

 Screening for carcinogens in general, and for the urinary bladder specifically, traditionally involves a two-year bioassay in rodents, the results of which often do not have direct relevance to humans with respect to mode of action (MOA) and/or dose response.
Screening for carcinogens in general, and for the urinary bladder specifically, traditionally involves a two-year bioassay in rodents, the results of which often do not have direct relevance to humans with respect to mode of action (MOA) and/or dose response.
Abstract
Screening for carcinogens in general, and for the urinary bladder specifically, traditionally involves a two-year bioassay in rodents, the results of which often do not have direct relevance to humans with respect to mode of action (MOA) and/or dose response. My proposal describes a multi-step short-term (90 day) screening process that characterizes known human urinary bladder carcinogens, and identifies those reported in rodent two-year bioassays. The initial step is screening for urothelial proliferation, by microscopy or by increased Ki-67 labeling index. If these are negative, the agent is not a urinary bladder carcinogen. If either of these is positive, an MOA and dose response analysis are performed. DNA reactivity is evaluated. If the chemical is non-DNA reactive, evaluation for cytotoxicity is performed. This involves examination of the urothelium and urine, the latter to identify the generation of urinary solids (e.g. calculi). If urinary solids are the cause of cytotoxicity, the MOA is not relevant to human cancer, but dose response becomes essential for evaluating potential toxicity to humans. If cytotoxicity occurs but no urinary solids are detected, urinary concentrations of the chemical and its metabolites are evaluated, and compared to in vitro cytotoxicity against rodent and human immortalized urothelial cell lines. Based on this process, a screen for urinary bladder carcinogenicity is reliable, and more importantly, can be based on MOA and dose response analyses useful in the overall risk assessment for possible human bladder cancer. The proposed procedure is shorter, less expensive and more relevant than the two-year bioassay.
Urinary bladder carcinogens
Cancer of the urinary bladder has been related to chemicals ever since the report by Rehn in 1895 associating development of cancer of the urinary bladder in workers of the German aniline dye industry with their exposure at work.1,2 This led to the eventual demonstration by Hueper et al.3 in 1937 that one of the active chemicals in the dye industry, 2-naphthylamine, induced bladder cancer in dogs by oral administration. This was the first demonstration of an aromatic amine as a carcinogen for the urinary bladder. In this study Hueper et al. also raised, for the first time, the issue of latency: induction of cancer takes time, often many years in humans. Tumors do not arise shortly after exposure to a chemical. This has been a mainstay of chemical carcinogenesis and cancer induction, in general, ever since this report by Hueper et al.
Modes of action
It is currently acknowledged that there are several modes of action by which a chemical may cause tumors. However, the traditional two-year bioassay was designed before the various modes of action were recognized, thus it is not planned for characterizing mode of action and/or dose–response, which are crucial for evaluation of human relevance. These modes of action for urinary bladder carcinogenesis are: DNA reactivity, mitogenicity, and cytotoxicity by formation of urinary solids or by chemical toxicity. Each of these modes of action is explained below.
DNA reactive carcinogens
The early observations by Hueper et al.3 led to intense investigations into the carcinogenicity of aromatic amines, including the metabolic activation of these chemicals, which is an important factor involved in the mode of action of tumor induction. Since Hueper et al.,3 numerous aromatic amines have been identified as human bladder carcinogens in occupational settings,4 including 4-aminobiphenyl (4-ABP), 2-naphthylamine, as well as benzidine and benzidine-based azo dyes. The chemical 2-acetylaminofluorene (2-AAF) was developed in the 1940s as a pesticide for commercialization, but was prevented from reaching the market following the discovery that it produced urinary bladder cancer in a number of species, including rats and mice.5 2-AAF became a model compound for investigating aromatic amines, and led the Miller5 to their discovery that aromatic amines were metabolically activated by N-hydroxylation, followed by N-esterification with sulfate or glucuronate, producing a reactive electrophilic metabolite, leading to DNA binding, DNA adduct formation, and ultimately mutation. The possibility that the reactive intermediate could be a free radical has also been investigated. The process of metabolic activation to reactive electrophiles which react with DNA was identified as a generalized phenomenon for a wide range of DNA-reactive chemicals.6
Numerous chemicals have been identified as DNA-reactive carcinogens in addition to aromatic amines and amides, including polycyclic aromatic hydrocarbons, N-nitrosamines, and aflatoxins. Several DNA reactive chemicals have been identified as urinary bladder carcinogens, such as aromatic amines and amides, phosphoramide mustards, such as cyclophosphamide, and alkylating agents such as thiotepa, and various chemicals related to these.1,2,7 Some of these chemicals, including 2-AAF, have also produced cancer in other organs in rodent assays, (e.g., liver, breast, and Zymbal's gland).5 A list of chemicals that are known to produce bladder cancer in humans by a DNA reactive mode of action are listed in Table 1. Of those chemicals that have been investigated in animal models, all have been carcinogenic, and the target organ is usually the urinary bladder.
Table 1. DNA reactive human urinary bladder carcinogens.
| 2-Acetylaminofluorene (2-AAF) |
| 2-Naphthylamine |
| 4-Aminobiphenyl |
| Benzidine |
| Benzidine-based azo dyes |
| Cyclophosphamide |
| Ifosphamide |
| Chlornaphazine |
| Phenacetin |
| o-Toluidine |
| 4,4′-Methylenebis(2-chloroaniline) (MOCA) |
| Methylenedianiline |
| Thiotepa |
| Aristolochic acid |
Although the mode of action for these chemicals involves metabolic activation to DNA reactive electrophiles, their dose response has generally not been linear, neither in animal models8,9 nor in humans.10 This non-linear dose response for carcinogenesis occurs despite the dose response for steady state levels for DNA adducts with these chemicals nearly always being linear. The ED01 mega-mouse experiment performed at the National Center for Toxicological Research (NCTR) in the 1970s demonstrated that using 2-AAF administered to mice as the model system resulted in a non-linear dose–response for tumors of the urinary bladder. This non-linearity was shown to be related to an increase in cell proliferation that occurred at higher doses with these chemicals.10
Principles of carcinogenesis
Cancer is clonal and occurs due to multiple mutations in a single stem cell (pluripotential cell).8,11,12 Carcinogens increase cancer risk in one of two ways: (a) by a direct damage to DNA every time DNA replicates (DNA reactive carcinogen), or (b) by increasing the number of DNA replications, leading to an increased opportunity for “spontaneous” mutations to occur (carcinogens that are not DNA reactive).8,11,12 In animal models, it appears that DNA reactivity by itself does not produce an increased incidence of tumors in the urinary bladder great enough to be detectible, whether there is a 10% detection limit (as with experiments involving 50–60 animals per group) or with a detection limit of 1% (as in the ED01 study).9 The use of modeling has demonstrated that although DNA adducts occur at low doses, the anticipated incidence at these low doses would be considerably below 1% over the lifetime of a rodent. At higher doses, cell proliferation rate and the number of target cells are increased (hyperplasia), resulting in a detectable incidence of bladder tumors. This dose response for DNA reactive carcinogens has practical significance for humans.
Cigarette smoking: DNA reactivity and increased cell proliferation
In addition to lung cancer, cigarette smoking has long been known to be associated with an increased risk of cancer of the lower urinary tract, including the kidney pelvis, ureters, and urinary bladder, with a relative risk of approximately 3 times the incidence that occurs in non-smokers.1,2,7 Numerous aromatic amines have been detected in cigarette smoke, including 4-ABP, and DNA adducts from these aromatic amines have been detected in the urothelium of animal models and of humans.13 However, the levels of exposure to the aromatic amines from cigarette smoke, and the level of DNA adducts formed from this exposure, are not sufficient to produce increased cell proliferation of the urothelium, and do not explain the increased incidence of urothelial cancer in smokers.10 In addition to DNA adducts, increased urothelial cell proliferation has been observed in smokers.14 It must be that other chemicals in cigarette smoke increase the cell proliferation. Cohen and Ellwein8,9 demonstrated that this increased proliferation acts in synergy with the DNA reactivity of the aromatic amines (and possibly other constituents) from cigarette smoke. A leading possibility for the substance in cigarette smoke that increases urothelial cell proliferation is nicotine, or possibly one or more nicotine metabolites.15
Numerous additional aromatic amines have been detected as environmental exposures of concern, including heterocyclic amines generated by heating foods.16 Many of these heterocyclic amines have been shown to be carcinogenic in rodent and dog models, but the level of exposure to these chemicals in the human diet appears to be at low levels of micrograms or lower. To date, there is no convincing evidence indicating an increased risk to humans from exposure to heterocyclic amines.17 Other amines to which humans are exposed include various tryptophan metabolites such as kynurenine, anthranilic acid, and their hydroxylated metabolites. In the 1950s and 60s there was some evidence that these might be associated with an increased risk of bladder cancer, based on the hypothesis that ortho-aminophenols could be carcinogenic.18 However, once the Millers5 demonstrated that it was N-hydroxylation that led to the induction of cancer by aromatic amines, this hypothesis could no longer be supported. Additionally, evidence accrued that administration of l-tryptophan to rats did not increase the risk of urinary bladder cancer, nor was there an increase in cell proliferation with l-tryptophan administration.19
Non-DNA reactive carcinogens
In addition to DNA reactive carcinogens, agents which are not associated with DNA reactivity have also been demonstrated to be associated with an increased risk of bladder cancer in humans. These compounds increase the risk of cancer by increasing cell proliferation by a mode of action involving either direct mitogenicity or cytotoxicity (cell death) with prolonged regenerative urothelial proliferation. Such agents in humans are inorganic arsenic,20 bacterial cystitis,2,7 and infection with the parasite Schistosoma mansoni (leading to schistosomiasis).21
Mode of action: increased cell proliferation
Increased cell proliferation can occur secondary to immunosuppression, which is associated with activation of various viruses known to produce cancer, such as Epstein–Barr virus (EBV), human papilloma virus (HPV), Kaposi's sarcoma virus (KSV or HHV-8), or hepatitis B and C viruses (HBV and HCV).6 However, none of these are known to be associated with bladder cancer.2,6,7 Furthermore, immunosuppressed patients, whether due to inherited disorders, to treatment with immunosuppressive chemicals such as those used in organ transplantation, cancer chemotherapy, or treatment of various autoimmune disorders, or to AIDS, do not have an increased risk of bladder cancer. Thus, immunosuppression does not appear to be a mode of action relevant to human or animal bladder cancer.
Another mode of action that can increase the risk of cancer in general is high levels of estrogenic activity.6 However, there is no evidence that this is relevant to urinary bladder carcinogenesis2,6,7 although this mode of action is associated with an increased risk of tumors of endometrium, breast, and to a more limited extent liver.2,6,7
Increased cell proliferation can be produced either by increasing cell births or decreasing cell deaths, which leads to an increase in the number of cells.32 The critical parameter for increased cell proliferation is the number of DNA replications (not the rate), although the two frequently occur together, especially in layered epithelia such as the urothelium. Increased cell births can be produced either by direct mitogenicity (usually involving hormones or growth factors) or by cytotoxicity and regenerative proliferation, although only one chemical, propoxur, has been associated with increased mitogenicity in the bladder urothelium.33 Raf inhibitors have recently been shown to increase urothelial proliferation and tumors in rats, but it is unknown whether this is due to direct mitogenicity or cytotoxicity.34 Decreased cell deaths can be produced either by decreasing apoptosis or decreasing differentiation. No agent has yet been identified which acts on the urothelium by decreasing cell deaths. Urothelial cytotoxicity with regenerative proliferation is by far and away the most common mode of action leading to increased cell proliferation for the urothelium, and this is true for both DNA reactive and non-DNA reactive agents. The cytotoxicity usually involves urothelial necrosis, but theoretically could be due to increased apoptosis, such as seen with fumonisin B1 and kidney tumors.35
Mode of action: urinary solids
Numerous chemicals have been identified that produce urothelial proliferation and carcinogenesis in the rodent model by increasing urinary solids, either precipitates, crystals, and/or calculi (Table 3).36–41 The urinary solids can be formed by the chemical itself (e.g. melamine),26 by a metabolite, or from crystallization of normal urinary constituents resulting from alteration of the urinary composition.38–42 An example of the latter is the decreased citrate levels induced by the dual peroxisome proliferator-activated receptor (PPAR) activator muraglitazar, leading to decreased urinary citrate concentrations and precipitation of calcium in the urine.40 Another example of urinary solids forming from normal urinary constituents is the formation of a calcium phosphate-containing precipitate in rats administered high doses of various sodium salts, such as saccharin and ascorbate (Table 4).41,42 This appears to occur only in rats and is not relevant to human toxicity or carcinogenicity.43–45 However, the precipitate must be identified to demonstrate this mode of action, as it cannot be assumed automatically that any sodium salt producing urothelial proliferation is acting by this mode of action. Sodium o-phenylphenol produces urothelial hyperplasia at high doses in rats, but does not produce this precipitate. Rather, it acts by chemical cytotoxicity, probably through formation of its quinone.46 Similarly, sodium cacodylate produces urothelial proliferation by cytotoxicity that is caused by a reactive metabolite while no urinary solids are formed.20
Table 3. Substances producing urinary calculi when administered to rodents and/or humans.
| Uracil | Oxamide | Salicylazosulfapyridine |
| Thymine | Acetazolamide | Disperse blue 1 |
| Fosetyl-al | Terephthalic acid | Triamterene |
| Melamine | Dimethylterephthalate | Sulfonamides |
| Urate | Polyoxyethylene-8-stearate | Ampicillin |
| Homocysteine | Glycine | Amoxicillin |
| Cysteine | Pyroxasulfone | Indinavir |
| Calcium oxalate | Sulfosulfuron | Glafenic acid |
| Calcium phosphate | Muraglitazar | Orotic acid |
| Calcium carbonate | Pioglitazone | Biphenyl |
| Diethylene glycol | 4-Ethylsulfonylnaphthalene-1-sulfonamide |
Table 4. Sodium salts administered orally at high doses to rats that produce urothelial proliferation.
| Saccharin | Ascorbate |
| Succinate | Erythorbate |
| Aspartate | Phosphate |
| Glutamate | Chloride |
| Bicarbonate | Citrate |
Considerable evidence has accumulated demonstrating that a mode of action involving urinary solids is not relevant to human cancer.38–45 Some chemicals, melamine for example, have been demonstrated to produce calculi in rats and mice, and urinary bladder tumors in rats if administered at a concentration sufficient to produce urinary tract calculi.26 This finding in rats and mice does not predict carcinogenicity in humans. However, exposure to sufficiently high levels of melamine in humans can lead to the formation of calculi, as was ultimately demonstrated in the unfortunate episode of adulteration of infant formula in China, where a large number of infants developed urinary tract calculi and had to be hospitalized.47 A small number died. The risk assessment for such chemicals as melamine is not related to its carcinogenicity in the animal model, but to the actual toxicity of the formation of calculi, which can occur in humans. Thus, if urinary solids formation is the mode of action concluded for a particular chemical, the risk assessment is related to the toxicity of urinary solid formation, not to carcinogenicity.
Mode of action: chemical cytotoxicity
Certain chemicals can produce urothelial cytotoxicity by a reactive metabolite, or by themselves, which is much less common. This mode of action is relevant to humans, but involves a threshold exposure, namely, the exposure necessary to produce a minimal urinary concentration of the metabolite(s) adequate to produce urothelial cytotoxicity. There is no risk of toxicity and no cancer risk when exposure to a chemical is below the level that can produce cytotoxicity. Similar considerations have been identified for chemicals in several other tissues, chloroform, for example, in the liver and kidney.48
The risk assessment for such a chemical must involve quantitation of the urinary levels of the metabolite(s) in the animal model that will enable evaluation of the threshold exposures in humans. If actual measurements are available from humans (e.g., for pharmaceuticals), those can be incorporated into the risk assessment.
Several chemicals have been identified that act by cytotoxicity of reactive metabolites (Table 5). For example, the pesticide diuron induces bladder tumors in the rat when administered in the diet at doses of 1250 ppm and 2500 ppm.49 Doses of 500 ppm leads to an increase in cytotoxicity and cell proliferation but no tumors. Investigations have shown that the urinary metabolites most likely involved with the cytotoxicity and tumor induction are N-(3,4-dichlorophenyl)urea and 4,5-dichloro-2-hydroxyphenylurea. The amounts required to produce a cytotoxic level of these metabolites in the urine are several orders of magnitude greater than would occur with possible human exposures.
Table 5. Chemicals producing urothelial proliferation in rodents that act by generating a cytotoxic metabolite in the urine.
| Inorganic arsenic |
| Dimethylarsinic acid |
| Pulegone |
| Diuron |
| Tributylphosphate |
| Sodium o-phenylphenol |
| Transfluthrin |
Screening for urinary bladder carcinogens
The generally accepted standard for screening chemicals for possible carcinogenicity in humans is the two-year bioassay in rodents, usually mice and rats. The chemicals that are known to increase the risk of bladder cancer in humans have generally been positive in the standard rodent two-year bioassay, with the notable exceptions of cigarette smoke22 and inorganic arsenic.20 However, the two-year bioassay has numerous problems:23,24 (1) the long time necessary for performing such an assay, (2) the high cost, (3) the large number of animals, (4) the frequently questionable extrapolation of the animal findings to humans with respect to the dose response, and often (5) the lack of relevance of the findings in animals to humans based on the mode of action.
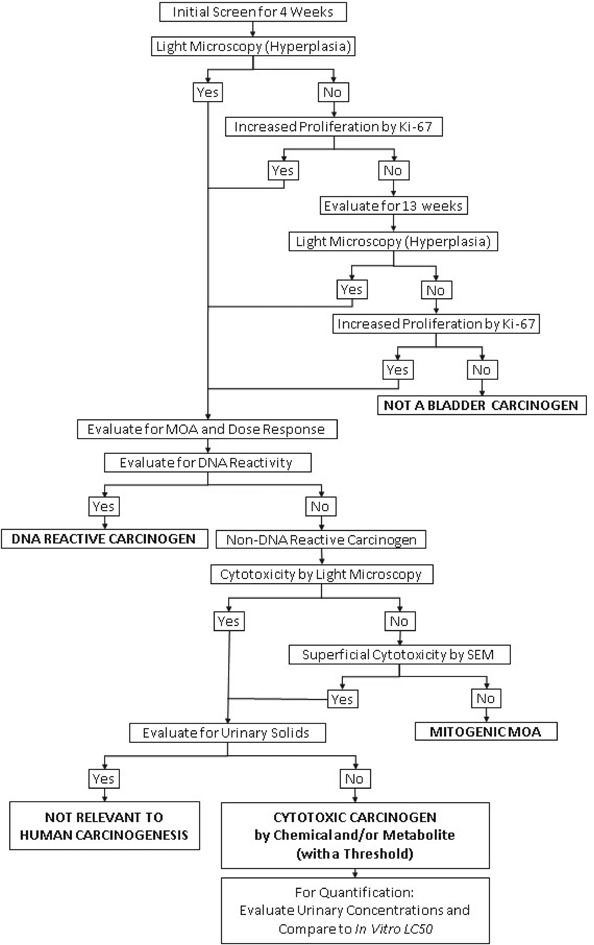
I have previously proposed an alternative to the two-year bioassay for screening chemicals for possible carcinogenicity in humans, with both a mode of action and dose response analysis that provides a better assessment of the risk to humans than a standard two-year bioassay.23,24 Furthermore, this proposal requires less time, less cost, fewer animals, and provides results that can be evaluated for human relevance both qualitatively and quantitatively. This screening proposal is a multi-step procedure based on the two possible generalized MOAs by which any agent can increase the risk of cancer: either directly damaging DNA, or increasing the number of DNA replications. It uses the mode of action/human relevance framework developed by the International Programme on Chemical Safety (IPCS), the U.S. Environmental Protection Agency (EPA), Health Canada and the International Life Sciences Institute (ILSI) as models to screen for possible carcinogens and evaluate modes of action and the dose response.25–29 The procedure proposed for the bladder is similar to the previous one described for the liver24 and can be applied to any tissue. The steps of the procedure are: (1) initial screen, (2) screen for DNA reactivity, (3) evaluation for increased cell proliferation, (4) evaluation for cytotoxicity, (5) evaluation for urinary solids, and (6) evaluation of cytotoxic chemicals. These steps are described in Fig. 1.
Fig. 1. A decision tree summarizing the proposed procedure for screening chemicals for urinary bladder carcinogenicity as described in detail in the text.

Step 1: Initial screen
The initial step in the evaluation is a screening assay for increased cell proliferation. Animals are administered the agent for 4 weeks and then the bladders are evaluated for increased cell proliferation. The evaluation includes light microscopic examination of the bladder epithelium for hyperplasia (Fig. 2). If the light microscopic evaluation is negative, the urothelium is evaluated for increased proliferation using a Ki-67 labeling index (avoiding the need for administration of an exogenous agent such as bromodeoxyuridine (BrdU)). The labeling index can be done on the same blocks of paraffin-embedded tissue used for the light microscopic examination of hematoxylin and eosin (H&E)-stained tissues. If the bladders are negative for hyperplasia and increased labeling index after 4 weeks of treatment, additional groups with the same treatment and started at the same time as the 4 weeks groups, are examined after 13 weeks. If the examination at 4 weeks is positive, the contingent 13 weeks animals can be terminated without further examination. If the examinations at 4 and 13 weeks are negative, the chemical is not a bladder carcinogen. This can be confidently concluded since all chemicals that are rodent bladder carcinogens in two-year bioassays are detected using hyperplasia or increased labeling index at 4 and/or 13 weeks. More importantly, all known human bladder carcinogens produce increased urothelial proliferation by 13 weeks of administration in rodents, including cigarette smoke by inhalation30 and inorganic arsenic administered in the drinking water or diet,20 which do not produce tumors in rodent in a standard two-year bioassay.20,22 This screening assay will detect DNA reactive as well as non-DNA reactive agents that increase the incidence of bladder cancer in rodents or in humans. If this initial screening assay is positive either at 4 or at 13 weeks, then additional analyses are performed to evaluate mode of action, dose response and relevance to humans.
Fig. 2. Normal urinary bladder urothelium (2A) compared to simple urothelial hyperplasia (2B), the initial abnormality to be evaluated in the proposed screening procedure. More severe hyperplasia (nodular and/or papillary hyperplasia) can be detected in this short term screen if the toxicity is sufficient to damage the full thickness of the epithelium (ulceration), usually accompanied by inflammation (original magnification ×200).

Step 2: Screen for DNA reactivity
DNA reactivity can generally be readily demonstrated by incorporating findings from structure–activity computerized models, in vitro Ames assay, and metabolism investigations evaluating the possibility of formation of a reactive electrophilic intermediate. Metabolic activation in rodents can be compared to humans, usually using in vitro methods and structure–activity analyses. If formation of DNA reactive metabolites is likely to occur in humans, then it is assumed that the agent will be a human carcinogen and likely carcinogenic for the urinary bladder. In addition to the DNA reactive urinary bladder carcinogens identified in humans which are listed in Table 1, numerous DNA reactive chemicals have been identified in rodent bioassays (Table 2).
Table 2. DNA reactive urinary bladder carcinogens identified in rodents.
| N,N-Dibutylnitrosamine |
| N-Butyl-N-(4-hydroxybutyl)nitrosamine (BBN) |
| N-Butyl-N-(3-carboxypropyl)nitrosamine |
| N-Methyl-N-octylnitroamine |
| N-Methyl-N-decylnitrosamine |
| N-Methyl-N-dodecylnitrosamine |
| N-Methyl-N-tetradecylnitrosamine |
| N-[4-(5-Nitro-2-furyl)-2-thiazolyl]formamide (FANFT) |
| N-[4-(5-Nitro-2-furyl)-2-thiazolyl]acetamide (Furium) |
| 2-Amino-4-(5-nitro-2-furyl)-thiazole |
| 4-Nitrodiphenyl |
| 4-Acetylaminobiphenyl |
| N-Hydroxy-2-acetylaminofluorene |
| 2-Acetylaminofluorene |
| o-Aminoazotoluene |
| Dichlorobenzidine |
| 3,2′-Dimethyl-4-aminobiphenyl |
| 2-Methoxy-3-aminodibenzofuran |
| 3-Methoxy-4-aminobiphenyl |
| 2,6-Dimethylaniline |
| 3,5-Dimethyaniline |
| 3-Ethylaniline |
| Anthraquinone |
| Ptaquiloside |
Step 3: Evaluation for increased cell proliferation
If the chemical is positive in the initial screen but is not DNA reactive, then the mode of action involves increased cell proliferation. Increased cell proliferation can be demonstrated by various in vitro methods, although more reliably it requires in vivo investigations.31 Immunosuppression and estrogenic activity can readily be evaluated by various in vitro and in vivo assays, but as explained above, these do not pertain to screening for urinary bladder carcinogens. Urinary bladder carcinogens are assessed by increased urothelial proliferation, which can be readily accomplished in animal models by histologic evaluation for hyperplasia in short term assays, usually as short as 4 weeks, but no longer than 13 weeks, or more sensitively, by incorporating a proliferation labeling index such as Ki-67 as described above.1,31,37 All of the known human and rodent urothelial carcinogens have shown evidence of increased cell proliferation in a 13 weeks or less assay. The results of the initial screen will determine if 4 weeks exposure of the animals is sufficient. Based on the results of the initial screening assay, the more sensitive sex will be used for mode of action and dose response investigations. If males and females are of equal sensitivity, either sex can be chosen for subsequent investigation. This greatly reduces the number of animals needed.
Step 4: Evaluation for cytotoxicity
The investigation for mode of action needs to involve enough groups of animals to broadly investigate the dose response of the increased cell proliferation effect, with at least one dose that will provide a no effect level. This follow up investigation needs to incorporate collection of urine specimens and examination of the urothelium by histology, and possibly scanning electron microscopy, and labeling index, either BrdU or Ki-67. We have found that proliferating cell nuclear antigen (PCNA) is not reliable for assessing cell proliferation for the urothelium, and BrdU requires administration exogenously, usually by intraperitoneal injection for assessment of the bladder. Details regarding collection of specimens and examination of both urine and urothelium have been described elsewhere.36,37
Examination of the urothelium by light microscopy can provide information regarding urothelial cytotoxicity if the agent is sufficiently potent to produce damage to the entire thickness of the urothelium, but it is not adequate for detection of toxicity limited to the superficial layer. The latter instance can be evaluated by examination with scanning electron microscopy (Fig. 3). Thus, half of the bladder can be processed for light microscopy and labeling index, and the other half should be kept for scanning electron microscopy.37 Scanning electron microscopy is only necessary if there is no evidence of toxicity by light microscopy.
Fig. 3. Scanning electron micrograph of the urinary bladder luminal surface showing toxicity and loss of the superficial (umbrella) cell layer (thick arrows), compared to the normal-appearing urothelium (thin arrows) with large, flat polygonal cells (white bar at bottom represents 200 μm.).

If no cytotoxicity is detected at 4 or 13 weeks, but there is an increase in labeling index, one can conclude that the agent is acting by a mitogenic mode of action. As indicated above, only one chemical, propoxur, so far has been demonstrated to act by direct mitogenicity.33 If cytotoxicity is detected, then examination of the urine for the possible presence of urinary solids (precipitate, crystals, or calculi) needs to be evaluated.
Step 5: Evaluation for urinary solids
For the assessment of urine, fresh void specimens are necessary. Usually, this should be within 2 hours of the lights being turned on in the animal room since the rat is a nocturnal animal. It is critical that the animals are not fasted. Adequate specimens can usually be collected from the rat (greater than 80% of the time) to provide sufficient material for evaluation of crystalluria, pH and normal constituents of the urine (e.g. calcium, protein, electrolytes). Further details regarding collection of urine have been described elsewhere.36,37
Evaluation for crystals can usually be performed utilizing light microscopy, but for some chemicals, crystalluria requires evaluation by scanning electron microscopy, with attached X-ray reflective spectroscopy for identification of the composition of the crystals.37
If urinary solids are identified, then the mode of action involves formation of the urinary solid with toxicity and regenerative proliferation. If urinary solids are not detected, then the cytotoxicity is being produced by chemical reactivity, either by the administered chemical itself (uncommon) or by one or more metabolites. As described above, if urinary solids are the mode of action, it is not relevant to human cancer risk.43–45
Step 6: Evaluation of cytotoxic chemicals
For substances that produce urothelial cytotoxicity but do not involve urinary solids, the identity of the metabolites in the animal model and in humans is most important. In some instances, the metabolite that is responsible for the cytotoxicity in the animal model may not be formed in humans. This has obvious implications for risk assessment.
The identity of the metabolites can often be made using in vitro metabolism models. When an in vitro model is not available, then collection of urine is needed for metabolite identification. In some instances, this can be done on fresh void specimens, but usually will require an overnight collection of urine, which can be accomplished during the course of the 4 weeks or 13 weeks investigation. Urine collection can be included in the protocol, and analysis performed if metabolite identification becomes essential. For extrapolation to humans, the urinary levels of the cytotoxic metabolites are critical for inter-species dose response comparisons, but not the blood levels.36,37 It has long been known that urothelial carcinogenesis occurs secondary to exposure to these chemicals in the urine, not through the blood.4
An estimation of the minimum urinary concentration required for cytotoxicity can be made based on measurement of urinary levels of the metabolites in vivo following administration of the chemical to the rat, compared with in vitro studies of cytotoxicity utilizing immortalized rodent and human urothelial cells. Since these immortalized urothelial cells do not differentiate and do not have the protective superficial (umbrella) cell layer, such in vitro assays will tend to be more sensitive to the toxic effects of the metabolites than would be expected in vivo. This provides adequate conservatism in the dose response risk assessment.
Conclusions
In summary, all known rodent and human urinary bladder carcinogens can be detected in a short term (13 weeks or less) assay incorporating microscopic evaluation for urothelial hyperplasia and increased DNA proliferation as determined by a BrdU or Ki-67 labeling index. Follow up studies can readily indicate the mode of action as being either DNA reactivity, direct mitogenicity or cytotoxicity with consequent regeneration. Cytotoxicity can be due to either the formation of urinary solids or chemical reactivity with regenerative proliferation. Direct mitogenicity is potentially relevant to humans, although the exact mechanism would have to be demonstrated. Formation of urinary solids is relevant to human toxicity, but not relevant to human cancer risk. Chemical reactivity is potentially relevant to humans, but is clearly dose dependent, with the dose required to produce urinary metabolite levels that can produce cytotoxicity being an actual threshold. Thus, this short term screening assay with relatively few follow-up short term studies can readily identify all potential human urothelial carcinogens, characterize their mode of action in the animal models and evaluate relevance to humans. If relevant to humans, the dose response can be determined, with estimates of urinary levels secondary to human exposures. In the future, possible molecular markers might be identified which could be used for screening, especially ones related to increased cell proliferation and genotoxicity. However, at the present time such molecular biomarkers have not been evaluated extensively for the wide variety of bladder carcinogens described in this paper, and the small amount of normal urothelium in the rodent bladder raises numerous technical issues.
As described elsewhere, with the availability of the proposed procedures the two-year bioassay is no longer necessary. In fact, for the urothelium, the proposed short term assay is more sensitive in detecting known human bladder carcinogens, such as cigarette smoking and inorganic arsenic, than the two-year bioassay, and offers more information with regard to mode of action and human relevance than the traditional two-year bioassay provides. We have found that the entire procedure can be completed in six to twelve months, with as few as 40 animals of one sex and species for the follow-up studies for chemicals detected as having an effect in the initial 28 or 90 day studies which are currently routinely performed. Thus, there is considerable savings in time, money, and use of animals compared to the two year bioassay. More importantly, the information is more useful for an evaluation of mode of action and human relevance than the two year bioassay. As stated elsewhere, it is time to discontinue performing the two-year bioassay.
Conflicts of interest
I have served on several committees involved in the development of the mode of action/human relevance framework, have received funding for my research from NIH and from industry sources and I consult for various companies, including ones that have had chemicals that produce urothelial changes in rodents.
Acknowledgments
I gratefully acknowledge the contributions of Lora Arnold and Dr Michal Eldan in reviewing this manuscript and providing insightful comments. Also, I am grateful for the expert assistance of Jeanne Bradford in the preparation of the manuscript.
Footnotes
†Special issue dedicated to Drs Cliff Elcombe and Iain Purchase.
References
- Cohen S. M. Toxicol. Pathol. 1998;26:121–127. doi: 10.1177/019262339802600114. [DOI] [PubMed] [Google Scholar]
- Johansson S. L., Cohen S. M. Semin. Surg. Oncol. 1997;13:291–298. doi: 10.1002/(sici)1098-2388(199709/10)13:5<291::aid-ssu2>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- Hueper W. C., Wiley F. H., Wolfe H. D. J. Ind. Hyg. Toxicol. 1938;20:46–84. [Google Scholar]
- Clayson D. B. and Cooper E. H., Cancer of the urinary tract, in Advances in Cancer Research, ed. G. K. a. S. Weinhouse, Academic Press, Inc., New York, 1970, vol. 13, pp. 271–381. [DOI] [PubMed] [Google Scholar]
- Miller E. C., Miller J. A. Cancer. 1981;47:2327–2345. doi: 10.1002/1097-0142(19810515)47:10<2327::aid-cncr2820471003>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Cohen S. M., Arnold L. L. Toxicol. Sci. 2011;120(Suppl 1):S76–S92. doi: 10.1093/toxsci/kfq365. [DOI] [PubMed] [Google Scholar]
- Cohen S. M., Shirai T., Steineck G. Scand. J. Urol. Nephrol. 2000;34(Suppl. 205):105–115. doi: 10.1080/00365590050509869. [DOI] [PubMed] [Google Scholar]
- Cohen S. M., Ellwein L. B. Science. 1990;249:1007–1011. doi: 10.1126/science.2204108. [DOI] [PubMed] [Google Scholar]
- Cohen S. M., Ellwein L. B. Toxicol. Appl. Pharmacol. 1990;104:79–93. doi: 10.1016/0041-008x(90)90284-2. [DOI] [PubMed] [Google Scholar]
- Cohen S. M., Boobis A. R., Meek M. E., Preston R. J., McGregor D. Crit. Rev. Toxicol. 2006;36:803–819. doi: 10.1080/10408440600977651. [DOI] [PubMed] [Google Scholar]
- Greenfield R. E., Ellwein L. B., Cohen S. M. Carcinogenesis. 1984;5:437–445. doi: 10.1093/carcin/5.4.437. [DOI] [PubMed] [Google Scholar]
- Moolgavkar S. H., Knudson A. G. J. J. Natl. Cancer Inst. 1981;66:1037–1052. doi: 10.1093/jnci/66.6.1037. [DOI] [PubMed] [Google Scholar]
- Talaska G., Schamer M., Skipper P., Tannenbaum S., Caporaso N., Unruh L., Kadlubar F. F., Bartsch H., Malaveille C., Vineis P. Cancer Epidemiol., Biomarkers Prev. 1991;1:61–66. [PubMed] [Google Scholar]
- Auerbach O., Garfinkel L. Cancer. 1989;64:983–987. doi: 10.1002/1097-0142(19890901)64:5<983::aid-cncr2820640502>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- Dodmane P. R., Arnold L. L., Pennington K. L., Cohen S. M. Toxicology. 2014;315:49–54. doi: 10.1016/j.tox.2013.11.002. [DOI] [PubMed] [Google Scholar]
- Sugimura T., Wakabayashi K., Nakagama H., Nagao M. Cancer Sci. 2004;954:290–299. doi: 10.1111/j.1349-7006.2004.tb03205.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustsson K., Skog K., Jagerstad M., Dickman P. W., Steineck G. Lancet. 1999;353:703–707. doi: 10.1016/S0140-6736(98)06099-1. [DOI] [PubMed] [Google Scholar]
- Bryan G. T. Am. J. Clin. Nutr. 1971;24:841. doi: 10.1093/ajcn/24.7.841. [DOI] [PubMed] [Google Scholar]
- Birt D. F., Julius A. D., St John M. K., Hasegawa R., Cohen S. M. Proc. Am. Assoc. Cancer Res. 1986;27:128. [Google Scholar]
- Cohen S. M., Arnold L. L., Beck B. D., Lewis A. S., Eldan M. Crit. Rev. Toxicol. 2013;43:711–752. doi: 10.3109/10408444.2013.827152. [DOI] [PubMed] [Google Scholar]
- Zheng Y.Y. L.L., Amr S., Saleh D. A., Dash C., Ezzat S., Mikhail N. N., Gouda I., Loay I., Hifnawy T., Abdel-Hamid M., Khaled H., Wolpert B., Abdel-Aziz M. A., Loffredo C. A. Cancer Epidemiol., Biomarkers Prev. 2012;213:537–546. doi: 10.1158/1055-9965.EPI-11-0589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, 2012, vol. 100E, pp. 43–211. [Google Scholar]
- Cohen S. M. Toxicol. Sci. 2004;80:225–229. doi: 10.1093/toxsci/kfh159. [DOI] [PubMed] [Google Scholar]
- Cohen S. M. Toxicol. Pathol. 2010;38:487–501. doi: 10.1177/0192623310363813. [DOI] [PubMed] [Google Scholar]
- Sonich-mullin C., Fielder R., Wiltse J., Baetcke K., Dempsey J., Fenner-Crisp P., Grant D., Hartley M., Knaap A., Kroese D., Mangelsdorf I., Meek E., Rice J. M., Younes M. Regul. Toxicol. Pharmacol. 2001;34:146–152. doi: 10.1006/rtph.2001.1493. [DOI] [PubMed] [Google Scholar]
- Meek M. E., Bucher J. R., Cohen S. M., Dellarco V., Hill R. N., Lehman-McKeeman L. D., Longfellow D. G., Pastoor T., Seed J., Patton D. E. Crit. Rev. Toxicol. 2003;33:591–653. doi: 10.1080/713608373. [DOI] [PubMed] [Google Scholar]
- Seed J., Carney E. W., Corley R. A., Crofton K. M., DeSesso J. M., Foster P. M., Kavlock R., Kimmel G., Klaunig J., Meek M. E., Preston R. J., Slikker Jr. W., Tabacova S., William G. M. Crit. Rev. Toxicol. 2005;35:663–672. doi: 10.1080/10408440591007133. [DOI] [PubMed] [Google Scholar]
- Boobis A. R., Cohen S. M., Dellarco V., McGregor D., Meek M. E., Vickers C., Willcocks D., Farland W. Crit. Rev. Toxicol. 2006;36:781–792. doi: 10.1080/10408440600977677. [DOI] [PubMed] [Google Scholar]
- Boobis A. R., Doe J. E., Heinrich-Hirsch B., Meek M. E., Munn S., Ruchirawat M., Schlatter J., Seed J., Vickers C. Crit. Rev. Toxicol. 2008;382:87–96. doi: 10.1080/10408440701749421. [DOI] [PubMed] [Google Scholar]
- Ohnishi T., Arnold L. L., He J., Clark N. M., Kawasaki S., Rennard S. I., Bioyer C. W., Cohen S. M. Toxicology. 2007;241:58–65. doi: 10.1016/j.tox.2007.08.088. [DOI] [PubMed] [Google Scholar]
- Wood C. E., Hukkanen R. R., Sura R., Jacobson-Kram D., Nolte T., Odin M., Cohen S. M. Toxicol. Pathol. 2015;436:760–775. doi: 10.1177/0192623315576005. [DOI] [PubMed] [Google Scholar]
- Cohen S. M. Drug Metab. Rev. 1998;30:339–357. doi: 10.3109/03602539808996317. [DOI] [PubMed] [Google Scholar]
- Cohen S. M., Cano M., Johnson L. S., St John M. K., Asamoto M., Garland E. M., Thyssen J. H., Sangha G. K., Van Goethem D. L. Carcinogenesis. 1994;15:2593–2597. doi: 10.1093/carcin/15.11.2593. [DOI] [PubMed] [Google Scholar]
- Wisler J. A., Afshari C., Fielden M., Zimmerman C., Taylor S., Carnahan J., Vonderfecht S. Toxicol. Pathol. 2011;39:809–822. doi: 10.1177/0192623311410442. [DOI] [PubMed] [Google Scholar]
- Dragan Y. P., Bidlack W. R., Cohen S. M., Goldsworthy T. L., Hard G. C., Howard P. C., Riley R. T., Voss K. A. Toxicol. Sci. 2001;61:6–17. doi: 10.1093/toxsci/61.1.6. [DOI] [PubMed] [Google Scholar]
- Cohen S. M. Food Chem. Toxicol. 1995;33:715–730. doi: 10.1016/0278-6915(95)00040-9. [DOI] [PubMed] [Google Scholar]
- Cohen S. M., Ohnishi T., Clark N. M., He J., Arnold L. L. Toxicol. Pathol. 2007;35:337–347. doi: 10.1080/01926230701197115. [DOI] [PubMed] [Google Scholar]
- Clayson D. B., Fishbein L., Cohen S. M. Food Chem. Toxicol. 1995;33:771–784. doi: 10.1016/0278-6915(95)00044-3. [DOI] [PubMed] [Google Scholar]
- Cohen S. M., Johansson S. L., Arnold L. L., Lawson T. A. Food Chem. Toxicol. 2002;40:793–799. doi: 10.1016/s0278-6915(02)00020-0. [DOI] [PubMed] [Google Scholar]
- Dominick M. A., White M. R., Sanderson T. P., Van Vleet T. R., Cohen S. M., Arnold L. L., Cano M., Tannehill-Gregg S., Moehlenkamp J. D., Waites C. R., Schilling B. E. Toxicol. Pathol. 2006;34:903–920. doi: 10.1080/01926230601072327. [DOI] [PubMed] [Google Scholar]
- Cohen S. M., Cano M., Garland E. M., St John M. K., Arnold L. Carcinogenesis. 1995;16:343–348. doi: 10.1093/carcin/16.2.343. [DOI] [PubMed] [Google Scholar]
- Cohen S. M., Arnold L. L., Cano M., Ito M., Garland E. M., Shaw R. A. Carcinogenesis. 2000;21:783–792. doi: 10.1093/carcin/21.4.783. [DOI] [PubMed] [Google Scholar]
- Rodent Bladder Carcinogenesis Working Group Food Chem. Toxicol. 1995;33:797–802. [PubMed] [Google Scholar]
- Cohen S. M., Calcium phosphate-containing urinary precipitate in rat urinary bladder carcinogenesis, International Agency for Research on Cancer, IARC Scientific Publications, 1999, vol. 147, pp. 175–189. [PubMed] [Google Scholar]
- IARC Working Group, Consensus Report, International Agency for Research on Cancer, IARC Scientific Publications, 1999, vol. 147, pp. 1–32. [Google Scholar]
- Smith R. A., Christenson W. R., Bartels M. J., Arnold L. L., St John M. K., Cano M., Wahle B. S., McNett D. A., Cohen S. M. Toxicol. Appl. Pharmacol. 1998;150:402–413. doi: 10.1006/taap.1998.8435. [DOI] [PubMed] [Google Scholar]
- Guan N., Fan Q., Ding J., Zhao Y., Lu J., Ai Y., Xu G., Zhu S., Yao C., Jiang L., Miao J., Zhang H., Zhao D., Liu X., Yao Y. N. Engl. J. Med. 2009;36011:1067–1074. doi: 10.1056/NEJMoa0809550. [DOI] [PubMed] [Google Scholar]
- Andersen M. E., Meek E., Boorman G. A., Brusick D. J., Cohen S. M., Dragan Y. P., Frederick C. B., Goodman J. I., Hard G. C., O'Flaherty E. J., Robinson D. E. Toxicol. Sci. 2000;53:159–172. doi: 10.1093/toxsci/53.2.159. [DOI] [PubMed] [Google Scholar]
- Da Rocha M. S., Arnold L. L., De Oliveira M. L., Catalano S. M. I., Cardoso A. P. F., Pontes M. G. N., Ferrucio B., Dodmane P. R., Cohen S. M., de Camargo J. L. V. Crit. Rev. Toxicol. 2014;44:393–406. doi: 10.3109/10408444.2013.877870. [DOI] [PubMed] [Google Scholar]
- Lijinsky W., Saavedra J. E., Reuber M. D. Cancer Res. 1981;41:1288–1292. [PubMed] [Google Scholar]
- Da Rocha M. S., Dodmane P. R., Arnold L. L., Pennington K. L., Anwar M. A., Adams B. R., Taylor S. V., Wermes C., Adams T. B., Cohen S. M. Toxicol. Sci. 2012;128:1–8. doi: 10.1093/toxsci/kfs135. [DOI] [PubMed] [Google Scholar]
- Arnold L. L., Christenson W. R., Cano M., St John M. K., Wahle B. S., Cohen S. M. Fundam. Appl. Toxicol. 1997;40:247–255. doi: 10.1006/faat.1997.2391. [DOI] [PubMed] [Google Scholar]
- Yokohira M., Arnold L. L., Lautraite S., Sheets L., Wason S., Stahl B., Eigenberg D., Pennington K. L., Kakiuchi-Kiyota S., Cohen S. M. Food Chem. Toxicol. 2011;49:1215–1223. doi: 10.1016/j.fct.2011.02.022. [DOI] [PubMed] [Google Scholar]