

 With the limited but ongoing usage of perfluorooctane sulfonate (PFOS), the health effects of both PFOS and its alternatives are far from being understood.
With the limited but ongoing usage of perfluorooctane sulfonate (PFOS), the health effects of both PFOS and its alternatives are far from being understood.
Abstract
With the limited but ongoing usage of perfluorooctane sulfonate (PFOS), the health effects of both PFOS and its alternatives are far from being understood. Long-term potentiation (LTP) was evaluated in rats after exposure to PFOS and its alternatives, aiming to provide some evidence about their potential to affect cognitive ability. Different dosages of PFOS and alternative chemicals, including perfluorohexane sulfonate (PFHxS), perfluorobutane sulfonate (PFBS) and chlorinated polyfluorinated ether sulfonate (Cl-PFAES), were given to rats via acute intracerebroventricular injection. The field excitatory postsynaptic potential (fEPSP) amplitude of the input/output functions, paired-pulse facilitations, and LTP in vivo were recorded. PFOS and its alternatives inhibited LTP in varying degrees, without significant effects on the normal synaptic transmission. In addition, PFHxS and Cl-PFAES exhibited comparable potential to PFOS in disturbing LTP. The results suggested that acute exposure to PFOS and its alternatives impaired the synaptic plasticity by a postsynaptic rather than a presynaptic mechanism. Besides, the fEPSP amplitude of the baseline was reduced by Cl-PFAES but not by other compounds, indicating that Cl-PFAES might act in a different mode. Providing some electrophysiological evidence and the potential mechanism of the neurotoxicity induced by PFOS and its alternatives, the present study addresses further evaluation of their safety and health risks.
Introduction
Perfluorooctane sulfonate (PFOS) is an eight-carbon fully fluorinated organic chemical, which is extremely stable and resistant to be degraded by biological metabolism and other physiochemical processes.1 Due to its physicochemical stability and oil- and water-resistance, PFOS has been extensively used in a variety of industrial processes and consumer applications, leading to its ubiquitous presence in various environmental matrices, even in human and wildlife.1–3 In 2009, PFOS was listed in Annex B of the Stockholm Convention on Persistent Organic Pollutants. According to the Stockholm Convention, although the ultimate goal is the elimination of PFOS-based substances, production of these chemicals may continue for limited purposes and 15 or more uses will be allowed, including uses that disburse PFOS directly into the environment, such as firefighting foams and pesticides.
Meanwhile, the replacement of PFOS by alternatives is undergoing a fast development. Possessing similar oleophobic and hydrophobic properties to PFOS, easier degradation and faster elimination from the body for the fluorinated compounds with shorter carbon chain length raise an expectation of lower toxicity and health risk. Therefore, perfluorohexane sulfonate (PFHxS) and perfluorobutane sulfonate (PFBS), with six and four perfluorinated carbon atoms, respectively, were regarded as the appropriate alternatives to PFOS.4 Correspondingly, the increasing temporal trends of PFHxS levels have been observed in primiparous women from Sweden during 1996–2010.5 And PFHxS was also extensively found in the breast milk collected from seven countries in Asia, at concentrations comparable to the report from Sweden.6 However, limited information is available about the toxicity of PFHxS and PFBS. Short carbon chain perfluorinated compounds were thought with lower bioaccumulation and toxicity, and the C4-based chemicals was reported neither to be bioaccumulative nor toxic in a battery of environmental and safety tests.4,7 However, recent studies showed that neonatal PFHxS exposure exhibited a similar potency to PFOS in altering both spontaneous behavior and neuroprotein levels.8–11 Moreover, chlorinated polyfluorinated ether sulfonate (Cl-PFAES, C8ClF16O4SK, locally called F-53B) has been used as the only available mist suppressant in Chinese electroplating industry before the emergence of PFOS related products.12 After phasing out of PFOS, Cl-PFAES might obtain a larger market share and potentially expand from the industries that use PFOS currently. However, this PFOS alternative has been overlooked for over 30 years until the first report of its toxicity, degradability and environmental presence by Wang et al.12 Cl-PFAES was classified as not readily degradable in the Closed Bottle Test, and its LC50 (96 h) was 15.5 mg L–1, which belonged to the same class as PFOS. Remarkably, Cl-PFAES was detected at high concentrations, 43–78 μg L–1 and 65–112 μg L–1 for the effluent and influent, respectively, in wastewater from the chrome plating industry in the city of Wenzhou, China.12 Moreover, Cl-PFAES was not successfully removed by the wastewater treatments in place and was found in the surface water at similar levels to PFOS, 10–50 ng L–1.12 Ruan et al.13 reported that Cl-PFAES was detected in the municipal sewage sludge samples collected around China, at relatively high levels following the PFOS levels. Most recently, it was also found to be bioaccumulated in crucian carp, with whole body bioaccumulation factors exceeding the regulatory bioaccumulation criterion and significantly higher than those of PFOS in the same datasets.14 Thus, it is of substantial significance to further evaluate the health effects of Cl-PFAES, as well as other PFOS alternatives.
The nervous system appears to be one of the most sensitive targets of environmental contaminants, which have been speculated as the possible reason for an increased prevalence and earlier occurrence of neurodegenerative diseases, such as Alzheimer's and Parkinson's disease.15 Several pieces of evidence suggest that PFOS can cross the blood–brain-barrier,16–18 and the neurotoxicity of PFOS has been studied at multiple biological levels during neural development.19 PFOS exposure was correlated with a reduction in learning and memory abilities exposed during the prenatal period, affecting the spontaneous behavior and habituation.16,20,21 In addition, PFOS presented adverse effects on the nervous system at the cellular level, inducing not only deficits in cell growth and viability, but also shifts in differentiation.22 PFOS also inhibited synaptogenesis and synaptic transmission, where the expression of postsynaptic density protein 95 (PSD95) in cultured neurons and synaptophysin in the hippocampus of neonatal mouse was repressed.10,23 Key factors in the induction of long-term potentiation (LTP) were identified by global gene expression in rats with prenatal and neonatal PFOS exposure.24 Furthermore, some other neurotoxicological findings of PFOS also suggest that PFOS possibly affects LTP including the calcium imbalance, the effects on Ca2+/calmodulin-dependent protein kinase II (CaMKII) and protein kinase C (PKC), and the interaction with glutamate receptors including N-methyl-d-aspartic acid (NMDA) receptors.25–28 Therefore, research concerning the mechanism related to synaptic plasticity would be valuable for a better understanding of the neurotoxicity of PFOS and its alternatives.
Long-term potentiation, as the physiological basis of learning and memory, is employed as the primary cellular and molecular model to evaluate synaptic plasticity.29 LTP can be initiated in certain areas of the central nervous system by a brief high frequency stimulus which is symbolized with a prolonged increase in synaptic responses. It is extensively studied in the neurotoxicity of environmental pollutants to evaluate the capacity for information processing and storage by the neural network. Polychlorinated biphenyl (PCB) 153 and decabrominated diphenyl ether (PBDE) 209 have been shown to block LTP in rats both in vitro and in vivo, leading to reduction in learning and memory abilities.30,31 Chronic lead (Pb) and aluminum (Al) exposure also impaired LTP in rats, which has been associated with cognitive dysfunction and neuronal diseases.32,33
The present study compared the neurotoxicity of PFOS and its alternatives by examining electrophysiological activity through acute intracerebroventricular (i.c.v.) administration. Intracerebroventricular administration is a fundamental method in the research of neurotoxicity and pharmacology, which can get the compounds across the “blood–brain” barrier and affect the central nervous system directly.34–36 Therefore, the i.c.v administration is valuable to avoid the underestimation of the neurotoxicity effects of PFOS and its alternatives, since the distribution of the target chemicals into the brain may be limited in the acute toxicity test. Furthermore, i.c.v. administration is also helpful in reducing the effects of the differences in the pharmacotoxicological kinetics among the chemicals. Input/output (I/O) functions, paired-pulse facilitations (PPF), and LTP in the hippocampus CA1 region of rat in vivo were monitored after exposure to PFOS, PFHxS, PFBS and Cl-PFAES. To the best of our knowledge, this is the first study on the LTP in vivo affected by exposure to PFOS and its alternatives. Based on these observations, some evidence is provided on the neurotoxicity and potential mechanisms of PFOS and its alternative compounds.
Results
Effects of PFOS and its alternatives on LTP
The raw data collected for LTP monitoring are shown in Fig. 1A. After the tetanic stimulation, the stable fEPSP amplitude increased up to 1.9–2.3 fold of the baseline, and then declined to a different degree with time. The amplitude of fEPSP in rats from the control group maintained above 140% of the baseline at 60 min (Fig. 1B). Exposure to PFOS and its alternatives induced obvious repression of LTP except Cl-PFAES at 10 μM (Fig. 1B). Fig. 1C presents fEPSP amplitude at 60 min after HFS. The fEPSP amplitude of the control group was 141% of the baseline. In the low dose treatment group, PFOS and PFHxS reduced the fEPSP amplitude of LTP, although the reduction did not reach statistical significance because of the large standard error. PFOS, PFHxS, and Cl-PFAES at 100 μM significantly lowered the fEPSP amplitude compared with control. Moreover, significant differences between low and high concentration treatment were observed for PFOS and Cl-PFAES. It seemed like PFBS also inhibited LTP as shown in Fig. 1B, but no significant change was observed at 60 min after HFS.
Fig. 1. Effects of exposure to PFOS and its alternatives on LTP in the hippocampus CA1 region of rat. (A) Representative raw data traces before and after induction of LTP. The solid line is the fEPSP amplitude before HFS, and the dashed line is the fEPSP of LTP at 60 min after titanic stimulation. (B) The pooled data of standardized fEPSP amplitude monitored before and after HFS. Each point represents the mean fEPSP amplitude of three responses of stimuli. (C) Pooled results of LTP at 60 min after HFS. a/A, b/B, c/C, d/D indicate the difference with control, PFOS, PFHxS and PFBS groups, respectively. The lowercase letters indicate significant difference at p < 0.05 among control and the low dose group of four compounds. The capital letters indicate significant difference at p < 0.05 among control and the high dose group of four compounds. Asterisks indicate significant difference at p < 0.05 between the low and high dose groups of the same compound.

After exposure to PFOS, PFHxS and PFBS by i.c.v. injection, no significant impacts on the fEPSP amplitude before HFS were observed (Fig. 1B). But Cl-PFAES injection decreased the fEPSP amplitude of the baseline, especially the high dose treatment. To further testify the observed effect of Cl-PFAES on the baseline, the baseline recording was prolonged to 90 min after Cl-PFAES injection. As shown in Fig. 2A, the inhibition on fEPSP amplitude induced by Cl-PFAES was irreversible and was still observed at 90 min after injection. A slight but statistically significant decrease in the baseline fEPSP was observed in the 10 μM Cl-PFAES group and a further depression was apparent in the 100 μM group (Fig. 2B).
Fig. 2. Effects of Cl-PFAES at 10 μM and 100 μM on the baseline of fEPSP amplitude. (A) Basal fEPSP amplitude recordings 30 min before Cl-PFAES injection and 90 min after injection. Each point represents the mean fEPSP amplitude of three responses of stimuli. (B) The averaged fEPSP amplitude before and after injection of 10 μM and 100 μM Cl-PFAES. Pre-injection averaged the fEPSP amplitude in 30 min before injection, and post-injection averaged the fEPSP amplitude in 90 min after injection. *: p < 0.05, **: p < 0.01.

Effects of PFOS and its alternatives on I/O curves and PPF
To test the effects of PFOS and its alternatives on basic synaptic transmission and short-term synaptic plasticity in the CA1 region, I/O curves and PPF were measured before induction of LTP. Fig. 3A illustrates the relationship between the stimulus current and fEPSP amplitude in rats from the control and treatment groups. There were no remarkable changes in the fEPSP amplitude at a stimulus current of 0.1–1.0 mA in 10 μM treatment groups compared with the control, with significant differences observed in few scattered points in 100 μM groups. As shown in Fig. 3B, all the groups exhibited a maximal facilitation at an inter-pulse interval of 60 ms, but neither 10 μM nor 100 μM of PFOS and its alternatives posed significant effects on the average peak facilitation compared with the control group.
Fig. 3. Effects of exposure to PFOS and its alternatives in 10 μM and 100 μM on I/O curves and PPF in the hippocampus CA1 region in vivo. (A) I/O curves of fEPSP amplitude at varying stimulus current of 0.1–1.0 mA. (B) PPF of the fEPSP amplitude at varying ISIs of 10–400 ms.

Discussion
The present study evaluated and compared the neurotoxic effects of PFOS, PFHxS, PFBS and Cl-PFAES in vivo on synaptic plasticity and elucidated the possible mechanism. To the best of our knowledge, this is the first study on LTP affected by perfluroalkyl compound (PFC) exposure in vivo. The findings added significant electrophysiological evidence that exposure to PFOS and its alternatives results in the impairment of synaptic plasticity.
The present findings about the impairment of LTP induced by PFOS and its alternatives provided some electrophysiological evidence of their neurotoxicity, consistent with the behavioral alterations reported in previous studies. Fuentes et al.20 reported that shortened retention in the water maze probe task was induced by the administration of 3 mg kg–1 PFOS per day via gavage for four consecutive weeks in adult mice. In the study of Johansson et al.,11 hyperactivity and the deficits in spontaneous behavior and habituation were observed in mice treated with a single-oral dose of PFOS on PND10. And our previous study further demonstrated that prenatal and postnatal PFOS exposure caused the prolonged escape latency in the water maze test of the rat pups, suggesting the decline in spatial learning and memory abilities.16 Although the relevance of LTP to some of these behavioral alternations is still unclear, our observations at minimum provide a possible cellular substrate for some of these alterations.
Until now, little information is available about the toxicity of PFOS alternatives. The present study found that PFHxS exhibited a comparable potency to PFOS in affecting LTP, consistent with a previous study that PFHxS exposure posed similar neurotoxic effects to PFOS in both behavior indicators and the neuroprotein levels of mammals.8,9 Viberg et al.8 reported that a single neonatal PFHxS dosage altered adult spontaneous behavior and cognitive function. Further, Lee and Viberg9 found that neonatal PFHxS exposure altered the neuroprotein levels, e.g. CaMKII, GAP-43, synaptophysin and tau, essential for normal brain development in mice. And these neurotoxic effects of PFHxS were similar to that observed for PFOS.10,11 These support the results from the present study and suggest that PFHxS and PFOS have a similar neurotoxic potency and mechanism of action. In contrast, the present electrophysiological examination found that PFBS exhibited a relatively lower potency to impair LTP than the other three target compounds. Similarly, only mild reduction in red blood cell count, hematocrit, and hemoglobin were observed in male rats given 600 mg kg–1 PFBS for 90 days via oral gavage, and no abnormal behaviors in motor activity and functional observation battery were noted.7 PFBS has a much lower potential for accumulation in human serum, and the minimal doses to elicit the same degree of hepatotoxicity was approximately 600 times lower than that of PFOS.7,37
The elimination kinetics has been regarded as a decisive factor leading to the difference of PFC homologues in their toxicity potency, where the rate of elimination is related to the carbon chain length.38 Olsen et al.37 reported that in human serum the geometric elimination half-life of PFOS was 1751 days, with 2662 days for PFHxS and 25.8 days for PFBS. Kudo et al.39 observed a tendency that perfluoroalkyl carboxylates (PFACs) with a longer carbon chain length were less eliminated in urine in both male and female rats. Although the elimination may contribute less to the difference among target compounds in LTP inhibition in the present acute exposed study, similar mechanism underlies the bioaccumulation potency and toxicity. The difference in the hydrophobicity of the PFC compounds and the corresponding bioavailability to the target cells may be an important reason.40 It had been demonstrated that C4–C6 PFCs is less hazardous than C7–C8 PFCs both in mammals and in aquatic organisms.41 Together with the findings in the present study that PFOS and PFHxS posed a higher potency to affect LTP, the concern is raised about the neurotoxicological potential of long carbon chain PFCs. Recently, Route et al.42 found that perfluorodecane sulfonate (PFDS) was the second abundant analyte, taking up 23% of the PFC amount in the blood plasma of the wild bald eagle in the upper Midwestern United States. Therefore, further toxicological evaluation of the long carbon chain PFCs is necessary.
The present study is the first about the neurotoxicity of Cl-PFAES. Different from PFOS, PFHxS and PFBS, Cl-PFAES showed the potency to inhibit the fEPSP amplitude of the baseline, indicating that Cl-PFAES might act in a different mode on synaptic transmission from perfluoroalkyl acids. Similar phenomena were observed when PCB153 and sodium valproate (VPA) was administered to hippocampal slices, when both the amplitude of the fEPSP of the baseline and LTP were decreased.30,43 PCB153 has widely been considered to be lacking in significant toxicity due to its poor activity with the Ah receptor. However, the findings about its effects on LTP suggest that it may not be the case.30 VPA was considered as an excitotoxicant which induced apoptotic neurodegeneration in the developing rat brain, lowered excitatory neurotransmission might be the reason for the inhibition of the baseline.44 Comparing the chemical structure with PFOS, Cl-PFAES with a larger molecular volume contained an ether group inside the carbon chain, which characterized an increasing hydrophobicity and better flexibility of the fluorinated chain making Cl-PFAES easier to be incorporated into the lipid bilayer of the cell membrane.45 As Wang et al.12 reported, the acute LC50 of Cl-PFAES is similar to that of PFOS, where the slope of the dose–response curve of Cl-PFAES was even higher than that of PFOS. Without human exposure assessment and the toxicokinetic data of Cl-PFAES in mammals and humans, it is impossible to estimate the health risk of Cl-PFAES. Therefore, the toxicity of Cl-PFAES needs further characterization, when the present study provides preliminary evidence of its potential effects on the nervous system.
I/O curves reflect the basal synaptic transmission competency. Thus, no effects of acute exposure to PFOS and its alternatives on I/O functions implied that the normal synaptic transmission at the Schaffer collateral-CA1 synapse was not interrupted. PPF is a short-term synaptic plasticity which is a sensitive indicator of the change in the transmitter release amount, or presynaptic connections.46,47 Neither PFOS nor its alternatives led to significant changes in PPF, hinting that PFOS and its alternatives might not exert effects on presynaptic cells after acute exposure. Besides, the quantity of PSD95 in dendrites decreased significantly when neurons were continuously treated with PFOS, which clarifies that the effects of PFOS mainly focus on postsynaptic cells.23 In the research of Xing et al.31 lactational PBDE 209 exposure from mother's milk did not affect I/O functions and PPF but decreased LTP, suggesting a weaker inhibition in synaptic plasticity compared with intragastric lactational exposure and exposure after weaning. Together with the findings in the present study that PFOS and its alternatives significantly affected the fEPSP amplitude of LTP, it is suggested that acute exposure to these compounds mainly acted in a postsynaptic rather than a presynaptic mechanism. On the other hand, acute exposure to the target compounds may pose relatively weak neural inhibitory effects. However, the chronic exposure to PFOS and its bioaccumulative alternatives, as well as the long carbon chain PFCs possibly pose stronger effects on the nervous system considering the bioaccumulation potency. Different from the present study, Liao et al.23 reported that 400 μM of PFOS could affect synaptic transmission in brain slices in rats. Besides the difference in the administration dose, the in vitro electrophysiological status also differs from the in vivo status, while the in vitro hippocampus slice is a valuable tool to elucidate the effects of pollutants on ion channel functions in central nervous system neurons.
The mechanisms underlying the impairment in LTP caused by PFOS and its alternatives might be related to several aspects. Firstly, the high concentrations of Ca2+ are necessary to induce LTP, with a number of Ca2+ sources available. The calcium imbalance induced by PFOS may cause the LTP deficit.48,49 Secondly, PFOS affected the Ca2+/calmodulin-dependent protein kinase II (CaMKII) and protein kinase C (PKC), which play dominant roles in the induction and maintenance of LTP.27,48 Thirdly, N-methyl-d-aspartic acid (NMDA) receptors were impaired by PFOS, while the activation of NMDA receptors and the consequent calcium flooding into postsynaptic cell is necessary for LTP induction.28,29 Moreover, the AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid)-type glutamate receptor might also be involved in the mechanism of the effects of PFOS and its alternatives, which is an important regulator of both LTP maintenance and the rise of the intracellular Ca2+ level.50 However, no information is available about the effects of PFOS on AMPA receptor regulation. Lastly, PFOS might act indirectly on learning and memory through disruption of thyroid function. LTP is known to be depressed under hypothyroid conditions in both animals and humans,51 while PFOS exposure significantly reduced the serum levels of free thyroxine in rats.52
Experimental
Animals and chemicals
All experiments were performed according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the School of Environmental Science and Technology, Dalian University of Technology (Dalian, China).
Adult male SD rats of clean grade weighing 200–240 g were provided by the Experimental Animal Center, Shanxi Medical University, China. Animals were acclimated in the lab for at least 7 days before experiments, with free accession to water and food. All experiments were performed at room temperature (25 ± 2 °C), with a 12 : 12 light/dark cycle.
PFOS, PFHXs, and PFBS were purchased from Sigma (USA) and Cl-PFAES was obtained from Shanghai Synica Co. (China), with a purity of higher than 98% (Table 1). The target chemicals were dissolved in 2% dimethyl sulfoxide (DMSO) and then diluted to 10 and 100 μM with physiological saline. Physiological saline with DMSO was administered in the same proportion to both treated and control groups. It was found that PFOS can accumulate up to 2–20 μM in some animal tissues.53 The doses (10, 100 μM) were administrated according to previous literature,11,23 representing the actual environmentally relevant and potential accumulated concentrations.
Table 1. PFOS and its alternatives.
| Product name | Chemical | CAS number | Chemical formula | Structure |
| PFOS | Potassium perfluorooctane sulfonate | 2795-39-3 | C8F17SO3K |
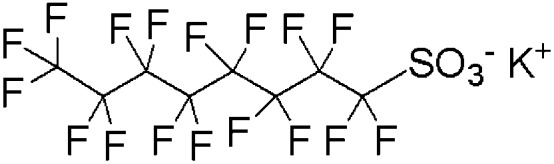
|
| PFHxS | Potassium perfluorohexane sulfonate | 3871-99-6 | C6F13SO3K |
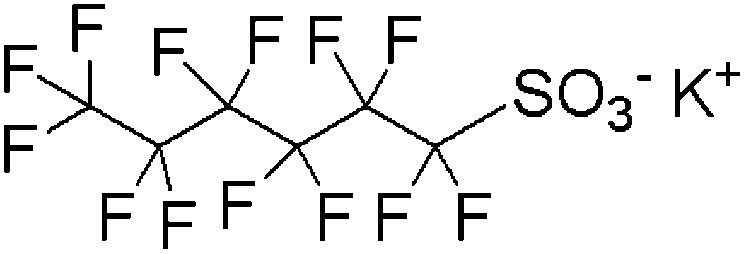
|
| PFBS | Potassium perfluorobutane sulfonate | 29420-49-3 | C4F9SO3K |
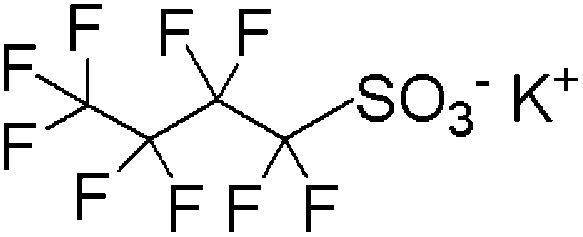
|
| Cl-PFAES | 2-[(6-Chloro-1,1,2,2,3,3,4,4,5,5,6,6-dodecafluorohexyl)oxy]-1,1,2,2,-tetrafluoroethanesulfonic acid potassium salt | 73606-19-6 | C8ClF16SO4K |
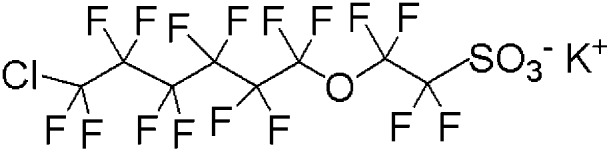
|
Animal treatment and electrophysiological recordings in vivo
Six animals were used for recording LTP in each group. The rats were deeply anesthetized with urethane (15 g per kg bw, Sigma) via intraperitoneal (i.p.) injection placed in a stereotaxic head holder (DMA-1511, Narishige, Japan) for surgery and recording. Skin and fascia were removed to expose the skull, and bregma and posterior fontanelle were kept at the same height. Small holes were drilled in the ipsilateral skull for the insertion of cannula, stimulating and recording electrode. A stainless steel cylindrical cannula (0.7 mm outer diameter) was inserted into the lateral ventricle (0.8 mm posterior to bregma, 1.3 mm lateral to midline, and 4.1 mm below skull) and fixed using acrylic dental cement for intracerebroventricular (i.c.v.) injection of chemicals. A concentric bipolar stimulating electrode (FHC, USA) was positioned at the Schaffer Collateral (4.2 mm posterior to bregma, 3.8 mm lateral to the midline) for LTP induction, and a monopolar recording electrode (FHC, USA) was placed at the CA1 region (3.8 mm posterior to bregma, 2.9 mm lateral to the midline) for field excitatory postsynaptic potential (fEPSP) recording.
The electrodes were slowly lowered with single test stimuli (0.033 Hz, interval of 30 s) until a stable and maximal fEPSP was monitored. The stimulus current was adjusted to yield about 50% of the maximum amplitude of fEPSP, and then began to record the baseline for 30 min. Targeted compound solution of 5 μL was slowly administered to the rats via i.c.v. injection in 5 min by using a micro-syringe. Thirty minutes of contact with the target compounds in the brain tissues were maintained after i.c.v. injection. Then the baseline was recorded for another 30 min, followed by I/O and PPF test. LTP was induced by a high-frequency stimulus (HFS) protocol composed of 3 trains of 20 pulses at 200 Hz at an interval of 30 s. After HFS, the amplitude of fEPSP was recorded for at least 60 min.
The input/output (I/O) curves reflect the relationship between the amplitude of fEPSP and the stimulus intensity, which were employed to evaluate the synaptic potency. I/O curves were generated by systematic variation of the stimulus current by steps of 0.1 mA (0.1–1.0 mA). Three responses were averaged at each current level. Paired-pulse facilitation (PPF), a form of short-lasting plasticity, was examined before HFS. The current was adjusted to yield about 50% of maximum amplitude of fEPSP, and pairs of stimuli were delivered with inter-stimulus intervals (ISI) of 10, 20, 40, 60, 80, 100, 120, 140, 160, 180, 200, 250, 300, 350 and 400 ms.47 Three responses were averaged at each ISI. PPF values were standardized at each ISI by fEPSP 2/fEPSP 1, comparing the peak facilitation with the control group.
Data analysis
The signals were recorded by A-M Systems (2100, USA), transferred through the amplifier (CED 1401, UK), and filtered by Spike 6 software (CED, UK). The amplitude of fEPSP was calculated by averaging the distance from the negative peak to the preceding and following positive peak. The fEPSP amplitude was standardized to pre-injection baseline values. The statistical analysis of the data was conducted by using Sigmaplot 10.0 and SPSS 16.0 software (USA). The comparisons between the groups were analyzed by one-way ANOVA, where probabilities less than 0.05 were considered as a significant difference.
Conclusions
In summary, the present study provides some electrophysiological evidence and the potential mechanism of the neurotoxicity of PFOS and its alternatives. Exposure to PFOS and its alternatives repressed LTP, and PFHxS and Cl-PFAES even exhibited a comparable potency to PFOS. The higher potency of PFHxS and PFOS than PFBS to inhibit LTP points to the possibly higher neurotoxicity potential of the long carbon chain perfluoroalkyl compounds. The absence of disruption of normal synaptic transmission suggested that acute exposure to the target compounds mainly acted in a postsynaptic rather than a presynaptic mechanism. Besides affecting LTP, Cl-PFAES also affected the baseline fEPSP, indicating a different action mode with the perfluoroalkyl acids. It should be noted that the present study is limited in the performance of acute exposure, and stronger effects on synaptic plasticity may occur when chronically exposed to PFOS, its bioaccumulative alternatives, as well as the long carbon chain perfluoroalkyl compounds. These findings present the fact that PFOS alternatives could impair synaptic plasticity, explore primarily the neurotoxic mechanism of PFOS alternatives by the neuroelectrophysiological method, and address the necessity of further toxicological evaluation of PFOS alternatives for improving their safety and health risk assessment.
The paper is to commemorate late Prof. Dr Yihe Jin (1959–2013), who has devoted his whole life to scientific research, and contributed greatly to the present research.
Acknowledgments
The study is financially supported by the National Natural Science Foundation of China (21177020, 81430078).
References
- D. Brooke, A. Footitt, T. Nwaogu and G. Britain, Environmental risk evaluation report: Perfluorooctane sulphonate (PFOS), 2004. [Google Scholar]
- Thompson J., Eaglesham G., Reungoat J., Poussade Y., Bartkow M., Lawrence M., Mueller J. F. Chemosphere. 2011;82:9–17. doi: 10.1016/j.chemosphere.2010.10.040. [DOI] [PubMed] [Google Scholar]
- Wang T., Wang P., Meng J., Liu S., Lu Y., Khim J. S., Giesy J. P. Chemosphere. 2015;129:87–99. doi: 10.1016/j.chemosphere.2014.09.021. [DOI] [PubMed] [Google Scholar]
- Renner R. Environ. Sci. Technol. 2006;40:12–13. doi: 10.1021/es062612a. [DOI] [PubMed] [Google Scholar]
- Tao L., Ma J., Kunisue T., Libelo E. L., Tanabe S., Kannan K. Environ. Sci. Technol. 2008;42:8597–8602. doi: 10.1021/es801875v. [DOI] [PubMed] [Google Scholar]
- Glynn A., Berger U., Bignert A., Ullah S., Aune M., Lignell S., Darnerud P. O. Environ. Sci. Technol. 2012;46:9071–9079. doi: 10.1021/es301168c. [DOI] [PubMed] [Google Scholar]
- Lieder P. H., Chang S. C., York R. G., Butenhoff J. L. Toxicology. 2009;255:45–52. doi: 10.1016/j.tox.2008.10.002. [DOI] [PubMed] [Google Scholar]
- Viberg H., Lee I., Eriksson P. Toxicology. 2013;304:185–191. doi: 10.1016/j.tox.2012.12.013. [DOI] [PubMed] [Google Scholar]
- Lee I., Viberg H. Neurotoxicology. 2013;37:190–196. doi: 10.1016/j.neuro.2013.05.007. [DOI] [PubMed] [Google Scholar]
- Johansson N., Eriksson P., Viberg H. Toxicol. Sci. 2009;108:412–418. doi: 10.1093/toxsci/kfp029. [DOI] [PubMed] [Google Scholar]
- Johansson N., Fredriksson A., Eriksson P. Neurotoxicology. 2008;29:160–169. doi: 10.1016/j.neuro.2007.10.008. [DOI] [PubMed] [Google Scholar]
- Wang S., Huang J., Yang Y., Hui Y., Ge Y., Larssen T., Yu G., Deng S., Wang B., Harman C. Environ. Sci. Technol. 2013;47:10163–10170. doi: 10.1021/es401525n. [DOI] [PubMed] [Google Scholar]
- Ruan T., Lin Y., Wang T., Liu R., Jiang G. Environ. Sci. Technol. 2015;49:6519–6527. doi: 10.1021/acs.est.5b01010. [DOI] [PubMed] [Google Scholar]
- Shi Y., Vestergren R., Zhou Z., Song X., Xu L., Liang Y., Cai Y. Environ. Sci. Technol. 2015;49:14156–14165. doi: 10.1021/acs.est.5b04299. [DOI] [PubMed] [Google Scholar]
- Brown R. C., Lockwood A. H., Sonawane B. R. Environ. Health Perspect. 2005;113:1250–1256. doi: 10.1289/ehp.7567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Liu W., Zhang Q., Zhao H., Quan X. Food Chem. Toxicol. 2015;76:70–76. doi: 10.1016/j.fct.2014.12.008. [DOI] [PubMed] [Google Scholar]
- Chang S. C., Ehresman D. J., Bjork J. A., Wallace K. B., Parker G. A., Stump D. G., Butenhoff J. L. Reprod. Toxicol. 2009;27:387–399. doi: 10.1016/j.reprotox.2009.01.005. [DOI] [PubMed] [Google Scholar]
- Bogdanska J., Borg D., Sundström M., Bergström U., Halldin K., Abedi-Valugerdi M., Bergman Å., Nelson B., DePierre J., Nobel S. Toxicology. 2011;284:54–62. doi: 10.1016/j.tox.2011.03.014. [DOI] [PubMed] [Google Scholar]
- Lau C., Anitole K., Hodes C., Lai D., Pfahles-Hutchens A., Seed J. Toxicol. Sci. 2007;99:366–394. doi: 10.1093/toxsci/kfm128. [DOI] [PubMed] [Google Scholar]
- Fuentes S., Vicens P., Colomina M. T., Domingo J. L. Toxicology. 2007;242:123–129. doi: 10.1016/j.tox.2007.09.012. [DOI] [PubMed] [Google Scholar]
- Mariussen E. Arch. Toxicol. 2012;86:1349–1367. doi: 10.1007/s00204-012-0822-6. [DOI] [PubMed] [Google Scholar]
- Slotkin T. A., MacKillop E. A., Melnick R. L., Thayer K. A., Seidler F. J. Environ. Health Perspect. 2008;116:716–722. doi: 10.1289/ehp.11253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao C., Li X., Wu B., Duan S., Jiang G. Environ. Sci. Technol. 2008;42:5335–5341. doi: 10.1021/es800018k. [DOI] [PubMed] [Google Scholar]
- Wang F., Liu W., Jin Y., Dai J., Yu W., Liu X., Liu L. Environ. Sci. Technol. 2010;44:1847–1853. doi: 10.1021/es902799f. [DOI] [PubMed] [Google Scholar]
- Liu X., Jin Y., Liu W., Wang F., Hao S. Toxicol. In Vitro. 2011;25:1294–1301. doi: 10.1016/j.tiv.2011.04.016. [DOI] [PubMed] [Google Scholar]
- Liu X., Liu W., Jin Y., Yu W., Wang F., Liu L. Arch. Toxicol. 2010;84:71–79. doi: 10.1007/s00204-009-0467-2. [DOI] [PubMed] [Google Scholar]
- Lee H., Lee Y. J., Yang J. Neurotoxicology. 2012;33:314–320. doi: 10.1016/j.neuro.2012.01.017. [DOI] [PubMed] [Google Scholar]
- Liao C. Y., Cui L., Zhou Q. F., Duan S. M., Jiang G. B. Environ. Toxicol. Pharmacol. 2009;27:338–344. doi: 10.1016/j.etap.2008.11.013. [DOI] [PubMed] [Google Scholar]
- Bliss T. V., Collingridge G. L. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Hussain R. J., Gyori J., DeCaprio A. P., Carpenter D. O. Environ. Health Perspect. 2000;108:827. doi: 10.1289/ehp.00108827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing T., Chen L., Tao Y., Wang M., Chen J., Ruan D. Y. Toxicol. Sci. 2009;110:401–410. doi: 10.1093/toxsci/kfp114. [DOI] [PubMed] [Google Scholar]
- Gilbert M. E., Lasley S. M. Neurotoxicol. Teratol. 2007;29:385–393. doi: 10.1016/j.ntt.2007.01.006. [DOI] [PubMed] [Google Scholar]
- Chen J., Wang M., Ruan D., She J. Neuroscience. 2002;112:879–887. doi: 10.1016/s0306-4522(02)00138-0. [DOI] [PubMed] [Google Scholar]
- Cook A. M., Mieure K. D., Owen R. D., Pesaturo A. B., Hatton J. Pharmacotherapy. 2009;29:832–845. doi: 10.1592/phco.29.7.832. [DOI] [PubMed] [Google Scholar]
- Wang J., Lin F., Cai F., Yan W., Zhou Q., Xie L. Chemosphere. 2013;93:223–229. doi: 10.1016/j.chemosphere.2013.04.069. [DOI] [PubMed] [Google Scholar]
- Song J., Liu Y., Zhang H. F., Zhang Q. L., Niu Q. Biomed. Environ. Sci. 2014;27:77–84. doi: 10.3967/bes2014.006. [DOI] [PubMed] [Google Scholar]
- Olsen G. W., Burris J. M., Ehresman D. J., Froehlich J. W., Seacat A. M., Butenhoff J. L., Zobel L. R. Environ. Health Perspect. 2007;115:1298–1305. doi: 10.1289/ehp.10009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conder J. M., Hoke R. A., Wolf W. D., Russell M. H., Buck R. C. Environ. Sci. Technol. 2008;42:995–1003. doi: 10.1021/es070895g. [DOI] [PubMed] [Google Scholar]
- Kudo N., Suzuki E., Katakura M., Ohmori K., Noshiro R., Kawashima Y. Chem.-Biol. Interact. 2001;134:203–216. doi: 10.1016/s0009-2797(01)00155-7. [DOI] [PubMed] [Google Scholar]
- Jones P. D., Hu W., De Coen W., Newsted J. L., Giesy J. P. Environ. Toxicol. Chem. 2003;22:2639–2649. doi: 10.1897/02-553. [DOI] [PubMed] [Google Scholar]
- Goecke Flora C. M., Reo N. V. Chem. Res. Toxicol. 1996;9:689–695. doi: 10.1021/tx950217k. [DOI] [PubMed] [Google Scholar]
- Route W. T., Key R. L., Russell R. E., Lindstrom A. B., Strynar M. J. Environ. Sci. Technol. 2014;48:6653–6660. doi: 10.1021/es501055d. [DOI] [PubMed] [Google Scholar]
- Zhang M. M., Xiao C., Yu K., Ruan D. Food Chem. Toxicol. 2003;41:1617–1623. doi: 10.1016/s0278-6915(03)00195-9. [DOI] [PubMed] [Google Scholar]
- Bittigau P., Sifringer M., Genz K., Reith E., Pospischil D., Govindarajalu S., Dzietko M., Pesditschek S., Mai I., Dikranian K. Proc. Natl. Acad. Sci. U. S. A. 2002;99:15089–15094. doi: 10.1073/pnas.222550499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawada H., Sumino E., Oue M., Baba M., Kira T., Shigeta S., Mitani M., Nakajima H., Nishida M., Moriya Y. J. Fluorine Chem. 1995;74:21–26. [Google Scholar]
- Bolshakov V. Y., Siegelbaum S. A. Science. 1995;269:1730–1734. doi: 10.1126/science.7569903. [DOI] [PubMed] [Google Scholar]
- Zucker R. S. Annu. Rev. Neurosci. 1989;12:13–31. doi: 10.1146/annurev.ne.12.030189.000305. [DOI] [PubMed] [Google Scholar]
- Malenka R. C. Cell. 1994;78:535–538. doi: 10.1016/0092-8674(94)90517-7. [DOI] [PubMed] [Google Scholar]
- Thibault O., Gant J. C., Landfield P. W. Aging Cell. 2007;6:307–317. doi: 10.1111/j.1474-9726.2007.00295.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derkach V. A., Oh M. C., Guire E. S., Soderling T. R. Nat. Rev. Neurosci. 2007;8:101–113. doi: 10.1038/nrn2055. [DOI] [PubMed] [Google Scholar]
- Niemi W. D., Slivinski K., Audi J., Rej R., Carpenter D. O. Neurosci. Lett. 1996;210:127–129. doi: 10.1016/0304-3940(96)12676-8. [DOI] [PubMed] [Google Scholar]
- Chang S. C., Thibodeaux J. R., Eastvold M. L., Ehresman D. J., Bjork J. A., Froehlich J. W., Lau C., Singh R. J., Wallace K. B., Butenhoff J. L. Toxicology. 2008;243:330–339. doi: 10.1016/j.tox.2007.10.014. [DOI] [PubMed] [Google Scholar]
- Giesy J. P., Kannan K. Environ. Sci. Technol. 2001;35:1339–1342. doi: 10.1021/es001834k. [DOI] [PubMed] [Google Scholar]