCR, Southern blotting should be performed to confirm gene deletion.
BASIC PROTOCOL 6: GENE SILENCING – Silencing vector system
M. circinelloides has a well-described gene silencing machinery that has become one of the most useful molecular tools to manipulate this fungus. Besides its molecular application, the RNAi mechanism of M. circinelloides also regulates endogenous gene expression that controls several biological processes such as growth, sexual and asexual reproduction, drug resistance, and autolysis by nutritional starvation (Calo et al., 2014; Ruiz-Vazquez et al., 2015; Torres-Martínez, 2017). This RNAi mechanism offers an alternative method to study functional genomics by silencing any target gene, including essential ones. The gene silencing vector for M. circinelloides is a self-replicative plasmid that contains a selectable marker and a silencing cassette. This cassette has two strong convergent promoters (Pzrt1 and Pgpd1) flanking a multiple cloning site (MCS) where any DNA fragment can be cloned. These promoters initiate transcription of the cloned DNA fragment in both directions, producing double-stranded RNA that triggers the gene silencing mechanism in the fungus.
The following protocol describes the use of the silencing vector pMAT1702 (Figure 5A) to generate a whole-genome silencing library and its application as a molecular tool for functional genomic screenings (Figure 5B). This strategy has allowed the identification of new virulence factors in the life-threatening fungal infection known as mucormycosis (Trieu et al., 2017).
Figure 5. Silencing vector system.
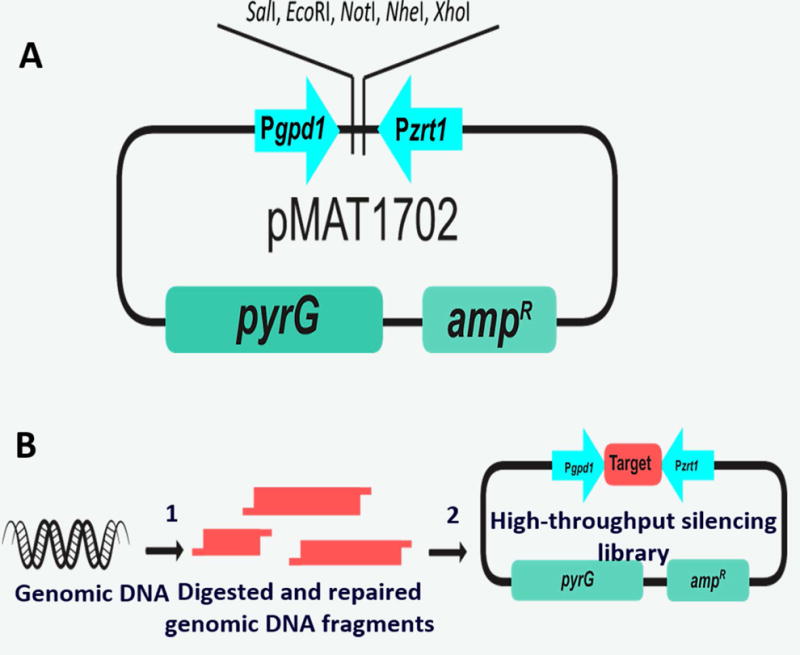
A) Diagram of the silencing vector pMAT1702. B) Diagram of the strategy used to build a genome-wide RNAi library: 1) Purified genomic DNA is digested with restriction enzymes and repaired to make compatible ends and avoid self-ligation, 2) Digested and repaired genomic DNA fragments are cloned into the digested vector (adapted from Trieu et al., (2017) RNAi-based functional genomics identifies new virulence determinants in mucormycosis. PLOS pathogens).
Materials
Silencing vector pMAT1702 (Trieu et al., 2017)
Restriction enzymes XhoI and Sau3AI (Thermo Scientific - Fermentas)
DNA Polymerase I Large (Klenow) Fragment Mini Kit (Promega)
GeneJET Gel Extraction Kit (Thermo Scientific) or equivalent
T4 DNA ligase (Thermo Scientific)
Escherichia coli competent cells obtained by calcium chloride procedure (Green & Sambrook, 2012)
Luria-Bertani (LB) liquid media (see reagents and solutions)
LB + ampicillin agar plates (see reagents and solutions)
Cell scrappers
GeneJET Plasmid Miniprep Kit (Thermo Scientific) or equivalent
Primers peuka-1 5′-CATGAAGTGTGAGACATTGCG-3′ and pzrt1 5′-GAGATGTGCCTTGATGATATGCTCT-3′.
Taq DNA polymerase kit (Sigma-Aldrich)
GeneJET PCR Purification Kit (Thermo Scientific) or equivalent
6.1. Whole-genome library
-
1)
Digest 3 μg of the silencing vector pMAT1702 with at least 3 units of the restriction enzyme XhoI in a buffered reaction mixture, as recommended by the supplier.
-
2)
Add dCTP-dTTP to fill in the cohesive ends to avoid self-ligation (Werner et al., 1997), using DNA Polymerase I Large (Klenow) Fragment Mini Kit and incubating the reaction for 30 minutes at 37°C, following the supplier recommendations.
-
3)
Extract M. circinelloides genomic DNA as described previously (Nicolas et al., 2007).
-
4)
Partially digest 20 μg of M. circinelloides genomic DNA with 20 units of Sau3AI in a buffered reaction mixture, as recommended by the supplier.
-
5)
Isolate the digested fragments of 0.5–4 kb by electrophoresis in an agarose gel with GeneJET Gel Extraction Kit.
-
6)
Fill in the purified genomic fragments with dGTP-dATP (following the same procedure as in step 2) to make compatible ends with the digested silencing vector.
-
7)
Ligate the digested genomic fragments with the silencing vector in a 3:1 molar ratio insert:vector with 1 unit of T4 DNA ligase in a total volume of 20 μl. Incubate reactions at 22°C for a minimum of 4 hours. After incubation, inactivate the ligase by heating the mixtures at 70°C for 5 minutes.
-
8)
Heat-shock transform 20 μl of the ligation mixtures into Escherichia coli competent cells to obtain a genome-wide RNAi library. The heat shock is performed by incubating cell mixtures at 42°C for 50 seconds, and then putting them immediately in ice for 2 minutes. After transformation, add 1 ml of LB liquid medium to the cells and incubate at 37°C for 1 hour before plating them in LB solid medium containing ampicillin (100 mg/l).
At least 83,000 clones are needed to obtain a library that represents M. circinelloides genome with a confidence level higher than 99%. To determine the coverage of M. circinelloides genome in the genomic library and its confidence level, the algorithm N = ln(1-P)/ln(1-f) was followed; where N is the required number of recombinants, P is the desired probability that any genomic sequence is represented in the library at least once, and f is the fractional proportion of the genome in a single recombinant; i.e. f = i/g, where i is the insert size and g is the genome size.
-
9)
Pool E. coli colonies from LB with ampicillin plates using a cell-scrapper.
-
10)
Purify plasmids from colony pools using GeneJET Plasmid Miniprep Kit.
-
11)
Transform M. circinelloides with 10 μg of the purified plasmid library (following basic protocol 5), as many times as needed to obtain a collection of transformants that represents the whole genome.
According to M. circinelloides genome size of 36.6Mbp (Corrochano et al., 2016), up to 50,000 silenced transformants are needed to ensure a 95% confidence level.
6.2. Functional genomic screening
The collection of M. circinelloides silenced transformants obtained with the genome-wide RNAi library can be screened under any restrictive condition to select the desired phenotypes. The genes silenced by the RNAi library in the selected transformants can be identified by the following protocol.
-
12)
Isolate M. circinelloides DNA from the selected transformants in the screenings. The silencing vector is purified together with the genomic DNA (Nicolas et al., 2007).
-
13)
PCR-amplify with Taq DNA polymerase the DNA fragment cloned into the silencing vector using primers peuka-1 and pzrt1 that anneal in the convergent promoters Pgpd1 and Pzrt1, respectively. The PCR samples are subjected to an initial denaturing step of 5 minutes at 95°C, and 30~35 cycles of denaturation (30 seconds at 95°C), annealing (30 seconds at 60°C) and elongation (for 4 minutes at 72°C).
-
14)
Purify the PCR products with GeneJET PCR Purification Kit.
-
15)
Sequence the PCR product by Sanger method using the primers above.
-
16)
Blast the obtained sequence against M. circinelloides genome database (http://genome.jgi.doe.gov/Mucci2/Mucci2.home.html).
The silencing cassette in the pMAT1702 can contain more than one gene fragment. To identify which gene is responsible for the selected phenotype a validation experiment is needed. A fragment of each gene must be cloned in the silencing vector and transformed in M. circinelloides independently. The silencing vector that reproduces the expected phenotype would harbor the responsible gene.
BASIC PROTOCOL 7: RELATIVE GENE EXPRESSION DATA USING REAL-TIME QUANTITATIVE PCR
Purification of total RNA and subsequent analysis by Northern blot techniques has been the traditional method used to study gene expression in M. circinelloides. However, the recent introduction of M. circinelloides as a model to study virulence in Mucorales has raised the requirement of analyzing gene expression from samples with a low amount of fungal RNA, for instance, infected tissues from in vivo experiments. In these cases, Real-Time quantitative PCR (RT-qPCR) is a useful method to quantify gene expression from samples that cannot be analyzed using traditional hybridization techniques. Relative quantitation describes changes in the expression level of a specific gene relative to some reference sample. Expression levels are normalized respect to an endogenous control gene, and the results are expressed as a fold-change or a fold-difference. The 18S rRNA expression levels are constant in all the morphological phases of Mucorales; therefore 18S rRNA can be an appropriate endogenous control gene in RNA samples of M. circinelloides (Pathan et al., 2017).
7.1 Reverse Transcription
Materials
High quality total RNA from M. circinelloides, usually purified with a silica-based kit.
Oligo dT(20) 50μM
RNase Inhibitor
Water RNase and DNase free
dNTPs 10 mM
DTT 100 mM
5x Expand Reverse Transcriptase buffer (Roche)
Expand Reverse Transcriptase (Roche)
-
1)Add reagents in the following order to a sterile RNase, DNase free tube:
- 1 μg of total RNA
- 1 μl of oligo dT(20) 50 μM
- 10 U of RNase Inhibitor
- Water RNase, DNase free to a final volume of 10.5 μl
-
2)
Incubate for 10 min at 65°C. Immediately cool on ice.
-
3)Add reagents in the following order to the same tube:
- 2 μl of dNTPs 10 mM
- 2 μl of DTT 100 mM
- 10 U (0.5 μl) de RNase Inhibitor
- 4 μl of 5x Expand Reverse Transcriptase buffer (Roche)
- 1 μl (50 U) of Expand Reverse Transcriptase (Roche)
- Total volume: 20 μl.
-
4)
Incubate PCR tube for 1 hr at 43°C.
-
5)
Heat inactivation of the enzyme by incubating at 95°C for 2 min.
-
6)
Stop reaction by placing the tube on ice.
7.2 Real-time quantitative PCR
Materials
2X SYBR® Green PCR Master Mix (Applied Biosystems)
Forward Primer (50–900 nM)
Reverse Primer (50–900 nM)
Water RNase and DNase free
Template (cDNA)
-
7)Add Thermal Cycling parameters:
- Step 1: AmpliTaq Gold® Polymerase Activation 95°C for 10 min
- Step 2: PCR 40 cycles of 95°C for 15 sec (denature) and 60°C for 1 min (anneal and extend)
-
8)
To confirm the absence of non-specific amplification non-template control and a melting curve should be included.
7.3 The Comparative CT Method
The comparative CT method also referred to as the ΔΔCT method, uses arithmetic formulas to achieve the result for relative quantitation (Livak & Schmittgen, 2001).
-
9)
Calculate the ΔCT value by: ΔCT = CT specific gene – CT endogenous
-
10)
Calculate the ΔΔCT value by: ΔΔCT = ΔCT test sample – ΔCT reference sample
-
11)
Calculate the standard deviation of the ΔΔCT value and incorporating into the fold-difference.
-
12)
The amount of specific gene, normalized to an endogenous reference and relative to a reference sample, is given by: 2–ΔΔCT
Relative quantitation studies require an optimally designed primer. Amplicons of 50–150 base pairs are strongly recommended, and Tm should be from 58 to 60 °C.
For a valid ΔΔCT calculation, the efficiency of the specific gene amplification and the efficiency of the endogenous control amplification must be approximately equal. To determine relative efficiencies, standard curves with template dilutions must be performed. The CT values generated from equivalent standard curve are used in the ΔCT calculation (CT specific gene – CT endogenous). These ΔCT values are plotted vs. log input amount to create a semi-log regression line. The absolute value of the slope of ΔCT vs. log input must be < 0.1.
REAGENTS AND SOLUTIONS
Minimal Media with Casamino acids (MMC) agar plates
In a flask add,
10 g casamino acids
0.5 g yeast nitrogen base w/o amino acids
20 g glucose
0.5 L deionized distilled water
Adjust the pH to 3.2 or 4.5
Sterilize by autoclaving
In another flask, dissolve 15 g agar in 0.5 L double distilled water, and sterilize by autoclaving. Mix both the flasks. Before pouring, add 1 ml of Niacin (1mg/ml) and Thiamine (1mg/ml) antibiotics. Plates can be stored at 4°C.
Note: One needs to prepare 2x water agar and 2x liquid MMC (pH 3.2 or 4.5) separately and, after autoclave, mix them to achieve 1x MMC agar (pH 3.2 or 4.5) to avoid acid hydrolysis of agar during autoclave.
MMC agar + 0.5 M sorbitol plates
Add 90.1 g of sorbitol per liter to MMC recipe before autoclaving.
Follow MMC agar preparation.
Yeast Peptone Glucose (YPG) media
3 g yeast extract
10 g peptone
20 g glucose
1L double distilled water
Adjust the pH to 4.5
Sterilize by autoclaving
For plates: In one flask, dissolve above mentioned quantities of YPG in 0.5 L of double distilled water (adjust pH and autoclave). In another flask, dissolve 20 g of agar in 0.5 L of double distilled water, and autoclave. After autoclaving, mix them. The plates can be stored at 4°C.
YPG + Sorbitol (YPGS) media
Add 90.1 g of sorbitol per liter to YPG recipe before autoclaving.
Follow YPG media preparation.
100X Uridine and Uracil (UU)
6.1 g uridine
5.6 g uracil
0.1 L double distilled water
Sterilize by autoclave
Store at 4°C
Yeast Peptone Dextrose (YPD) media
10 g yeast extract
20 g peptone
20 g dextrose
1L double distilled water
Sterilize by autoclaving
Preparation of PS buffer
Step 1 – preparation of sodium phosphate buffer (0.1 M, pH 6.5)
1.42 g Na2HPO4 in 100 ml of double distilled water (solution 1)
1.38 g NaH2PO4 in 100 ml of double distilled water (solution 2)
To solution 2 add solution 1 until pH is 6.5. Store at room temperature.
Step 2 – preparation of PS buffer
18.22 g sorbitol
Dissolve sorbitol in 20 ml of sodium phosphate buffer (step 1). Makeup to 200 ml with double distilled water. Filter the resulting buffer using 0.22 μM filter. Store at room temperature.
Preparation of chitosanase
Thaw and briefly centrifuge the chitosanase vial purchased from the manufacturer and dilute it using PS buffer. Make aliquots such that each vial of 100 μl should yield a final concentration of 0.15 U, when added to the mixture containing 4 ml M. circinelloides pellet and 1 ml lysing enzyme (see Basic Protocol 5.1, step 7–11). All the aliquots should be stored at -80°C.
Luria- Bertani (LB) media
10 g tryptone
10 g sodium chloride
5 g yeast extract
1L double distilled water
Sterilize by autoclaving
LB agar plates
Add 15 g of agar per liter to LB recipe before autoclaving.
Add 100 mg of ampicillin after autoclaving if needed.
COMMENTARY
Background Information
Mucor circinelloides is a member of Mucorales belonging to the phylum Mucoromycota (Spatafora et al., 2016). This fungus is filamentous by nature and is most commonly found in soil, plants and decaying fruits. M. circinelloides is associated with both biotechnology industry and human health (Carvalho et al., 2015; Lee et al., 2014).
Besides growing on a wide range of carbon sources (McIntyre et al., 2002), M. circinelloides is rich in polyunsaturated fats and stores lipids, which favors its use for biodiesel production (Wynn et al., 1999). Previous studies have shown that 93% of single cell oil can be converted to ethyl esters, suggesting that M. circinelloides are a good single-cell oil producer (Carvalho et al., 2015). Studies have also shown that the quality of biodiesel produced using M. circinelloides met the specifications set by American and European standards (Vicente et al., 2009). This suggests that M. circinelloides can be possibly used as an alternative to plant oils as a feedstock for biodiesel production. M. circinelloides is also used to produce enzymes such as proteases, which is integral for industrial applications (Andrade et al., 2002; Sathya et al., 2009).
M. circinelloides poses a serious threat to public health. M. circinelloides is one of the common causative agent of mucormycosis, a potentially fatal infection in immunocompromised humans and animals (Dizbay et al., 2009; Ghuman & Voelz, 2017). The mortality rates associated with mucormycosis can be as high as 96%, depending on site of infection (Petrikkos et al., 2012). It can contaminate and spoil foods, causing food poisoning (Brouillet et al., 2005; Hollmann et al., 2008; Lazar et al., 2014; Snyder et al., 2016).
M. circinelloides is also used for studying biological processes such as light sensing. Three photoreceptor genes have been identified and characterized in M. circinelloides - mcwc-1a, mcwc-1b and mcwc-1c (Silva et al., 2006). While mcwc-1c regulates the light transduction pathway that induces carotene biosynthesis, mcwc-1a mainly controls positive phototropism. These mechanisms are required for activation of caratenogenesis, and production of asexual spores (See Basic Protocol 1).
Along with light, maintenance of other physical (temperature) and chemical (pH, nutrient composition) parameters is also essential for growth of the M. circinelloides. As mentioned in the Introduction section when using strains such as R7B and MU402 the growth medium should be supplemented with additional nutrients such as leucine alone or with uracil.
Gene knockout is an important tool for understanding the fundamental biology of M. circinelloides. We have previously used this tool to identify the function of calcineurin, the key regulator of dimorphic transitions (Lee et al., 2013; Lee et al., 2015). We have also a developed a recyclable marker system that allows using a single marker to delete a series of genes (Garcia et al., 2017). Recently, a plasmid-free CRISPR/Cas9 system has been developed to manipulate genes in M. circinelloides (Nagy et al., 2017). This approach involves direct insertion of guide RNA and Cas9 enzyme into the fungal organism. A template DNA can also be introduced to facilitate homology-mediated repair. M. circinelloides serves as a model organism to study RNAi-mediated silencing (Nicolas & Ruiz-Vazquez, 2013; Nicolas et al., 2009; Nicolas et al., 2015; Trieu et al., 2015). This early-diverging fungus combines the basic components of eukaryotic RNAi machinery with some novel elements to generate sRNAs that induce gene silencing. M. circinelloides presents two functional RNA-dependent RNA polymerases (RdRPs), RdRP-1, which produces dsRNAs from transgene transcripts, and RdRP-2, which amplifies the silencing signal by producing new dsRNA molecules, using the processed target mRNA as a template (Calo et al., 2012). These dsRNA molecules are then processed by the major Dicer protein in M. circinelloides, Dcl-2 (de Haro et al., 2009), into 21- and 25-nt long siRNAs (Nicolas et al., 2003). The siRNAs are loaded into Ago-1 to guide the degradation of target transcripts by complementary base pairing (Cervantes et al., 2013). M. circinelloides takes advantage of this mechanism to defend its genome against transposon-derived invasive nucleic acids (Nicolas et al., 2010); but, more importantly, it exploits the RNAi machinery to regulate its own gene expression (Nicolas et al., 2015). Together, genetic manipulation facilitates understanding of pathogenesis and drug resistance mechanisms of M. circinelloides (see Basic Protocol 5–6).
Low amounts of RNA in M. circinelloides poses a fundamental challenge to perform analysis at post-transcription level. Basic Protocol 7 provides the directions for analyzing steady state gene expression using RT-qPCR.
We anticipate that the protocols provided in this unit will allow new researchers to not only grow and maintain M. circinelloides in the lab effortlessly but will also permit them to manipulate genetics to generate new knowledge.
Critical Parameters and Troubleshooting
Following the transfer of frozen M. circinelloides spores to the solid plate, if no growth is observed, it is possible that the spores have lost their viability. Hence, all the frozen stock should be maintained at -80°C to prevent viability loss. We also recommend creating duplicate stocks of frequently used strains as back up. Also, the M. circinelloides requires light for metabolism, therefore, the incubator used for solid culture must be equipped with a light source. Following inoculation of spores on the solid plate (Basic Protocol 1), the plates must be taped on two sides to prevent the escape of fungal spores, and the plates must be placed in an incubator with the lid facing upwards such that the spores receive direct light. It is possible that one can use a benchtop at room temperature to grow M. circinelloides with continuous exposure to light. It is recommended that the culture plates need to be separated to avoid cross-contamination.
We recommend using freshly collected spores for the protoplast preparation part (Basic Protocol 5.1) of gene knockout procedure for optimum results. Also, following the treatment with the chitosanase and lysing enzyme, the digested germ tubes are very frail, thus handling them with care is recommended.
In the Basic Protocol 5.2, we recommended using 1 μg of DNA for circular plasmids and 3 μg for linear DNA fragments for transformation into M. circinelloides. It is important to limit the volume of DNA to 10μl, we observed that volume exceeding 10 μl negatively impacts protoplast.
Although M. circinelloides is multi-nucleated, following transformation of gene deletion cassette, the sub-population of spores with knockout construct inserted at the right position are uni-nucleated. The uni-nucleated spores are known to be about 3 μm in size while multi-nucleated spores are much larger. Thus, filtering out the collected spores will allow us to enrich uni-nucleated spores. The filtered spores are grown in MMC (pH 3.2) media, and then junction PCR and Southern blotting are used to identify true transformants, as explained in Basic Protocol 5.3.
In the Basic Protocol 6, M. circinelloides multi-nucleated spores are also a decisive factor to obtain fully silenced transformants. The vector pMAT1702 that harbors the library is self-replicative; therefore only a proportion of the asexual spores produced by the first transformant colonies may contain the plasmid. To maintain the plasmid and avoid the loss of the gene silencing in the following vegetative cycles, the transformant colonies must be grown under selective media. Since the vector pMAT1702 contains the auxotrophy marker pyrG, the silenced transformants should be plated in media without uridine, thus selecting the Ura+ colonies.
Anticipated results
Basic Protocol 1 describes the growth of M. circinelloides on solid plates. Maintenance of plates 26°C with light is sufficient for the plates to reach maximum confluence in 4 days. Basic Protocol 2 describes the methodology for collection of spores. A confluent plate should provide at least 107 - 108 spores/ml. A suspension culture of about 18 hrs (overnight; Basic Protocol 3) is sufficient for the spores to germinate into hyphae form. Basic Protocol 4 describes the steps involved in the preparation of freezer stocks. Reinoculation of frozen spores on solid plates should restore normal growth into hyphae. Basic Protocol 5 describes the procedure for knocking out a target gene in M. circinelloides. Depending on genes, about 5–30% of colonies obtained post-transformation have the selection marker inserted at the required position, the rest of the colonies are ectopic. In Basic Protocol 6 the vector pMAT1702 with genomic DNA fragments from 0.5–4 kb ensures a silencing frequency near 90%. By using Basic Protocol 7, the relative expression of a target gene can be determined.
Time Considerations
The fresh spores are ready to be collected from a solid plate within 3–4 days after being transferred from a frozen stock or previously collected spores (Basic Protocol 1). The collected spores can be stored for up to six months at 4°C.
During the protoplast preparation (Basic Protocol 5.1), occasionally 3–4 hrs of growth is not enough time for the spores to germinate. In that case, allow the spores to grow for a longer time and monitor the hyphal growth every 15 min until it reaches 75% confluence. In Basic Protocol 5.2 and 5.3, if no growth is observed after plating of transformants on MMC agar containing 0.5 M sorbitol for 3 days in dark, then grow the plates for another 2 days in the presence of light.
Plasmid DNA aliquots of each pooled library can be stored at -20°C and used later to transform M. circinelloides. The spores collected from the silencing transformants can be stored in PBS at 4°C to perform further phenotypic screenings.
SIGNIFICANCE STATEMENT.
The current article provides the essential protocols for laboratory growth, maintenance, and genetic manipulation of the fungus Mucor circinelloides. M. circinelloides belongs to the early diverged fungal lineage Mucoromycota. M. circinelloides serves as a model system to understand pathogenesis in mucormycosis a fatal fungal infection caused by Mucorales fungi, to elucidate light sensing system in fungi, and to study RNAi based gene silencing. This fungal system, however, is understudied compared to other fungal systems in dikarya. Hence, by outlining the fundamental protocols, we encourage scientists to study this versatile fungal system.
Acknowledgments
This work is supported by NIH/NIAID R03 AI11917 and UTSA Research funds to S.C.L.; and “Ministerio de Economía y Competitividad, Spain” (BFU2015-65501-P and BFU2015-62580-ERC; co-financed by FEDER) and “Fundación Séneca, Murcia, Spain” (19339/PI/14) to F.E.N., V.G., L.M., M.I.N.M. and C.P.A. Particularly, M.I.N.M. and C.P.A. were co-funded by “Ministerio de Educación, Cultura y Deporte” (FPU-14/01832 and FPU-14/01983, respectively) and F.E.N. was co-funded by “Ministerio de Economía y Competitividad, Spain” (RYC-2014-15844).
LITERATURE CITED
- Andrade VS, Sarubbo LA, Fukushima K, Miyaji M, Nishimura K, Campos-Takaki GMd. Production of extracellular proteases by Mucor circinelloides using D-glucose as carbon source/substrate. Braz J Microbiol. 2002;33:106–110. [Google Scholar]
- Bartnicki-Garcia S, Nickerson WJ. Induction of yeast-like development in Mucor by carbon dioxide. J Bacteriol. 1962;84:829–840. doi: 10.1128/jb.84.4.829-840.1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouillet E, Jacquard C, Bizat N, Blum D. 3-Nitropropionic acid: a mitochondrial toxin to uncover physiopathological mechanisms underlying striatal degeneration in Huntington’s disease. J Neurochem. 2005;95(6):1521–1540. doi: 10.1111/j.1471-4159.2005.03515.x. [DOI] [PubMed] [Google Scholar]
- Calo S, Nicolas FE, Vila A, Torres-Martinez S, Ruiz-Vazquez RM. Two distinct RNA-dependent RNA polymerases are required for initiation and amplification of RNA silencing in the basal fungus Mucor circinelloides. Mol Microbiol. 2012;83(2):379–394. doi: 10.1111/j.1365-2958.2011.07939.x. [DOI] [PubMed] [Google Scholar]
- Calo S, Shertz-Wall C, Lee SC, Bastidas RJ, Nicolas FE, Granek JA, Mieczkowski P, Torres-Martinez S, Ruiz-Vazquez RM, Cardenas ME, Heitman J. Antifungal drug resistance evoked via RNAi-dependent epimutations. Nature. 2014;513(7519):555–558. doi: 10.1038/nature13575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho AKF, Rivaldi JD, Barbosa JC, de Castro HF. Biosynthesis, characterization and enzymatic transesterification of single cell oil of Mucor circinelloides – A sustainable pathway for biofuel production. Bioresour Technol. 2015;181(Supplement C):47–53. doi: 10.1016/j.biortech.2014.12.110. [DOI] [PubMed] [Google Scholar]
- Cervantes M, Vila A, Nicolas FE, Moxon S, de Haro JP, Dalmay T, Torres-Martinez S, Ruiz-Vazquez RM. A single argonaute gene participates in exogenous and endogenous RNAi and controls cellular functions in the basal fungus Mucor circinelloides. PLoS One. 2013;8(7):e69283. doi: 10.1371/journal.pone.0069283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrochano LM, Kuo A, Marcet-Houben M, Polaino S, Salamov A, Villalobos-Escobedo JM, Grimwood J, Alvarez MI, Avalos J, Bauer D, Benito EP, Benoit I, Burger G, Camino LP, Canovas D, Cerda-Olmedo E, Cheng JF, Dominguez A, Elias M, Eslava AP, Glaser F, Gutierrez G, Heitman J, Henrissat B, Iturriaga EA, Lang BF, Lavin JL, Lee SC, Li W, Lindquist E, Lopez-Garcia S, Luque EM, Marcos AT, Martin J, McCluskey K, Medina HR, Miralles-Duran A, Miyazaki A, Munoz-Torres E, Oguiza JA, Ohm RA, Olmedo M, Orejas M, Ortiz-Castellanos L, Pisabarro AG, Rodriguez-Romero J, Ruiz-Herrera J, Ruiz-Vazquez R, Sanz C, Schackwitz W, Shahriari M, Shelest E, Silva-Franco F, Soanes D, Syed K, Tagua VG, Talbot NJ, Thon MR, Tice H, de Vries RP, Wiebenga A, Yadav JS, Braun EL, Baker SE, Garre V, Schmutz J, Horwitz BA, Torres-Martinez S, Idnurm A, Herrera-Estrella A, Gabaldon T, Grigoriev IV. Expansion of signal transduction pathways in fungi by extensive genome duplication. Curr Biol. 2016;26(12):1577–1584. doi: 10.1016/j.cub.2016.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Haro JP, Calo S, Cervantes M, Nicolas FE, Torres-Martinez S, Ruiz-Vazquez RM. A single dicer gene is required for efficient gene silencing associated with two classes of small antisense RNAs in Mucor circinelloides. Eukaryot Cell. 2009;8(10):1486–1497. doi: 10.1128/EC.00191-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dizbay M, Adisen E, Kustimur S, Sari N, Cengiz B, Yalcin B, Kalkanci A, Gonul II, Sugita T. Fungemia and cutaneous zygomycosis due to Mucor circinelloides in an intensive care unit patient: case report and review of literature. Jpn J Infect Dis. 2009;62(2):146–148. [PubMed] [Google Scholar]
- Garcia A, Adedoyin G, Heitman J, Lee SC. Construction of a recyclable genetic marker and serial gene deletions in the human pathogenic mucorales Mucor circinelloides. G3 (Bethesda) 2017;7(7):2047–2054. doi: 10.1534/g3.117.041095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghuman H, Voelz K. Innate and adaptive immunity to Mucorales. J Fungi. 2017;3(3):48. doi: 10.3390/jof3030048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green MR, Sambrook J. Molecular cloning: a laboratory manual. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 2012. [Google Scholar]
- Hollmann M, Razzazi-Fazeli E, Grajewski J, Twaruzek M, Sulyok M, Bohm J. Detection of 3-nitropropionic acid and cytotoxicity in Mucor circinelloides. Mycotoxin Res. 2008;24(3):140–150. doi: 10.1007/BF03032341. [DOI] [PubMed] [Google Scholar]
- Lazar SP, Lukaszewicz JM, Persad KA, Reinhardt JF. Rhinocerebral Mucor circinelloides infection in immunocompromised patient following yogurt ingestion. Del Med J. 2014;86(8):245–248. [PubMed] [Google Scholar]
- Lee SC, Billmyre RB, Li A, Carson S, Sykes SM, Huh EY, Mieczkowski P, Ko DC, Cuomo CA, Heitman J. Analysis of a food-borne fungal pathogen outbreak: Virulence and genome of a Mucor circinelloides isolate from yogurt. mBio. 2014;5(4):e01390–01314. doi: 10.1128/mBio.01390-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SC, Li A, Calo S, Heitman J. Calcineurin plays key roles in the dimorphic transition and virulence of the human pathogenic zygomycete Mucor circinelloides. PLoS Pathog. 2013;9(9):e1003625. doi: 10.1371/journal.ppat.1003625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SC, Li A, Calo S, Inoue M, Tonthat NK, Bain JM, Louw J, Shinohara ML, Erwig LP, Schumacher MA, Ko DC, Heitman J. Calcineurin orchestrates dimorphic transitions, antifungal drug responses, and host-pathogen interactions of the pathogenic mucoralean fungus Mucor circinelloides. Mol Microbiol. 2015;97(5):844–865. doi: 10.1111/mmi.13071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li CH, Cervantes M, Springer DJ, Boekhout T, Ruiz-Vazquez RM, Torres-Martinez SR, Heitman J, Lee SC. Sporangiospore size dimorphism is linked to virulence of Mucor circinelloides. PLoS Pathog. 2011;7(6):e1002086. doi: 10.1371/journal.ppat.1002086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- McIntyre M, Breum J, Arnau J, Nielsen J. Growth physiology and dimorphism of Mucor circinelloides (syn. racemosus) during submerged batch cultivation. Appl Microbiol Biotechnol. 2002;58(4):495–502. doi: 10.1007/s00253-001-0916-1. [DOI] [PubMed] [Google Scholar]
- Nagy G, Szebenyi C, Csernetics Á, Vaz AG, Tóth EJ, Vágvölgyi C, Papp T. Development of a plasmid free CRISPR-Cas9 system for the genetic modification of Mucor circinelloides. Sci Rep. 2017;7(1):16800. doi: 10.1038/s41598-017-17118-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolas FE, de Haro JP, Torres-Martinez S, Ruiz-Vazquez RM. Mutants defective in a Mucor circinelloides dicer-like gene are not compromised in siRNA silencing but display developmental defects. Fungal Genet Biol. 2007;44(6):504–516. doi: 10.1016/j.fgb.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Nicolas FE, Moxon S, de Haro JP, Calo S, Grigoriev IV, Torres-Martinez S, Moulton V, Ruiz-Vazquez RM, Dalmay T. Endogenous short RNAs generated by Dicer 2 and RNA-dependent RNA polymerase 1 regulate mRNAs in the basal fungus Mucor circinelloides. Nucleic Acids Res. 2010;38(16):5535–5541. doi: 10.1093/nar/gkq301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolas FE, Ruiz-Vazquez RM. Functional diversity of RNAi-associated sRNAs in fungi. Int J Mol Sci. 2013;14(8):15348–15360. doi: 10.3390/ijms140815348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolas FE, Torres-Martinez S, Ruiz-Vazquez RM. Two classes of small antisense RNAs in fungal RNA silencing triggered by non-integrative transgenes. EMBO J. 2003;22(15):3983–3991. doi: 10.1093/emboj/cdg384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolas FE, Torres-Martinez S, Ruiz-Vazquez RM. Transcriptional activation increases RNA silencing efficiency and stability in the fungus Mucor circinelloides. J Biotechnol. 2009;142(2):123–126. doi: 10.1016/j.jbiotec.2009.04.003. [DOI] [PubMed] [Google Scholar]
- Nicolas FE, Vila A, Moxon S, Cascales MD, Torres-Martinez S, Ruiz-Vazquez RM, Garre V. The RNAi machinery controls distinct responses to environmental signals in the basal fungus Mucor circinelloides. BMC Genomics. 2015;16:237. doi: 10.1186/s12864-015-1443-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasteur L. Etudes sur la Biere. Paris, France: Gauthier-Villars; 1876. [Google Scholar]
- Pathan EK, Ghormade V, Deshpande MV. Selection of reference genes for quantitative real-time RT-PCR assays in different morphological forms of dimorphic zygomycetous fungus Benjaminiella poitrasii. PLoS One. 2017;12(6):e0179454. doi: 10.1371/journal.pone.0179454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrikkos G, Skiada A, Lortholary O, Roilides E, Walsh TJ, Kontoyiannis DP. Epidemiology and clinical manifestations of mucormycosis. Clin Infect Dis. 2012;54(suppl_1):S23–S34. doi: 10.1093/cid/cir866. [DOI] [PubMed] [Google Scholar]
- Roncero MI, Zabala C, Cerda-Olmedo E. Mutagenesis in multinucleate cells: the effects of N-methyl-N′ -nitro-N-nitrosoguanidine on Phycomyces spores. Mutat Res. 1984;125(2):195–204. doi: 10.1016/0027-5107(84)90069-1. [DOI] [PubMed] [Google Scholar]
- Ruiz-Vazquez RM, Nicolas FE, Torres-Martinez S, Garre V. Distinct RNAi pathways in the regulation of physiology and development in the fungus Mucor circinelloides. Adv Genet. 2015;91:55–102. doi: 10.1016/bs.adgen.2015.07.002. [DOI] [PubMed] [Google Scholar]
- Sathya R, Pradeep BV, Angayarkanni J, Palaniswamy M. Production of milk clotting protease by a local isolate of Mucor circinelloides under SSF using agro-industrial wastes. Biotechnol Bioprocess Eng. 2009;14(6):788–794. [Google Scholar]
- Silva F, Torres-Martinez S, Garre V. Distinct white collar-1 genes control specific light responses in Mucor circinelloides. Mol Microbiol. 2006;61(4):1023–1037. doi: 10.1111/j.1365-2958.2006.05291.x. [DOI] [PubMed] [Google Scholar]
- Snyder AB, Churey JJ, Worobo RW. Characterization and control of Mucor circinelloides spoilage in yogurt. Int J Food Microbiol. 2016;228:14–21. doi: 10.1016/j.ijfoodmicro.2016.04.008. [DOI] [PubMed] [Google Scholar]
- Spatafora JW, Chang Y, Benny GL, Lazarus K, Smith ME, Berbee ML, Bonito G, Corradi N, Grigoriev I, Gryganskyi A, James TY, O’Donnell K, Roberson RW, Taylor TN, Uehling J, Vilgalys R, White MM, Stajich JE. A phylum-level phylogenetic classification of zygomycete fungi based on genome-scale data. Mycologia. 2016;108(5):1028–1046. doi: 10.3852/16-042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres-Martínez S, Ruiz-Vázquez RM. The RNAi universe in fungi: A varied landscape of small RNAs and biological functions. Annu Rev Microbiol. 2017;71:371–391. doi: 10.1146/annurev-micro-090816-093352. [DOI] [PubMed] [Google Scholar]
- Trieu TA, Calo S, Nicolas FE, Vila A, Moxon S, Dalmay T, Torres-Martinez S, Garre V, Ruiz-Vazquez RM. A non-canonical RNA silencing pathway promotes mRNA degradation in basal Fungi. PLoS Genet. 2015;11(4):e1005168. doi: 10.1371/journal.pgen.1005168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trieu TA, Navarro-Mendoza MI, Perez-Arques C, Sanchis M, Capilla J, Navarro-Rodriguez P, Lopez-Fernandez L, Torres-Martinez S, Garre V, Ruiz-Vazquez RM, Nicolas FE. RNAi-based functional genomics identifies new virulence determinants in mucormycosis. PLoS Pathog. 2017;13(1):e1006150. doi: 10.1371/journal.ppat.1006150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicente G, Bautista LF, Rodríguez R, Gutiérrez FJ, Sádaba I, Ruiz-Vázquez RM, Torres-Martínez S, Garre V. Biodiesel production from biomass of an oleaginous fungus. Biochem Eng J. 2009;48(1):22–27. [Google Scholar]
- Werner E, Patel K, Holder AA. Construction of a library for sequencing long regions of malarial genomic DNA. Biotechniques. 1997;23(1):20, 22, 24. doi: 10.2144/97231bm02. [DOI] [PubMed] [Google Scholar]
- Wynn JP, bin Abdul Hamid A, Ratledge C. The role of malic enzyme in the regulation of lipid accumulation in filamentous fungi. MicroSoc. 1999;145(Pt 8):1911–1917. doi: 10.1099/13500872-145-8-1911. [DOI] [PubMed] [Google Scholar]