

 Valproic acid (VPA) is one of the representative compounds of histone deacetylase inhibitors (HDACis) and is used widely for the clinical treatment of epilepsy and other convulsive diseases.
Valproic acid (VPA) is one of the representative compounds of histone deacetylase inhibitors (HDACis) and is used widely for the clinical treatment of epilepsy and other convulsive diseases.
Abstract
Valproic acid (VPA) is one of the representative compounds of histone deacetylase inhibitors (HDACis) and is used widely for the clinical treatment of epilepsy and other convulsive diseases. Current reports indicate that HDACis may also be an attractive radiosensitizer for some tumor cells; however, it is unknown whether the safe blood concentration of VPA (0.3–0.8 mM) used for the treatment of epilepsy can also induce radiosensitivity in breast cancer cells. In addition, the mechanism by which VPA may induce radiosensitivity in breast cancer cells is yet to be determined. Our results clearly indicated that VPA at a safe dose (0.5 mM) could significantly increase the radiosensitivity of MCF7 breast cancer cells and result in more accumulation of DNA double strand breaks in response to DNA damage. After VPA treatment, the frequencies of homologous recombination (HR) and non-homologous end joining (NHEJ) tested by recombination substrates, pDR-GFP and EJ5-GFP, were dramatically decreased in the cells without the change of the cell cycle profile. It was further found that VPA could inhibit the recruitment of key repair proteins to DNA break areas, such as Rad51, BRCA1, and Ku80. Thus, our results demonstrated that a safe dose of VPA causes radiosensitivity in breast cancer cells through disrupting the molecular mechanisms of both BRCA1-Rad51-mediated HR and Ku80-mediated NHEJ pathways.
Introduction
Increasing evidence shows that most cells in multicellular organisms have undergone epigenetic modifications caused by histone deacetylases (HDACs) and DNA methyltransferase, which are associated with cancer development and tumor progression.1,2 It was found that HDACs are overexpressed in breast, colon, prostate and other cancers, indicating that HDACs may be an attractive anticancer target.3–6 HDAC inhibitors (HDACis) have recently emerged as novel anti-cancer drugs that are toxic to malignant cells, but show minimal toxicity towards normal cells,7 HDACis act by targeting HDAC activity and non-histone proteins directly, and may also play a crucial role in the regulation of gene transcription leading to cell-cycle arrest, differentiation, ROS generation, autophagy and inhibition of tumor angiogenesis.2,8
Valproic Acid (VPA) is a short chain fatty acid containing eight carbon atoms and one of the representative compounds of HDACis. To date, VPA is mainly used for the treatment of epilepsy patients in the clinic.9–12 Further studies on the biological effects of VPA demonstrated that this drug not only suppresses the growth of some tumor and transforming cells in various tissues,11,13 but also increases the radiosensitivity of tumor cells at high concentrations (2 or 5 mM),9,14,15 thus suggesting that VPA may be a potential radiosensitizer to cancer cells. However, it needs to be determined whether the clinical dose of VPA used for epilepsy patients could also be used to enhance the radiosensitivity of breast cancer cells.
Some reports demonstrated that the mechanism of HDACis-induced radiosensitization may be related to apoptosis, autophagy or the DNA damage repair function.2,3,16,17 Several studies indicate that disruption of DNA repair activity may be associated with HDACis-mediated radiosensitization.1,18,19 It is widely known that mammalian cells rely mainly on the homologous recombination (HR) and non-homologous end joining (NHEJ) mechanisms to repair DNA double strand breaks (DSBs),20–22 and studies demonstrate that HDACis can inhibit DNA repair by downregulating the activity of DNA repair proteins, such as Rad51 and DNA-PKcs, in cancer cells.23,24 This evidence indicates that there may be a close relationship between the DNA repair function and VPA-mediated radiosensitization.
The safe blood concentration of VPA for the treatment of epilepsy is 50–100 mg per 1 L of blood, which is equal to 0.3–0.8 mM.12 In this study, a safe dose of 0.5 mM and a critical safe dose of 1 mM were chosen to explore the effect of VPA on the radiosensitivity and its mechanism in breast cancer MCF7 cells. Our results suggest that VPA at the safe dose and critical safe dose causes the accumulation of DNA DSBs in the nucleus of cells in response to DNA damage. In addition, VPA-induced radiosensitization was associated with the disruption of HR and NHEJ through targeting the activity of DNA repair proteins, such as BRCA1 (breast cancer susceptibility gene 1), Rad51 and Ku80. Finally, apoptosis of MCF7 cells was not induced due to the lack of apoptosis related gene-caspase 3, and VPA at the safe concentration had no effect on the cell cycle, which suggests that the inhibition effect on DNA repair activity might be the key mechanism for VPA-induced radiosensitivity in breast cancer cells.
Results
VPA at safe dose can cause DNA DSBs in the cells in response to DNA damage
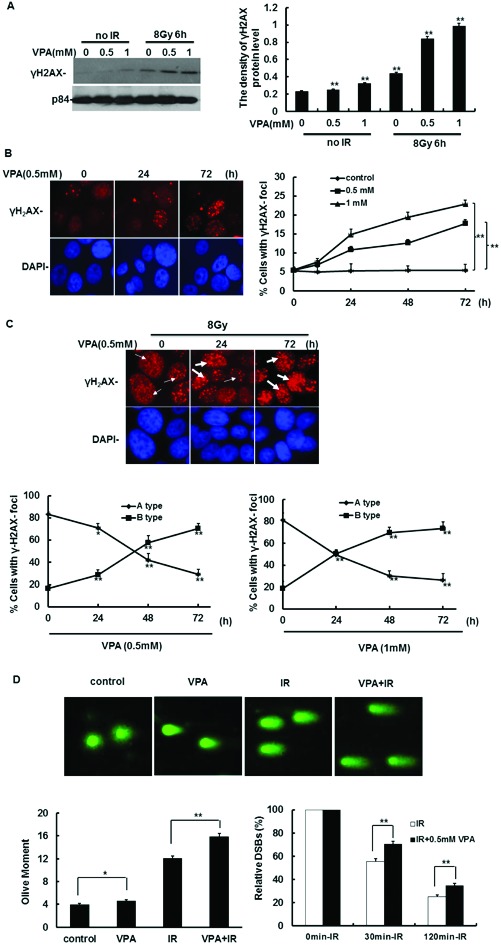
DSBs generated by ionizing radiation (IR) are the most dangerous DNA lesions to the cells. It was previously demonstrated that DSBs result in the phosphorylation of H2AX on serine 139 to form γH2AX,25–27 which is commonly considered to represent DNA DSBs. Thus, γH2AX was used as a DSBs marker to determine whether VPA can induce or enhance IR-induced DNA DSBs. At first, γH2AX expression was detected for this idea. The results by immunoblotting showed that VPA at the concentration of 0.5 mM and 1 mM for 24 h could induce γH2AX expression as compared with the cells without VPA treatment (Fig. 1A). After MCF7 cells pretreated with VPA were further irradiated by 8 Gy, at 6 h post-IR, the VPA pretreatment with 0.5 mM and 1 mM for 24 h significantly increased the γH2AX expression as compared with the single IR exposure group (Fig. 1A), suggesting that VPA can accumulate unrepaired IR-associated DSBs in MCF7 cells. Next, these results were confirmed by γH2AX foci formation via immunofluorescence staining. After treatment with VPA alone, the percentage of cells with γH2AX foci formation in MCF7 cells showed a significant increase in a time-dependent manner (Fig. 1B). After MCF7 cells were treated with 0.5 mM VPA for 24, 48 and 72 h, the percentage of cells with γH2AX foci formation was increased by 5.30% (P < 0.05), 7.11% (P < 0.01) and 12.24% (P < 0.01), respectively, as compared with the percentage of 5.51% in the control group without VPA treatment (Fig. 1B). The percentage of cells with γH2AX foci formation in the group of 1 mM VPA presented the same tendency as the group of 0.5 mM VPA, which was increased by 9.33% (P < 0.05), 13.91% (P < 0.01) and 17.38% (P < 0.01), respectively (Fig. 1B), compared to the control group with the percentage of 5.51% (Fig. 1B), indicating that VPA alone at safe and critical safe doses is able to induce DSBs in unirradiated breast cancer cells.
Fig. 1. The effect of VPA on γH2AX activity in MCF7 cells before and after IR treatment. (A) γH2AX expression in the cells treated with 0.5 or 1 mM VPA and in combination with IR was detected by western blotting; p84 was used as a loading control (left). The density of the γH2AX expression in A (left) was analyzed by “Image J” (right). (B) The images represent γH2AX foci formation in the cells treated with 0.5 mM VPA (left). The percentage of cells with γH2AX foci formation was calculated after VPA treatment (right panel, the cell with >10 foci was called positive and counted). (C) The images represent IR (8 Gy)-induced γH2AX foci formation in the cells pretreated with 0.5 mM VPA, thin and thick arrows separately exhibited Type A and B of γH2AX foci formation (upper). The percentage of cells with Type A and B of IR-induced γH2AX foci formation was calculated in the cells treated with the combination of IR with 0.5 mM and 1 mM VPA (lower). (D) The 0.5 mM VPA-treated and -untreated cells before and after 8 Gy treatment are presented in the images from comet assay (upper), and the olive moment was further analyzed (lower left); after correcting the data, the relative olive moment at 0 min, 30 min and 60 min was exhibited in the cells (lower right). DAPI was used for nuclear staining. Each data point in the graphs was from three independent experiments (mean ± SD). P-values were calculated by Student's t-test (*P < 0.05, **P < 0.01).

We further analyzed the effect of VPA on IR-associated γH2AX foci in MCF7 cells. At 6 h post-8 Gy, we found that γH2AX foci formation was present in all the MCF7 cells (Fig. 1C upper). However, according to the form of the foci, the cells were divided into two types, A and B:28 Type A cells contain smaller, darker γH2AX foci, which represents slighter DNA damage (Fig. 1C upper, thin arrow), and Type B cells contain only larger and brighter γH2AX foci or two kinds of smaller and larger foci, which represents severe DNA damage (Fig. 1C upper, thick arrows). The pattern of γH2AX foci formation in MCF7 cells treated with IR alone is mainly Type A cells, and its percentage was around 83.49% but the frequency of Type B was only 16.51%. After the cells were pretreated with VPA and then irradiated with 8 Gy IR, at 6 h post-IR, the percentage of Type B cells is obviously increased as compared with the single IR exposure group (Fig. 1C lower). Moreover, the percentage of Type B cells was obviously enhanced following the prolonged pretreatment time of VPA, which was carried out in a time-dependent manner (Fig. 1C lower). After MCF7 cells were pretreated with 0.5 mM VPA for 24, 48 and 72 h, followed by 8 Gy treatment, the percentage of Type B cells was increased by 12.43% (P < 0.05), 41.46% (P < 0.01) and 54.25% (P < 0.01) respectively, as compared with the single IR exposure group rate of 16.51% (Fig. 1C lower left); the frequency of Type A and B cells treated with the combination of 1 mM VPA with IR was similar to the combination of 0.5 mM VPA and IR, the percentage of Type B cells was increased by 31.75% (P < 0.05), 51.19% (P < 0.01) and 54.92% (P < 0.01), respectively (Fig. 1C lower right). Here, the neutral comet assay was used to detect DSBs in VPA-treated cells in order to confirm the above results. Before IR treatment, VPA can obviously enhance the olive moments as compared to the control group (P < 0.05, Fig. 1D upper and lower left). At 0 min post-8 Gy, the olive moments in 0.5 mM VPA pretreated cells were significantly increased as compared with VPA-untreated cells (P < 0.01, Fig. 1D upper and lower left). After the data from the groups of IR alone or the combination of IR with VPA were corrected by the results of each corresponding group at 0 min post-IR respectively, it was further found that at both 30 min and 120 min post-IR the relative olive moments in VPA-pretreated cells were also obviously higher than in the cells without VPA treatment (P < 0.01, Fig. 1D lower right), which was consistent with the results of testing γH2AX activity. Thus, through two different assays, our data implied that VPA at both safe and safe critical doses not only induces DSBs accumulation but also has the ability to result in IR-induced DSBs retention in breast cancer cells.
The effect of VPA at safe dose on the radiosensitivity of breast cancer cells
Since the data demonstrated that VPA at safe and safe critical doses can increase DNA DSBs accumulation, it was assessed whether radiosensitivity in MCF7 cells could be influenced by VPA. At first, the MTT assay was used to test the dynamic change of cell survival; the results from Fig. 2A showed that the survival of cells treated with 0.5 mM or 1 mM VPA alone can be suppressed after 5 days (P < 0.05), and 4Gy of IR can significantly increase cell sensitivity at the indicated time points (Fig. 2A, P < 0.01). Importantly, after the cells were pretreated with 0.5 or 1 mM VPA for 24 h and then irradiated with 4 Gy, at 4 days post-IR treatment the percentage of cell survival obviously decreased as compared with the single IR group (Fig. 2A, P < 0.01), suggesting that VPA can sensitize breast cancer cells to irradiation (the percentage of cell survival for the combination group was corrected by VPA alone). Next, to confirm this result, the cell survival in breast cancer cells treated with VPA was further assessed by the clonogenic survival assay, and colony survival was determined 21 days later. Fig. 2B shows that VPA alone at the safe dose significantly reduced cell survival by 30% relative to cells without VPA treatment (P < 0.01). After the cells were pretreated with 0.5 mM of VPA for 24 h and then exposed to different doses of IR (2, 4, 6 Gy), a significant reduction in 4 Gy and 6 Gy-induced clonogenic survival in the cells was shown as compared with IR alone (Fig. 2C, P < 0.05 or 0.01, the survival fraction for the combination group was corrected by VPA alone), which is consistent with the results from the MTT assay. Therefore, our data obtained by two different approaches suggested that VPA at a safe concentration can inhibit cell survival and enhance the radiosensitization of breast cancer cells, which may be associated with VPA-induced DSBs accumulation.
Fig. 2. Radiosensitivity was detected in MCF7 cells after the treatment with VPA or the combination with IR. The cell growth was tested by the MTT assay in the cells in VPA alone or the combination treatment of 0.5 mM or 1 mM VPA with 4 Gy of IR (A). The clonogenic survival assay was used to detect the plating efficiency in the cells treated with 0.5 mM alone (B) and surviving fraction in the cells treated with both 0.5 mM and different doses of 0, 2, 4 or 6 Gy (C). Each data point in the graphs was from three independent experiments (mean ± SD). P-values were calculated by Student's t-test (*P < 0.05, **P < 0.01).

VPA at safe dose can lead to the dysfunction of HR repair pathway
Based on the above results, it would be very interesting to know the mechanism of VPA-induced radiosensitization in breast cancer cells. Since VPA can lead to the accumulation of DSBs in the cells, it is possible that VPA can also influence the activity of DNA repair. To detect this possibility, the effect of VPA on the time it takes for IR-induced γH2AX foci to disappear was observed by immunofluorescence staining. As shown in Fig. 1B, the percentage of cells with γH2AX foci in the control group has no significant change during 0 h–72 h. At 6 h post-IR, in all of the cells with or without VPA pretreatment there was γH2AX foci formation (Fig. 3A, Fig. 1C). However, at 24 h post-IR, we found that the percentage of irradiated cells with γH2AX foci has decreased to 26.27% (Fig. 3A), indicating that most of the DNA damage caused by irradiation was repaired under these conditions. In contrast, at this time point, the proportion of γH2AX foci in irradiated cells pretreated with 0.5 mM or 1 mM VPA for 24 h was decreased to 45.63% and 52.11% (Fig. 3A), respectively, which was significantly higher than cells with IR exposure alone (Fig. 3A, P < 0.01), indicating that VPA may lead to the delay of IR-induced DNA DSBs repair; in other words, the DNA repair capacity in breast cancer cells was inhibited by VPA at a safe dose.
Fig. 3. The effect of VPA on HR activity. (A) The dynamic change of IR-induced γH2AX foci formation in the cells treated with 0.5 and 1 mM VPA was observed after 8 Gy treatment. The images represent γH2AX foci formation in the VPA-treated cells at 6 h and 24 h post-8 Gy, DAPI was used for nuclear staining (left). The percentage of γH2AX foci formation in A (left) was evaluated (right). (B) The working schematic of the recombination substrate, pDR-GFP, was presented. (C) MCF7 cells with pDR-GFP expression were transfected with plasmid of I-SceI or GFP and then treated with 0.5 mM or 1 mM VPA for 24 h. GFP was used as a positive control to represent transfection efficiency. The images showed that the results of HR frequency in VPA-treated cells were measured by flow cytometry. (D) The relative HR frequency was analyzed, and the percentage of each phase in the cell cycle was tested by flow cytometry. Each data point in the graphs was from three independent experiments (mean ± SD). P-values were calculated by Student's t-test (**P < 0.01).

Since mammalian cells rely mainly on HR and NHEJ mechanisms to repair DSBs, the effect of VPA on the frequency of HR and NHEJ was measured in our working system. First, we employed breast cancer cell line MCF7 to express the pDR-GFP recombination reporter for the HR frequency assay (Fig. 3B). Prescreening results demonstrated that the HR frequency can be tested by flow-cytometry after the generation of I-SceI-induced DSBs (Fig. 3C). It was found that cells treated with 0.5 mM VPA for 24 h can dramatically decrease HR frequency by 42.59% as compared to the cells without VPA treatment (Fig. 3D, P < 0.01). Also, 1 mM VPA treatment for 24 h showed a significant reduction of 37.58% in HR relative to the VPA-untreated group (Fig. 3D, P < 0.01), but there was no difference in the HR function between the doses of VPA at 0.5 mM and 1 mM (P > 0.05), indicating that VPA at a safe dose is sufficient to cause the disruption of the HR pathway. Since HR functions mainly in the S and G2 phases of the cell cycle, where the identical sister-chromatid is available for HR repair as a template,29–31 cell cycle profiling was tested in VPA-treated cells by flow cytometry. The results displayed that the percentage of cells at the G1, S and G2 phases in VPA-treated cells did not alter as compared with untreated cells (Fig. 3D), indicating that the change of HR is not associated with the cell cycle. In addition, we discovered that a higher dose of VPA (5 mM) can also dramatically reduce the HR frequency to 40%, but most of the cells (around 80%) accumulated at the G1 phase under this situation (data not shown). This suggests that a high dose of VPA can trigger the secondary effect to influence the cell function, but a low dose of VPA (such as 0.5 mM) can only exhibit the primary mechanism.
VPA at a safe dose can suppress the Rad51 and BRCA1 activity before and after IR treatment
Our data demonstrated that VPA can affect the HR function in response to DNA damage, and it was reported that a number of DNA repair proteins are involved in the HR process for repairing damaged DNA. One such protein, recombinase Rad51, plays a central role in the HR mechanism, thus we investigated whether VPA influences the Rad51-mediated HR pathway. The results from immunoblotting displayed that 0.5 mM and 1 mM VPA treatment for 24 h did not affect the Rad51 protein level in breast cancer cells before irradiation (Fig. 4A). At 6 h post-IR, the Rad51 protein level underwent around 50% reduction in the cells pretreated with 0.5 mM and 1 mM VPA (Fig. 4A right). Some reports indicate that Rad51 foci still form in response to DNA damage even when there is a lower protein level of Rad51 in the cells,32,33 so we determined whether IR-induced Rad51 foci formation was inactivated by VPA. The results from the immunofluorescence assay showed that the percentage of the cells with Rad51 foci formation in the 0.5 mM VPA-treated group was slightly decreased as compared with the control group (P > 0.05) and 1 mM VPA treatment reduced Rad51 foci by 2.03% (P < 0.05) as compared with the control group (4.33%) (Fig. 4B). In response to DNA damage, MCF7 cells pretreated with 0.5 or 1 mM VPA for 24 h before 8 Gy treatment showed a more significant decrease in the percentage of cells with Rad51 foci formation, which were reduced by 6.64% (P < 0.01) and 6.59% (P < 0.01) respectively, as compared with the single IR exposure group (12.55%) (Fig. 4B). This indicated that VPA at a safe dose impaired Rad51 activity after irradiation, and the VPA-inhibited HR pathway is Rad51-dependent.
Fig. 4. The effect of VPA on HR associated proteins. (A) Rad51 protein level was detected by western blotting in VPA-treated cells before and after 8 Gy treatment, p84 was used as a loading control (left). The density of the Rad51 protein level in A (left) was analyzed by “Image J” (right). (B) IR-induced Rad51 foci formation in 0.5 mM VPA-treated cells was exhibited in the images (left). The percentage of the cells with Rad51 foci formation was evaluated in VPA-treated cells at 6 h post-IR (right). (C) The BRCA1 protein level was detected by western blotting in VPA-treated cells before and after 8 Gy treatment (left). The density of the BRCA1 protein level in C (left) was measured by “Image J” (right), p84 was used as a loading control. (D) IR-induced BRCA1 foci formation in VPA-pretreated cells was observed in the images at 6 h post-8 Gy (left), the percentage of IR-induced BRCA1 foci formation in 0.5 mM VPA-pretreated cells was calculated (right). (E) The colocalization of Rad51 and BRCA1 foci formation was presented at 6 h post-8 Gy irradiation. DAPI was used for nuclear staining in the images. Each data point in the graphs was from three independent experiments (mean ± SD). P-values were calculated by Student's t-test (*P < 0.05 **P < 0.01).

BRCA1 is another important protein that mediates DNA repair and regulates HR via interaction with Rad51,34 so we also determined whether BRCA1 activity was influenced by VPA. The BRCA1 protein level was slightly decreased in non-irradiated cells treated with 0.5 mM VPA, however, 1 mM VPA caused more reduction of the BRCA1 protein level (Fig. 4C). At 6 h post-IR, it was found that the protein levels of BRCA1 significantly decreased (Fig. 4C), which is consistent with the pattern of change of VPA-induced Rad51 protein expression mentioned in Fig. 4B. Also, the immunofluorescence assay was used to determine BRCA1 foci formation in VPA-treated cells. After MCF7 cells were treated with 0.5 mM VPA for 24 h and 48 h, there was no difference in the percentage of cells with BRCA1 foci formation as compared with the control group; however, the percentage of the cells with BRCA1 foci formation at 72 h in VPA-treated cells was significantly reduced by 3.84% (P < 0.01) as compared with 5.13% in the control group (positive cells with ≥10 foci, Fig. 4D). MCF7 cells pretreated with 0.5 VPA for 24, 48 and 72 h before 8 Gy IR showed a more significant decrease in the percentage of cells with BRCA1 foci formation, which were reduced by 3.48% (P > 0.05), 23.57% (P < 0.01) and 30.83% (P < 0.01) respectively, as compared with 38.45% in the single IR exposure group (Fig. 4D). IR-induced BRCA1 foci formation in the cells treated with 1 mM VPA exhibited the same tendency as 0.5 mM VPA (data not shown). Therefore, VPA can impair the recruitment of BRCA1 to DNA breaks, thus inhibiting the DNA damage repair pathway.
It was reported that BRCA1 regulates the Rad51-mediated HR repair pathway in response to DNA damage, and both proteins can occur in the same DNA break areas. Here we investigated the colocalization of BRCA1 and Rad51 foci formation in our working system. In response to DNA damage, it was observed that BRCA1 foci can perfectly colocalize with Rad51 foci at DNA break areas (Fig. 4E), and 0.5 mM VPA can decrease the association of both the proteins in IR-treated cells, implying that VPA maybe inhibit the BRCA1-Rad51-mediated HR repair pathway, thus sensitizing breast cancer cells to irradiation treatment.
The effect of VPA at safe dose on NHEJ repair pathway
NHEJ is another important mechanism to repair DNA DSBs. To test the possibility that VPA may influence the NHEJ function in our working system, we used U2OS cells expressing the EJ5-GFP reporter to measure the NHEJ frequency35,36 (Fig. 5A). Prescreening results demonstrated that the NHEJ frequency can be tested by flow-cytometry after the generation of I-SceI-induced DSBs (Fig. 5B). It was discovered that the NHEJ frequency dramatically decreased by 40% in the cells treated with 0.5 mM VPA for 24 h as compared with untreated cells (Fig. 5C, P < 0.01). Cells treated with 1 mM VPA for 24 h showed a significant reduction of around 45% in NHEJ relative to the control group (Fig. 5C, P < 0.01). There was no difference between 0.5 mM and 1 mM VPA groups (Fig. 5C, P > 0.05), indicating that VPA at a safe dose also leads to the disruption of the NHEJ pathway. Here cell cycle profiling was tested in VPA-treated U2OS cells by flow cytometry, and the results demonstrated that the proportion of cells at the G1, S and G2 phases in VPA-treated cells had no change as compared with control cells (Fig. 5C). Thus, VPA at a safe dose can suppress the NHEJ repair capacity.
Fig. 5. The effect of VPA on NHEJ activity. (A) The working schematic of the EJ5-GFP reporter for the NHEJ assay is presented. (B) U2Os cells expressing EJ5-GFP were transfected with the plasmid of I-SceI or GFP and then treated with 0.5 mM or 1 mM VPA for 24 h. GFP was used as a positive control to represent transfection efficiency. The results of NHEJ frequency in VPA-treated cells as measured by flow cytometry are shown in the images. (C) The relative NHEJ frequency was analyzed in 0.5 mM and 1 mM VPA-treated cells, and the percentage of each phase in the cell cycle was evaluated by flow cytometry. (D) The NHEJ-associated proteins, such as DNA-PKcs, Ku70 and Ku80, in VPA-pretreated cells were detected at 6 h before and after 8 Gy treatment. Actin was used as a loading control. Each data point in the graphs was from three independent experiments (mean ± SD). P-values were calculated by Student's t-test (**P < 0.01).

A number of proteins are involved in the NHEJ repair pathway, such as DNA-PKcs, Ku70, Ku80, etc. Since our data demonstrated that VPA had a suppressive effect on NHEJ, it would be reasonable to detect whether VPA can influence the major NHEJ-associated proteins. The results from the immunoblotting assay demonstrated that the DNA-PKcs and Ku70 protein level had no significant change between the cells with or without VPA treatment before and after 8 Gy IR. However, the protein expression of Ku80 in the cells treated with 0.5 mM VPA for 24 h before and after irradiation was significantly decreased as compared with the control and IR alone groups, potentially declaring that VPA may suppress NHEJ via targeting Ku80 protein expression.
Discussion
It has been reported that overexpression of HDACs was discovered in many cancers.2,17 HDACs can inhibit histone acetylation and result in transcriptionally inactive chromatin.3,4,8 HDACis is able to increase the histone acetylation level via suppression of HDACs activity and affect chromatin structure and gene regulation.2,3,6,17 VPA, a representative compound of HDACis and a drug used for anticonvulsant treatment in the clinic, was investigated in this study. Our data here have indicated that 0.5 mM VPA used for the treatment of epilepsy in the clinic can also inhibit breast cancer cell proliferation and induce radiosensitivity by disrupting BRCA1-Rad51-mediated HR and Ku80-mediated NHEJ repair pathways, which are associated with the accumulation of IR-induced DSB cells. This indicates that VPA at a safe dose may be a promising anti-cancer drug and radiosensitizer (Fig. 6).
Fig. 6. The working model of the effect of VPA at safe and critical doses on anti-cancer radiosensitization through inhibiting BRCA1-Rad51-mediated HR and Ku80-mediated NHEJ pathways.

Increasing evidence has shown that a number of HDACis not only have potential anticancer effects but also are radiosensitizers for some tumors at cellular and animal levels;1,4,10,19,24 however, the molecular mechanism of HDACis-induced sensitivity to IR remains unknown. In the present study, besides the effect of VPA on DNA damage repair, it was also found that both 0.5 mM and 1 mM VPA did not affect cell cycle profiling. Only a higher dose of VPA at 5 mM can cause the accumulation of the cells in G1 phase, indicating that lower dose VPA-induced cell death is independent of the cell cycle control. It was also reported that apoptosis resistance in MCF-7 breast carcinoma cells subject to IR was independent of p53 and cell cycle control since MCF7 cells carry defective caspase-3,37 suggesting that IR could not induce apoptosis in MCF7 cells. Based on the study, it is clearly indicated that the disruption of the DNA repair pathway caused by VPA at a safe dose is the key mechanism for its effects of radiosensitization and anticancer in breast cancer. The following reasons strongly support the close relationship between VPA-mediated radiosensitization and the disruption of DNA repair. Firstly, our and other reports obviously demonstrated that VPA and other HDACis can enhance accumulation of DSBs in breast cancer cells (Fig. 1) in response to DNA damage,10 and prolong the resolution of DNA DSBs (Fig. 3A),4,10,38 indicating that DNA repair activity was damaged in HDACis-treated cells. Secondly, the inhibition of DNA repair is directly associated with the inactivation of both HR and NHEJ measured by the recombination reporters, pDR-GFP and EJ5-GFP (Fig. 3B–D and 5A–C). However, there is a contrasting report that HDACis targeting drugs (such as SAHA or TSA) did not alter the HR frequency but induced significant increase in NHEJ activity in HeLa cells.19,23 Therefore, it is indicated that different HDACis may initiate different mechanisms for radiosensitization, and HDACis-triggered mechanisms may also rely on the type of cancer cells. Thirdly, the key question to be answered is how DNA repair-associated proteins play a role in VPA-treated cancer cells. The activity of crucial proteins in HR, BRCA1 and Rad51, were evidently down-regulated and were not efficiently recruited to DNA DSB areas after VPA treatment (Fig. 4). In addition, other HDACis (such as Vorinostat and PCI-24781) can also decrease Rad51 and BRCA1.24,39,40 In terms of NHEJ, only Ku80 was found to be down-regulated in VPA-treated cells (Fig. 5D), indicating that the recruitment of the Ku70/80 heterodimer to DSBs to activate the classical mechanisms of NHEJ was suppressed. This is consistent with other reports indicating that HDACis (such as Vorinostat, TSA etc.) attenuated up-regulation of Ku80, DNA-PKcs etc. in prostate and colon cancer cells.41 Most importantly, the mechanisms by which VPA or other HDACis are able to target DNA repair proteins and affect repair pathways in cancer cells require further verification.
Currently radiotherapy remains one of the most common forms of cancer therapy, even though the pre-clinical study of VPA in combination with radiation for some cancer treatments has been carried out,42 there is little evidence for the combination of VPA with radiation in the treatment of breast cancer. The safe blood concentration of VPA in the treatment of epilepsy is 0.3–0.8 mM, and the safe dose (0.5 mM) of VPA in the present study successfully resulted in the radiosensitization in breast cancer cells (Fig. 2), which provide reliable evidence for additional studies. The urgent studies would be done at the animal level and probably preclinical stage since the pharmacological activities of VPA at a safe dose in the clinic are well-known. Given that the inhibition of DNA repair activity is the key mechanism for VPA-induced radiosensitization, it would be very interesting to identify whether HR- or NHEJ-associated proteins could be used as efficient biomarkers to predict those patients who would benefit from either VPA therapy alone or in combination with radiotherapy.
Materials and methods
Cell culture
MCF7 and U2OS cell lines (American type culture collection) were maintained in DMEM (Gibco) supplemented with 10% FBS (Gibco) under a humidified atmosphere of 5% CO2 at 37 °C.
VPA and irradiation treatment
Cells with 0.5 or 1 mM VPA (Sigma) pretreatment for 24, 48 or 72 h were irradiated by different doses of X-ray with a Primus linear accelerator (Siemens, 6 MV X-ray, absorbed dose rate: 2.33 Gy per min, field size: 40 cm × 40 cm).
Immunoblotting and Immunofluorescence analysis
The methods are mentioned in our published paper.43 The primary antibodies used for immunoblotting analysis are mouse anti-BRCA1 (D9, Santa Cruz Technology), anti-Rad51 (Ab-2, Calbiochem), anti-DNA-PKcs (Abcam), anti-Ku70 (Santa Cruz Biotechnology), anti-Ku80 (Santa Cruz Biotechnology), anti-γH2AX (Ser139, clone JBW301, Millipore), anti-P84 (Abcam), and anti-β-actin (Sigma). Secondary antibodies used were the goat anti-rabbit IgG horseradish peroxidase conjugated, goat anti-mouse IgG-horseradish peroxidase conjugated (Pierce, Rockford, IL), AlexaFluor 594-labeled goat anti-mouse IgG, and AlexaFluor 488-labeled chicken anti-rabbit (Molecular Probe).
Comet assay
The neutral comet assay was performed for detection of DSBs by using the Comet Assay kit (Trevigen, Gaithersburg, MD) following the manufacturer's instructions. Olive Moment was analyzed using CometScore software (TriTek, Sumerduck, VA).
Clonogenic survival assay
The method was described in our previous paper.43 The number of cell colonies (≥50 cells per clone) was counted and the ability of cell survival was presented by the survival fraction (SF), SF = (the number of clones/seeded cells)/plating efficiency (PE).
MTT assay
MTT solution (5 mg ml–1, Sigma) was added to the treated cells and incubated for 4 h. Then the medium was replaced with dimethyl sulfoxide. The absorbance of the solution was measured using an enzyme immunoassay analyzer at 490 nm.
HR assay
MCF7 cell line carrying a pDR-GFP reporter was used for this assay to measure the HR frequency.43 Cells were transfected with I-SceI, GFP, pcDNA3 plasmids and subjected to flow cytometry for analysis of GFP-positive cells.
NHEJ assay
U2OS cell line with the expression of End Joining reporter (EJ5-GFP) in the genome was applied to measure the NHEJ frequency.36 The nucleofector (Amaxa, USA) was used for cell transfection with I-SceI, GFP or pcDNA3 plasmids. The NHEJ frequency was analyzed by flow cytometry.
Cell cycle analysis
The cells were collected and fixed with 70% cold ethanol overnight and stained with prodidium iodide solution for cell cycle analysis by flow cytometry as mentioned in our previous paper.43
Disclosure of potential conflicts of interest
The authors declared no conflict of interest.
Grant support
This work was supported by the Natural Science Foundation of China [81172527, 81472800], and Shandong University of Science and Technology of Shandong Province [2013GGE27052, 2014GGH218010].
Acknowledgments
We thank Dr STARK J M for kindly providing the U2OS cell line with the expression of End Joining reporter in the genome (EJ5-GFP), Dr Jasin M for providing the recombination substrate, pDR-GFP plasmid, and Dr Callie Baker at Washington University at St Louis for correcting the manuscript.
References
- Groselj B., Sharma N. L., Hamdy F. C., Kerr M., Kiltie A. E. Br. J. Cancer. 2013;108:748–754. doi: 10.1038/bjc.2013.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks P. A., Xu W. S. J. Cell. Biochem. 2009;107:600–608. doi: 10.1002/jcb.22185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks P. A. Biochim. Biophys. Acta. 2010;1799:717–725. doi: 10.1016/j.bbagrm.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He G., Wang Y., Pang X., Zhang B. Tumour Biol. 2014;35:1003–1011. doi: 10.1007/s13277-013-1134-z. [DOI] [PubMed] [Google Scholar]
- Sharma N. L., Groselj B., Hamdy F. C., Kiltie A. E. BJU Int. 2013;111:537–542. doi: 10.1111/j.1464-410X.2012.11647.x. [DOI] [PubMed] [Google Scholar]
- Blattmann C., Oertel S., Ehemann V., Thiemann M., Huber P. E., Bischof M., Witt O., Deubzer H. E., Kulozik A. E., Debus J., Weber K. J. Int. J. Radiat. Oncol., Biol., Phys. 2010;78:237–245. doi: 10.1016/j.ijrobp.2010.03.010. [DOI] [PubMed] [Google Scholar]
- Kelly W. K., Marks P. A. Nat. Clin. Pract. Oncol. 2005;2:150–157. doi: 10.1038/ncponc0106. [DOI] [PubMed] [Google Scholar]
- Dokmanovic M., Clarke C., Marks P. A. Mol. Cancer Res. 2007;5:981–989. doi: 10.1158/1541-7786.MCR-07-0324. [DOI] [PubMed] [Google Scholar]
- Karagiannis T. C., Kn H., El-Osta A. Epigenetics. 2006;1:131–137. doi: 10.4161/epi.1.3.2896. [DOI] [PubMed] [Google Scholar]
- Kawano T., Akiyama M., Agawa-Ohta M., Mikami-Terao Y., Iwase S., Yanagisawa T., Ida H., Agata N., Yamada H. Int. J. Oncol. 2010;37:787–795. doi: 10.3892/ijo_00000728. [DOI] [PubMed] [Google Scholar]
- Kuendgen A., Gattermann N. Cancer. 2007;110:943–954. doi: 10.1002/cncr.22891. [DOI] [PubMed] [Google Scholar]
- Duenas-Gonzalez A., Candelaria M., Perez-Plascencia C., Perez-Cardenas E., de la Cruz-Hernandez E., Herrera L. A. Cancer Treat. Rev. 2008;34:206–222. doi: 10.1016/j.ctrv.2007.11.003. [DOI] [PubMed] [Google Scholar]
- Lee J. H., Choy M. L., Ngo L., Foster S. S., Marks P. A. Proc. Natl. Acad. Sci. U. S. A. 2010;107:14639–14644. doi: 10.1073/pnas.1008522107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harikrishnan K. N., Karagiannis T. C., Chow M. Z., El-Osta A. Cell Cycle. 2008;7:468–476. doi: 10.4161/cc.7.4.5405. [DOI] [PubMed] [Google Scholar]
- Sha K., Winn L. M. Birth Defects Res., Part B. 2010;89:124–132. doi: 10.1002/bdrb.20236. [DOI] [PubMed] [Google Scholar]
- Zhang F., Zhang T., Teng Z. H., Zhang R., Wang J. B., Mei Q. B. Cancer Biol. Ther. 2009;8:823–831. doi: 10.4161/cbt.8.9.8143. [DOI] [PubMed] [Google Scholar]
- Giannini G., Cabri W., Fattorusso C., Rodriquez M. Future Med. Chem. 2012;4:1439–1460. doi: 10.4155/fmc.12.80. [DOI] [PubMed] [Google Scholar]
- Pont L. M., Naipal K., Kloezeman J. J., Venkatesan S., van den Bent M., van Gent D. C., Dirven C. M., Kanaar R., Lamfers M. L., Leenstra S. Cancer Lett. 2015;356:525–535. doi: 10.1016/j.canlet.2014.09.049. [DOI] [PubMed] [Google Scholar]
- Koprinarova M., Botev P., Russev G. DNA Repair. 2011;10:970–977. doi: 10.1016/j.dnarep.2011.07.003. [DOI] [PubMed] [Google Scholar]
- Krejci L., Altmannova V., Spirek M., Zhao X. Nucleic Acids Res. 2012;40:5795–5818. doi: 10.1093/nar/gks270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman J. R., Taylor M. R., Boulton S. J. Mol. Cell. 2012;47:497–510. doi: 10.1016/j.molcel.2012.07.029. [DOI] [PubMed] [Google Scholar]
- Moynahan M. E., Jasin M. Nat. Rev. Mol. Cell Biol. 2010;11:196–207. doi: 10.1038/nrm2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith S., Fox J., Mejia M., Ruangpradit W., Saberi A., Kim S., Choi Y., Oh S., Wang Y., Choi K., Li L., Hendrickson E. A., Takeda S., Muller M., Myung K. PLoS One. 2014;9:e87203. doi: 10.1371/journal.pone.0087203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnaiyan P., Vallabhaneni G., Armstrong E., Huang S. M., Harari P. M. Int. J. Radiat. Oncol., Biol., Phys. 2005;62:223–229. doi: 10.1016/j.ijrobp.2004.12.088. [DOI] [PubMed] [Google Scholar]
- Kinner A., Wu W., Staudt C., liakis I. G. Nucleic Acids Res. 2008;36:5678–5694. doi: 10.1093/nar/gkn550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogakou E. P., Boon C., Redon C., Bonner W. M. J. Cell Biol. 1999;146:905–916. doi: 10.1083/jcb.146.5.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogakou E. P., Pilch D. R., Orr A. H., Ivanova V. S., Bonner W. M. J. Biol. Chem. 1998;273:5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- Chen H., Ma Z., Vanderwaal R. P., Feng Z., Gonzalez-Suarez I., Wang S., Zhang J., Roti Roti J. L., Gonzalo S. Radiat. Res. 2011;175:214–224. doi: 10.1667/rr2323.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodarzi A. A., Jeggo P., Lobrich M. DNA Repair. 2010;9:1273–1282. doi: 10.1016/j.dnarep.2010.09.013. [DOI] [PubMed] [Google Scholar]
- Iliakis G., Wang H., Perrault A. R., Boecker W., Rosidi B., Windhofer F., Wu W., Guan J., Terzoudi G., Pantelias G. Cytogenet. Genome Res. 2004;104:14–20. doi: 10.1159/000077461. [DOI] [PubMed] [Google Scholar]
- Kostyrko K., Bosshard S., Urban Z., Mermod N. Cell Cycle. 2015;14:2853–2861. doi: 10.1080/15384101.2015.1049784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alagpulinsa D. A., Ayyadevara S., Shmookler Reis R. J. Front. Oncol. 2014;4:289. doi: 10.3389/fonc.2014.00289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Powell S. N. Mol. Cancer Res. 2005;3:531–539. doi: 10.1158/1541-7786.MCR-05-0192. [DOI] [PubMed] [Google Scholar]
- Vaclova T., Gomez-Lopez G., Setien F., Bueno J. M., Macias J. A., Barroso A., Urioste M., Esteller M., Benitez J., Osorio A. Breast Cancer Res. Treat. 2015;152:271–282. doi: 10.1007/s10549-015-3459-3. [DOI] [PubMed] [Google Scholar]
- Wu L., Shao L., Li M., Zheng J., Wang J., Feng W., Chang J., Wang Y., Hauer-Jensen M., Zhou D. Radiat. Res. 2013;179:160–170. doi: 10.1667/RR3034.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunn A., Stark J. M. Methods Mol. Biol. 2012;920:379–391. doi: 10.1007/978-1-61779-998-3_27. [DOI] [PubMed] [Google Scholar]
- Essmann F., Engels I. H., Totzke G., Schulze-Osthoff K., Janicke R. U. Cancer Res. 2004;64:7065–7072. doi: 10.1158/0008-5472.CAN-04-1082. [DOI] [PubMed] [Google Scholar]
- Chen X., Wong P., Radany E., Wong J. Y. Cancer Biother. Radiopharm. 2009;24:689–699. doi: 10.1089/cbr.2009.0629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstantinopoulos P. A., Wilson A. J., Saskowski J., Wass E., Khabele D. Gynecol. Oncol. 2014;133:599–606. doi: 10.1016/j.ygyno.2014.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adimoolam S., Sirisawad M., Chen J., Thiemann P., Ford J. M., Buggy J. J. Proc. Natl. Acad. Sci. U. S. A. 2007;104:19482–19487. doi: 10.1073/pnas.0707828104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kachhap S. K., Rosmus N., Collis S. J., Kortenhorst M. S., Wissing M. D., Hedayati M., Shabbeer S., Mendonca J., Deangelis J., Marchionni L., Lin J., Hoti N., Nortier J. W., DeWeese T. L., Hammers H., Carducci M. A. PLoS One. 2010;5:e11208. doi: 10.1371/journal.pone.0011208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krauze A. V., Myrehaug S. D., Chang M. G., Holdford D. J., Smith S., Shih J., Tofilon P. J., Fine H. A., Camphausen K. Int. J. Radiat. Oncol., Biol., Phys. 2015;92:986–992. doi: 10.1016/j.ijrobp.2015.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C., Zhang F., Luo Y., Wang H., Zhao X., Guo G., Powell S. N., Feng Z. DNA Repair. 2015;33:60–69. doi: 10.1016/j.dnarep.2015.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]