

 Polymeric nanomaterials, hybridized with lipid components, e.g. phosphocholine or fatty acids, are currently being explored for efficient nano-platforms for hydrophobic drugs.
Polymeric nanomaterials, hybridized with lipid components, e.g. phosphocholine or fatty acids, are currently being explored for efficient nano-platforms for hydrophobic drugs.
Abstract
Polymeric nanomaterials, hybridized with lipid components, e.g. phosphocholine or fatty acids, are currently being explored for efficient nano-platforms for hydrophobic drugs. However, their toxicology and toxicokinetics need to be established before enabling their clinical potential. The aim of this study was to investigate the toxicological profile of thiomer enveloped hybrid nanoliposomes (ENLs) and bare nanoliposomes (NLs), loaded with docetaxel (DTX) hydrophobic drug, biocompatible nano-carriers for therapeutic cargo. The in vitro toxicity of hybrid ENLs and NLs was evaluated towards the HCT-116 colon cancer cell line. Biocompatibility was explored against macrophages and acute oral toxicity was examined in mice for 14 days. The anticancer IC50 for ENLs was 0.148 μg ml–1 compared with 2.38 μg ml–1 for pure docetaxel (DTX). The human macrophage viability remained above 65% and demonstrated a high level of biocompatibility and safety of ENLs. In vivo acute oral toxicity showed slight changes in serum biochemistry and haematology but no significant toxicities were observed referring to the safety of DTX loaded hybrid ENLs. On histological examination, no lesions were determined on the liver, heart and kidney. These studies showed that hybrid ENLs can serve as a safe and biocompatible platform for oral delivery of hydrophobic drugs.
1. Introduction
During the last decade, unique physico-chemical properties and biomedical advances in the fabrication of nano-carriers (NCs) have provided remarkable applications in targeted drug delivery, diagnostics and patient compliance.1–3 Despite the numerous breakthroughs in nanostructured drug delivery systems, their clinical applications are still limited by the balance between biocompatibility and toxicity.4,5 Toxicity arises because of the distinctive biodistribution profile, and cellular and molecular interactions of NCs at unwanted sites.6 The cellular toxicity of nano-carriers depends on the type of biomaterial, size, shape, composition, surface charge, and surface chemistry.7 To advance the healthcare system with nano-scale drug delivery platforms by optimizing synthetic procedures, it is highly desirable to functionalize the surface to enhance biocompatibility, and coupling with complementary targeting molecules for specific tissue distribution.8 Immune cells in the blood stream and tissues have a tendency to engulf and eliminate the nanoparticles thus affecting their distribution.9
Nanoliposomes (NLs) are among the most promising NC systems that can carry different moieties simultaneously, including drug and targeting moieties, and deliver them to the target site overcoming the barriers associated with oral administration, intestinal absorption and the erratic pharmacokinetic profile.10,11 These NLs have been much explored for cancer therapeutics using hydrophobic drugs.12
Thiomers are thiolated polymers emerging as a novel class of biomaterial as well as pharmaceutical excipients, which offer improved intracellular uptake, mucoadhesion, and P-gp inhibition to avoid efflux and thus result in improved oral bioavailability and enhanced pharmacokinetics.13 Fabricating NLs and thiomer enveloped NLs can provide a better therapeutic profile of hydrophobic anticancer drugs; however, the subsequent systemic toxicity and fate of NLs still need to be investigated. Furthermore, NLs are occasionally found to be responsible for initiating hypersensitivity reactions owing to the use of surfactants in their synthesis.14 Following oral administration, the interaction of polymeric NCs with mucus during penetration and with enterocytes during circulation may affect the normal physiology and could turn into toxic manifestations. To date, most of the toxicological evaluations of NLs and thiomer based NCs have been carried out by in vitro studies using cell culture techniques resulting in in-consistent results of in vivo studies, where the protein corona and organ distribution substantially influence the pharmacology and pharmacokinetics of NCs.7,15
In this study, we have explored the tissue distribution and tissue specific toxicity of thiomer enveloped nanoliposomes (ENLs), as potential NCs of docetaxel after oral administration. ENLs and NLs were evaluated for their in vitro cellular interaction for anti-cancer drug delivery to in vivo toxicology, particle distribution and the effect on different organs in animal models in the context of cytotoxicity, clinical biochemistry, haematology, histopathological examination of major storage organs and genotoxicity.
2. Experimental methods
2.1. Materials
Chitosan (LMW, degree of deacetylation 75–85%), thioglycolic acid (TGA) 99%, 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDAC), hydroxylamine, hydrogen peroxide, sodium hydroxide, 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), egg yolk choline, cholesterol, disodium dihydrogen phosphate, sodium dihydrogen phosphate, glucose, sodium chloride, sodium borohydride, potassium chloride, magnesium chloride, trehalose, foetal bovine serum (FBS), penicillin, streptomycin, sulforhodamine B and dialysis membrane (cut-off value 12 kDa) were purchased from Sigma-Aldrich (Germany). Docetaxel (DTX) was received as a gift from NovaMed Pharmaceuticals Pvt. Ltd. All the solvents used were of analytical and HPLC grade.
2.2. Methods
2.2.1. Synthesis of nanoliposomes (NLs) and enveloped hybrid nanoliposomes (ENLs)
The synthesis of NLs and ENLs was achieved in a three step process.16 Briefly, in the first step, thiolated chitosan (Chito-TGA) was synthesized through EDAC coupling,17 followed by the grafting of folic acid through EDAC coupling to chito-TGA18 resulting in folate grafted thiolated chitosan (FA-Chito-TGA). In the second step, docetaxel (DTX) loaded NLs, composed of choline, DPPC, oleic Acid and cholesterol (4 : 2 : 2 : 2, w/w), were prepared by the thin film rehydration technique. The prepared NLs were coated with FA-Chito-TGA to synthesize ENLs.
2.2.2. Physicochemical characterization of NLs and hybrid ENLs
The synthesized NLs and hybrid ENLs were characterized for their encapsulation efficiency, particle size, zeta potential and surface morphology. Drug loading in NLs and ENLs was determined through HPLC. The hydrodynamic size, poly-dispersity (PDI) and surface zeta potential were measured through a zetasizer nano (Malvern, Nano ZSP, UK). The surface morphology of the NLs and hybrid ENLs was examined through a scanning electron microscope (SEM) equipped with a transmission electron detector (FEI Nova NanoSEM 450, USA).19
2.2.3. In vitro cytotoxicity
In vitro cytotoxicity of NLs and ENLs was screened through the sulforhodamine B (SRB) assay using a colon cancer (HCT-116) cell line.20 Briefly, HCT-116 cells were seeded in a 96-well optiplate at a density of 3000 cells per well, suspended in 10% fetal bovine serum (FBS) and incubated for 24 h in a humidified incubator with 5% CO2. Thereafter, cells were fixed with 10% trichloroacetic acid representing the cell population at the time of treatment. The cells were treated with a vehicle control (0.1% DMSO), DTX suspension and different concentrations of NLs and hybrid ENLs equivalent to 10 μg, 5 μg, 2.5 μg, 1.25 μg, 0.625 μg, 0.312 μg and 0.156 μg of DTX for 48 h. Blank NLs and hybrid ENLs were used as the control. After incubation, the cells were again fixed with 10% trichloroacetic acid followed by staining with sulforhodamine B (0.4%, w/v) in 1% acetic acid solution. Excess SRB was removed by using 1% acetic acid solution and dye containing cells were lysed with 10 mmol Trizma base. Absorbance was measured at 490 nm using a multi-plate reader (PerkinElmer, USA). Untreated cells with 100% viability were taken as the control and the cells without addition of SRB were used as the blank to calibrate the instrument. IC50 values for each formulation were calculated using Graphpad Prism 6.02 software. The results were obtained in triplicate and expressed as mean ± SD.21
2.2.4. In vitro toxicity against human macrophages
Macrophages, from fresh human blood (with volunteer consent under the approved protocol from departmental ethical committee under the protocol no. DFBS/216-266/BEC-FBS-QAU-21), were separated using the ficoll–percoll purification technique.22,23 Briefly, macrophage isolation was achieved using the ficoll–gastrografin gradient (density 1.070 g ml–1). Ficoll solution was prepared by dissolving 5.6 g ficoll in 9.5 ml deionized water and 5 ml gastrografin to achieve a density of 1.070 g ml–1. The fresh human blood (5 ml) was diluted three times with Hank's buffer salt solution (HBSS) and carefully layered over ficoll–gastrografin solution and centrifuged for 5 min at 400 G to separate the macrophage layer. Afterwards, purification with the percoll gradient was carried out. Percoll density (1.064 g ml–1) was achieved using deionized water and 10× HBSS. The separated cells were suspended in RPMI medium (10% FBS, 100 U ml–1 penicillin, 0.1 mg ml–1 streptomycin and 25 mM HEPES) and incubated in 5% CO2. Viable cells were seeded in a 96-well plate (1 × 105 cells per well) and treated with different concentrations of NLs, hybrid ENLs and vehicle control. Empty NLs and hybrid ENLs served as the negative control and Triton X (1%) served as the positive control to check the cytotoxicity of treatment. After 24 h incubation, the cell viability was assessed with trypan blue. The number of viable cells was counted and IC50 was calculated using graph pad prism software (version 6.02).
2.2.5. In vitro haemolysis assay
Fresh human blood was used for in vitro haemolysis assay as reported.24,25 The fresh blood (with volunteer consent) was withdrawn and washed thrice with sterile Dulbecco phosphate buffer saline (PBS). The RBCs were pelleted out after each washing at 150 G for 5 min and the supernatant was discarded. The final pellet was diluted 9 times (v/v) with sterile PBS. Afterwards, 100 μL of RBC suspension per well was seeded in a 96-well plate. RBCs were treated with different concentrations of NLs and ENLs. Empty NLs and hybrid ENLs served as the negative control and Triton X (1%) served as the positive control to check haemolysis induced by the formulations. After 24 h incubation, the absorbance was measured at 570 nm using a multiplate reader (PerkinElmer, USA). Haemolysis percent induced was calculated using the formula: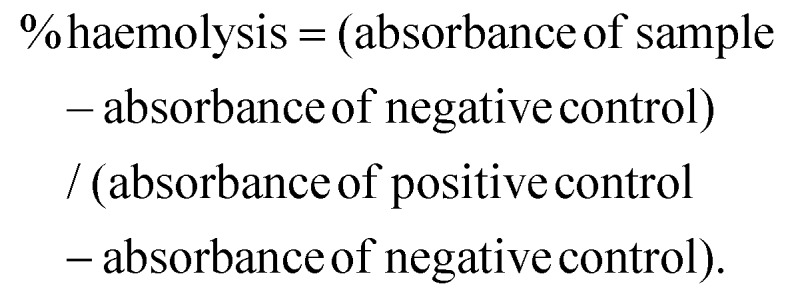
2.2.6. In vitro micronucleus assay
Fresh peripheral blood (with volunteer consent) was collected in heparinized sterile vials (BD Vacutainer). Triplicate blood cultures were set up by diluting 0.6 ml blood in 9.4 ml RPMI media containing 10% FBS, penicillin, streptomycin and HEPES buffer solution. Phytohemagglutanin (PHA) solution (4%) was added to the culture and incubated for 48 h at 37 °C with gentle shaking to stimulate lymphocyte growth. After incubation for 48 h, cultures were treated with NLs and ENLs (500 μg ml–1) followed by further 24 h incubation. Binucleated lymphocytes were arrested by replacing the supernatant with fresh RPMI media having cytochalasin-B (cyt-B) at a final concentration of 6 μg ml–1. After 4 h of incubation, the culture was centrifuged at 300 G for 10 min and the supernatant was carefully replaced with pre-warmed (37 °C) mild hypotonic solution (0.075 KCl) and incubated for 4 min. Thereafter, the lymphocytes were harvested using ice-cold Carony's fixative (methanol : glacial acetic acid; 3 : 1). The culture was centrifuged at 300 G and the pellet was re-suspended in Carnoy's medium and gently mixed. The process was repeated twice and the suspension was refrigerated for 3 h prior to slide preparation. The slides were prepared by placing a single drop of suspension and air dried for 1 h, followed by staining with Giemsa (4% in PBS) and acridine orange (0.125 mg mL–1 PBS) for 10 min. After that, the slides were washed with PBS and air-dried.26 The prepared slides were stored for in vitro micronucleus assay by observing 1000 bi-nucleated cells on each slide blindly.
2.2.7. Acute oral toxicity
Acute oral toxicity of NLs and hybrid ENLs was evaluated in mice for 14 days following OECD 425 guidelines. The in vivo studies were proceeded as per the approved guidelines of the Bio-Ethical Committee of Quaid-i-Azam University Islamabad, Pakistan (Protocol No. DFBS/216-266/BEC-FBS-QAU-21). Healthy, female Swiss albino mice, weighing 30 ± 5 g and 8–10 weeks aged, were obtained from an animal house. Mice were divided into 5 groups, each having 6 mice and were kept under standard conditions of food and water in a controlled environment. The group 1 was administered with a DTX suspension, group 2 with NLs, group 3 with hybrid ENLs, group 4 with empty hybrid ENLs and group 5 with normal saline, which served as a control. The dose (20 mg kg–1) was administered orally through gavage. The mice were kept under observation for 24 h for any change in weight and visual observations for mortality, behaviour pattern (fur and skin, consistency of faeces, urination colour, salivation, eyes, respiration, sleep pattern, mucous membrane, convulsions, and coma), physical appearance changes and signs of illness were conducted daily throughout the week. The same procedure was repeated to complete three cycles of oral administration.27 After 14 days, the mice were sacrificed for serum biochemistry and tissue histology studies.28
2.2.7.1. Serum biochemistry analysis
After 14 days, serum biochemistry analysis was carried out to assess the toxicity induced by NLs and hybrid ENLs. Blood from each mouse was drawn through cardiac puncture into a sterile vial. The blood was centrifuged at 1200 G for 10 min to separate plasma. The supernatant was carefully removed and stored at –20 °C. Liver function tests (LFTs) including ALP, SGPT, SGOT and bilirubin, renal functions tests (RFTs) including urea and creatinine, serum electrolytes (Na, Mg, Ca and P), glucose, and total protein were carried out using the serum.
2.2.7.2. Haematology analysis
Haematology analysis was performed on the other part of blood collected in a heparinized vial through cardiac puncture from each mouse. Haematology parameters i.e. the number of red blood cells (RBCs), red cell distribution width (RDW), and haemoglobin distribution width (HDW), mean corpuscular volume (MCV), mean corpuscular haemoglobin (MCH), mean corpuscular haemoglobin concentration (MCHC), packed cell volume (PCV), haemoglobin concentration (Hb), hematocrit (HCT), platelet count (PLT), and mean platelet volume (MPV) were measured. In addition, the number and percentage of neutrophils, monocytes, lymphocytes, eosinophils, and basophils were also measured using a haematology autoanalyzer, Mindrey, BC 2800VET.29
2.2.7.3. Organ to body ratio
Change in organ weight was measured for toxicity evaluation of the test formulation after exposure for a specific time. The vital organs (heart, kidneys and liver) were removed from mice after being sacrificed, washed with normal saline and weighed individually. The weights of organs from treated groups were compared with the control group and the body mass index was calculated using the formula.27,30

2.2.7.4. Histopathology of vital organs
The washed vital organs (liver, kidney and heat) were macroscopically examined for any abnormalities or lesions against the control. Afterwards, the organs were stored in 10% formalin solution. The organs were fixed in paraffin blocks and sections (0.5 μm) were cut carefully using a rotary microtome and fixed on a glass slide followed by staining with hematoxylin and eosin periodic acid Schiff (PAS). The sections were microscopically examined for any toxic effects produced by the nano-cargo.31
2.2.7.5. Tissue distribution analysis
Tissue distribution of DTX loaded NLs and hybrid ENLs was analysed using tissue homogenates from different groups. Briefly, a weighed amount of chopped organ (liver, kidneys and heart) was mixed with 1 ml normal saline (0.9% w/v) and homogenized. To this, 1 ml mobile phase was added to extract the drug from tissues and the mixture was further sonicated for 15 min followed by centrifugation at 5000 G for 10 min. The supernatant was carefully separated and analysed using the HPLC method previously developed for DTX quantification in plasma samples.
2.2.8. Statistical analysis
All the results were generated using two-way analysis of variance (ANOVA) to compare the results of different treatments with NLs, hybrid ENLs and DTX. The results were further analyzed with Tukey's test and student's t-test with the level of significance p < 0.05. All the results are presented as mean ± standard deviation (S.D.).
3. Results and discussion
3.1. Synthesis and physicochemical characterization of NLs
NLs and ENLs were synthesized and loaded with DTX through thin film rehydration followed by coating of FA-Chito-TGA on NLs through electrostatic interaction between the anionic NLs and cationic polymer (FA-Chito-TGA). This was further confirmed by the change in the zeta potential from –24.40 ± 3.55 mV to +19.77 ± 5.18 mV, after coating. The hydrodynamnic size of NLs calculated was 227.39 ± 7.88 nm, which slightly increased to 306.50 ± 8.44 nm. The polydispersity of both formulations was observed below 0.2, indicating the fairly uniform size of the particles. This uniformity was also evident through SEM analysis (Fig. 1a & b). The encapsulation efficiency of the formulations was found to be 84% for hybrid ENLs.
Fig. 1. (a) SEM images of the synthesized ENLs and NLs.

Charge density and polarity play an important role in inducing cytotoxicity.32 Cationic particles induce cytotoxicity via membrane damage, whereas anionic particles cause intracellular damage after uptake.33 Generally, cationic particles are considered more toxic compared to the anionic particles of the same size and chemistry. Phagocytic cells preferentially interact with anionic particles and engulf them as foreign particles e.g. bacteria that have negative charge. This results in higher cytotoxicity of anionic particles compared to the cationic particles.34 Free amino groups of polymers play a vital role in the magnitude of toxicity produced by cationic nanoparticles.35 Primary amino groups of chitosan were neutralized due to covalently attached thioglycolic acid and folic acid, which reduced the toxicity of cationic ENLs.36
3.2. In vitro cytotoxicity
The in vitro cytotoxicity of NLs and ENLs against human colon cancer cells HCT-116 was compared with that due to pure DTX at different concentrations to scan for IC50. HCT-116 cells have over-expressed folate receptors,37 which could be targeted through folate decorated ENLs. SRB assay was performed using HCT-116 and the results are shown in Fig. 2. Pure DTX being hydrophobic in nature has difficulties in crossing the cell membrane and showed a relatively higher IC50 value of 2.38 μg ml–1. NLs and ENLs showed improved cell interaction and a lower IC50i.e. 0.532 and 0.148 μg ml–1, respectively. ENLs possessed the highest cytotoxicity owing to the surface grafting of folic acid, which facilitated particle attachment and subsequent internalization. The ENL control (without the drug) remained unreacted toward HCT-116 with no significant cytotoxicity at almost every concentration.
Fig. 2. In vitro toxicity studies of NLs and ENLs loaded with DTX against the HCT-116 cell line using SRB assay. The ENL control was used to check the toxic potential of formulations alone. IC50 values were calculated to be 0.532 and 0.148 μg ml–1, respectively, for NLs and ENLs.

3.3. In vitro haemolysis assay
In vivo haemolysis may result in jaundice, anemia or other pathological conditions.38 Thus, blood compatibility is of prime concern for nanoparticles because of their initial interaction with blood components and blood cells resulting in toxic haemolysis.39In vitro haemolysis assay was performed on human blood to study the haemolytic profile of NLs and ENLs. Fresh human RBCs were treated with different concentrations of DTX suspension, NLs, ENLs and ENL control (without the drug) to examine the concentration dependent response on percentage haemolysis. The results indicated that the pure drug was highly toxic even at the lowest concentration used (Fig. 3a). In contrast, the ENLs reduced the haemolytic effect of DTX at all concentrations indicating the improved biocompatibility with RBCs resulting in decreased haemolysis. The ENL control showed that formulations along with all the ingredients were biocompatible. IC50 for ENLs and ENL control was calculated to be 164.2 and 487.3 μg ml–1, respectively, that demonstrated a high therapeutic window with these NLs.
Fig. 3. In vitro biocompatibility studies of NLs and ENLs at different concentrations to determine toxicity against (a) red blood cells via the haemolysis assay and (b) macrophages via the MTT assay.

3.4. In vitro toxicity against human macrophages
Nanoparticles, having plasma proteins adsorbed on them, are readily engulfed and cleared by the immune system as they reach systemic circulation. However, this clearance is highly dependent on certain properties like the particle size, surface charge and hydrophilicity/hydrophobicity.40 These characteristics of nanoparticles influence the interaction with plasma proteins which, after being adsorbed on the surface, form bridges between particles and immune cells. Thus, particles with high surface potential and polarity have a long circulating life and the least interaction with macrophages.41 To assess the compatibility and potential toxicity of DTX loaded NLs and ENLs, an in vitro assay with human macrophages was performed with different concentrations of DTX and DTX loaded NLs and ENLs. The results (Fig. 3b) demonstrated that at higher concentrations, the NLs and ENL control were able to reduce the cytotoxicity of DTX towards macrophages. At higher concentrations (<150 μg), NLs and ENLs showed toxicity towards macrophages due to their engulfment and increased DTX internalization. ENLs showed the least cytotoxicity, owing to surface modification with folic acid labelled thiomers. The ENL control was used to evaluate that the carrier only induced cytotoxicity. The results showed that at the highest concentrations, the cell viability remained above 65% indicating the biocompatibility of all the materials used and ENLs themselves (Fig. 3b). The LD50 values of ENLs and ENL control were found to be 113.4 and 341.2 μg ml–1, respectively. The high IC50 of hybrid ENLs showed their higher biocompatibility and safety.
3.5. Tissue distribution
Tissue distribution and drug accumulation in organs are key factors in producing off-target effects. NPs have the ability to penetrate deep into tissues, which is highly dependent on the particle size and surface properties. As reported earlier, the mean plasma concentration–time profiles of NLs and ENLs with DTX in mice were quite similar to those previously reported,16 both indicating that NLs and ENLs produced higher plasma concentration than pure DTX in 2–3 h followed by oral administration. Moreover, they remained in plasma for >96 h after a single dose, which indicated the presence of NLs and ENLs in tissues and attached to plasma proteins with sustained release of DTX for up to 96 h. The similar distribution patterns for both formulations demonstrated that thiomer coating did not alter the tissue distribution behavior of DTX in mice significantly. Followed by administration of NLs and ENLs, the amount of DTX was the highest in the liver between 3–4 h and then it dropped quickly at 4–6 h due to the rapid metabolism of DTX by the liver microsome implicated cytochrome P450 enzyme system. DTX was quantified in vital organs using HPLC (Fig. 4a), which showed the least amount of drug in the liver and kidney with ENLs compared to NLs and DTX, which showed the maximum amount of drug.
Fig. 4. (a) Organ drug quantification using HPLC analysis showing the DTX concentration after 14 days, (b) organ to body weight index of control, NLs and ENLs treated mice after 14 days of oral treatment.

3.6. Acute oral toxicity
The in vivo toxic potential of NLs and ENLs was evaluated in female Swiss albino mice. Formulations were orally administered at a relatively higher concentration of 50 mg kg–1. The mice showed normal signs for the skin, fur, behavioural patterns and digestion during the first 24 h of administration that prevailed throughout the week. No mortality and significant change in body weight was observed throughout the course of the study. After 14 days, the blood was collected in sterilized vials depending upon the analysis to be performed and the mice were euthanized to collect different organs for further studies.
3.6.1. Organ to body index
After 14 days of treatment, the organ to body index was calculated for vital organs including the kidney, liver and heart. The organs were carefully removed from the euthanized mice and washed with normal saline.
The relative organ to body index of each organ was compared with the control (Fig. 4b). The decrease in liver weight was observed with all the treatments in which DTX showed a slightly increased effect compared to NLs and ENLs, whereas the ENL control showed a little change in the weight of the liver. The kidneys showed no significant weight change after treatments. The heart remained unaffected with all the treatments showing no toxic effects of formulations towards cardiac muscles.
3.6.2. Serum biochemistry and haematology
The effect of the DTX suspension, DTX loaded NLs, ENLs and ENL control on serum biochemistry and haematology was assessed. Liver function tests (LFTs) shown in Fig. 5a describe its functionality through the albumin level which was not influenced by treatment. Cellular integrity of the liver is depicted in transaminases (SGPT), which are produced within liver cells.42 The higher blood level of SGPT indicates cell damage or necrosis leading to leakage of this enzyme in blood. An increased level of SGPT was observed with DTX and DTX loaded ENLs compared to the control that remained within the acceptable limits. However, it was less affected by the ENL control. Cellular integrity and its link with the bile duct are represented by ALP, which is one of the characteristic findings of cholestatic liver disease. The ALP level was increased with all the treatments suggesting some obstruction in the bile duct. Hybrid ENLs decreased the ALP level compared to the control. The bilirubin level did not represent any significant changes. The liver is the prime source of all serum proteins and any change in total protein is an indicator of liver abnormality. The total protein content, however, remained unaffected with NLs and ENLs. The effect of the ENLs and ENL control on LFTs was significant (P < 0.005) compared to DTX, revealing the compatibility of ENLs.43,44
Fig. 5. Serum biochemistry analysis of mice plasma after acute oral treatment with DTX, NLs and ENLs compared with the control to monitor changes in (a) LFTs; (b) RFTs; (c) electrolytes and (d) glucose, cholesterol and total protein, induced after treatment due to the metabolism of formulations or drug. The results are presented as mean ± S.D. of triplicate.

The effect on the kidney was assessed through RFTs. The results in Fig. 5b showed no significant deviation from the reference values of RFTs. The creatinine level remained unaffected with all the treatments. However, BUN was increased with NLs compared to the control, which remained within the reference range. Serum electrolytes (Na, Ca, Mg and P) were assessed to investigate any other toxicity induced during the treatments. The results (Fig. 5c) revealed a slight increase in Na level with the maximum increase in the case of NLs compared to the control but the values were below the reference range (140–160 mEq L–1).
The levels of Ca, Mg and phosphate were slightly decreased compared to the control yet they were within the limits. The influence on serum glucose and cholesterol was also assessed. The results showed a decrease in cholesterol and glucose levels in all groups compared to the control (Fig. 5d).
The critical task in drug nanocarrier systems is to ensure their biocompatibility once they come into contact with blood/organs. Inflammatory response is most likely to occur, depending upon the level of their incompatibility.45 To check the biocompatibility of NLs and ENLs with blood and its components, complete blood count (CBC) was performed and the results are shown in Table 1.
Table 1. The effect of DTX, NLs, ENLs and ENL control on the CBC of mice. The results are presented as mean ± S.D. of triplicate.
| Blood parameter | Control | DTX | NLs | ENLs-control | ENLs |
| RBC (1012/L–1) | 9.02 ± 2.13 | 7.42 ± 2.75 | 8.35 ± 2.54 | 7.95 ± 2.79 | 8.96 ± 2.51 |
| MCV(fL) | 56.41 ± 5.21 | 53.57 ± 5.94 | 55.46 ± 6.76 | 54.59 ± 5.83 | 54.84 ± 4.76 |
| MCH (pg) | 15.53 ± 2.65 | 15.54 ± 2.76 | 15.44 ± 3.65 | 18.69 ± 4.33 | 15.94 ± 3.65 |
| PCV (%) | 48.02 ± 7.53 | 47.71 ± 3.76 | 47.88 ± 8.32 | 48.17 ± 6.72 | 47.93 ± 5.77 |
| Hb (g dL–1) | 14.15 ± 3.65 | 12.87 ± 2.65 | 14.63 ± 3.52 | 13.66 ± 3.65 | 13.94 ± 3.65 |
| WBC (109/L) | 15.56 ± 4.21 | 14.45 ± 3.65 | 14.49 ± 3.76 | 15.01 ± 2.54 | 14.85 ± 4.23 |
| Platelets 109/L | 753.33 ± 34.54 | 702.66 ± 65.87 | 733.66 ± 56.29 | 737.33 ± 65.82 | 741.66 ± 68.66 |
| RDW (%) | 16.07 ± 3.12 | 17.58 ± 3.54 | 16.69 ± 4.28 | 17.46 ± 3.65 | 17.34 ± 3.55 |
| MPV (fL) | 6.75 ± 1.67 | 7.45 ± 1.43 | 6.63 ± 1.69 | 7.49 ± 1.65 | 7.33 ± 2.43 |
The results revealed that the DTX suspension destroyed RBCs resulting in a decreased RBC count, which in turn led to a decreased Hb level. DTX loaded NLs and ENLs showed haemolysis but that was less compared to pure DTX. Moreover, the ENL-control showed negligible haemolysis. Neutropenia is one of the side effects of DTX that was mitigated by encapsulating DTX inside NLs and ENLs. The ENL control didn't show a significant effect on WBCs. The other parameters (MCV, MCH, PCV, PDW %) were also monitored. Previously, similar results were observed in the case of lipid emulsified DTX.46 There were slight changes in the values but no significant change was observed declaring the safety of DTX loaded ENLs.
3.6.3. Tissue histology
The functional biochemical analysis was coupled with tissue histological studies as they are helpful in the anatomical localization of toxicity induced by the treatment. The histological slides of the heart, liver and kidney were prepared through microtomy and stained slides were examined for structural changes and lesions in tissues. The liver cells in the control were observed to be round and polygonal with a clear nucleus as shown in Fig. 6b. Slight changes in cellular morphology with the appearance of fatty globules were observed in liver slides (Fig. 6, b1–3) treated with formulations.
Fig. 6. Microscopic examination of the tissue histology of vital organs (liver, kidney and heart) to examine any necrosis or histological changes compared to the control for these organs after treatment with the formulations; (a) heart tissue of control; (1a) treated with NLs, (2a) treated with ENLs and (3a) treated with ENL-control; (b) liver tissue of control, (1b) treated with NLs, (2b) treated with ENLs and (3b) treated with ENL-control; (c) kidney tissue of control, (1c) treated with NLs, (2c) treated with ENLs and (3c) treated with ENL-control.

No changes were observed in heart histology compared to the control as shown in Fig. 6, a1–3. The histological examination of kidneys (Fig. 6, c1–3) did not show any evident change in the cellular morphology supporting the safety of ENLs due to renal clearance.
3.7. Genotoxicity studies
Genotoxicity can be assessed with the end point of chromosomal anomaly using rodent micronucleus (MN) assay. Both the in vitro and in vivo micro-nucleus assays are used for this purpose, whereas the in vivo micro-nucleus assay is most widely used to check the genotoxic potential of chemicals in the animal body. In the current study, genotoxicity was expressed in percent of micronuclei per 1000 binucleated cells and is presented in Table 2. For the cytokinesis blocking MN assay, 1000 bi-nucleated cells per slide with a well-preserved cytoplasm were examined against each treatment. The results in Table 2 showed that the means of MN produced by ENLs compared to the vehicle control and positive control were very low and the results were statistically significant (p < 0.05) in revealing that the ENLs were non-genotoxic. Cells were stained with acridine orange (Fig. 7(a & b)) showing no effect of ENLs compared to the positive control (Fig. 7c).
Table 2. Results of the in vitro MN assay. The number of micronuclei counted in 1000 binucleated cells on slides is shown.
| Formulation | Cells with 1 MNs | Cells with 2 MNs | Cells with 3 MNs |
| Vehicle control | 4 ± 2 | 0 | 0 |
| ENLs | 143 ± 8 | 38 ± 5 | 14 ± 1 |
| ENL-control | 12 ± 3 | 3 ± 1 | 0 |
| Positive control | 82 ± 5 | 27 ± 3 | 9 ± 1 |
Fig. 7. Fluorescence images of acridine orange stained slides of micronucleus assay.

4. Conclusion
The toxicity profile of nanomaterials is one of the crucial concerns to address and thus unlock their clinical potential. The present work revealed that both NLs and ENLs possessed limited cellular toxicity at lower doses compared to higher doses. In the rodent animal model, vital organs including the kidney and heart remained significantly unaffected with all the tested formulations, but the ENL control showed a slight effect on the liver. The functional biochemical and haematology analysis data also confirmed the biocompatible potential of ENLs. The ENL control (the carrier) displayed 6.8-fold decreased genotoxicity (p < 0.05) in the human lymphocyte cells compared to the positive control. These encouraging cellular and rodent animal data indicate that ENLs are expected to be promising biocompatible nano-cargo for future clinical applications.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgments
The authors thankfully acknowledge the support provided by Veterinary Research Institute (VRI) for the permission to conduct the animal studies in their animal house and the Services Institute of Medical Sciences (SIMS) for provision of analytical support in biochemical and histological analyses.
References
- Liu J., Huang Y., Kumar A., Tan A., Jin S., Mozhi A., Liang X.-J. Biotechnol. Adv. 2014;32:693–710. doi: 10.1016/j.biotechadv.2013.11.009. [DOI] [PubMed] [Google Scholar]
- Zhang Q., Wang X., Li P. Z., Nguyen K. T., Wang X. J., Luo Z., Zhang H., Tan N. S., Zhao Y. Adv. Funct. Mater. 2014;24:2450–2461. [Google Scholar]
- Sun T., Zhang Y. S., Pang B., Hyun D. C., Yang M., Xia Y. Angew. Chem., Int. Ed. 2014;53:12320–12364. doi: 10.1002/anie.201403036. [DOI] [PubMed] [Google Scholar]
- Shi J., Votruba A. R., Farokhzad O. C., Langer R. Nano Lett. 2010;10:3223–3230. doi: 10.1021/nl102184c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo J.-W., Irvine D. J., Discher D. E., Mitragotri S. Nat. Rev. Drug Discovery. 2011;10:521–535. doi: 10.1038/nrd3499. [DOI] [PubMed] [Google Scholar]
- Daglar B., Ozgur E., Corman M., Uzun L., Demirel G. RSC Adv. 2014;4:48639–48659. [Google Scholar]
- Zoroddu M. A., Medici S., Ledda A., Nurchi V. M., Lachowicz J. I., Peana M. Curr. Med. Chem. 2014;21:3837–3853. doi: 10.2174/0929867321666140601162314. [DOI] [PubMed] [Google Scholar]
- Naahidi S., Jafari M., Edalat F., Raymond K., Khademhosseini A., Chen P. J. Controlled Release. 2013;166:182–194. doi: 10.1016/j.jconrel.2012.12.013. [DOI] [PubMed] [Google Scholar]
- Rothen-Rutishauser B. M., Schürch S., Haenni B., Kapp N., Gehr P. Environ. Sci. Technol. 2006;40:4353–4359. doi: 10.1021/es0522635. [DOI] [PubMed] [Google Scholar]
- Marasini N., Giddam A. K., Ghaffar K. A., Batzloff M. R., Good M. F., Skwarczynski M., Toth I. Nanomedicine. 2016;11:1223–1236. doi: 10.2217/nnm.16.36. [DOI] [PubMed] [Google Scholar]
- Javed I., Hussain S. Z., Ullah I., Khan I., Ateeq M., Shahnaz G., ur Rehman H., Razi M. T., Shah M. R., Hussain I. J. Mater. Chem. B. 2015;3:8359–8365. doi: 10.1039/c5tb01258a. [DOI] [PubMed] [Google Scholar]
- Nho T. D. T., Ly H. T., Vo T. S., Nguyen H. D., Phung T. T. H., Zou A., Liu J. Adv. Nat. Sci.: Nanosci. Nanotechnol. 2017;8:015008. [Google Scholar]
- Saremi S., Atyabi F., Akhlaghi S. P., Ostad S. N., Dinarvand R. Int. J. Nanomed. 2011;6:119–128. doi: 10.2147/IJN.S15500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khosravi-Darani K., Mozafari M. Int. J. Biomed. Nanosci. Nanotechnol. 2010;6:3–13. [Google Scholar]
- Yang Y., Qin Z., Zeng W., Yang T., Cao Y., Mei X., Kuang Y. Nanotechnol. Rev. 2016:1–11. [Google Scholar]
- Sohail M. F., Javed I., Hussain S. Z., Sarwar S., Akhtar S., Nadhman A., Batool S., Irfan Bukhari N., Saleem R. S. Z., Hussain I., Shahnaz G. J. Mater. Chem. B. 2016;4:6240–6248. doi: 10.1039/c6tb01348a. [DOI] [PubMed] [Google Scholar]
- Iqbal J., Shahnaz G., Perera G., Hintzen F., Sarti F., Bernkop-Schnürch A. Eur. J. Pharm. Biopharm. 2012;80:95–102. doi: 10.1016/j.ejpb.2011.09.010. [DOI] [PubMed] [Google Scholar]
- Wan A., Sun Y., Li H. Int. J. Biol. Macromol. 2008;43:415–421. doi: 10.1016/j.ijbiomac.2008.07.016. [DOI] [PubMed] [Google Scholar]
- Javed I., Hussain S. Z., Shahzad A., Khan J. M., Rehman M., Usman F., Razi M. T., Shah M. R., Hussain I. Colloids Surf., B. 2016;141:1–9. doi: 10.1016/j.colsurfb.2016.01.022. [DOI] [PubMed] [Google Scholar]
- Huang H.-L., Lee H.-Y., Tsai A.-C., Peng C.-Y., Lai M.-J., Wang J.-C., Pan S.-L., Teng C.-M., Liou J.-P. PLoS One. 2012;7:e43645. doi: 10.1371/journal.pone.0043645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain A., Thakur K., Kush P., Jain U. K. Int. J. Biol. Macromol. 2014;69:546–553. doi: 10.1016/j.ijbiomac.2014.06.029. [DOI] [PubMed] [Google Scholar]
- de Almeida M. C., Silva A. C., Barral A., Barral Netto M. Mem. Inst. Oswaldo Cruz. 2000;95:221–223. doi: 10.1590/s0074-02762000000200014. [DOI] [PubMed] [Google Scholar]
- Nadhman A., Nazir S., Khan M. I., Arooj S., Bakhtiar M., Shahnaz G., Yasinzai M. Free Radicals Biol. Med. 2014;77:230–238. doi: 10.1016/j.freeradbiomed.2014.09.005. [DOI] [PubMed] [Google Scholar]
- Malagoli D. Invert. Surviv. J. 2007;4:92–94. [Google Scholar]
- Nadhman A., Nazir S., Khan M. I., Ayub A., Muhammad B., Khan M., Shams D. F., Yasinzai M. Int. J. Nanomed. 2015;10:6891. doi: 10.2147/IJN.S91666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huerta I., Barasoain M., Télez M., longa M., Muga J., Barrenetxea G., Ortiz-Lastra E., González J., Criado B., Arrieta I. Mutat. Res., Genet. Toxicol. Environ. Mutagen. 2014;767:1–7. doi: 10.1016/j.mrgentox.2014.04.005. [DOI] [PubMed] [Google Scholar]
- Saleem U., Ahmad B., Ahmad M., Erum A., Hussain K., Irfan Bukhari N. Drug Chem. Toxicol. 2015:1–5. doi: 10.3109/01480545.2015.1092040. [DOI] [PubMed] [Google Scholar]
- Singh T., Sinha N., Singh A. Indian J. Pharmacol. 2013;45:61. doi: 10.4103/0253-7613.106437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandebriel R. J., Tonk E. C., de la Fonteyne-Blankestijn L. J., Gremmer E. R., Verharen H. W., van der Ven L. T., van Loveren H., de Jong W. H. Part. Fibre Toxicol. 2014;11:1. doi: 10.1186/1743-8977-11-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatasubbu G. D., Ramasamy S., Gaddam P. R., Kumar J. Int. J. Nanomed. 2015;10:137. doi: 10.2147/IJN.S79991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautam M., Goel R., J. Toxicol., 2014, 2014 , , 135654 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaeublin N. M., Braydich-Stolle L. K., Schrand A. M., Miller J. M., Hutchison J., Schlager J. J., Hussain S. M. Nanoscale. 2011;3:410–420. doi: 10.1039/c0nr00478b. [DOI] [PubMed] [Google Scholar]
- Asati A., Santra S., Kaittanis C., Perez J. M. ACS Nano. 2010;4:5321–5331. doi: 10.1021/nn100816s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita Y., Rikimaru-Kaneko A., Hashiguchi K., Shirotake S. Immunopharmacol. Immunotoxicol. 2011;33:730–737. doi: 10.3109/08923973.2011.565345. [DOI] [PubMed] [Google Scholar]
- Naha P. C., Davoren M., Lyng F. M., Byrne H. J. Toxicol. Appl. Pharmacol. 2010;246:91–99. doi: 10.1016/j.taap.2010.04.014. [DOI] [PubMed] [Google Scholar]
- Fröhlich E. Int. J. Nanomed. 2012;7:5577–5591. doi: 10.2147/IJN.S36111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaszewski R., Khan A., Sarkar F. H., Kucuk O., Tobi M., Zagnoon A., Dhar R., Kinzie J., Majumdar A. P. Am. J. Physiol., Cell Physiol. 1999;277:C1142–C1148. doi: 10.1152/ajpcell.1999.277.6.C1142. [DOI] [PubMed] [Google Scholar]
- Dobrovolskaia M. A., Clogston J. D., Neun B. W., Hall J. B., Patri A. K., McNeil S. E. Nano Lett. 2008;8:2180. doi: 10.1021/nl0805615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fornaguera C., Calderó G., Mitjans M., Vinardell M. P., Solans C., Vauthier C. Nanoscale. 2015;7:6045–6058. doi: 10.1039/c5nr00733j. [DOI] [PubMed] [Google Scholar]
- Ma Y., Fuchs A. V., Boase N. R., Rolfe B. E., Coombes A. G., Thurecht K. J. Eur. J. Pharm. Biopharm. 2015;94:393–403. doi: 10.1016/j.ejpb.2015.06.014. [DOI] [PubMed] [Google Scholar]
- Laine A.-L., Gravier J., Henry M., Sancey L., Bejaud J., Pancani E., Wiber M., Texier I., Coll J.-L., Benoit J.-P. J. Controlled Release. 2014;188:1–8. doi: 10.1016/j.jconrel.2014.05.042. [DOI] [PubMed] [Google Scholar]
- Agbaje E., Adeneye A., Daramola A. Afr. J. Tradit., Complementary Altern. Med. 2009;6(3):241–254. doi: 10.4314/ajtcam.v6i3.57162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozer J., Ratner M., Shaw M., Bailey W., Schomaker S. Toxicology. 2008;245:194–205. doi: 10.1016/j.tox.2007.11.021. [DOI] [PubMed] [Google Scholar]
- Thapa B., Walia A. Indian J. Pediatr. 2007;74:663–671. doi: 10.1007/s12098-007-0118-7. [DOI] [PubMed] [Google Scholar]
- Simak J., Nanotoxicity: From in vivo and in vitro models to health risks, John Wiley & Sons, Chichester, 2009, pp. 191–225. [Google Scholar]
- Zhao M., Su M., Lin X., Luo Y., He H., Cai C., Tang X. Pharm. Res. 2010;27:1687–1702. doi: 10.1007/s11095-010-0180-0. [DOI] [PubMed] [Google Scholar]