

 This study investigated the molecular mechanism(s) of the protective effects of a C-alkylated flavonoid, viscosine on an animal model of CCl4-induced hepatotoxicity.
This study investigated the molecular mechanism(s) of the protective effects of a C-alkylated flavonoid, viscosine on an animal model of CCl4-induced hepatotoxicity.
Abstract
This study investigated the molecular mechanism(s) of the protective effects of a C-alkylated flavonoid, viscosine on an animal model of CCl4-induced hepatotoxicity. Viscosine at 20, 50 and 100 mg kg–1 was orally administered in a dose dependent manner per day for 3 days before the CCl4 (1 : 1 v/v in olive oil, 1 ml kg–1) treatment and 2 days after the treatment. Hepatoprotection was assessed in terms of reduction in serum enzyme activities (ALT, AST, and ALP) that occur after CCl4 injury, and by histopathology and immunohistochemistry. The rise in serum levels of alanine aminotransferase (ALT), aspartate aminotransferase (AST), and alkaline phosphatase (ALP) in CCl4-intoxicated rats was markedly suppressed by viscosine in a concentration dependent manner. The decrease in the activity of hepatic antioxidant enzyme, SOD, was significantly prevented by viscosine, likewise gradually the levels of MDA and GSH were also normalized compared to silymarin. Viscosine also reduced the CCl4-induced damaged area from 2% to 0% as assessed by histopathology and prevented the mixed inflammatory infiltrate. Viscosine attenuated the inflammation in the liver around the injured central vein region by downregulating the CCl4 induced activation of hepatic CD68+ macrophages, thereby reducing their number as well. The expression of inducible nitric oxide synthase (iNOS) was more potentially suppressed by viscosine compared to the FDA approved positive control silymarin. The results of this study indicate that viscosine could be effective in protecting the liver from acute CCl4-induced injury. The hepatoprotective mechanisms of viscosine may be related to the free radical scavenging and attenuation of oxidative stress, as well as to the inhibition of inflammatory response in the liver. Here, we are proposing a novel mechanism of action of viscosine and suggesting that it may be a safe and better in vivo antioxidant.
1. Introduction
The liver is a vital organ that metabolizes all foreign compounds, drugs and toxic chemicals and hence, it is very susceptible to injury.1 Worldwide, inflammation of the liver or hepatitis is a very serious health problem with a staggering incidence of 550 million.2 Hepatitis is more common in developing countries such as Pakistan where ∼35 million people are believed to be infected with various hepatitis viruses.3 Complications of hepatitis such as cirrhosis are life threatening, and have no specific treatment available and lead to mortality around the world. Many drugs cause liver injury and upon its continuous insult lead to fulminant hepatitis and mortality in most cases.4
The uses of synthetic or conventional drugs are mostly inadequate to treat these liver diseases and can have serious adverse effects. A number of drugs induce liver injury; one of them is acetaminophen, the most popular selling analgesic drug in the US and the world.5,6 Although it is safe at therapeutic doses, overdose can cause severe liver injury manifested as centrilobular necrosis.7 Acetaminophen associated toxicity generates reactive metabolites that leads to adduct formation causing severe oxidative stress. In rodents it causes mitochondrial dysfunction and nuclear fragmentation from the necrosis of hepatocytes.7 Isoniazid (INH) is another widely used antituberculous drug associated with idiosyncratic liver injury in susceptible patients. Most of the anti-TB drugs (INH, rifampicin and pyrazinamide) have been found to be potentially hepatotoxic.8,9 Up to 20% of INH treated patients reportedly showed elevated levels of alanine aminotransferase whereas hepatotoxicity can occur in up to 2% of patients.10,11 Most cases of liver biopsies from patients with severe INH induced hepatotoxicity are indistinguishable from the pathology of viral induced hepatitis which is characterized by necrosis, inflammation and infiltration of eosinophils.12 Many natural products contain polyphenolic compounds with antioxidant properties that prevent the deleterious effects of toxic agents either by scavenging free radicals or modulating the inflammatory response13,14 and thus, protect from liver diseases.15 Silymarin is a very well-known standardized extract, containing a mixture of flavonolignans consisting of silibinin, isosilibinin, silicristin, silidianin, and others isolated from the herb milk thistle (Silybum marianum L.) which is traditionally used for the treatment of liver disorders.16 The mechanism of the hepatoprotective action of silymarin is proposed to be due to its antioxidant, anti-inflammatory, immunomodulatory17 and antifibrotic properties.18 In experimental models of hepatotoxicity, including the CCl4-induced hepatitis, the injured hepatocytes release different soluble inflammatory mediators.19 In the hepatocytes, cytochrome P450 activates CCl4 to form its trichloromethyl radical, (CCl3–). Then the CCl3– radicals bind to cellular components such as nucleic acids, proteins and lipids, impairing cellular processes like lipid metabolism and leading to fatty degeneration. The formation of CCl3– radical adducts with DNA is thought to be an initiator of liver cancer.20 The CCl3– radicals in turn react with cellular oxygen forming the trichloromethylperoxy radical CCl3OO– species. The CCl3OO– initiates a chain of reactions which attacks and destroys polyunsaturated fatty acids causing lipid peroxidation. The adverse effects of lipid peroxidation result in leaking of the plasma membrane and the membranes of the intracellular organelles causing the loss of intracellular calcium and subsequent cell damage.21 The breakdown products of fatty acids are reactive aldehydes, which can form binding interactions with the functional groups of proteins and thus halt enzymatic activities. CCl4 intoxication also causes hypomethylation that leads to inhibition of protein synthesis and may also inhibit the secretion of lipoproteins. CCl4 at a molecular level activates a number of factors such as TNF-α, nitric oxide and TGF-α and TGF-β in the cell which cause hepatocyte destruction and fibrosis.22 TNF-α is one of the main key mediators of hepatitis,23 an early rise of its level induces expression of pro-inflammatory genes of inducible nitric oxide synthase (iNOS), cyclooxygenase-2 (COX-2) and resident macrophages of the liver.19 TNF-α initiates the process of apoptosis while TGFs direct towards liver fibrosis. Processes involved in CCl4 toxicity can specifically interrupt cellular methylation, and calcium levels causing membrane damage24 which consequently induce hepatic injury, inflammation, necrosis and apoptosis.25
Dodonaea viscosa, belongs to the family sapindaceae, and the genus Dodonaea consists of 60 species. Different classes of major secondary metabolites were reported from Dodonaea viscosa like diterpenoids, triterpenoid saponins, flavonoids, viscosine,26 hautriwaic acid,27 methylenebissantin,28 tannins and sterols.29Dodonaea viscosa is widely used in folk medicine for the treatment of a number of diseases including diabetes,30 ulcer31 and hepatitis.32
Viscosine, a naturally occurring C-alkylated flavonoid (4′,5,7-trihydroxy-3,6-dimethoxyflavone) was isolated previously by our research group from Dodonaea viscosa.33 Viscosine has been shown to possess antinociceptive and lipoxygenase inhibitory activities both in vivo and in vitro.26,34 Recently their pharmacokinetic profiles35 as well as pharmacological effects on the GABA receptors have also been reported.36 We have previously reported the mechanism of hepatoprotection of hautriwaic acid, one of the bioactive constituents of Dodonaea viscosa.37 Viscosine is structurally related to the CNS-active flavonoids, 2′-MeO6MF and 3-OH′2 MeO6MF.38 Taking into account our interest in the pharmacological and therapeutic significance of the bioactive compounds from Dodonaea viscosa, the present study aims to investigate the possible mechanism of hepatoprotection by viscosine by downregulating the activation of Kupffer cells and iNOS expression in a chemical-induced liver injury model.
2. Materials and methods
2.1. Chemicals and antibodies
CCl4 was obtained from Sigma Chemical Co., St Louis, MO, USA. Silymarin was purchased from MP Biomedicals Rue Geiler de Kaysersberg lllkirch Cedex 67402, France. Viscosine was isolated from Dodonaea viscosa according to the methods described.33 Bovine serum albumin (BSA), Cu/Zn superoxide dismutase (Cu/Zn SOD), and glutathione (GSH) were purchased from Sigma–Aldrich (Taufkirchen, Germany). Diagnostic kits for the plasma alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were purchased from Roche Diagnostics GmbH, Mannheim, Germany. Mono-basic and dibasic sodium phosphate and Triton X-100 were obtained from Fisher Scientific (Fair Lawn, NJ). Rabbit polyclonal anti-iNOS antibodies (ab3523), and mouse monoclonal anti-CD68 [ED1] antibody (ab31630) were purchased from Abcam (Cambridge, UK). Glass slides, cover slip and Roti-immunoblock, for immunohistochemistry were from Carl Roth, Karlsruhe, Germany. All other chemicals were of the highest grade available.
2.2. Animals
Male Wistar rats weighing 180 to 200 g were housed in individual cages which were kept at 22 °C to 26 °C under 12-hour light/dark cycles, with free access to standard laboratory chow and tap water ad libitum. All animals received humane care and all protocols involving the animals were in compliance with the guidelines approved (Animal study protocol#2013-006) by the Institutional Ethics Committee of ICCBS, Karachi University adhering to the guidelines of the Institutional Animal Care and Use Committee (IACUC) for animal studies.
3. Experimental design
3.1. CCl4-induced liver injury
The animals were randomly divided into six groups each having six rats as described below. Group 1 (normal control) was injected with vehicle only (1 ml per kg body weight olive oil); Group 2 (hepatitis model) was injected with intraperitoneal injection of CCl4 (1 ml kg–1) with 1 : 1 olive oil; Group 3 (positive control) was injected with an intraperitoneal injection of CCl4 (1 ml kg–1) with 1 : 1 olive oil and also received silymarin (200 mg kg–1, oral), per day for 3 days before the CCl4 treatment and 2 days after the treatment; Groups 4–6 were injected with intraperitoneal injection of CCl4 (1 ml kg–1) with 1 : 1 olive oil but also received viscosine at a dose of 20, 50 and 100 mg per kg body weight per day for 3 days before the CCl4 treatment and 2 days after the treatment.
3.2. Determination of oxidative stress markers
The livers were homogenized (Polytron homogenizer) in 50 mM phosphate buffer saline (PBS), pH 7.4 (Kinematica, Lucerne, Switzerland). A Beckman L7–65 Ultracentrifuge (Beckman, Fullerton, USA) was used to separate the supernatants at 15 000g for 20 min, at 4 °C. The supernatants were used for determination of the Cu/Zn SOD activity, MDA and GSH content. The Cu/Zn SOD activity was measured at 550 nm by using a UV-VIS spectrophotometer from the decrease in cytochrome c reduction by superoxide radicals as described previously.39 Likewise, the GSH and MDA contents were determined according to a reported study.40 To deproteinize, the supernatant was centrifuged for 8 minutes at 4500g with 1.2 M metaphosphoric acid (Rotina 420R, Andreas Hettich GmbH, Tuttlingen, Germany). Then, a solution consisting of 700 μl of 0.3 mM NADPH in phosphate buffer saline, 25 μl of the deproteinized sample and water, and 100 μl of 6 mM DTNB, were mixed to a final volume of 1.0 ml in a cuvette. After that, 10 μl of glutathione reductase (50 U ml–1) was added to the mixture and absorbance was monitored at 405 nm for 30 min. The glutathione content in the samples was determined from the standard curve using a series of dilutions of glutathione stock solution. Bradford's method was used for protein content in the liver homogenates.41
3.3. Cytotoxicity analysis using 3T3, CC-1 and MDBK cell lines
The cytotoxicity of viscosine was evaluated using 3T3, CC-1 and MDBK cell lines. Briefly 3T3 cells were grown in DMEM supplemented with 10% FBS, CC1 cells were cultured in EMEM added with 2 mM l-glutamine, 1% NEAA, 20 mM HEPES, and 10% FBS, whereas MDBK cells were grown in RPMI-1640 supplemented with 5% FBS in 75 cm2 flasks, and incubated in a 5% CO2 incubator at 37 °C until they attained 70% confluence. The cytotoxicity of viscosine was evaluated using 6 × 104 cells per mL by the MTT colorimetric assay as described.34
4. Histopathological examination and quantification of the liver damage
Liver tissues were rapidly removed and fixed in 10% neutral buffered formalin, dehydrated through a graded series of alcohol, embedded in paraffin, and cut into 6 μm thick sections. The liver tissues were stained with Hematoxylin–Eosin (H&E) and Periodic Acid Schiff's (PAS) stains. The tissues were then examined under bright field and multichannel fluorescence microscope at different magnifications using a Nikon 90i microscope. Histopathological analysis of the liver under different conditions was carried out and the necrotic area was measured in 30 different sections of the liver using the NIS-elements software from Nikon, Japan. Then the damaged area was quantified using the same software and the necrotic area was expressed as percent damage compared to the whole area of the section.
4.1. Immunohistochemistry analysis
For immunohistochemistry, 6 μm thick liver sections were used, as described.27 Briefly, the slides were deparaffinized in xylene and dehydrated in graded alcohol. The liver sections were incubated for 1 hour with primary antibodies for liver macrophages, clone ED1 (diluted 1 : 100), and anti-iNOS (diluted 1 : 50). After thoroughly washing with PBS, the sections were then incubated with the secondary antibody, Texas Red-conjugated goat anti-mouse IgG (1 : 100) for 45 min. Then the slides were counterstained with DAPI, and mounted, while the expression profile and cellular localization of liver macrophages and iNOS were analyzed by multichannel fluorescence microscopy (Nikon 90i, Japan).
5. Statistical analysis
The data were analyzed using the SPSS software. Differences among groups were assessed by one-way ANOVA followed by t-test. Values in the text are means ± SD, standard deviation. Differences with p < 0.05 were considered to be statistically significant.
6. Results
6.1. Histopathology
A histopathological study of the normal control liver showed a typical central vein lined with endothelial cells and normal hepatocytes making hepatic cords of cells with clear cell boundaries and sinusoidal spaces (Fig. 1A and B). The CCl4 induced liver injury group was characterized by an increase in inflammatory infiltrate around the central vein and the hepatocytes showed ballooning degeneration in the central vein region (Fig. 1C and D). The silymarin treated group showed some protection against this injury but still inflammation was present around the central vein region. Treatment with silymarin reduced the mixed inflammatory cells at the site of injury (Fig. 1E and F). However, viscosine (20 mg kg–1, 50 mg kg–1 and 100 mg kg–1), dose dependently decreased the abnormality of the liver architecture induced by CCl4 (Fig. 1G and H) and showed hepatic cells with a well-preserved cytoplasm, nucleus, nucleolus and central vein (Fig. 1I–L) as compared to the silymarin treated group. Viscosine at a dose of 100 mg kg–1 completely abolished the damage caused by CCl4-produced reactive oxygen species. Therefore, it appears that one of the reasons for hepatoprotection by viscosine is its strong scavenging capabilities of reactive oxygen species and inhibition of the formation of adducts.
Fig. 1. Liver histology after CCl4 intoxication. Histopathology of the liver showing normal (green arrows) central vein, hepatic cords and sinusoids in the untreated normal control (normal, A, B); pale necrotic areas (red arrows) after CCl4 treatment in the control group (CCl4, C, D); protection by the positive control 200 mg kg–1 silymarin (CCl4 + silymarin E, F); protection by viscosine (20 mg kg–1) (CCl4 + viscosine, G, H), 50 mg kg–1 (CCl4 + viscosine, I, J); protection by the viscosine treated group (100 mg kg–1) (CCl4 + viscosine, K, L), chemical structure of viscosine (M). Note that the hepatoprotection by viscosine at a dose of 100 mg kg–1 is better than that of silymarin or viscosine at a low dose of 20 mg kg–1. Note that the viscosine protection is better than that of silymarin. Scale bar for A, C, E, G, I and K is 250 μm and is shown in G. Scale bar for B, D, F, H, J and L is 25 μm and is shown in H.

6.2. Effects of viscosine on serum levels of ALT, AST, and AP
Treatment with CCl4 caused elevated blood levels of ALT, AST, and AP representing membrane damage due to oxidative damage to the hepatocytes (Fig. 2A). The AP elevated levels were due to increased damage to the bile duct cells which are located at the portal triad region. Silymarin treatment showed protection by decreasing the levels of ALT, AST and AP (Fig. 2A) which correlated with the quantification of pathological changes in histology (Fig. 2B). The elevated levels of ALT, AST, and ALP were reduced despite CCl4 treatment in the animals treated with viscosine, 20 mg kg–1 (Fig. 2A). Treatment with viscosine (50 mg per kg and 100 mg per kg body weight) showed significant hepatoprotective activity that was superior to silymarin (200 mg kg–1). Quantification of the histopathological data showed 0% pathology in the normal control group (Fig. 2B). CCl4 administration caused huge pathological changes (50% damage) in the model hepatitis group (Fig. 2B). However, treatment with viscosine at 20 mg per kg, 50 mg per kg and 100 mg per kg body weight restricted (p < 0.001) the pathological changes to 2%, 1% and 0% respectively. Therefore, viscosine was better at hepatoprotection compared to silymarin which showed 10% damage (Fig. 2B).
Fig. 2. Quantification of the effects of viscosine on CCl4-induced liver injury. (A) Serum ALT, AST and ALP levels as markers of liver injury under various conditions. Note that viscosine showed complete protection against CCl4-induced liver injury as compared to CCl4 (P < 0.001) or CCl4 + silymarin (P < 0.001). (B) Percent damage of the liver as assessed by histology under various conditions. Note the slight damage (3%) in the silymarin group whereas viscosine showed no damage (0.1%).

6.3. Periodic acid Schiff's staining of liver treated with viscosine
Light microscopic evaluation of liver tissues showed normal hepatic architecture (Fig. 3A) in the control group. The usual glycogen deposits were present in the hepatocyte cytoplasm in the untreated group (Fig. 3A). After 24 h of CCl4 treatment, necrotic changes of hepatocytes were evident along with decreased glycogen content (Fig. 3B). In the silymarin treated group, inflammatory cell infiltration and slight vacuolar degeneration around the central veins and sinusoids were detected which was also associated with decreased glycogen content (Fig. 3C). Treatment with viscosine did not restore the glycogen deposits compared to the CCl4 control or silymarin treated group (Fig. 3D), probably due to the high-energy demand of the hepatocytes for the possible repair processes in CCl4-given animals. In all other aspects, viscosine completely preserved the hepatic architecture of hepatocytes like normal hepatocytes.
Fig. 3. Periodic-acid Schiff's (PAS) staining of liver sections of rats treated with viscosine. Photomicrographs of liver sections of rats showing the normal distribution of glycogen in the hepatocytes of the control rat (A). Depletion of glycogen in the hepatocytes of CCl4 treated group (B). Glycogen in the hepatocytes of rat liver treated with silymarin (C) or viscosine (100 mg kg–1) (D). Scale bar is 25 μm.

6.4. Effect of viscosine on oxidative stress
In order to investigate the antioxidant effects of viscosine against CCl4-induced oxidative stress in the liver of different experimental groups, markers of oxidative stress such as malondialdehyde, glutathione, and superoxide dismutase (MDA, GSH, and SOD) were measured. Fig. 4 shows the dose dependent effects of viscosine on CCl4-induced oxidative stress. In the CCl4-intoxication group, the MDA level was significantly elevated (P < 0.01). However, hepatic MDA levels were significantly reduced with 20, 50, and 100 mg per kg body weight doses of viscosine (P < 0.05). On the other hand, CCl4 dramatically reduced the level of GSH compared with the normal control group in the liver of rats (P < 0.01). However, its level was increased significantly by pretreatment with viscosine (P < 0.05). The hepatic antioxidant enzyme (SOD) activities were decreased significantly in the liver of rats treated with CCl4 alone compared to the normal control group (P < 0.01). However, co-administration of viscosine significantly (P < 0.05) attenuated changes produced by CCl4 in hepatic SOD in a dose dependent manner.
Fig. 4. Effects of viscosine on the hepatic oxidative system. MDA (A), GSH (B), and SOD (C), in CCl4-intoxicated rats. Data are expressed as the mean ± SD, n = 10. ++P < 0.01, when compared to the normal control; *P < 0.05, **P < 0.01, when compared to the CCl4 model control. Group I: normal control; Group II: CCl4 model control; Group III: 200 mg kg–1 silymarin + CCl4; Group IV: 20 mg kg–1 viscosine + CCl4; Group V: 50 mg kg–1 viscosine + CCl4; Group VI: 100 mg kg–1 viscosine + CCl4.

6.5. Cytotoxicity assay
The cell lines 3T3 (mouse embryo, fibroblast cells), CC1 (rat, epithelial, liver cells) and MDBK (bovine kidney cells) were purchased from ATCC. Cytotoxicity of pure viscosine at different concentrations was evaluated using these cell lines to ensure that it was not toxic. Viscosine was incubated with 3T3, CC1 and MDBK cell lines for 24 hours, we found that 0.5, 5 or 50 μg mL–1 has very little to no cytotoxic effects (Fig. 9B). From the data it is clear that viscosine is non-cytotoxic.
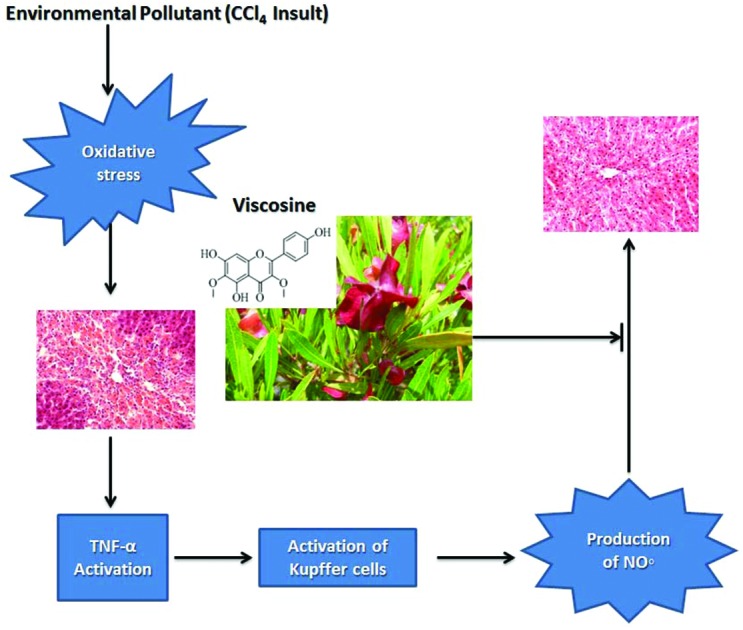
Fig. 9. (A) Possible proposed mechanisms of hepatoprotective activity of viscosine. Upon exposure of liver hepatocytes to CCl4 oxidative stress is induced due to its metabolism into highly reactive free radicals. CCl4 induced oxidative stress can trigger the release of TNF-α from injured hepatocytes and Kupffer cells, which can further activate NFκB allowing its nuclear translocation. Consequently, NFκB stimulates the expression of iNOS and COX-2. The final product of iNOS, COX-2 and NO˙, contributes to oxidative stress and on the other hand, initiates the cascade of inflammatory responses in the injured liver. Inflammation, in turn, is associated with the release of highly reactive oxygen and nitrogen species from inflammatory cells, further exacerbating the oxidative stress. Viscosine prevents oxidative damage, as indicated by the decrease in lipid peroxidation, and improves the antioxidant status. Furthermore, viscosine suppresses the inflammatory response by downregulating the proinflammatory cascade initiated by TNF-α, and attenuates oxidative stress by iNOS inhibition. (B) Cytotoxicity effects of the test materials on 3T3 mice fibroblast cell line, CC1 (rat, epithelial, liver cells) and MDBK (bovine kidney cells). Cells were incubated with viscosine at different concentrations for 48 h then MTT was added for 4 h followed by addition of DMSO and the absorbance was read at 540 nm using a 96-well plate reader. Results are expressed as mean ± SD of three replicates. Positive control is normal fibroblasts without any treatment.

7. Immunohistochemistry of Kupffer cells (KCs) of the liver
Liver inflammation is associated with the activation of CD68+ macrophages and migration of macrophages into hepatic cords where these macrophages secrete proinflammatory cytokines such as TNF-α and IL-6.42 Slender-looking sinusoidal-lining CD68+ immunoreactive cells with distinct enlarged nuclei were present in the normal control group (Fig. 5A–C) in a small number around the central vein (P < 0.001, Fig. 7). The DAPI stain was useful in identifying the elongated shaped nucleus of the macrophages as shown in Fig. 5B. From double and triple channel fluorescence microscopy it is evident that the resident macrophage cells exhibit a characteristic elongated shape with some processes while they are in the sinusoidal spaces (Fig. 6A and B). Upon CCl4 induced liver injury, the CD68+ macrophages were more densely stained and were more numerous in number (Fig. 5D and 6C, D) especially in the injured area around the central vein. There was a huge mixed inflammatory cell infiltrate around the central vein region which was clear from the DAPI staining (Fig. 5E). CD68+ cells were present in a larger number (P < 0.001, Fig. 7) compared to the normal control in the central vein region (Fig. 7). It is obvious from Fig. 5G that silymarin treatment decreased the number of activated macrophages (P < 0.001, Fig. 8) around the central vein compared to the CCl4 control group. From the DAPI staining (Fig. 5H) it is clear that the positive control silymarin did not completely reduce the number or morphology of the activated macrophages in the injured region (Fig. 6E and F). Interestingly viscosine significantly decreased (P < 0.001, Fig. 7) the number of CD68+ macrophages around the central vein (Fig. 5J) to levels very similar to that of the normal control group while the DAPI staining revealed a normal morphology of the nuclei (Fig. 5K) compared to the normal control as well as the CCl4 control group. Therefore, viscosine treatment limited the number and activity of the hepatic sinusoidal macrophages (P < 0.001, Fig. 7), despite the CCl4 treatment as evidenced from the double and triple channeled immunohistochemistry (Fig. 6G and H).
Fig. 5. Effects of viscosine on CD68+ Kupffer cells. Immunohistochemistry for CD68+ cells of liver showed normal central vein (A) and hepatocyte nucleus stained with DAPI (B) or in DIC image (C) of the control (A, B, C) group and increased migration of CD68+ cells (D) at the site of injury caused by CCl4 treatment (D, E, F). Silymarin treatment (G, H, I) slightly reduced the activation of CD68+ cells (G). Normal distribution of CD68+ cells treated with 100 mg kg–1 viscosine (J, K, L), CD68+ cells (J). DAPI (nucleus) is represented by green. Scale bar is 25 μm.

Fig. 7. Quantification of the CD68+ cells to elucidate the effects of viscosine treatment on the CCl4-induced liver injury. CCl4-induced liver injury showed increase in the number of CD68+ cells (P < 0.001) as compared to the normal control in the central vein region (P < 0.001). Silymarin treatment (P < 0.001) somewhat reduced this number while viscosine (P < 0.001) reduced this number to control levels. Note that 100 mg kg–1 viscosine reduced the number of CD68+ cells around the central vein region in a dose dependent manner more than the positive control silymarin (P < 0.001).

Fig. 6. Viscosine reduced the number of CD68+ cells. double fluorescence for CD68 and DAPI in normal control rat liver (A, B) and in the CCl4 induced liver injury showed numerous CD68+ cells along the sinusoids around the central vein (C, D). Silymarin positive control decreased the number of CD68+ cells to some extent (E, F), whereas the 100 mg kg–1 viscosine treated group showed CD68+ immunocytochemistry similar to that of the normal control (G, H). Scale bar is 25 μm.

Fig. 8. Immunohistochemical detection of iNOS. iNOS immunoreactivity was almost absent in the livers of the normal control group (A, B, C). Strong iNOS immunoreactivity in the livers of CCl4-treated group (D, E, F) was observed. The CCl4 treated group showed strong iNOS immunopositive hepatocyte nuclei around the central vein region. Presence of immunopositive cells for iNOS in the silymarin treated group (G, H, I). In the 100 mg kg–1 viscosine treated group only minute traces of iNOS immunopositivity around the central vein hepatocytes and endothelial cells (J, K, L) were seen.

8. Immunohistochemistry of inducible nitric oxide synthase (iNOS)
As expected, no apparent expression of iNOS was detected in the livers of the normal control group (Fig. 8A and B) which is further confirmed in Fig. 8C. CCl4-intoxication induced strong expression of iNOS in the liver and the expression of iNOS were localized in the cytoplasm and surrounding the nuclei of hepatocytes (Fig. 8D), which were further confirmed from the double channel fluorescence of iNOS and DAPI (Fig. 8F). Pretreatment and posttreatment with viscosine at 20, 50 and 50 mg kg–1, dose-dependently reduced the iNOS immunopositivity (Fig. 8J), which were further confirmed and much more evident from the double channel fluorescence of iNOS with DAPI (Fig. 8L). In the silymarin treated group iNOS immunopositive hepatocytes were still present around the central vein region. The iNOS expression was negligible in normal treatment groups (Fig. 8A and C), whereas iNOS expression was decreased in the viscosine treated group at 50 mg kg–1. The hepatocyte nuclei as well as the Kupffer cells were immunonegative for iNOS in the normal control as well as the viscosine treated group.
9. Discussion
CCl4-induced acute hepatic injury is widely used as a model for the screening of hepatoprotective drugs. The main goal of this study was to determine a possible mechanism for the hepatoprotective potential of viscosine in the CCl4-intoxicated model. Our results suggest that viscosine can prevent acute CCl4 induced hepatotoxicity, by attenuation of the oxidative and the inflammatory response as well as reduction of the expression of iNOS in the liver. Liver macrophage population was highly reduced in the viscosine treated group compared to CCl4 and the positive control silymarin treated group. The dose dependent decrease in serum AST, ALT and ALP activity by viscosine in CCl4-intoxicated rats indicates that viscosine preserves the hepatocellular membrane structural integrity, which was further confirmed and supported by the histological findings. Furthermore, viscosine seems to possess a considerably stronger activity against CCl4-induced liver damage than the positive control silymarin which is widely recognized as a potent hepatoprotective agent. Based on our results and previous studies, we propose a mechanism of viscosine-mediated hepatoprotection which is summarized in Fig. 9.
Oxidative stress is associated with increased production of free radicals which have been implicated in the pathogenesis of acute hepatic disorders, including CCl4-intoxication.43 CCl4 metabolism via the cytochrome P450 leads to the formation of CCl3– free radicals leading to lipid peroxidation24 and impairment of the antioxidant status of hepatocytes which ultimately results in hepatocyte degeneration and liver fibrosis.42 These oxidative injuries of hepatocytes were successfully prevented by viscosine. Our results suggest that the hepatoprotective effects of viscosine may be due to its antioxidant properties. The results of this study showed the hepatoprotective and anti-inflammatory effects of viscosine in the CCl4-induced liver injury similar to the reported compound berberine.43
The resident hepatic macrophages carry out very important functions in modulating the severity of the hepatic inflammation.42,44 It has been suggested that in acute liver injury, the injured hepatocytes and Kupffer cells produce a variety of proinflammatory cytokines, such as tumor necrosis factor-α (TNF-α) and monocyte chemoattractant protein (MCP), and this subsequently contributes to hepatic injury.45,46 Their expression level rises in acute liver injury following exposure to hepatotoxic chemicals,47,48 contributing to the hepatotoxicity induced by CCl4.49,50 TNF-α stimulates the process of inflammation and liver fibrosis but does not directly cause hepatocyte necrosis in CCl4-induced hepatotoxicity.48 The results of current research agree well with the already reported studies involving berberine51 and other alkaloids.52 Our study revealed that viscosine effectively suppressed the infiltration of proinflammatory cells (Fig. 2C and D) around the central vein and inhibited the expression of TNF-α, and its downstream mediators, iNOS and COX-2, which are involved in the process of inflammation. The downregulation of TNF-α, iNOS, and CD68 expression by viscosine suggests its important role in the attenuation of the CCl4-induced inflammatory cascade in liver diseases.
TNF-α also induces the expression of iNOS which produces nitric oxide (NO˙) causing a chemically induced stress.53 In the mitochondria, NO˙ may react with superoxide (O2˙–) to produce peroxynitrite (ONOO–), which are mediators of cellular dysfunction. In both acute and chronic liver injury overexpression of iNOS has been reported,54 but the role played by NO˙ in causing tissue damage is still controversial. It seems that iNOS-derived NO˙ could regulate expression of proinflammatory genes, contributing to inflammatory liver injury.36 It has been shown in various experimental models of acute liver injury that hautriwaic acid downregulates activated CD68+ macrophages,27 while berberine suppressed the downstream pathway of proinflammatory mediators such as iNOS and COX-2.54 In this study, increased CD68+ cells and iNOS expression around the injured central vein region in CCl4-intoxicated rats indicate enhanced production of nitric oxide and nitrosative stress, as a response to liver injury. Our results suggest that activated CD68+ cells and iNOS expression in acute CCl4-induced liver injury and its inhibition by viscosine exert beneficial effects in the prevention of acute hepatic damage. Most importantly, the results obtained from this study suggest the possibility of therapeutic application of viscosine in patients with liver injury, although further studies, in a more chronic model of liver fibrosis, are required to explore its mechanism of action in detail.
Our study and others regarding viscosine suggest that it could be a potent inhibitor of enzymes involved in the inflammatory process such as iNOS or lipoxygenase26 in addition to its anti-oxidant activity. Although viscosine has structural similarity to apigenin, quercetin or resveratrol in terms of its polyphenol and flavonoid structure, it is different from them in having the methoxy groups at positions 3 and 6 of the flavonoid which have been shown to enhance the nitric oxide production activity.55 The clinical trial failure of quercetin or resveratrol has been suggested to be due to poor bioavailability resulting from rapid deactivation of the hydroxyl groups on aromatic rings in the intestine and liver—a phenomenon which has been shown to be countered by methylated flavonoids which can accumulate in vivo to 150 fold more than their non-methylated counterparts.56 This would make viscosine a better anti-oxidant compared to apigenin, quercetin or resveratrol—all of which lack the protective methyl groups. Altogether it appears that viscosine could be a better flavonoid in vivo because of its anti-inflammatory and other activities in addition to its long lasting antioxidant activity.
Conflict of interest statement
The authors declare that there are no conflicts of interest.
We wish to confirm that there are no known conflicts of interest associated with this publication and there has been no significant financial support for this work that could have influenced its outcome.
Abbreviations
- CCl4
Carbon tetrachloride
- iNOS
Inducible nitric oxide synthase
- ALT
Alanine aminotransferase
- AST
Aspartate aminotransferase
- ALP
Alkaline phosphatase
- MDA
Malondialdehyde
- SOD
Superoxide dismutase
- GSH
Glutathione
- FDA
Food and drug administration
References
- Bissell D. M., Gores G. J., Laskin D. L., Hoofnagle J. H. Hepatology. 2001;33:1009–1013. doi: 10.1053/jhep.2001.23505. [DOI] [PubMed] [Google Scholar]
- Alter M. J. J. Hepatol. 2006;44:S6–S9. doi: 10.1016/j.jhep.2005.11.004. [DOI] [PubMed] [Google Scholar]
- André F. Vaccines. 2000;18:S20–S22. [Google Scholar]
- Ghabril M., Chalasani N., Björnsson E. Curr. Opin. Gastroenterol. 2010;26:222–226. doi: 10.1097/MOG.0b013e3283383c7c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman D. W., Kelly J. P., Rosenberg L., Anderson T. E., Mitchell A. A. J. Am. Med. Assoc. 2002;3:337–344. doi: 10.1001/jama.287.3.337. [DOI] [PubMed] [Google Scholar]
- Wilcox C. M., Cryer B., Triadafilopoulos G. J. Rheumatol. 2005;11:2218–2224. [PubMed] [Google Scholar]
- McGill M. R., Williams C. D., Xie Y., Ramachandran A., Jaeschke H. Toxicol Appl. Pharmacol. 2012;3:387–394. doi: 10.1016/j.taap.2012.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park W. B., Kim W., Lee K. L., Yim J. J., Kim M., Jung Y. J., Kim N. J., Kim D. H., Kim Y. J., Yoon J. H., Oh M. D., Lee H. S. J. Infect. 2010;61:323–329. doi: 10.1016/j.jinf.2010.07.009. [DOI] [PubMed] [Google Scholar]
- Sharifzadeh M., Rasoulinejad M., Valipour F., Nouraie M., Vaziri S. Pharmacol. Res. 2005;51:353–358. doi: 10.1016/j.phrs.2004.10.009. [DOI] [PubMed] [Google Scholar]
- Boelsterli U. A., Lee K. K. J. Gastroenterol. Hepatol. 2014;29:678–687. doi: 10.1111/jgh.12516. [DOI] [PubMed] [Google Scholar]
- Metushi I. G., Cai P., Zhu X., Nakagawa T., Uetrecht J. P. Clin. Pharmacol. Ther. 2011;89:911–914. doi: 10.1038/clpt.2010.355. [DOI] [PubMed] [Google Scholar]
- Bjornsson E., Kalaitzakis E., Olsson R. Aliment. Pharmacol. Ther. 2007;25:1411–1421. doi: 10.1111/j.1365-2036.2007.03330.x. [DOI] [PubMed] [Google Scholar]
- Grimble R. F. New Horiz. 1994;2:175–185. [PubMed] [Google Scholar]
- Domitrović R., Jakovac H., Milin C., Radosevic-Stasić B. Exp. Toxicol. Pathol. 2009;61:581–589. doi: 10.1016/j.etp.2008.12.005. [DOI] [PubMed] [Google Scholar]
- Sreelatha S., Padma P., Umadevi M. Food Chem. Toxicol. 2009;47:702–708. doi: 10.1016/j.fct.2008.12.022. [DOI] [PubMed] [Google Scholar]
- Tamayo C., Diamond S. Integr. Cancer Theory. 2007;6:146–157. doi: 10.1177/1534735407301942. [DOI] [PubMed] [Google Scholar]
- Luper S. Altern. Med. Rev. 1998;3:410–421. [PubMed] [Google Scholar]
- Mata-Santos H. A., Lino F. G., Rocha C. C., Paiva C. N., Castelo Branco M. T., Pyrrho Ados S. Parasitol Res. 2010;107:1429–1434. doi: 10.1007/s00436-010-2014-8. [DOI] [PubMed] [Google Scholar]
- Panuganti S. D., Khan F. D., Svensson C. K. J. Pharmacol. Exp. Ther. 2006;8:26–34. doi: 10.1124/jpet.106.100933. [DOI] [PubMed] [Google Scholar]
- Boll M., Weber L. W., Becker E., Stampfl A. Z. Naturforsch., C: 2001;56(7–8):649–659. doi: 10.1515/znc-2001-7-826. [DOI] [PubMed] [Google Scholar]
- Masuda Y. Yakugaku Zasshi. 2006;126(10):885–899. doi: 10.1248/yakushi.126.885. [DOI] [PubMed] [Google Scholar]
- Huang H. L., Wang Y. J., Zhang Q. Y., Liu B., Wang F. Y., Li J. J., Zhu R. Z. World J. Gastroenterol. 2012;18(45):6605–6613. doi: 10.3748/wjg.v18.i45.6605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schümann J., Tiegs G. Toxicology. 1999;138:103–126. doi: 10.1016/s0300-483x(99)00087-6. [DOI] [PubMed] [Google Scholar]
- Weber L. W., Boll M., Stampfl A. Crit. Rev. Toxicol. 2003;33:105–136. doi: 10.1080/713611034. [DOI] [PubMed] [Google Scholar]
- Lin B. R., Yu C. J., Chen W. C., Lee H. S., Chang H. M., Lee Y. C., Chien C. T., Chen C. F. J. Biomed. Sci. 2009;25:16–35. doi: 10.1186/1423-0127-16-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan A. Z., Mohammad A., Iqbal Z., Anis I., Shah M. R., Nadeem S., Rabnawaz M., Shahidullah A., Khan H., Khan I. Bangladesh J. Pharmacol. 2013;8:36–39. [Google Scholar]
- Ali H., Kabir N., Muhammad A., Shah M. R., Musharraf S. G., Iqbal N., Nadeem S. Phytomedicine. 2014;21:131–140. doi: 10.1016/j.phymed.2013.08.019. [DOI] [PubMed] [Google Scholar]
- Muhammad A., Anis I., Ali Z., Awadelkarim S., Khan A., Khalid A., Shah M. R., Galal M., Khan I. A., Choudhary M. I. Bioorg. Med. Chem. Lett. 2012b;22:610–612. doi: 10.1016/j.bmcl.2011.10.072. [DOI] [PubMed] [Google Scholar]
- Wagner H., Ludwig C., Grotjahn L., Khan M. S. Phytochemicals. 1987;26:697–701. [Google Scholar]
- Veerapur V. P., Prabhakar K. R., Kandadi M. R., Srinivasan K. K., Unnikrishnan M. K. Pharm. Biol. 2010;48:1137–1148. doi: 10.3109/13880200903527736. [DOI] [PubMed] [Google Scholar]
- Arun M., Asha V. J. Ethnopharmacol. 2008;118:460–465. doi: 10.1016/j.jep.2008.05.026. [DOI] [PubMed] [Google Scholar]
- Muhammad A., Anis I., Khan A., Marasini B. P., Choudhary M. I., Shah M. R. Arch. Pharmacal Res. 2012;35:431–436. doi: 10.1007/s12272-012-0305-6. [DOI] [PubMed] [Google Scholar]
- Khan A. Z., Mohammad A., Iqbal Z., Anis I., Shah M. R., Nadeem S. Bangladesh. J. Pharmacol. 2012;8:36–39. [Google Scholar]
- Khan A., Akhter M., Anis I., Iqbal Z., Muhammad R. S., Khan I. J. Chem. Soc. Pak. 2014a;36:1150–1152. [Google Scholar]
- Karim N., Gavande N., Wellendorph P., Johnston G. A., Hanrahan J. R., Chebib M. Biochem. Pharmacol. 2011;82:1971–1983. doi: 10.1016/j.bcp.2011.09.002. [DOI] [PubMed] [Google Scholar]
- Ali H., Musharraf S. G., Iqbal N., Adhikari A., Abdalla O. M., Mesaik M. A., Kabir N. Int. Immunopharmacol. 2015;28:235–243. doi: 10.1016/j.intimp.2015.06.009. [DOI] [PubMed] [Google Scholar]
- Karim N., Irshad S., Khan I., Mohammad A., Anis I., Shah M. R., Khan I., Chebib M. Pharmacol., Biochem. Behav. 2015;136:64–72. doi: 10.1016/j.pbb.2015.07.006. [DOI] [PubMed] [Google Scholar]
- Karim N., Curmi J., Gavande N., Johnston G. A., Hanrahan J. R., Tierney M. L. Br. J. Pharmacol. 2012;165:880–896. doi: 10.1111/j.1476-5381.2011.01604.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domitrović R., Jakovac H., Blagojević G. Toxicology. 2011;280:33–43. doi: 10.1016/j.tox.2010.11.005. [DOI] [PubMed] [Google Scholar]
- Anderson M. E. Methods Enzymol. 1985;113:548–555. doi: 10.1016/s0076-6879(85)13073-9. [DOI] [PubMed] [Google Scholar]
- Bradford M. M. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Kiso K., Ueno S., Fukuda M. I., Kobayashi I. K., Sakai T., Fukui K., Kojo S. Biol. Pharm. Bull. 2012;35:980–983. doi: 10.1248/bpb.35.980. [DOI] [PubMed] [Google Scholar]
- Slater T. F. Biochem. J. 1984;222:1–15. doi: 10.1042/bj2220001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domitrović R., Jakovac H., Tomac J., Sain I. Toxicol. Appl. Pharmacol. 2009;241:311–321. doi: 10.1016/j.taap.2009.09.001. [DOI] [PubMed] [Google Scholar]
- Edwards M., Keller B., Kauffman F., Thurman R. Toxicol. Appl. Pharmacol. 1993;119:275–279. doi: 10.1006/taap.1993.1069. [DOI] [PubMed] [Google Scholar]
- Friedman S. L. J. Clin. Invest. 2005;115:29–32. doi: 10.1172/JCI23928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehring S., Dickson E. M., SanMartin M. E., van Rooijen N., Papa E. F., Harty M. W., Tracy Jr. T. F., Gregory S. H. Gastroenterol. 2006;130:810–822. doi: 10.1053/j.gastro.2005.11.015. [DOI] [PubMed] [Google Scholar]
- Friedman S. L. J. Biol. Chem. 2000;275:2247–2250. doi: 10.1074/jbc.275.4.2247. [DOI] [PubMed] [Google Scholar]
- Luster M. I., Simeonova P. P., Gallucci R., Matheson J. Crit. Rev. Toxicol. 1999;29:491–511. doi: 10.1080/10408449991349258. [DOI] [PubMed] [Google Scholar]
- Simeonova P. P., Gallucci R. M., Hulderman T., Wilson R., Kommineni C., Rao M., Luster M. I. Toxicol. Appl. Pharmacol. 2001;177:112–120. doi: 10.1006/taap.2001.9304. [DOI] [PubMed] [Google Scholar]
- Czaja M. J., Xu J., Alt E. Gastroenterology. 1995;108:1849–1854. doi: 10.1016/0016-5085(95)90149-3. [DOI] [PubMed] [Google Scholar]
- Morio L. A., Chiu H., Sprowles K. A., Zhou P., Heck D., Gordon M. K., Laskin D. L. Toxicol. Appl. Pharmacol. 2001;172:44–51. doi: 10.1006/taap.2000.9133. [DOI] [PubMed] [Google Scholar]
- Sass G., Koerber K., Bang R., Guehring H., Tiegs G. J. Clin. Invest. 2001;107:439–447. doi: 10.1172/JCI10613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussler A. K., Billiar T. R. J. Leukocyte Biol. 1993;54:171–178. [PubMed] [Google Scholar]
- Matsuda H., Morikawa T., Ando S., Toguchida I., Yoshikawa M. Bioorg. Med. Chem. 2003;11:1995–2000. doi: 10.1016/s0968-0896(03)00067-1. [DOI] [PubMed] [Google Scholar]
- Wen X., Walle T. Drug Metab. Dispos. 2006;34:1786–1792. doi: 10.1124/dmd.106.011122. [DOI] [PubMed] [Google Scholar]