

BCL11B is a transcriptional regulator of various developmental processes, and a BCL11B mutation has previously been reported in a single patient with syndromic immunodeficiency. Lessel et al. describe ten patients with mutations in BCL11B, all with global developmental delay, speech impairment and intellectual disability, but with no signs of immunodeficiency.
Keywords: BCL11B, developmental delay, intellectual disability, type 2 innate lymphoid cells, neurodevelopment
Abstract
The transcription factor BCL11B is essential for development of the nervous and the immune system, and Bcl11b deficiency results in structural brain defects, reduced learning capacity, and impaired immune cell development in mice. However, the precise role of BCL11B in humans is largely unexplored, except for a single patient with a BCL11B missense mutation, affected by multisystem anomalies and profound immune deficiency. Using massively parallel sequencing we identified 13 patients bearing heterozygous germline alterations in BCL11B. Notably, all of them are affected by global developmental delay with speech impairment and intellectual disability; however, none displayed overt clinical signs of immune deficiency. Six frameshift mutations, two nonsense mutations, one missense mutation, and two chromosomal rearrangements resulting in diminished BCL11B expression, arose de novo. A further frameshift mutation was transmitted from a similarly affected mother. Interestingly, the most severely affected patient harbours a missense mutation within a zinc-finger domain of BCL11B, probably affecting the DNA-binding structural interface, similar to the recently published patient. Furthermore, the most C-terminally located premature termination codon mutation fails to rescue the progenitor cell proliferation defect in hippocampal slice cultures from Bcl11b-deficient mice. Concerning the role of BCL11B in the immune system, extensive immune phenotyping of our patients revealed alterations in the T cell compartment and lack of peripheral type 2 innate lymphoid cells (ILC2s), consistent with the findings described in Bcl11b-deficient mice. Unsupervised analysis of 102 T lymphocyte subpopulations showed that the patients clearly cluster apart from healthy children, further supporting the common aetiology of the disorder. Taken together, we show here that mutations leading either to BCL11B haploinsufficiency or to a truncated BCL11B protein clinically cause a non-syndromic neurodevelopmental delay. In addition, we suggest that missense mutations affecting specific sites within zinc-finger domains might result in distinct and more severe clinical outcomes.
Introduction
BCL11B (RefSeq NM_138576.3, MIM 606558) is a lineage-specific, Krüppel‐like C2H2 zinc-finger-containing transcriptional regulator of different physiological processes including apoptosis, cell proliferation, and differentiation (Lennon et al., 2017). Biallelic loss of Bcl11b leads to perinatal lethality in mice, accompanied by defects in the development of the CNS (Arlotta et al., 2005; Simon et al., 2012), the epidermis (Golonzhka et al., 2009a), the teeth (Golonzhka et al., 2009b), as well as in the development and maintenance of the T cell lineage (Wakabayashi et al., 2003). Indeed, Bcl11b-deficient mice show an arrest at the CD4−CD8− double-negative stage of thymocyte development, resulting in a loss of Tαβ (but not Tγδ cells) and reprogramming to natural killer (NK)-like cells (Wakabayashi et al., 2003; Li et al., 2010). Recently, BCL11B has been shown to control lineage specification and function of type 2 innate lymphoid cells (ILC2) in mice (Califano et al., 2015; Walker et al., 2015; Yu et al., 2015, 2016), an innate counterpart of Th2 lymphocytes. In addition to its function in the immune system, BCL11B plays a pivotal role in murine neurogenesis, i.e. in the development of corticospinal motor neurons (Arlotta et al., 2005), differentiation of striatal medium spiny neurons (Arlotta et al., 2008), and the development and maintenance of the dentate gyrus by regulation of progenitor cell proliferation (Simon et al., 2012, 2016). Notably, loss of Bcl11b in neurons of the murine dentate gyrus results in impaired spatial learning and memory and the formation of hippocampal circuitry (Simon et al., 2012, 2016). Given these pleiotropic functions in mice, BCL11B constitutes an excellent candidate gene for human neurodevelopmental and/or immunological disorders. Indeed, a recent study reported severe combined immunodeficiency (SCID), severe developmental delay, craniofacial abnormalities, absence of corpus callosum, and erythematous psoriasiform dermatitis in a single individual with a de novo BCL11B missense alteration (Punwani et al., 2016). Moreover, genetic alterations in BCL11A, another member of the BCL11 family, have been associated with monogenic intellectual disability and persistency of foetal haemoglobin (Basak et al., 2015; Dias et al., 2016). Further, Bcl11a deficiency results in the lack of B lymphocytes and plasmacytoid dendritic cells (Liu et al., 2003; Ippolito et al., 2014). Interestingly, recent studies have shown that neurons in mucosal tissues react to environmental changes by producing the neuropeptide neuromedin U, which upon interaction with its receptor on ILC2s promotes type 2 cytokine production and tissue inflammation (Cardoso et al., 2017; Klose et al., 2017; Wallrapp et al., 2017). These findings indicate that neuroimmune sensory units act in concert to regulate mucosal tissue homeostasis and may have evolved together.
Here we report developmental delay and intellectual disability in 13 individuals bearing heterozygous alterations in BCL11B. Remarkably, all analysed individuals exhibited a severe reduction of peripheral ILC2s and impaired T cell development, but no overt immune deficiency.
Materials and methods
Patients and samples
All biological samples and images were obtained following written informed consent from the parents of the affected individuals. The study was performed in accordance with the Declaration of Helsinki protocols and approved by the ethics committees of the respective institutions. Some of the investigators presenting patients in this study were connected through GeneMatcher, a web-based tool for researchers and clinicians working on identical genes (Sobreira et al., 2015).
Genetic analysis
Whole-exome sequencing (WES) experiments were performed in seven different centres with slightly different procedures. Briefly, trio-WES in Families A and B and single-WES in Patient D:I-1 was performed with a SureSelect Human All Exon 50 Mb V5 Kit (Agilent Technologies), and sequenced on a HiSeq2500 system (Illumina), as described before (Hempel et al., 2015; Lessel et al., 2017). Trio-WES in Family C was performed using the SureSelect XT Human All Exon V5 kit (Agilent Technologies) and sequenced in rapid run mode on the HiSeq2500 sequencing system (Illumina), as described before (Hempel et al., 2015). Trio-WES in Family E was performed by Genomics Platform at the Broad Institute of Harvard and MIT (Broad Institute, Cambridge, MA, USA), as previously described (Lek et al., 2016). Trio-WES in Family F was performed using the SureSelect Target Enrichment System (Agilent) and sequenced on the Illumina HiSEQ 2500 platform, as described before (de Bruin et al., 2016). Single-WES sequencing in Patient J:II-1 was performed at the CHU Sainte-Justine CIGCP genomics platform using SureSelect V4 (XT) exome capture kit (Agilent) and paired-end sequencing on a HiSeq2500 (Illumina), as described before (Gauthier et al., 2018). Trio-WES in Family K was performed using Agilent Clinical Research Exome kit (Agilent Technologies), as described previously (Tanaka et al., 2015). Trio-WES in Family L was performed on a NextSeq 500 Sequencing System (Illumina), with a 2 × 150 bp high output sequencing kit after enrichment with Seq Cap EZ MedExome kit (Roche), according to manufacturer’s specifications. All putative de novo variants were validated and confirmed by Sanger sequencing, as described previously (Hempel et al., 2015). For Family G, duo-WES was performed using Agilent SureSelectXT Clinical Research Exome capture kit (Agilent Technologies) and sequenced on a NexSeq500 system (Illumina) as described previously (Louie et al., 2017). Further, a phenotype-driven analysis did not yield any variants of clinical significance. To identify rare variants in uncharacterized genes, a phenotype-independent inheritance-driven analysis was performed to identify rare variants (below 0.01 allele frequency in the public SNP databases) that were shared between the affected proband and her mother, resulting in 56 variants. Of the 56 variants, two were deemed potentially causative based on pathway analysis and or animal studies. Sanger sequencing was performed under standard PCR conditions to confirm the findings. Primer pairs for the amplification are available on request. Karyotyping was performed by standard procedures.
Genomic regulatory blocks
Genomic regulatory blocks were called using human:mouse conserved non-coding elements, with minimal conservation of 96%, over a minimum of 50 bp, as described previously (Harmston et al., 2017).
FANTOM5 permissive enhancers were obtained from http://fantom.gsc.riken.jp/5/datafiles/latest/extra/Enhancers/human_permissive_enhancers_phase_1_and_2.bed.gz.
Breakpoint mapping by whole genome sequencing
Blood DNA was extracted with the QIAamp® DNA Blood Midikit (Qiagen) according to the manufacturer’s instructions. Whole genome libraries were prepared following the Illumina TruSeq protocol (Illumina) with 3 µg DNA. Libraries of 350-bp fragments were sequenced on an Illumina NextSeq 500 as paired-end 101-bp reads. The sequencing depth was 7.85× and 10.47×. For each sample, an alignment of the reads against the hg19 version of the human genome hg19 was done using BWA-MEM v.0.7.10. The reads were then sorted using Samtools v.1.3.1(Li et al., 2009), and the duplicates removed by PicardTools v.1.138 (picard.sourceforge.net). Then, the structural variants were detected using BreakDancer v.1.4.5 (Chen et al., 2009), and annotated using an in-house script, mainly for the purpose of filtering them on the basis of their occurrence in a local database. Integrative Genomics Viewer v.2.3 (Thorvaldsdottir et al., 2013) was used for structural variant visualization. Primer pairs were selected on each side of the breakpoint region delimited by WGS (primers sequence available on request). Junction fragments were amplified using the Taq DNA Core kit 25 (MP Biomedicals). DNA from a control not carrier of chromosomal rearrangement was amplified as a negative control. PCR products were verified on LabChip GX (PerkinElmer). Then specific products corresponding to the junction fragment were sequenced by the Sanger method (Genoscreen).
Quantitative real-time PCR
Total RNA was extracted from blood cells collected on PAXgene tubes according to the manufacturer’s instructions (Qiagen). Reverse transcription was performed with 0.5 µg RNA from patients and sex-matched controls using random primers and SuperScript® II Reverse Transcriptase (Invitrogen). Quantitative PCR for the BCL11B gene was performed in triplicate with 2 µl of cDNA diluted to 1/20 using the QuantiTect® SYBR® Green PCR kit (Qiagen) on a LightCycler 2000 (Roche). ACTB was used for normalization. Relative quantification was performed according to the 2−ΔΔCt method (Livak and Schmittgen, 2001).
Ex utero electroporation and hippocampal slice culture
All mouse experiments were carried out in compliance with German law and approved by the respective government offices in Tübingen. Ex utero electroporation and organotypic slice cultures of Bcl11bflox/flox; Emx-1Cre as well as control embryonic brains were carried out as described previously (Simon et al., 2012, 2016; Venkataramanappa et al., 2015). Briefly, up to 9 µg DNA was electroporated into the prospective dentate gyrus area of embryonic brains at embryonic Day 15.5 using five pulses at 50 V, brains were cut into 250 µm slices and kept in culture up to in vitro Day 11. To determine the proliferation rate of dentate gyrus cells, BrdU was added to the culture medium for the first 20 h after electroporation. For the rescue experiments, brains were electroporated with either empty vector pIRES2 EGFP or pIRES2 EGFP Bcl11b-hu (containing wild-type human BCL11B cDNA), pIRES2 Bcl11b-hu dup (human BCL11B cDNA containing the c.2449_2456dupAGCCACAC variant), or with the corresponding mouse cDNA constructs.
Immunophenotyping
Fifty microlitres of blood were stained with specific antibodies (BioLegend or BD Biosciences), lysed with BD FACS Lysing Solution, fixed in 1% paraformaldehyde and measured on a LSR Fortessa (BD Biosciences). ILC2s were identified in the lymphocyte gate as CD45+, lin− (CD3, CD19, CD14, CD34 and CD94), HLA-DR−, CD127+ CD161+ CRTH2+ and c-kit+/−. Recent thymic emigrants were defined as CD45RA+ CD31+ cells in the CD4+ subset. Tγδ cells were positively identified with antibodies against CD3 and pan-Tγδ (clone 11F2), Vd1 (TS-1), Vd2 (123R3) and Vg9 (IMMU_360). Other markers used for immune profiling of T cells were: CD4, CD8a, CD8b, Va7.2, CCR7, CD45RA, CD45RO, CD28, CD27, CD95, CD57, HLA-DR, CD69, CD38, CD39, CD73, CD25, CD127, CCR4, CCR6, CD161, CXCR3, CCR10 and CRTh2, CXCR5. For intracytoplasmic staining, peripheral blood mononuclear cells were first isolated by Ficoll gradient and subsequently stimulated with PMA/ionomycin in the presence of Brefeldin A for 5 h. After stimulation, cells were incubated with surface markers for CD3, CD4, CD8 and Tγδ, permeabilized and stained for intracellular cytokines IFNγ, TNFα, IL-17, IL-4, IL-10, IL-8 and IL-2. All other immune cell populations were identified according to commonly-used immune cell surface markers. Data were analysed using the software FlowJo 10.0.8 (TreeStar).
T cell receptor repertoire analysis by next generation sequencing
Peripheral blood mononuclear cells were stained with following antibodies: dead/alive (DAPI), hCD45 (APC-Vio770), hCD3 (PE-Cy7), hTCR αβ (FITC), hTCRγδ (PE), hVγ9 (PC5), hCD4 (PerCP), hCD8 (VioGreen), hCD25 (BrilliantViolet605) and hCD127 (APC). They were sorted into CD8+ and CD4+ conventional T cells, as well as CD4+ CD25+ CD127− regulatory T cells using a FACS Aria Fusion flow cytometer. Messenger RNA was extracted using the RNeasy® Plus Micro Kit (Qiagen) and then reverse-transcribed into cDNA according to the SMARTer® RACE 5′-3′ PCR Kit (Clontech) manual. Combined amplification of the T-cell receptor β CDR3 region and Illumina adaptor sequences was performed with the Advantage 2 PCR Kit (Clontech). After amplicon DNA size confirmation by gel electrophoresis, bands were extracted using the Gel Extraction Kit (QIAGEN). Indexing of the samples was performed with Nextera Primer Kit (Illumina) in an additional Advantage 2 PCR reaction and the product was purified with the Agencourt AMPure® XP Kit. DNA concentration was measured via the Qubit 2.0 fluorometer, samples were pooled and the pool concentration was set to 4 nM. Denaturation and dilution of the pool was performed according to the Illumina MiSeq Dilution and Denaturation Guide. Finally, next generation sequencing was completed using the Illumina MiSeq System. Finally, FastQ files were annotated at IMGT/HighV-Quest database and processed with tcR-package and VDJtools.
Statistics
Analysis was performed using Prism 6 (GraphPad). Significance was determined with two-tailed unpaired Student’s t-test. P-values of <0.05 were considered significant. Data represent mean ± standard error of the mean (SEM) unless otherwise indicated.
Data availability
The raw whole-exome sequencing data that support the findings in patients cannot be made publicly available for confidentiality reasons. Qualified researchers may apply for access to these data, pending institutional review board approval. All other data generated or analysed during this study are included herein and in the Supplementary material.
Results
Clinical characteristics of patients
We studied 13 patients affected by global developmental delay and intellectual disability with speech impairment. Autistic features were observed in four patients. Partially overlapping facial dysmorphisms were observed in all patients. Brain MRI gave normal results in all patients except for Patient H:II-1, who had a moderate ectopia of amygdala, and Patient J:II-1, who had hypoplasia of the globus pallidus. Refractive error was observed in five patients. Small teeth, oligodontia and/or enamel defects were present in five patients. One patient presented with congenital erosive dermatitis, and one with multiple café au lait spots. Concerning the immune system, the individual bearing a missense mutation (Patient E:II-1) had low T cell receptor excision circles at birth, though T cell measurements at later time points revealed T cell counts close to standard values. Eight patients showed exacerbated type 2 responses, namely eosinophilia (4/10) and allergies or asthma (7/12) (Fig. 1, Table 1 and Supplementary material).
Figure 1.

Images of patients with BCL11B associated disorder. Facial images of Patient A:II-3 at age of 2 11/12 and 3 11/12 years (A); Patient B:II-2 at the age of 8 11/12 and 15 1/12 years (B); Patient C:II-2 at the age of 1 8/12 years (C); Patient D:II-1 at the age of 1 6/12 years (D); Patient E:II-1 at the age of 1 month and 2 1/12 years (E); Patient F:II-2 at the age of 5, 7 and 17 years (F); Patient H:II-1 at 11 years (G); and Patient J:II-1 at 6 2/12 years (H).
Table 1.
Clinical characteristics of patients with BCL11B alterations
| Patient | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Clinical findings | A:II-3 | B:II-2 | C:II-2 | D:II-1 | E:II-1 | F:II-2 | G:III-1 | H:II-1 | I:II-2 | J:II-1 | K:II-1 | L:II-2 |
| Sex | Female | Male | Male | Female | Male | Male | Female | Male | Male | Female | Male | Male |
| Ethnicity | Caucasian | Caucasian | Caucasian | Arab | Caucasian | Brazilian | Caucasian | Caucasian | Caucasian | Caucasian | Caucasian | North Africa |
| Age at last examination (years) | 3 11/12 | 15 1/12 | 1 8/12 | 1 10/12 | 2 3/12 | 17 7/12 | 13 | 11 | 29 | 6 6/12 | 9 11/12 | 9 9/12 |
| Cognitive and motor development | ||||||||||||
| Intellectual disability | + | + | + | + | + | + | + | + | + | + | + | + |
| Speech impairment | + | + | + | + | + | + | + | + | + | + | + | + |
| Delay in motor development | + | + | + | + | + | + | + | + | − | + | + | + |
| Autistic features | + | − | + | − | − | − | + | + | − | − | − | − |
| Dysmorphic features | ||||||||||||
| Myopathic facial appearance | + | − | + | + | + | − | + | − | − | + | − | − |
| Thin eyebrows | + | + | + | − | − | − | + | − | − | − | − | + |
| Small palpebral fissures | + | + | + | − | + | − | + | − | + | + | − | − |
| Hypertelorism | + | − | + | + | + | − | + | − | − | − | + | + |
| Prominent nose | + | + | + | − | + | + | + | + | + | − | − | + |
| Long philtrum | + | + | + | + | − | − | + | − | + | + | + | + |
| Thin upper lip vermilion | + | + | + | + | + | + | + | + | + | − | + | + |
| Other | ||||||||||||
| Refractive error | Hyperopia | − | Hyperopia | − | − | Myopia | − | − | − | Myopia | Exotropia | − |
| Dental anomalies | + | + | − | − | + | + | − | − | − | − | − | + |
| Feeding difficulties | − | − | + | − | + | − | − | − | − | + | − | − |
| Immune system function | ||||||||||||
| Immune response | − | Frequent infectionsa | Frequent infectionsa | − | Low TREC at birth | Frequent/atypical infections | − | − | − | − | − | Frequent infectionsa |
| Allergy/asthma | − | − | + | − | + | + | − | + | − | + | + | + |
| BCL11B alteration | p.Gly820 Alafs*27 | p.Gly649Ala fs*67 | p.Ala891Pro fs*106 | p.Thr502His fs*15 | p.Asn807Lys | p.Cys81Leu fs*76 | p.Asp534Thr fs*29 | 46,XY,t(4;14) (p15;q32.1) | 46,XY,t(4;14) (q31.1;q32.2) | p.Glu499* | p.Tyr455* | p.Arg518Alafs*45 |
+ = present; − = absent; TREC = T cell receptor excision circles.
aReported by the family.
Genetic studies
Using WES, we identified heterozygous germline mutations in BCL11B in nine unrelated patients, namely six frameshift, two nonsense and one missense mutation (Fig. 2A, Table 1 and Supplementary Table 1), none of which was present in either of the parents of a given patient, thus proving that these mutations occurred de novo. In addition, we identified an inherited heterozygous frameshift mutation, p.(Asp534Thrfs*29), transmitted from an affected mother with intellectual disability, who did not consent to further detailed clinical or laboratory investigations in this study, to her similarly affected daughter. One of the frameshift mutations [p.(Cys81Leufs*76)] is located in exon 2, predicted to activate the nonsense-mediated mRNA decay and to result in haploinsufficiency. Seven mutations [p.(Tyr455*), p.(Glu499*), p.(Thr502Hisfs*15), p.(Arg518Alafs*45), p.(Asp534Thrfs*29), p.(Gly649Alafs*67) and p.(Gly820Alafs*27)] are located in the last exon (exon 4) and are thus predicted to escape nonsense-mediated mRNA decay and probably result in a protein with loss of, at least, the last two C-terminal DNA-binding zinc-finger domains. One further single base-pair deletion is predicted to change the reading frame and to remove the physiological stop codon, with a concomitant extension of the protein for 103 further erroneous amino acids [p.(Ala891Profs*106)], probably triggering non-stop mRNA decay (Hamby et al., 2011). The missense alteration, c.2421C>G, p.(Asn807Lys), affects the alpha-helix containing DNA recognition site within the zinc finger domain ZnF4_C2H2. This domain—including the mutated asparagine residue—is perfectly conserved in BCL11B and BCL11A vertebrate orthologues (Supplementary Fig. 1A). Recently, an independent deleterious de novo BCL11B missense mutation, c.1323T>G, p.(Asn441Lys), was identified in a single patient affected by syndromic immunodeficiency (Punwani et al., 2016). Notably, both missense mutations affect one of the four ZnF_C2H2 ‘specificity residues’ (Wolfe et al., 2000) of the DNA-contacting alpha-helix within the ZnF2_C2H2 and ZnF4_C2H2, respectively. Using ChIP-seq analysis, Punwani et al. (2016) demonstrated that the p.(Asn441Lys) mutation results not only in impaired BCL11B binding to known target DNA sites, but also promotes binding to novel DNA binding sites. Given that the prediction algorithm ‘Zinc Finger Recognition Code’ (Najafabadi et al., 2015) suggests that both missense mutations are predicted to bind to different alternative genomic sequences (Supplementary Fig. 1B), we hypothesize that the here-identified missense mutation may have comparable effects, and thus may cause the severe clinical phenotype by additional gain-of-function mechanism.
Figure 2.

Genetic data of patients with BCL11B associated disorder. (A) Schematic protein structure of BCL11B: the position of the mutations identified in this study are marked with vertical arrows and shown in red, and the recently identified missense mutation (Punwani et al., 2016) is shown in black. C = C terminus; N = N terminus; ZnF = zinc-finger C2H2 domain. (B) Genomic context of the translocation breakpoints in Patients H:II-1 and I:II-2. Top: BCL11B genomic regulatory block (GRB) model. Middle: The position of the breakpoints in chromosome 14 is shown in blue. The yellow bar shows the position of the T cell-specific enhancer (Li et al., 2013). Bottom (in pink): Location of permissive enhancers according to the FANTOM5 algorithm. The legend at the bottom describes the symbols used.
None of the here-identified BCL11B mutations were present in dbSNP, ExAC or gnomAD browsers, precluding that they represent rare polymorphisms. In addition, the residual variation intolerance (RVI) score of BCL11B, which is based on the ExAC sequencing data and quantifies gene intolerance to functional mutations (Petrovski et al., 2013), lies on the 11th percentile, which is even lower than the average RVI score for genes known to be involved in developmental disorders (20th percentile). Further, the probability of loss-of-function intolerance (pLI) for BCL11B is 0.93, suggesting strong intolerance, and BCL11B is ranked 48 of 18 000 analysed genes by its missense Z-score of 6.42 (Lek et al., 2016), which is even higher than the average Z-score for genes involved in developmental disorders (Samocha et al., 2014). Taken together, these findings corroborate the deleterious effect of the identified germline mutations.
In Patients H:II-1 and I:II-2, karyotype analysis revealed de novo novel balanced translocations (Supplementary Fig. 2) which were further characterized by WGS. For Patient H:II-1, the breakpoints were mapped to chr4:24,332,309–24,332,315 (hg19) and chr14: 98,758,657–98,758,653 (hg19). For Patient I:II-2, the breakpoints were mapped to chr4:148,252,852–148,252,854 (hg19) and chr14: 99,097,976–99,097,980 (hg19). None of the rearrangements disrupted a gene, but the breakpoint in chromosome 14 was localized in both cases in close vicinity to the 3′ end of BCL11B, at a distance of 877 kb and 538 kb, respectively (Fig. 2B). Both breakpoints are located within a genomic regulatory block of BCL11B that covers a 2.95 Mb region around the gene. FANTOM5 defines 70 permissive enhancers within this genomic regulatory block; the ones involved in long-range regulation in the region, i.e. not targeting the closest gene, are predicted to regulate the activity of BCL11B through looping events. Moreover, both breakpoints are located between BCL11B and its previously reported T cell specific enhancer (Li et al., 2013). Therefore, it is likely that, by moving BCL11B out of its native regulatory landscape, the spatiotemporal precision of gene expression might be disrupted. We therefore assessed the relative amount of BCL11B mRNA in blood cells and indeed found approximately half the amount of BCL11B mRNA in both patients as compared to controls, thus confirming a positional regulatory effect (Supplementary Fig. 2C).
Pathogenic nature of the C-terminally located frameshift mutations
To gain further insight into the molecular effects of the four premature termination codon mutations likely escaping NMD [i.e. p.(Tyr455*), p.(Glu499*), p.(Thr502Hisfs*15), p.(Arg518Alafs*45), p.(Asp534Thrfs*29), p.(Gly649Alafs*67) and p.(Gly820Alafs*27)] we turned to a previously-established mouse model (Simon et al., 2012; Venkataramanappa et al., 2015) and analysed the influence of the most C-terminally located frameshift mutation p.(Gly820Alafs*27) on hippocampal neurogenesis. As expected, hippocampal slice cultures derived from Bcl11b homozygous mutant mice displayed severely reduced progenitor cell proliferation at 11 days in vitro (Simon et al., 2012). This phenotype was completely rescued upon reintroduction of a cDNA construct (pIRES2-EGFP) into the Bcl11b mutant hippocampus containing either wild-type BCL11B (human) or Bcl11b (murine), but not by an equimolar amount of cDNA bearing the mutation as identified in Patient A:II-3 (Fig. 3 and Supplementary Fig. 3). Moreover, immunohistological analysis demonstrated that, in contrast to the wild-type constructs, no stable BCL11B protein was expressed from the mutation-containing cDNAs in dentate neurons (Fig. 3 and Supplementary Fig. 3). These data suggest that these premature termination codon mutations in BCL11B result in a functional null allele and recapitulate the hippocampal phenotype observed in Bcl11b mutant mice (Simon et al., 2012).
Figure 3.
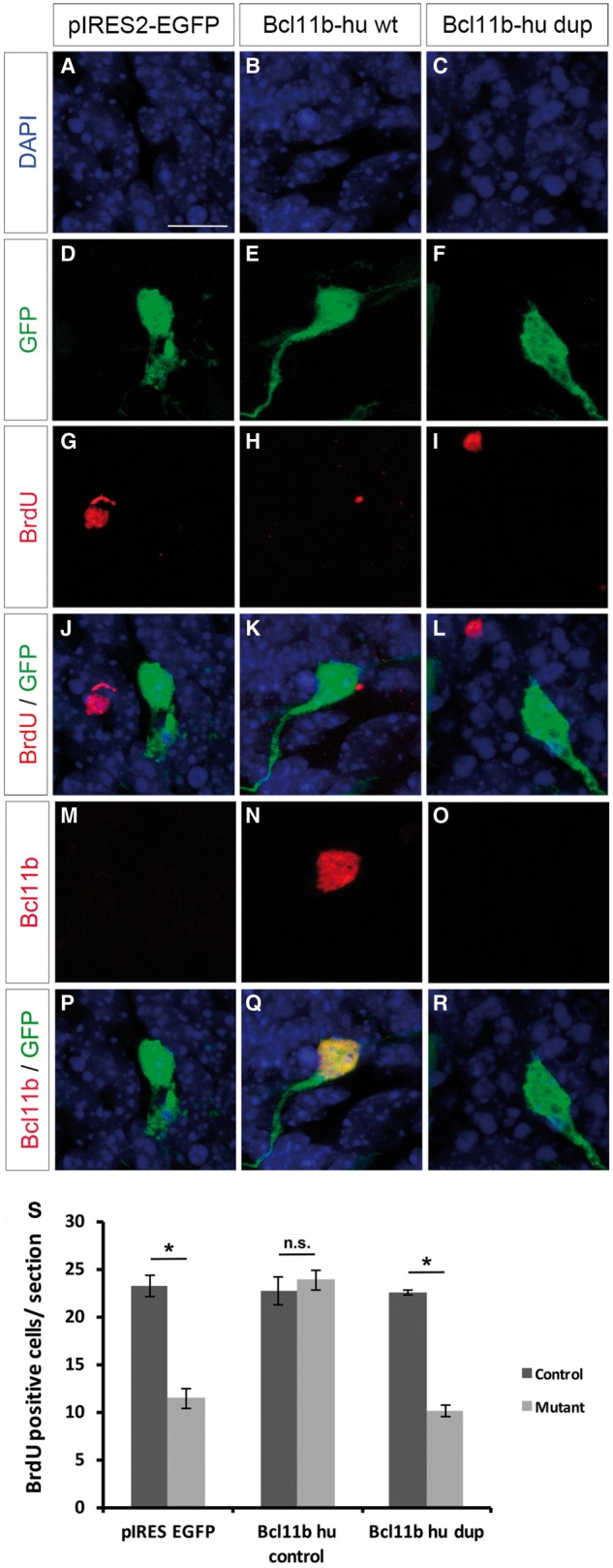
Functional analysis of the human p.Gly820Alafs*27 BCL11B mutation. (A–R) Immunohistological analysis of Bcl11bflox/flox;Emx1-Cre hippocampal slice cultures after in vitro Day 11 electroporation. Animals were electroporated with pIRES-EGFP (A, D, G, J, M and P), pIRES2-EGFP Bcl11b-hu wt (wild-type human BCL11B cDNA) (B, E, H, K, N and Q) as well as pIRES2-EGFP Bcl11b-hu dup (human BCL11B cDNA containing the c.2449_2456dupAGCCACAC, p.Gly820Alafs*27) (C, F, I, L, O and R). DAPI (blue) as morphological marker, GFP (green) and BrdU (red) as marker for cell proliferation as well as Bcl11b (red). Images were taken at 63× magnification, 2× zoom. (S) Statistical analysis of BrdU-positive cells in Bcl11bflox/flox;Emx1-Cre and control hippocampal slice cultures. [t-test, *P < 0.0005; control n = 4, mutant (Bcl11bflox/flox; Emx1-Cre n = 3)]. Scale bar = 10 µm (A).
Patients with BCL11B mutations show impaired T cell development and a severe reduction in peripheral type 2 innate lymphoid cells
Given the importance of BCL11B in T cell development and in the specification of the ILC2 lineage in mice (Wakabayashi et al., 2003; Li et al., 2010; Califano et al., 2015; Walker et al., 2015; Yu et al., 2015), and the findings in the previously published patient (Punwani et al., 2016), we evaluated the immune compartment of eight available patients, including six bearing frameshift mutations, one with a balanced translocation and the patient with the novel missense mutation (Supplementary Table 2). Haematocrit and differential blood counts were normal in all individuals, except for the eosinophil counts, which were high in three cases. The percentage of T cells was low in two individuals, who in turn showed higher relative frequencies of B cells. NK cells were present in normal numbers in all patients. We next performed exhaustive immune profiling of the T cell compartment, encompassing the usage of T cell receptor chains, frequencies of classical and innate-like T lymphocytes and regulatory cells, frequencies of naïve, memory and effector subsets, expression of activation markers, and production of inflammatory cytokines. In a 2D t-distributed stochastic neighbour embedding (t-SNE) plot that analyses frequencies of 102 T cell subpopulations, patients with alterations in BCL11B clearly cluster apart from healthy children (Fig. 4A). The traits accounting for the differences in the T cell compartment between healthy donors and patients with mutations in BCL11B can be traced to: (i) an abnormally low percentage of CD4+ recent thymic emigrants (Fig. 4B); (ii) an overrepresentation of cells of the Tγδ lineage and TCR Vδ1 bias in detriment of the most common Vδ2γ 9 chains (Fig. 4D); and (iii) changes in the frequency of effector and cytokine producing T cells (Supplementary Table 2).
Figure 4.

Impaired T cell development in patients with BCL11B associated disorder. (A) Two-dimensional t-distributed stochastic neighbour embedding (t-SNE) plot showing the clustering of seven BCL11B patients (red circles) and 14 healthy controls aged under 16 (grey circles) according to 102 T cell traits. (B) Frequency of recent thymic emigrants (RTE) (CD4+ CD45RA+ CD31+) in relation to age in control donors (black circles) and BCL11B patients (red circles). (C) Shannon Diversity Index of the TCRαβ-repertoire in CD4 and CD8 T cells in unaffected donors aged 15 (HD) and in individuals with BCL11B mutations (mut). (D) Percentage of T-γδ cells and usage of TCRδ chains in unaffected donors aged under 16 (HD) and in BCL11B patients (mut). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
The involvement of murine Bcl11b in the lineage specification of ILC2s prompted us to analyse the ILC compartment in these patients. We found that the frequency and absolute number of ILCs in peripheral blood of our patients were similar to healthy donors. However, in line with the previously reported findings in mice (Califano et al., 2015; Walker et al., 2015; Yu et al., 2015, 2016) we observed a severe reduction, both in frequency and absolute numbers, of ILC2s in the peripheral innate lymphoid cell compartment (Fig. 5). In contrast to Bcl11b-deficient mice (Walker, 2016), however, ILC3 cells were not elevated. Therefore, our findings rather support a role for BCL11B in the development of ILC2s (Walker, 2015; Yu, 2015), and not merely in lineage maintenance and function.
Figure 5.

Drastic reduction in peripheral ILC2 in patients with BCL11B associated disorder. (A) Dot plots corresponding to the gating of ILC2s in an unaffected individual (left) and in Patient C:II-2 (right). The plots show events in the CD45+ lineage− HLA-DR− gate (left plots) and after selection of the CD127+ cells (right plots). (B) Frequency and absolute numbers of ILCs in unaffected individuals and in individuals with BCL11B mutations. *P < 0.05, **P < 0.01. Significant outliers (Grubb’s test P < 0.05) are displayed in parentheses, but were excluded from statistical analysis.
In agreement with the previously reported repressive function of BCL11B on Th2 differentiation (Califano et al., 2014), exacerbated Th2 responses were clinically recognized in five patients, including two with asthma (Table 1 and Supplementary Table 2). Although several studies suggested that ILC2s may be crucial players in allergic airway responses (van Rijt et al., 2016), these cells are also required for restoring epithelial integrity and airway remodelling upon lung inflammation (Monticelli et al., 2011; Kubo, 2017). Thus, a combination of exacerbated Th2 immunity and the lack of ILC2 may contribute to development of asthma in our patients.
Discussion
A de novo missense mutation in BCL11B has recently been reported in a single patient affected by a leaky form of severe combined immunodeficiency (SCID), severe developmental delay, craniofacial abnormalities, absence of corpus callosum and erythematous psoriasiform dermatitis (Punwani et al., 2016). Here we describe 13 patients bearing heterozygous BCL11B mutations, all of whom are affected by global developmental delay, with speech impairment and mild to moderate intellectual disability, mild facial dysmorphisms, accompanied by impaired development of the immune system, but without overt signs of immunodeficiency. These findings add this zinc-finger transcription factor to the rapidly growing list of monogenic disease genes for human developmental disorders.
Of the seven identified frameshift mutations, [p.(Cys81Leufs*76) and p.(Ala891Profs*106)] are predicted to result in haploinsufficiency, as we have shown for both chromosomal rearrangements in Patients H:II-1 and I:II-2. On the other hand, [p.(Tyr455*), p.(Glu499*), p.(Thr502Hisfs*15), p.(Arg518Alafs*45), p.(Asp534Thrfs*29), p.(Gly649Alafs*67) and p.(Gly820Alafs*27)] are predicted to result in a protein with loss of the last C-terminal DNA-binding zinc-finger domains. To evaluate the pathogenicity of the latter seven, we analysed the impact of the most C-terminal one, [p.(Gly820Alafs*27)], on hippocampal neurogenesis. Indeed, we could show that the introduction of this mutation fails to rescue the severe progenitor cell proliferation defect in hippocampal slice cultures of Bcl11b-deficient mice.
The neurodevelopmental findings, impaired T cell development and drastic reduction in ILC2s recapitulate the previously published mouse phenotype (Wakabayashi et al., 2003; Simon et al., 2012; Walker et al., 2015; Yu et al., 2015). Moreover, all seven analysed patients cluster clearly apart from healthy control subjects in an unsupervised analysis based on over 100 T-cell subpopulations, altogether strongly supporting the causality of the here-identified mutations. Further evidence for their pathogenic nature comes from large-scale sequencing studies suggesting high intolerance of BCL11B to germline mutations (Lek et al., 2016).
However, although the majority of the patients are affected by a non-syndromic neurodevelopmental disorder (we do not regard the common ILC2 reduction, which is just identified by a specific laboratory investigation, as an additional clinically overt symptom), it is worth noting that two of the here-identified patients, Patients D:II-1 and E:II-1, developed symptoms not observed in the others. Patient D:II-1 additionally suffered epileptic seizures, requiring combined anticonvulsive therapy. In addition to a de novo frameshift mutation in BCL11B, this patient however bears a de novo nonsense mutation affecting the C-terminus of RELN. Heterozygous RELN mutations have been previously associated with isolated lateral temporal lobe epilepsy (Dazzo et al., 2015), and we therefore suggest that the dysfunction of both genes contribute to her condition. Noteworthy, besides de novo mutations in BCL11B, in some of the here presented patients (Patients A:II-3, B:II-3, F:II-2, G:III-1 and L:II-2) we additionally identified further de novo alterations in various other genes (Supplementary Table 1), which is not unexpected according to the mean number of de novo coding variants in any individual. However, as (i) none of these genes has been associated with a monogenic human disease before; (ii) all these genes either have a low pLI (CAMSAP1, pLI = 0.75, and DMAP1, pLI = 0.34) or a low missense Z-score (BAI3, z = 1.22, and NYAP1, z = 0.85), or the exact alteration is found in gnomAD [CHD5: p.(Lys68Thr)]; and (iii) we did not observe major phenotypic differences between the patients, we do not believe that variants in these genes have significantly contributed to the observed phenotype, although a minor modifying contribution cannot be excluded. Strikingly, and in contrast to many other Mendelian disorders, the recently published patient affected by a syndromic immunodeficiency (Punwani et al., 2016) and Patient E:II-1, both bearing BCL11B missense mutations, were more severely affected than any of the other patients with frameshift/nonsense mutations/and translocations. Indeed, Patient E:II-1 was the only patient with suspected immunodeficiency diagnosed upon newborn screening, and this patient also developed severe congenital erosive dermatitis, consistent with the role of BCL11B in epidermal development and homeostasis (Golonzhka et al., 2009a) and dermatitis pathogenesis (Wang et al., 2012). Notably, both missense mutations affect one of the four ZnF_C2H2 ‘specificity residues’ of the DNA-contacting alpha-helix within the ZnF2 and ZnF4, respectively. Using ChIP-seq analysis, Punwani et al. (2016) demonstrated that the p.(Asn441Lys) mutation results not only impaired BCL11B binding to known target DNA sites, but also promotes binding to novel DNA binding sites. We hypothesize that unlike the other here-identified mutations, which are predicted to result in a loss of DNA-binding, the here-identified missense mutation may also result in acquisition of novel DNA-binding regions. As BCL11B acts as both transcriptional activator and repressor (Kominami, 2012), the differential binding to novel genomic regions might thus explain the observed clinical differences. Similarly, we propose that other missense mutations affecting the DNA-recognition interface within ZnF_C2H2 domains might also result in distinct, probably more severe clinical outcomes either through the change of affinity for genomic regions, or altered DNA binding kinetics due to steric effects of the mutated amino acid in the alpha-helix containing DNA recognition site. Distinguishing between the differential outcomes of BCL11B mutations, especially the elucidation of putative novel target genes induced by p.(Asn807Lys) will require further work and will be the focus of future studies. However, the possibility that these two patients bear additional pathogenic mutations in regions that were either not properly covered by WES or are located within deep intronic or even intragenic regions cannot be completely excluded. Identification of further patients bearing missense mutations in ZnF_C2H2 domains is necessary to delineate both the phenotypic spectrum and their underlying mechanisms.
Taken together, we show that mutations resulting in haploinsufficiency or a truncation of the BCL11B protein mainly cause a non-syndromic neurodevelopmental disorder (NS-NDD). This also means that there are probably no clinical reasons to opt for a specific testing of BCL11B (unless one would know about a concomitant ILC2 reduction), but that rather diagnostic tests using massively parallel sequencing, i.e. either gene panels, WES or WGS, are essential for clinical diagnosis of a BCL11B-associated disorder. The latter especially holds true for identifying patients with missense mutations that may actually present with a multitude of additional symptoms depending on the way the ZnF_C2H2 domain is affected. Additionally, in light of the balanced translocations identified in our study, it is tempting to speculate that alterations affecting BCL11B enhancers may also result in an NS-NDD. WGS studies in previously WES-negative NS-NDD patients might therefore concentrate also on this region on chromosome 14. Interestingly, the extremely low frequency of recent thymic emigrants, together with the high frequencies of Tγδ cells suggests that, in addition to Patient E:II-1, other patients might also have had low Tαβ cell counts at birth. Homeostatic proliferation driven by lymphopaenia, expansion of Tαβ cells, and possibly antigen-driven clonal proliferation—as indicated by the high frequencies of effector cells in four patients—have probably contributed to the normal T cell counts in most patients at the time of analysis. Even though the families subjectively reported more infections than usual in four of the cases, none of the patients was considered immune deficient at the time of analysis, indicating a functionally sufficient diversity of the T cell receptor repertoire, as we could show in the two cases available for testing (Fig. 4C). Notably, we did not find increased frequencies of NK cells or of NK markers on T cells, as has been reported for Bcl11b-deficient mice (Wakabayashi et al., 2003; Li et al., 2010).
In summary, our data establish disruptions of BCL11B as a monogenic cause of a neurodevelopmental disorder, underscoring a central role for BCL11B in the development of the human neural systems. Moreover, our data provide first direct evidence that BCL11B plays a role in ILC2 development in humans. Finally, we propose that missense mutations affecting the DNA-recognition interfaces of BCL11B may result in more severe clinical outcomes than mutations resulting in haploinsufficiency or a truncation of the BCL11B protein.
Supplementary Material
Acknowledgements
We are thankful to the family members for participation. We thank R. Hackbusch for expert technical help, L. Glau for preparing the t-SNE plots and Dr A. Gieras for critical reading of the manuscript.
Glossary
Abbreviations
- ILC2
type 2 innate lymphoid cells
- WES
whole-exome sequencing
Funding
This work was supported by the German Research Foundation DFG TO-235, KFO296 (to E.T.) and DFG BR-2215 (to S. Britsch), Studienstiftung des Deutschen Volkes (to C.G.), the German Ministry of Research and Education 01GS08167 (to D.W.) and 01GS08163 (to T.M.S.) as part of the National Genome Research Network, the São Paulo Research Foundation FAPESP 2013/03236–5 and 2013/02162-8 (to A.A.L.J.), by NIH/NIAMS 1R01AR068429‐01 and NICHD/NHGRI/NIH U19HD077671 (to P.B.A.l), and by the French Ministry of Health (DGOS) and the French National Agency for Research (ANR) (PRTS 2013 grant to C.S-B.). We thank the Broad Center for Mendelian Genomics for providing sequencing assistance in Family E, supported in part by National Institutes of Health UM1 HG008900 to D.M. and H.R.
Supplementary material
Supplementary material is available at Brain online.
References
- Arlotta P, Molyneaux BJ, Chen J, Inoue J, Kominami R, Macklis JD. Neuronal subtype-specific genes that control corticospinal motor neuron development in vivo. Neuron 2005; 45: 207–21. [DOI] [PubMed] [Google Scholar]
- Arlotta P, Molyneaux BJ, Jabaudon D, Yoshida Y, Macklis JD. Ctip2 controls the differentiation of medium spiny neurons and the establishment of the cellular architecture of the striatum. J Neurosci 2008; 28: 622–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basak A, Hancarova M, Ulirsch JC, Balci TB, Trkova M, Pelisek M et al. BCL11A deletions result in fetal hemoglobin persistence and neurodevelopmental alterations. J Clin Invest 2015; 125: 2363–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Califano D, Cho JJ, Uddin MN, Lorentsen KJ, Yang Q, Bhandoola A et al. Transcription factor Bcl11b controls identity and function of mature type 2 innate lymphoid cells. Immunity 2015; 43: 354–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Califano D, Sweeney KJ, Le H, VanValkenburgh J, Yager E, O'Connor W Jr et al. Diverting T helper cell trafficking through increased plasticity attenuates autoimmune encephalomyelitis. J Clin Invest 2014; 124: 174–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoso V, Chesne J, Ribeiro H, Garcia-Cassani B, Carvalho T, Bouchery T et al. Neuronal regulation of type 2 innate lymphoid cells via neuromedin U. Nature 2017; 549: 277–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Wallis JW, McLellan MD, Larson DE, Kalicki JM, Pohl CS et al. BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat Methods 2009; 6: 677–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dazzo E, Fanciulli M, Serioli E, Minervini G, Pulitano P, Binelli S et al. Heterozygous reelin mutations cause autosomal-dominant lateral temporal epilepsy. Am J Hum Genet 2015; 96: 992–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bruin C, Finlayson C, Funari MF, Vasques GA, Lucheze Freire B, Lerario AM et al. Two patients with severe short stature due to a FBN1 mutation (p.Ala1728Val) with a mild form of acromicric dysplasia. Horm Res Paediatr 2016; 86: 342–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias C, Estruch SB, Graham SA, McRae J, Sawiak SJ, Hurst JA et al. BCL11A haploinsufficiency causes an intellectual disability syndrome and dysregulates transcription. Am J Hum Genet 2016; 99: 253–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier J, Meijer IA, Lessel D, Mencacci NE, Krainc D, Hempel M et al. Recessive mutations in VPS13D cause childhood-onset movement disorders. Ann Neurol 2018, doi: 10.1002/ana.25204. [DOI] [PubMed] [Google Scholar]
- Golonzhka O, Liang X, Messaddeq N, Bornert JM, Campbell AL, Metzger D et al. Dual role of COUP-TF-interacting protein 2 in epidermal homeostasis and permeability barrier formation. J Invest Dermatol 2009a; 129: 1459–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golonzhka O, Metzger D, Bornert JM, Bay BK, Gross MK, Kioussi C et al. Ctip2/Bcl11b controls ameloblast formation during mammalian odontogenesis. Proc Natl Acad Sci USA 2009b; 106: 4278–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamby SE, Thomas NS, Cooper DN, Chuzhanova N. A meta-analysis of single base-pair substitutions in translational termination codons ('nonstop' mutations) that cause human inherited disease. Hum Genomics 2011; 5: 241–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmston N, Ing-Simmons E, Tan G, Perry M, Merkenschlager M, Lenhard B. Topologically associating domains are ancient features that coincide with Metazoan clusters of extreme noncoding conservation. Nat Commun 2017; 8: 441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hempel M, Cremer K, Ockeloen CW, Lichtenbelt KD, Herkert JC, Denecke J et al. De Novo mutations in CHAMP1 cause intellectual disability with severe speech impairment. Am J Hum Genet 2015; 97: 493–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ippolito GC, Dekker JD, Wang YH, Lee BK, Shaffer AL III, Lin J et al. Dendritic cell fate is determined by BCL11A. Proc Natl Acad Sci USA 2014; 111: E998–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klose CSN, Mahlakoiv T, Moeller JB, Rankin LC, Flamar AL, Kabata H et al. The neuropeptide neuromedin U stimulates innate lymphoid cells and type 2 inflammation. Nature 2017; 549: 282–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kominami R. Role of the transcription factor Bcl11b in development and lymphomagenesis. Proc Jpn Acad Ser B Phys Biol Sci 2012; 88: 72–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo M. Innate and adaptive type 2 immunity in lung allergic inflammation. Immunol Rev 2017; 278: 162–72. [DOI] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016; 536: 285–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennon MJ, Jones SP, Lovelace MD, Guillemin GJ, Brew BJ. Bcl11b-A critical neurodevelopmental transcription factor-roles in health and disease. Front Cell Neurosci 2017; 11: 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessel D, Schob C, Kury S, Reijnders MRF, Harel T, Eldomery MK et al. De Novo missense mutations in DHX30 impair global translation and cause a neurodevelopmental disorder. Am J Hum Genet 2017; 101: 716–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N et al. The sequence alignment/map format and SAMtools. Bioinformatics 2009; 25: 2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Zhang JA, Dose M, Kueh HY, Mosadeghi R, Gounari F et al. A far downstream enhancer for murine Bcl11b controls its T-cell specific expression. Blood 2013; 122: 902–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Burke S, Wang J, Chen X, Ortiz M, Lee SC et al. Reprogramming of T cells to natural killer-like cells upon Bcl11b deletion. Science 2010; 329: 85–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Keller JR, Ortiz M, Tessarollo L, Rachel RA, Nakamura T et al. Bcl11a is essential for normal lymphoid development. Nat Immunol 2003; 4: 525–32. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 2001; 25: 402–8. [DOI] [PubMed] [Google Scholar]
- Louie RJ, Tan QK, Gilner JB, Rogers RC, Younge N, Wechsler SB et al. Novel pathogenic variants in FOXP3 in fetuses with echogenic bowel and skin desquamation identified by ultrasound. Am J Med Genet A 2017; 173: 1219–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monticelli LA, Sonnenberg GF, Abt MC, Alenghat T, Ziegler CG, Doering TA et al. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat Immunol 2011; 12: 1045–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najafabadi HS, Mnaimneh S, Schmitges FW, Garton M, Lam KN, Yang A et al. C2H2 zinc finger proteins greatly expand the human regulatory lexicon. Nat Biotechnol 2015; 33: 555–62. [DOI] [PubMed] [Google Scholar]
- Petrovski S, Wang Q, Heinzen EL, Allen AS, Goldstein DB. Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet 2013; 9: e1003709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punwani D, Zhang Y, Yu J, Cowan MJ, Rana S, Kwan A et al. Multisystem anomalies in severe combined immunodeficiency with mutant BCL11B. N Engl J Med 2016; 375: 2165–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samocha KE, Robinson EB, Sanders SJ, Stevens C, Sabo A, McGrath LM et al. A framework for the interpretation of de novo mutation in human disease. Nat Genet 2014; 46: 944–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon R, Baumann L, Fischer J, Seigfried FA, De Bruyckere E, Liu P et al. Structure-function integrity of the adult hippocampus depends on the transcription factor Bcl11b/Ctip2. Genes Brain Behav 2016; 15: 405–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon R, Brylka H, Schwegler H, Venkataramanappa S, Andratschke J, Wiegreffe C et al. A dual function of Bcl11b/Ctip2 in hippocampal neurogenesis. EMBO J 2012; 31: 2922–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobreira N, Schiettecatte F, Valle D, Hamosh A. GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum Mutat 2015; 36: 928–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka AJ, Cho MT, Millan F, Juusola J, Retterer K, Joshi C et al. Mutations in SPATA5 are associated with microcephaly, intellectual disability, seizures, and hearing loss. Am J Hum Genet 2015; 97: 457–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 2013; 14: 178–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rijt L, von Richthofen H, van Ree R. Type 2 innate lymphoid cells: at the cross-roads in allergic asthma. Semin Immunopathol 2016; 38: 483–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkataramanappa S, Simon R, Britsch S. Ex utero electroporation and organotypic slice culture of mouse hippocampal tissue. J Vis Exp 2015; doi: 10.3791/52550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakabayashi Y, Watanabe H, Inoue J, Takeda N, Sakata J, Mishima Y et al. Bcl11b is required for differentiation and survival of alphabeta T lymphocytes. Nat Immunol 2003; 4: 533–9. [DOI] [PubMed] [Google Scholar]
- Walker JA, Oliphant CJ, Englezakis A, Yu Y, Clare S, Rodewald HR et al. Bcl11b is essential for group 2 innate lymphoid cell development. J Exp Med 2015; 212: 875–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallrapp A, Riesenfeld SJ, Burkett PR, Abdulnour RE, Nyman J, Dionne D et al. The neuropeptide NMU amplifies ILC2-driven allergic lung inflammation. Nature 2017; 549: 351–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Zhang LJ, Guha G, Li S, Kyrylkova K, Kioussi C et al. Selective ablation of Ctip2/Bcl11b in epidermal keratinocytes triggers atopic dermatitis-like skin inflammatory responses in adult mice. PLoS One 2012; 7: e51262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe SA, Nekludova L, Pabo CO. DNA recognition by Cys2His2 zinc finger proteins. Annu Rev Biophys Biomol Struct 2000; 29: 183–212. [DOI] [PubMed] [Google Scholar]
- Yu Y, Tsang JC, Wang C, Clare S, Wang J, Chen X et al. Single-cell RNA-seq identifies a PD-1hi ILC progenitor and defines its development pathway. Nature 2016; 539: 102–6. [DOI] [PubMed] [Google Scholar]
- Yu Y, Wang C, Clare S, Wang J, Lee SC, Brandt C et al. The transcription factor Bcl11b is specifically expressed in group 2 innate lymphoid cells and is essential for their development. J Exp Med 2015; 212: 865–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw whole-exome sequencing data that support the findings in patients cannot be made publicly available for confidentiality reasons. Qualified researchers may apply for access to these data, pending institutional review board approval. All other data generated or analysed during this study are included herein and in the Supplementary material.