

 Eriodictyol exerts inhibitory effects on DMH-induced colon carcinogenesis, which means it can act as an anticarcinogenic agent.
Eriodictyol exerts inhibitory effects on DMH-induced colon carcinogenesis, which means it can act as an anticarcinogenic agent.
Abstract
Eriodictyol, one of the strong flavonoids extracted from Eriodictyon californicum, is known for its antioxidant and anticarcinogenic properties. We estimated the chemopreventive effect of eriodictyol on 1,2 dimethylhydrazine (DMH)-induced experimental colon carcinogenesis in male albino Wistar rats. The rats were randomized into six groups. Our results evaluated the effect of eriodictyol supplementation (200 μg per kg b.w.) on DMH (20 mg per kg b.w)-induced rats (Groups 4–6). The incidence of polyps, aberrant crypt foci (ACF) and the lipid peroxidation levels were significantly decreased as compared to those in the DMH-alone treated rats (Group 2). In eriodictyol-supplemented DMH-treated rats, we observed increased activity of enzymatic and non-enzymatic antioxidants in the circulatory system, liver, and colon. The bacterial enzymes activities of mucosa and faecal were significantly decreased in the group with treatment of eriodictyol on DMH-induced rats. Moreover, in the eriodictyol-supplemented DMH-exposed rats, we observed reduced malignant glands of a histopathological appearance in both liver and colon tissue. Furthermore, we also observed reduced AgNORs counts of eriodictyol supplemented to the DMH-exposed rats. Therefore, we can conclude that eriodictyol can be used as an effective chemopreventive agent against DMH-induced colon carcinogenesis in experimental animal models.
Introduction
Colorectal cancer (CRC) is the third most common malignancy worldwide with high mortality, high morbidity and nearly 1.4 million new cases diagnosed in 2012.1 Despite developments made in the diagnosis and treatment2 of colorectal cancer, the five-year survival rate for CRC is still only 67%.3 The constant increase in life anticipation, increasing urbanization, and the subsequent changes in environmental conditions, including lifestyle and dietary habits, make CRC an acquired health problem throughout the world. Etiological agents include the heavy drinking of alcohol, the consumption of red meat, low fibre in the diet, high fats in foods and the absence of physical activities. These agents induce oxidative stress, leading to DNA damage and mutations in colonic genes, while cell cycle arrest regeneration increases the risk of developing colon cancer.4
Oxidative stress is an imbalance between the production of reactive oxygen species (ROS) and a decrease in the power of the antioxidant defence. Importantly, free radicals are reported and the biological sources of origin disputed,5 together with the action of the major antioxidant defence mechanisms, such as catalase (CAT), superoxide dismutase (SOD), glutathione peroxidase (GPX), reduced glutathione (GSH), glutathione-S-transferase (GST) and glutathione reductase (GR).6 Recently, there have been many advances in the comprehension of colon cancer epidemiology, pathogenesis, pathology, chemoprevention and therapeutic options, which appear to have been emerged from continuing basic and clinical research.
Host genetic factors play a censorious role in the pathophysiology of most human cancers. Rodent models have many desired attributes and they share an extensive variety of features with the human model that have been demonstrated to be important in understanding many complex molecular surfaces of colon cancer.7 They also act as an invaluable tool in the evolution of recent chemotherapeutic drugs. This has made laboratory rats as one of the most agreeable entities in oncologic research.8 Earlier studies showed DMH-induced colonic tumours were closely similar to the human colon carcinoma.9
A potent procarcinogen and an alkylating agent of DMH is metabolized by the liver in the active form of azoxymethane and methylazoxymethanol, which are actively transported subsequently via bile and blood into the colon.10 After being metabolized in the liver, the active methyl diazonium ions capable of methylating DNA, RNA or protein of colonic epithelial cells, inducing more oxidative stress, results in an overproduction of ROS, which leads to the destruction of complex cellular molecules, such as fat, carbohydrates and proteins and also induces DNA damage and mutation in tumour suppressor genes.11 To study the pathogenesis of human colon cancer, this type of procarcinogen could thus deliver an excellent experimental model.
The development of colon cancer induced by DMH involves a multistep process of pathological alterations, ranging from detached microscopic mucosal lesions of Aberrant Crypt Foci (ACF) to malignant tumours.12 ACF are a cluster form of abnormal tube-like glands in the lining of the colon and rectum. ACF form before colorectal polyps and are involved in one of the rapid changes that can be optically discerned in the colon that may lead to cancer. The development of colonic crypts was visualized as a larger, thicker and darker staining than normal crypts when visualized with methylene blue by treating rats with the DMH-induced carcinogen.13 Biochemical, genetic and morphological studies have shown that ACF and colonic tumours share homogeneous alterations, further criteria fortifying the hypothesis that ACF is a precursor of colorectal cancer.14
A high fat diet consumption could lead to the formation of a colonic adenoma through inflammation, metabolic abnormalities and increased cell proliferation in a DMH-induced experimental model.15 The imbalanced diet and high fat utilization alter the preventive metabolism of the intestinal microflora, and reduce the host immune response by restoring the bacterial enzyme activity.16,17 In that context, the produced DMH metabolites MAM undergoes glucuronidation in the liver and the MAM glucuronide is transported from the liver to the colon via bile. Furthermore, it is deconjugated by the effect of gut microbial enzymes and was released as a carcinogen in the colonic epithelium to induce colon cancer in experimental rats.18 The enzymatic hydrolysis of cellulose is a major restriction in lignocellulose's conversion to glucose, as an activity of β-glucosidase causes an accumulation of aglycones and mucinase bacterial enzyme acts on the demolition of mucins or their prosthetic polysaccharides produced in the intestinal lumen.19 Nitroreductase is complicit in the reduction of nitrogen-containing substances, including nitro functional group members, such as dinitrotoluene, nitrobenzenes and nitropyrenes, to amines. These proteins are reported as oxidoreductases and are primarily found in bacterial lineages that reveal carcinogenic activities in animal models.20
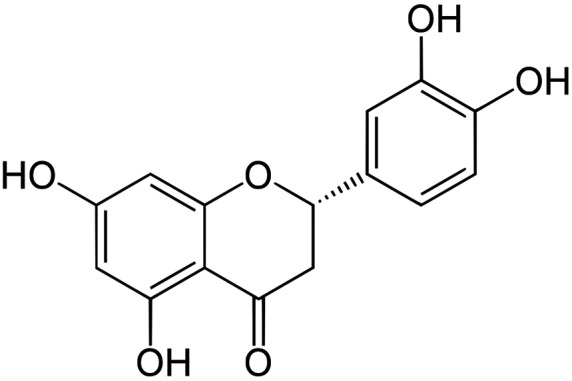
Natural products possess biological activity that can be of preventive and therapeutic benefit in various diseases, including cancer, and can protect against oxidative stress, potent antioxidants and can neutralize toxins and reactive oxygen species.21 The natural compounds have the most promising pharmacological properties to exert their anticarcinogenic action by regulating lipid peroxidation (LPO) and antioxidant status.22 Earlier evidence has suggested that flavonones can be extensively used in the chemoprevention of colorectal cancer and studies have elucidated their significant antiproliferative potential. Among such flavonones, naringin, eriodictyol, naringenin and hesperitin are reported to possess significant antioxidant potential, which makes them more antiproliferative against tumours both in vitro and in vivo.23 Eriodictyol (Fig. 1) is one such flavanone and can exert a protective effect against lipid peroxidation and oxidative stress. Besides its anti-tumour properties, many studies have shown that eriodictyol has a variety of other pharmacological effects, including anti-allergic, antioxidant, anti-inflammatory,24 antimicrobial and anticancer25,26 activities, as shown from in vitro studies.
Fig. 1. Chemical structure of eriodictyol.
To date, no studies have demonstrated the astounding pharmacological properties of eriodictyol using an in vivo model of colon cancer. This study thus focused on the scientific evidence for the chemopreventive effect of eriodictyol against DMH-induced rat colon carcinogenesis. The main objective of the study was to provide substantial evidence for the chemopreventive potential of eriodcityol by assessing its action on lipid peroxidation, biotransforming enzymes profiles, bacterial enzymes, ACF, the incidence of colonic polyps, histopathological observation and scores of AgNORs in DMH-induced experimental rat colon carcinogenesis models.
Material and methods
Chemicals
Eriodictyol, 1,2-dimethylhydrazine hydrochloride (DMH), DTNB (5,5′-dithiobis (2-nitrobenzoic acid)), CDNB (1′-chloro-2,4-dinitrobenzene), GSH (reduced glutathione) and NBT (nitroblue tetrazolium) were purchased from Sigma-Aldrich, Bangalore, India. All the other chemicals and solvents were of analytical grade.
Animals and diet
Male Wistar albino rats weighing 100–110 g were into 6 groups of 8 rats each. All the rats were maintained in polypropylene cages with 12 h light/dark cycles under constant temperature and humidity. All the experiments were performed in accordance with the guidelines of the Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA) and were approved by the Committee of Ethics in Periyar University Institutional Animal Ethics Committee (PUIAEC) and Central Animal House Registration No. 1085/AC/07, Periyar University, Salem, Tamil Nadu, India (Approval No. 1085/ac/07/PUIAEC/OCT-2012/18). Commercial pellet diet containing 4.2% fat (Sri Venkateshwara Enterprises, Bangalore, India) were powdered and mixed with 15.8% peanut oil, making a total of 20% fat, which the high fat diet fed to all the rats ad libitum (Table 1). The total caloric intake of the rats in all the groups was adjusted to be the same.
Table 1. Composition of the experimental diet.
| Nutrients | Commercial diet (84.2%) | Peanut oil (15.8%) | Total (100%) |
| Protein | 17.7 | — | 17.7 |
| Fat | 4.2 | 15.8 | 20.0 |
| Carbohydrate | 50.5 | — | 50.5 |
| Fibre | 3.4 | — | 3.4 |
| Minerals | 6.7 | — | 6.7 |
| Vitamins | 1.7 | — | 1.7 |
Treatment schedule
The rats were randomized into 6 groups of 8 animals each, after 2 weeks of acclimatization. Group 1 rats received the high fat diet daily for 30 weeks. Group 2 rats received the high fat diet plus DMH once a week subcutaneously for the first 15 weeks. Group 3 received eriodictyol via intragastric intubation every day for the entire study period. Group 4 received DMH + eriodictyol via intragastric intubation every day starting alongside DMH injection till the 16th week (initiation-I). Group 5 received DMH + eriodictyol via intragastric intubation every day of the 17th week till the 30th week (post-initiation – PI). Group 6 received DMH once a week subcutaneously for the first 15 weeks + eriodictyol via intragastric intubation every day for the entire study period (entire period, EP). The experimental design of the control and experimental animals is shown in Fig. 2.
Fig. 2. Schematic representation of the experimental design.

Colon cancer induction
DMH was dissolved in 1 mM EDTA just prior to use and the pH was adjusted to 6.5 with 1 mM NaOH to ensure the stability of the chemical. The experimental rats received subcutaneous injections of DMH in the groin at a dose of 20 mg per kg body weight for the first 15 weeks.27
Preparation of eriodictyol
Eriodictyol was dissolved in 5% Tween 80, 20% polyethylene glycol and 75% saline (0.9% NaCl) and each animal belonging to the four different groups received 1.0 ml of eriodictyol suspension at a daily dose of 200 μg per kg (ref. 28) body weight every day respectively by intragastric intubation.29
Body weight changes
A careful record of the body weight changes of the control, DMH- and eriodictyol-treated rats was kept throughout the study. The rats were weighed at the beginning of the experiment and subsequently once a week and finally before sacrifice.
Macroscopic evaluation of the incidence of polyps
At the end of the experimental period of 30 weeks, the rats were anaesthetized by ketamine hydrochloride (30 mg per kg body weight)30 and the animals were sacrificed and the colons removed and flushed with physiological saline. The colons were cut opened longitudinally not disturbing the polyps, which were carefully counted through visual macroscopic examination and later verified with histopathological examination.
Preparation of haemolysate and tissue homogenate
Blood was collected in heparinized tubes and plasma was separated by centrifugation at 2000g for 10 min. After the separation of plasma, the buffy coat was removed and packed cells (RBCs) were washed three times with cold physiological saline. To determine the activity of RBC antioxidant enzymes, RBC lysate was prepared by lysing a known volume of RBCs with hypotonic phosphate buffer, pH 7.4. Centrifuging at 3000g for 10 min at 2 °C separated the haemolysate. Tissue samples were immediately transferred to ice-cold containers, then weighed and homogenized using the appropriate buffer in a tissue homogenizer.
Faecal and colonic mucosal tissue processing
Fresh faecal pellets were collected for assay of the bacterial enzymes. Mucosa from the colon was collected by scraping with a slide. Faecal pellets and colonic mucosa were homogenized using phosphate-buffered saline, centrifuged at 2000g for 10 min at 4 °C and then the supernatant collected for measuring the activity of the faecal and colonic mucosal bacterial enzymes.
Biochemical estimations
Estimation of lipid peroxidation
Lipid peroxidation was estimated by measuring the level of thiobarbituric acid reactive substances (TBARS) in the plasma using a previously reported method.31 The pink chromogen produced was measured at 532 nm. The level of conjugated dienes was assessed using a previously reported method.32 This method was based on the arrangement of the double bonds in polyunsaturated fatty acids (PUFA) to form conjugated dienes with an absorbance maximum at 233 nm. The lipid hydroperoxide contents were measured using a previously reported method.33 Hydroperoxides were detected by their ability to oxidize ferrous iron leading to the formation of a chromophore with an absorbance maximum at 560 nm.
Assay of bacterial enzymes in mucosal and faecal content
Fresh faecal pellets were collected for assay of the faecal bacterial enzymes. The mucosa of the colon was collected by scraping with a slide. The faecal pellets and colonic mucosa were homogenized using phosphate-buffered saline, then centrifuged at 2000g for 10 min at 4 °C and the supernatant collected for assay of the activity of faecal and colonic mucosal bacterial enzymes. β-Glucosidase (EC. 3.2.1.21), β-glucuronidase (EC. 3.2.1.31) and β-galactosidase (EC. 3.2.1.23) activity were measured using a previously reported method.34 Mucinase (EC. 4.2.2.1) activity was measured using a previously reported method.35 Sulfatase (EC. 3.1.6.1) activity was measured using a previously reported method.36 Nitroreductase (EC. 1.6.99.7) activity was measured using a previously reported method.37
Assay of enzymatic and non-enzymatic antioxidant activity
Reduced glutathione (GSH) content was determined by a previously reported method.38 Glutathione peroxidase (GPx, EC.1.11.1.9) activity was assayed using a previously reported method.39 The activity of glutathione-S-transferase (GST, EC. 2.5.1.18) was determined using a previously reported method.40 Glutathione reductase (GR, EC.1.6.4.2) activity was assayed using a previously reported method.41 Superoxide dismutase (SOD, EC.I.15.1.1) was assayed using a previously reported method.42 The activity of catalase (CAT, EC.1.1 1.1 6) was determined using a previously reported method.43
Determination of ACF
The fixation, staining and counting of ACF were adopted by using a previously reported method.44 Briefly, the colons were flushed with PBS (0.1 M, pH 7.2) and were split open longitudinally and placed on strips of filter paper with their luminal surface open and exposed. Another strip of filter paper was placed on top of the luminal surface. The colon was fixed and set up in a tray containing 10% buffered formalin overnight. Each segment was placed in a petri dish and stained with 0.2% methylene blue solution for 2 min. The sections were then transferred to another petri dish containing wash buffer to remove excess stains. The segments were examined using a light microscope at low magnification to score the total number of ACF as well as the number of crypts per focus.
Histopathological changes
The colons from the experimental animals were microscopically examined for the presence of tumours or other pathological lesions. Tissues with an abnormal morphology were fixed in 10% buffered formalin and embedded in paraffin blocks. Histological sections stained with haematoxylin and eosin were used to confirm the presence and type of ACF and tumours by histopathological examination, which was performed by a pathologist unaware of the experimental codes.
Argyrophilic nucleolar organizer region (AgNOR) staining
The AgNOR staining was performed using a one-step staining technique measured using a previously reported method.45 In brief, hydrated sections were incubated in a freshly prepared solution of one part 2 g dL–1 solution of gelatin in 1 g per dL formic acid to two parts 50 g per dL aqueous silver nitrate for 40 min at room temperature (37 °C) and counterstained with haematoxylin for 30 s. The AgNORs in 200 nuclei of the basal and lower spinous cells were counted, and the average number of AgNOR per nucleus was estimated for each.
Statistical analysis
All the grouped data were evaluated using SPSS version 16.0 software. The hypothesis testing methods included one-way analysis of variance (ANOVA) followed by the least significant difference (LSD) test. P values of less than 0.05 (p < 0.05) were considered to indicate statistical significance. All the results are expressed herein as the mean ± SD for the 8 animals in each group.46
Results
Effect of eriodictyol on body weight changes in the control and experimental rats
In the initial weeks to final weeks of the total experimental period, the body weight changes of the control and experimental rats were recorded and are shown in Fig. 3. At the end of the study period, no changes were observed in the body weights of the control rats (Group 1) and the mean weight in the DMH-alone treated rats (Group 2) were significantly decreased as compared with the control rats (Group 1). The eriodictyol-treated rats (Group 3) and the DMH-with-eriodictyol-treated rats in the initiation (Group 4), post-initiation (Group 5) and over the entire period (Group 6) were significantly increased as compared with the DMH-alone treated rats (Group 2). During the entire study period, there were no signs of toxicity observed in any of the experimental groups.
Fig. 3. Body weight gain changes of the control, DMH-induced and eriodictyol-treated experimental rats.

Effects of eriodictyol and DMH on the incidence of colon polyps
In the DMH-treated rats (Group 2), the incidence of polyps per tumour was 100% (Table 2). With supplementation with eriodictyol (200 μg per kg b.w.) to the DMH-exposed rats (Groups 4–6) the incidence of polyps was significantly reduced as compared to the DMH-alone treated rats (Group 2). No specific changes were noticed between the control and eriodictyol-alone supplemented rats (Groups 1 and 3).
Table 2. Effect of eriodictyol on the incidence of colonic polyps in the colon of control and experimental rats.
| Groups | Number of rats examined | Number of tumour-bearing rats | Total number of tumours/polyps | Percentage incidence of polyps a | Percentage inhibition of polyps | Average number of polyps-bearing rats b |
| Control | 8 | 0 | Nil | Nil | — | Nil |
| DMH | 8 | 8 | 19 | 100 | 0 | 2.38 |
| Eriodictyol | 8 | 0 | Nil | Nil | — | Nil |
| Initiation | 8 | 6 | 10 | 75 | 25 | 1.67 |
| Post-initiation | 8 | 5 | 4 | 62.5 | 37.5 | 0.80 |
| Entire period | 8 | 3 | 2 | 37.5 | 62.5 | 0.67 |
a(Number of polyps-bearing rats/total number of rats examined in each group) × 100.
bTotal number of polyps/number of polyps tumour-bearing rats in each group.
Eriodictyol suppresses cell proliferation
Colonic tissue was stained with methylene blue; normal crypts were shown in the control rats. DMH-induced colonic crypts were stereologically detached from normal crypts as indicated by their darker staining, larger size, ovate shape, thicker epithelial lining and larger pericryptal zone (Fig. 4). The DMH-alone treated rats (Group 2) had a significantly excessive number of aberrant crypts compared with the control rats (62.37 ± 9.79). Daily supplementation with eriodictyol to the DMH-treated rats (Groups 4–6) led to a significant reduction in the frequency of ACF (52.58 ± 8.83, 29.52 ± 4.83 and 17.26 ± 2.97). There was a total reduction of 75% in the incidence of ACF in the eriodictyol-administered rats (Table 3).
Fig. 4. Topographical view of normal crypts and ACF (arrows) in the colonic mucosa stained with methylene blue of rats treated with DMH and eriodictyol. (A) Control group shows normal crypt, (B) DMH-induced rat group shows two aberrant crypt foci having more than 7 crypts, (C) positive control rat group showing normal crypts, (D) initiation rats group showing small aberrant crypt foci having two or three crypts, (E) post-initiation rats group showing two aberrant crypt foci having more than 10 crypts, (F) entire period rats group showing no aberrant crypt.

Table 3. Effect of eriodictyol on aberrant crypt foci formation, crypt multiplicity and percentage inhibition in the colon of control and experimental rats.
| Groups | No. of rats/no. of ACF-bearing rats | Total number of ACF | Crypt multiplicity |
% Inhibition | ||
| 1 crypt | 2 crypts | >3 crypts | ||||
| Control | 8/0 | Nil | Nil | Nil | Nil | — |
| DMH | 8/8 | 62.37 ± 9.79a | 28.21 ± 4.21a | 12.53 ± 2.12a | 21.63 ± 3.46a | — |
| Eriodictyol | 8/0 | Nil | Nil | Nil | Nil | — |
| Initiation | 8/8 | 52.58 ± 8.83b | 25.32 ± 4.01b | 10.98 ± 1.96b | 16.28 ± 2.86b | 9.97 |
| Post-initiation | 8/8 | 29.52 ± 4.83c | 20.45 ± 3.26c | 9.07 ± 1.57c | — | 32.85 |
| Entire period | 8/8 | 17.26 ± 2.97d | 17.26 ± 2.97d | — | — | 45.11 |
Changes in the levels of lipid peroxidation in the circulatory system and liver
The circulatory system and hepatic tissue levels of TBARS, lipid hydroperoxides and conjugated dienes are shown in Tables 4 and 5 The levels of TBARS, hydroperoxides and conjugated dienes were significantly increased in the DMH-alone treated rats (Group 2) as compared to the control rats (Group 1). Eriodictyol treatment to the DMH-treated rats at initiation, post-initiation and over the entire period stages (Groups 4, 5 and 6) of DMH treatment significantly decreased the levels of TBARS, lipid hydroperoxides and conjugated dienes in the circulatory system and liver to the near normal levels displayed by the untreated control rats.
Table 4. Effect of eriodictyol on circulatory lipid peroxidation of control and experimental rats.
| Groups | TBARS a | Lipid hydro peroxides b | Conjugated dienes c |
| Control | 2.24 ± 0.28a | 4.60 ± 0.32a | 0.74 ± 0.12a |
| DMH | 4.51 ± 0.14b | 6.52 ± 0.26b | 1.76 ± 0.17b |
| Eriodictyol | 2.22 ± 0.17a | 4.52 ± 0.44a | 0.78 ± 0.14a |
| Initiation | 3.71 ± 0.12c | 5.89 ± 0.15c | 1.06 ± 0.14b |
| Post-initiation | 3.10 ± 0.06d | 5.75 ± 0.26c | 0.95 ± 0.08c |
| Entire period | 3.08 ± 0.13d | 5.40 ± 0.23d | 0.88 ± 0.56c |
anmoles ml–1.
bmmoles ml–1.
cμmoles ml–1.
Table 5. Effect of eriodictyol on hepatic tissue lipid peroxidation of control and experimental rats.
| Groups | TBARS a | Lipid hydro peroxides a | Conjugated dienes a |
| Control | 0.91 ± 0.08c | 55.35 ± 0.02b | 49.54 ± 0.22b |
| DMH | 2.81 ± 0.31a | 72.14 ± 0.16a | 68.36 ± 0.07a |
| Eriodictyol | 0.88 ± 0.16c | 54.82 ± 0.44b | 48.96 ± 0.24b |
| Initiation | 1.53 ± 0.22b | 63.22 ± 0.05c | 56.44 ± 0.04c |
| Post-initiation | 1.10 ± 0.08d | 61.25 ± 0.36c,d | 55.35 ± 0.18c |
| Entire period | 1.08 ± 0.14d,e | 59.16 ± 0.13b | 52.28 ± 0.16c,d |
ammoles per mg tissue.
Changes in the levels of lipid peroxidation in colonic tissue
Table 6 shows the colonic tissue levels of TBARS, lipid hydroperoxides and conjugated dienes in the control and experimental animals. The levels of TBARS, hydroperoxides and conjugated dienes were significantly decreased in the proximal colon, distal colon and intestines of the DMH-treated rats (Group 2) as compared to the control rats (Group 1). Eriodictyol treatment to the DMH-treated rats at initiation, post-initiation and over the entire period (Groups 4, 5 and 6) were significantly restored the levels of TBARS, lipid hydroperoxides and conjugated dienes of the colon and intestines showed near normal levels of tissue lipid peroxidation markers.
Table 6. Effect of eriodictyol on colonic tissue lipid peroxidation of control and experimental rats.
| Groups | Control | DMH | Eriodictyol | Initiation | Post-initiation | Entire period |
| TBARS a | ||||||
| Proximal colon | 2.56 ± 0.06a | 0.75 ± 0.17b | 2.12 ± 0.13a | 0.99 ± 0.11c | 1.05 ± 0.06d | 1.09 ± 0.12d |
| Distal colon | 2.61 ± 0.12a | 0.79 ± 0.12b | 2.15 ± 0.06a | 1.02 ± 0.16c | 1.08 ± 0.14d | 1.11 ± 0.27e |
| Intestine | 2.42 ± 0.07a | 0.71 ± 0.13b | 2.09 ± 0.18a | 0.92 ± 0.06c | 1.03 ± 0.04d | 1.05 ± 0.21d |
| LOOH a | ||||||
| Proximal colon | 81.61 ± 0.25a | 52.32 ± 0.11b | 75.12 ± 0.17c | 60.12 ± 0.13d | 64.33 ± 0.17d | 71.25 ± 0.07c |
| Distal colon | 82.22 ± 0.09a | 53.91 ± 0.18b | 76.21 ± 0.29c | 61.55 ± 0.43d | 66.20 ± 0.07d | 71.95 ± 0.41e |
| Intestine | 80.23 ± 0.44a | 50.54 ± 0.14b | 72.45 ± 0.24c | 59.22 ± 0.18d | 62.61 ± 0.49d | 69.28 ± 0.38d |
| CD a | ||||||
| Proximal colon | 83.14 ± 0.11a | 60.12 ± 0.07b | 77.18 ± 0.24c | 65.21 ± 0.14b | 69.45 ± 0.16d | 73.08 ± 0.04c |
| Distal colon | 85.01 ± 0.15a | 62.14 ± 0.02b | 78.02 ± 0.07c | 66.17 ± 0.05b | 73.41 ± 0.12d | 74.58 ± 0.24d |
| Intestine | 81.51 ± 0.21a | 58.13 ± 0.15b | 74.32 ± 0.46c | 63.27 ± 0.08d | 67.41 ± 0.32d | 70.08 ± 0.37e |
ammoles per mg tissue.
Changes in the circulatory system and hepatic tissue levels of enzymatic and non-enzymatic antioxidants
Tables 7 and 8 show the circulatory system and hepatic tissue levels of these antioxidant enzymes that were significantly decreased in the DMH-alone treated rats (Group 2) as compared to the control rats (Group 1). Eriodictyol administration (Group 3) significantly increased the activities of the above enzymes when compared to the DMH-alone treated rats. Eriodictyol administration during initiation, post-initiation and over the entire period of carcinogenesis (Group 4, 5 and 6) significantly increased the levels of the antioxidant enzymes in the circulatory system and hepatic tissue levels as compared with the DMH-alone treated rats.
Table 7. Effect of eriodictyol on circulatory enzymatic and non-enzymatic antioxidants.
| Group | SOD a | CAT b | GPX c | GST d | GSH e | GR f |
| Control | 4.12 ± 0.14a | 3.12 ± 0.25a | 31.24 ± 0.14a | 3.56 ± 0.03a | 42.21 ± 0.06a | 57.22 ± 0.14a |
| DMH | 1.98 ± 0.08b | 1.75 ± 0.09b | 19.21 ± 0.01b | 2.02 ± 0.01b | 30.12 ± 0.12b | 39.32 ± 0.25b |
| Eriodictyol | 4.56 ± 0.12a | 3.37 ± 0.14c | 32.01 ± 0.02a | 3.68 ± 0.12a | 43.65 ± 0.08a | 59.02 ± 0.16a |
| Initiation | 3.32 ± 0.24c | 3.05 ± 0.03c | 26.12 ± 0.09c | 2.72 ± 0.15c | 32.24 ± 0.12c | 43.11 ± 0.02c |
| Post-initiation | 3.74 ± 0.14c | 3.12 ± 0.11d | 27.32 ± 0.05c | 2.84 ± 0.22c | 38.02 ± 0.04c | 44.21 ± 0.07c |
| Entire period | 4.01 ± 0.09d | 3.10 ± 0.15d | 30.24 ± 0.02d | 3.25 ± 0.23d | 41.36 ± 0.08d | 49.65 ± 0.23d |
aEnzyme required for 50% inhibition of NBT reduction per min per mg of Hb.
bμmol H2O2 utilized per min per mg of Hb.
cμmoles of GSH utilized per min per mg Hb.
dnmoles of CDNB-GSH conjugate formed per min per mg Hb.
emmol per mg of HB.
fμmoles of NADPH oxidized per min per mg Hb.
Table 8. Effect of eriodictyol on hepatic tissue enzymatic and non-enzymatic antioxidants.
| Group | SOD a | CAT b | GPX c | GST d | GSH e | GR f |
| Control | 8.03 ± 0.11a | 36.28 ± 0.42a | 6.08 ± 0.17a | 3.69 ± 0.09a | 3.74 ± 0.62a | 35.16 ± 0.07a |
| DMH | 2.68 ± 0.24b | 18.95 ± 0.23b | 2.46 ± 0.12b | 1.02 ± 0.26b | 0.58 ± 0.05b | 26.81 ± 0.34b |
| Eriodictyol | 9.01 ± 0.36c | 37.16 ± 0.38a | 6.11 ± 0.43a | 4.03 ± 0.11a | 3.99 ± 0.06c | 41.07 ± 0.02c |
| Initiation | 5.38 ± 0.09d | 32.46 ± 0.09c | 4.84 ± 0.17c | 2.54 ± 0.15c | 2.46 ± 0.02d | 32.52 ± 0.16d |
| Post-initiation | 5.98 ± 0.14d | 32.52 ± 0.21c | 5.06 ± 0.22d | 2.95 ± 0.74d | 3.31 ± 0.42e | 32.71 ± 0.62d |
| Entire period | 6.25 ± 0.08e | 34.78 ± 0.39c,d | 5.78 ± 0.39d | 3.78 ± 0.19c | 3.94 ± 0.25f | 39.79 ± 0.38c,d |
aEnzyme required for 50% inhibition of NBT reduction per min per mg of protein.
bμmoles H2O2 utilized per min per mg of protein.
cμmoles of GSH utilized per min per mg protein.
dnmoles of CDNB-GSH conjugate formed per min per mg protein.
emmol per mg of protein.
fμmoles of NADPH oxidized per min per mg protein.
Changes in the colonic tissue levels of enzymatic and non-enzymatic antioxidants
Table 9 represents the levels of colonic tissue enzymatic antioxidants (SOD and CAT) in the control and experimental animals. The levels of SOD and CAT in the proximal colon, distal colon and intestines were significantly decreased with DMH treatment (Group 2) as compared to the control rats (Group 1), whereas the tissue levels of SOD and CAT were significantly increased in the tissues of rats supplemented with eriodictyol (initiation, post-initiation and over the entire period, i.e. in Groups 4, 5 and 6) as compared to the untreated DMH-administered rats (Group 2). The activities of the enzymatic antioxidants (SOD and CAT) were significantly elevated in the colon and intestines of the control rats treated with eriodictyol (Group 2) as compared to untreated control rats (Group 1). Table 10 represents the levels of tissue glutathione and its dependent enzymes (GPx, GST, GSH and GR) in the control and experimental animals. The levels of glutathione and its dependent enzymes in the proximal colon, distal colon and intestines were significantly decreased with DMH treatment (Group 2) as compared to the control rats (Group 1), whereas the tissue levels of GPx, GST, GSH and GR were significantly increased in the tissues of rats treated with eriodictyol (initiation, post-initiation and over the entire period, i.e. Groups 4, 5 and 6) as compared to the untreated DMH-administered rats (Group 2). The levels of glutathione and glutathione-dependent enzymes were decreased in the colon and intestines of the control rats treated with eriodictyol (Group 3) as compared to the untreated control rats (Group 1).
Table 9. Effect of eriodictyol on colonic tissue levels of SOD and CAT in control and experimental rats.
| Parameters and samples | Groups |
|||||
| Control | DMH | Eriodictyol | Initiation | Post-initiation | Entire period | |
| SOD a | ||||||
| Proximal colon | 10.23 ± 0.11a | 5.42 ± 0.03b | 11.12 ± 0.04a | 7.52 ± 0.12c | 7.85 ± 0.34c | 8.74 ± 0.18d |
| Distal colon | 10.61 ± 0.21a | 5.49 ± 0.08b | 11.19 ± 0.13c | 7.51 ± 0.05d | 7.95 ± 0.11d | 8.77 ± 0.36e |
| Intestine | 10.20 ± 0.14a | 5.39 ± 0.17b | 11.09 ± 0.22c | 7.49 ± 0.27d | 7.84 ± 0.24d | 8.71 ± 0.05e |
| CAT b | ||||||
| Proximal colon | 38.35 ± 0.14a | 22.42 ± 0.33b | 39.12 ± 0.18a | 28.33 ± 0.01c | 29.16 ± 0.11c | 30.91 ± 0.13d |
| Distal colon | 38.21 ± 0.45a | 22.84 ± 0.08b | 40.42 ± 0.34c | 28.57 ± 0.04d | 29.49 ± 0.06d | 31.31 ± 0.61e |
| Intestine | 37.81 ± 0.34a | 21.89 ± 0.63b | 39.14 ± 0.64c | 28.08 ± 0.17d | 28.61 ± 0.42d | 29.62 ± 0.37c,d |
aEnzyme required for 50% inhibition of NBT reduction per min per mg of protein.
bμmoles of H2O2 utilized per mg protein.
Table 10. Effect of eriodictyol on glutathione and glutathione-dependent enzymes in the colonic tissue levels of control and experimental rats.
| Parameters and samples | Groups |
|||||
| Control | DMH | Eriodictyol | Initiation | Post-initiation | Entire period | |
| GPX a | ||||||
| Proximal colon | 7.21 ± 0.03a | 3.55 ± 0.02b | 8.11 ± 0.21c | 5.34 ± 0.17d | 6.38 ± 0.18e | 6.72 ± 0.16e |
| Distal colon | 7.25 ± 0.17a | 3.54 ± 0.48b | 8.09 ± 0.26c | 5.45 ± 0.35d | 5.61 ± 0.41d | 6.81 ± 0.71e |
| Intestine | 7.20 ± 0.44a | 3.24 ± 0.07b | 8.04 ± 0.32c | 5.29 ± 0.37d | 5.33 ± 0.04d | 6.01 ± 0.08e |
| GST b | ||||||
| Proximal colon | 4.03 ± 0.18a | 1.99 ± 0.04b | 4.11 ± 0.14a | 2.88 ± 0.14c | 3.14 ± 0.09d | 3.52 ± 0.05d |
| Distal colon | 4.07 ± 0.05a | 1.98 ± 0.43b | 4.12 ± 0.04a | 2.90 ± 0.43c | 3.14 ± 0.08d | 3.55 ± 0.09e |
| Intestine | 3.95 ± 0.52a | 1.98 ± 0.09b | 4.08 ± 0.14c | 2.80 ± 0.31d | 3.11 ± 0.02e | 3.52 ± 0.11e |
| GSH c | ||||||
| Proximal colon | 4.12 ± 0.08a | 1.85 ± 0.15b | 4.26 ± 0.36a | 3.21 ± 0.42c | 3.86 ± 0.41d | 4.09 ± 0.65e |
| Distal colon | 4.16 ± 0.22a | 1.84 ± 0.51b | 4.29 ± 0.13a | 3.21 ± 0.05c | 3.85 ± 0.37d | 4.12 ± 0.11e |
| Intestine | 4.09 ± 0.20a | 1.80 ± 0.39b | 4.23 ± 0.08a | 3.19 ± 0.62c | 3.82 ± 0.07d | 4.02 ± 0.38e |
| GR d | ||||||
| Proximal colon | 41.62 ± 0.47a | 29.54 ± 0.83b | 43.67 ± 0.42a | 36.32 ± 0.46c | 37.75 ± 0.62c | 40.72 ± 0.34d |
| Distal colon | 42.13 ± 0.08a | 28.42 ± 0.63b | 43.18 ± 0.44a | 36.62 ± 0.81c | 38.22 ± 0.34c | 39.12 ± 0.05d |
| Intestine | 41.30 ± 0.42a | 28.03 ± 0.19b | 42.65 ± 0.18a | 36.23 ± 0.09c | 37.33 ± 0.15c | 37.62 ± 0.12c,d |
aμmoles of GSH utilized per min per mg protein.
bμmoles of CDNB-GSH conjugate formed per min per mg protein.
cmmol per mg tissue protein.
dμmoles of NADPH oxidized per min per mg protein.
Changes in the levels of bacterial enzymes in mucosal and faecal content
The activities of β-glucuronidase, β-glucosidase, nitroreductase, mucinase, β-galactosidase and sulfatase in the faecal and colonic mucosal contents of rats subjected to the different treatment schedules are shown in Tables 11 and 12. The activities of these enzymes were found to be significantly increased in the DMH-alone treated rats (Group 2) as compared with the control rats. Eriodictyol treatment with DMH during the initiation, post-initiation and over the entire period (Groups 4, 5 and 6) stages of carcinogenesis significantly decreased the levels of these enzymes in the faecal and colonic mucosal enzymes as compared with the DMH-alone treated rats (Group 2).
Table 11. Effect of eriodictyol on bacterial enzymes of colonic mucosa of control and experimental rats.
| Group | β-Glucuronidase a | β-Glucosidase a | Nitroreductase b | β-Galactosidase a |
| Control | 19.23 ± 0.13a | 27.36 ± 0.02a | 13.28 ± 0.09a | 28.33 ± 0.11a |
| DMH | 27.46 ± 0.11b | 68.74 ± 0.06b | 30.54 ± 0.10b | 79.22 ± 0.05b |
| Eriodictyol | 20.88 ± 0.15c | 29.15 ± 0.08a | 12.05 ± 0.09a | 29.43 ± 0.02a |
| Initiation | 23.17 ± 0.18d | 59.44 ± 0.03b | 26.28 ± 0.08c | 66.71 ± 0.13c |
| Post-initiation | 20.65 ± 0.14c | 45.32 ± 0.02c | 19.34 ± 0.07d | 55.30 ± 0.08d |
| Entire period | 19.53 ± 0.16a | 39.24 ± 0.06d | 16.78 ± 0.02e | 30.64 ± 0.07e |
amg of p-nitrophenol liberated per min per g protein.
bμmol of p-aminobenzoic liberated per min per g protein.
Table 12. Effect of eriodictyol on faecal bacterial enzymes of control and experimental rats.
| Group | β-Glucuronidase a | β-Glucosidase a | Nitroreductase d | Mucinase b | β-Galactosidase a | Sulfatase c |
| Control | 75.21 ± 0.04a | 56.12 ± 0.21a | 18.38 ± 0.03a | 46.22 ± 0.15a | 32.57 ± 0.12a | 3.61 ± 0.02a |
| DMH | 110.63 ± 0.18b | 121.34 ± 0.22b | 34.57 ± 0.13b | 75.27 ± 0.08b | 73.29 ± 0.25b | 8.19 ± 0.04b |
| Eriodictyol | 74.91 ± 0.14a | 54.25 ± 0.28a | 19.53 ± 0.05a | 47.33 ± 0.18a | 30.42 ± 0.04a | 3.46 ± 0.12a |
| Initiation | 98.46 ± 0.24c | 96.44 ± 0.14c | 28.41 ± 0.04c | 72.15 ± 0.16c | 68.44 ± 0.12c | 4.18 ± 0.13c |
| Post-initiation | 83.31 ± 0.20d | 77.32 ± 0.15d | 24.66 ± 0.07c | 57.36 ± 0.05d | 57.82 ± 0.09d | 4.06 ± 0.02c |
| Entire period | 79.62 ± 0.17e | 58.41 ± 0.19e | 19.73 ± 0.05d | 49.54 ± 0.14e | 49.61 ± 0.02e | 3.78 ± 0.14d |
amg of p-nitrophenol liberated per min per g protein.
bmg of glucose liberated per min per mg protein.
cμmol of p-nitrocatechol liberated per min per g protein.
dμmol of p-aminobenzoic liberated per min per g protein.
Determination of pathological alterations in hepatic tissue
Fig. 5 shows the results of the histological examination of liver sections of the control and experimental groups. The control and drug-control rat (Groups 1 and 3, A and C) livers show a normal architecture. It can be seen in Fig. 5(B) that the DMH-induced (Group 2) rat livers show a loss of architecture with more irregular nuclei and numerous mitotic figures. Groups 4, 5 and 6 (D, E and F), referring to the initiation post-initiation and over the entire period treated rats, show few neoplastically transformed cells or hepatocytes and show fewer neoplastic cells, a near-normal architecture and a significant improvement in liver histopathology.
Fig. 5. Effect of eriodictyol treatment with DMH-induced rats in hepatic tissue. (A) Normal histological appearance of the liver (Group 1). (B) Showing micro and macrovesicular types of fatty changes, Kupffer cell hyperplasia, nuclear pleomorphism and inflammatory cell infiltrates in the portal triad. (C) Positive control rats group showing normal crypts, initiation, post-initiation and entire-period (D, E and F) treatment in DMH-induced rat livers showing less neoplastic cells, a near-normal architecture and significant improvement in liver histopathology.

Determination of pathological alteration in colonic tissue
Fig. 6 constitutes the histological analysis of the control and experimental colon carcinogenesis rats: (A) colon of a control rat showing a normal mucosal fold with a normal histological appearance; (B) colon of a DMH-treated rat showing the mucin produce adenocarcinoma with a dysplastic zone, mucosa infiltrating up to the submucosa and dilation of the goblet cells; (C) colon of a control + eriodictyol-treated rat showing the normal mucosal gland with infiltration in the porphyria region; (D) colon of an initiation group (Group 4) rat showing intramucosal cancerous change with dilated mucosal glands; (E) colon of a post-initiation group (Group 5) rat showing crypt aggregates in a colonic lumen that appears to resist the tumour invading deeper structures; (F) colon of an entire period group (Group 6) rat showing reduced malignant glands with lymphoid aggregates in the sub mucosa and an histological appearance similar to that of the control rat.
Fig. 6. Effect of eriodictyol treatment with DMH-induced rats in the distal colon. (A) Normal histological appearance of the colon (Group 1) (B) DMH-treated rats showing tubular adenocarcinoma compared to the control rats. (C) Positive control rats group showing normal crypts, (D) initiation, (E) post-initiation, (F) entire period groups showing reduced malignant glands.

Eriodictyol suppresses cell proliferation
The silver staining method for argyrophilic nucleolar organizer region-associated proteins (AgNORs) was used to visualize the NORs in chromosomes and nucleoli. Fig. 7 shows the colonic crypts of the control and experimental rats. The DMH-induced group (Group 2) shows a significantly increased count of AgNORs, whereas Groups 4, 5 and 6 have significantly reduced AgNORs counts.
Fig. 7. Silver staining of argyrophilic nucleolar organizing regions in the colonic tissue (190 μm). (A and C) normal colonocytes lack silver positive black dots. (B) Exhibiting irregular glands of intense silver positive black dots. (D, E, F and G) AgNORs was significantly decreased upon treatment with eriodictyol during the initiation, post-initiation and entire period.

Discussion
Our results obtained in this study indicate that eriodictyol supplementation brings about intense alterations in tissue lipid peroxidation, bacterial enzyme activities, antioxidant status and the incidence of aberrant crypt foci (ACF) in a chemically induced rat colon carcinogenesis model.
The colon specific carcinogen of DMH by itself is not mutagenic, but when it is metabolized in the liver it is converted to azoxymethane (AOM), which ultimately leads to the conversion of methyldiazonium ions and carbonium ions, which are active carcinogenic electrophiles that are secreted through bile into the intestines.47 DMH induction is known to increase the incidence of DMH-induced colon cancer. ACF are benign adenomatous polyps and preneoplastic lesions of colorectal carcinoma, which are effective intermediate biomarkers to evaluate the modifying effects of specific natural and synthetic compounds on DMH-induced carcinogenesis.48 Eriodictyol supplementation has been shown to decrease the number of crypts multiplicity and to improved morphological and structural changes, along with a few dysplastic colonocytes and congested sinusoids. The treatment of eriodictyol for the entire period of the experiment significantly decreased the occurrence of ACF as observed at the end of the study. It has been reported that β-glucuronidase inhibitor suppresses ACF incidence in azoxymethane (AOM)-induced carcinogenesis in rat colon.49–51 These findings suggest that the suppression of the formation of ACF is strongly influenced by a decrease in the level of β-glucuronidase activity in the colon. Our results also correlate with the above findings showing that eriodictyol supplementation significantly reduces the activity of β-glucuronidase and also lowers the number of ACF in DMH-treated rats.
Lipid peroxidation plays a major role in the carcinogenicity pathway in different cancers, including CRC, since lipid peroxidation is mutagenic at the end of the pathways and so it may be probably involved in cell proliferation, which can lead to the development of colon cancer cells.52,53 Hence, a decreased level of colonic lipid peroxidation in DMH-induced rats could be due to increased cell proliferation. Therefore, malignant tissues are observed to be less efficient and more resistant to free radical attack, and consequently, lipid peroxidation is less intense.54 Previous studies have proven that rats on DMH treatment for a long period have shown reduced rates of colonic lipid peroxidation as compared to the control rats,55,56 which indicates an apparent inverse relationship between lipid peroxidation and the rates of cell proliferation. Our results showed that DMH-alone treated rats had significantly decreased lipid peroxidation levels in colonic tissues. Eriodictyol supplementation to DMH-treated rats throughout the experimental period (Groups 4–6) restored the TBARS, LOOH and CD levels as compared to DMH-alone treated rats (Group 2). From our results, it was noted that eriodictyol can protect cells from the DMH-induced loss of their oxidative capacity, which may be also due to the antiproliferative activity of eriodictyol. Hence, we suggest that eriodictyol prevents chemically induced colon carcinogenesis through its antiproliferative effect.
The primary defence antioxidant enzymes SOD and CAT act against both endogenous and exogenous toxic compounds, such as ROS and chemical carcinogens. These enzymes are involved in the direct elimination of reactive oxygen species.57,58 Increased exposure to radicals or impaired regulation of these protective enzymes leads to diseases, including cancer.59 In our study, we observed that the decreased activities of SOD and CAT in the DMH-treated rats could be due to the dangerous increase in the levels of ROS and thus the enhanced oxidative stress, leading to the proliferation of malignant colonocytes. Eriodictyol, due to its ability to scavenge free radicals and toxins and carcinogenic electrophiles, may substitute the antioxidant enzymes, which could lead to enhanced SOD and CAT levels in the colonic tissues. GSH, GST and GR predominantly participate in the detoxification of environmental carcinogens,60,61 free radicals and peroxides by conjugating toxic substances with GSH, eventually protecting cells and organs against carcinogen-induced toxicity. GPx plays a very important protective role by removing hydrogen peroxide and lipid hydroperoxides.62 These enzymes constitute an important defence mechanism in protecting cells against oxygen free radicals. Decreased lipid peroxidation associated with enhanced GSH and glutathione-dependent enzymes in the colon are a well-known phenomenon in experimental colon carcinogenesis.63 In our study, we found increased levels of GSH, GPx, GST and GR in the colonic cells of rats treated with DMH, which may be due to the elevated cell proliferation, expressed in greater amounts in the neoplastic cells and conferring a selective growth advantage in the pathogenesis of DMH-induced colon cancer.64,65 Eriodictyol rejuvenated and restored these antioxidant enzymes, thus acting as an ideal chemopreventive agent.
The bacterial metabolic products of colonic cells are associated with damaging effects on the host and in particular an induced initiation and promotion of tumourigenesis.66,67 β-Glucuronidase, a bacterial enzyme, is believed to be predominantly responsible for the hydrolysis of β-glucuronic acid and liberates unconjugated isoforms in the final activation of DMH metabolites to its toxic carcinogen on the walls of colonic epithelial cells.68 The increased activity of β-glucuronidase observed in DMH-induced colorectal tumours and also faecal β-glucuronidase activity shows the metabolic degradation of carcinogens. At this point, the β-glucuronidase activity was found to be decreased upon eriodcityol treatment, which may be due to the anti-inflammatory property of eriodictyol. The higher faecal β-glucuronidase activity could be related to excessive tumour incidence and further, we assumed that the microflora may have been modified by the continuous high fat feeding and DMH injections. Our findings state that eriodictyol treatment to DMH-treated rats significantly decreased the β-glucuronidase activity as compared to in the DMH-alone treated group.
β-Glucosidase plays an important role in metabolic pathways that occur in the cytosol in animals, such as glycolysis and gluconeogenesis, but it is mostly found in the liver, kidney and intestines and efficiently hydrolyzes β-d-glucoside.69 The hydrolysis of glycosides by β-glucosidase in the gut can deliver toxic mutagenic and carcinogenic aglycones upon its increased activity as in the case of the DMH-alone treated rats. Our results demonstrated that the activity of β-glucosidase could be reduced on eriodictyol treatment to DMH-induced rats. The decreased activity of β-glucosidase in the colon might be relative to the inhibitory effect of eriodictyol in DMH-induced colon carcinogenesis.70
Beta-galactosidase is a lysosomal exoglycosidase involved in the degradation of glycoconjugates by the sequential release of β-linked terminal galactosyl residues.71 β-Galactosidase enzymes may act on short chain oligosaccharides released from pectin galactans. The increased levels of β-galactosidase were brought back to a near normal level in the eriodictyol-treated rats as the tumours completely regressed. Mucinase is an enzyme present in the intestinal microflora, which hydrolyses the protective mucins in the colon. Increased mucus shedding by an enzyme capable of destroying mucin, with a contribution from multiple enzymes, is an important factor elaborating the degradation of the mucosal layer on the intestine and represents a potential target on the colon epithelium, which permits measurement of mucin degradation by the release of reducing sugars.72 The results of mucinase demonstrated that the treatment of eriodictyol was found to reduce the activity of mucinase by decreasing the colonic mucosal cell microsomal enzyme activity that plays a role in the metabolism of many drugs by inhibiting the transformation of the colonic cells.73 Moreover, decreased mucinase activity is correlated with the lowered incidence of the colon tumour. The activity of mucinase was significantly decreased in the eriodictyol-treated to DMH-induced groups as compared to the unsupplemented DMH-induced groups in our study.
Being a flavonoid, eriodictyol is also considered to inhibit sulfo-conjugations leading to decreased activities of the nitroreductase sulfatase enzymes. Increased levels of sulfatase and nitroreductase were found in the DMH-treated groups due to the bacterial lineages and elevated colon cancer risk.74 The findings of the current study clearly suggest that eriodictyol treatment reduces the faecal activity of sulfatase and nitroreductase, which may be due to the suppressive activity of eriodictyol through the inhibition of pro-oncogenic growth factors. Finally, we assumed that the increased activity of bacterial enzymes by DMH-induced groups failed to proceed as eriodictyol plays an important role in interrupting the energy requirement of tumour tissues, leading to the suppression of tumour growth and producing an inhibitory mechanism against enhanced bacterial activity in the colon.
In addition, we revealed the observation of histopathological changes in various ranges of the pathological abnormalities in the colon and liver of DMH-treated rats. Our results indicated that supplementation with eriodictyol could decrease the pathological alterations in the colon, thereby reducing the degree of dysplastic cells in the DMH-treated rats. However, eriodictyol treatment to DMH-exposed rats effectively reduced hyperplasia, anaplasia and inflammatory cell infiltration in the liver and colon tissues, validating eriodictyol as a promising anticarcinogenic and hepatoprotective agent for colon carcinogenesis.75,76
The uncontrolled and rapid cell proliferation of epithelial cells is considered an indicator of colon cancer risk. AgNORs scores may be important not only in the diagnosis of a potentially malignant lesion but also in defining the margins of an abnormal tissue.77 It has been revealed that variations in the number, size and shape of AgNORs per nucleus reflect the status of tumour cell proliferation.78 Based on the above findings, we observed that the area of AgNORs was significantly increased in carcinogen-intoxicated rats, indicating enhanced cell maturation on eriodictyol treatment, where the AgNORs/nucleus was significantly reduced in the colonic tissues of DMH-induced rats. This result indicates eriodictyol has beneficial effects against tumour cell proliferation, which proves its antiproliferative activity. Overall, the results of the present study demonstrate that eriodictyol has a beneficial effect on chemically induced colonic preneoplastic progression in rats induced colon cancer by DMH. A schematic representation of eriodictyol activities in DMH-induced colon cancer is shown in Fig. 8. Overall, the findings suggest that the preventive effect of eriodictyol against rat colon tumorigenesis was more detectable when eriodictyol was supplemented during the entire period of the study.
Fig. 8. Schematic of eriodictyol activities in DMH-induced colon cancer.

Conclusions
Our study proves that eriodictyol exerts inhibitory effects on DMH-induced colon carcinogenesis, which means it can act as an anticarcinogenic agent. Eriodictyol exhibits a protective role by inhibiting as well as detoxifying the carcinogen, modulation of LPO markers, by regulation of the biotransforming enzymes and by maintaining the antioxidant defence enzymes. Finally, eriodictyol significantly inhibits the incidence of ACF, having a modulatory effect on preneoplastic lesions and leading to a reduction of cell proliferation in AgNORs staining in the DMH-induced rat colon carcinogenesis. Further studies are in progress to evaluate eriodictyol, which has shown promise as a chemotherapeutic agent of colon cancer.
Conflict of interest
There are no conflicts of interest to declare.
Acknowledgments
This study was supported by a grant from the Science and Engineering Research Board, Department of Science & Technology, Government of India. (SERB File No.SB/FT/LS-215/2012 dated 26.04.2013).
References
- Li B., Chen P., Chang Y., Qi J., Fu H., Guo H. Biochem. Biophys. Res. Commun. 2016;478:739–745. doi: 10.1016/j.bbrc.2016.08.018. [DOI] [PubMed] [Google Scholar]
- Wang J., Wu H. F., Shen W., Xu D. Y., Ruan T. Y., Tao G. Q., Lu P. H. Gene. 2016;586:41–47. doi: 10.1016/j.gene.2016.03.051. [DOI] [PubMed] [Google Scholar]
- Przygodzka P., Papiewska-Pajak I., Bogusz H., Kryczka J., Sobierajska K., Kowalska M. A., Boncela J. Biochim. Biophys. Acta. 2016;1860:2445–2453. doi: 10.1016/j.bbagen.2016.07.012. [DOI] [PubMed] [Google Scholar]
- Manju V., Nalini N. Clin. Chim. Acta. 2005;358:60–67. doi: 10.1016/j.cccn.2005.02.018. [DOI] [PubMed] [Google Scholar]
- Betteridge D. J. Metabolism. 2000;49:3–8. doi: 10.1016/s0026-0495(00)80077-3. [DOI] [PubMed] [Google Scholar]
- Amérand A., Mortelette H., Belhomme M., Moisan C. Respir. Physiol. Neurobiol. 2017;235:40–44. doi: 10.1016/j.resp.2016.10.001. [DOI] [PubMed] [Google Scholar]
- Taketo M. M., Edelmann W. Gastroenterology. 2009;136:780–798. doi: 10.1053/j.gastro.2008.12.049. [DOI] [PubMed] [Google Scholar]
- Hawk E. T., Levin B. J. Clin. Oncol. 2005;23:378–391. doi: 10.1200/JCO.2005.08.097. [DOI] [PubMed] [Google Scholar]
- Tsuda H., Futakuchi M., Fukamachi K., Shirai T., Imaida K., Fukushima S., Tatematsu M., Furukawa F., Tamano S., Ito N. Toxicol. Pathol. 2010;38:182–187. doi: 10.1177/0192623309356451. [DOI] [PubMed] [Google Scholar]
- Ha-Na N., Hoonjeong K., You-Gyoung P., Choong-III C., Jong-Sug P., Taesun P., Okezie I. A., Mi-Kyung S. Nutr. Res. 2007;27:659–664. [Google Scholar]
- Kinjo T., Suzui M., Morioka T., Nabandith V., Inamine M., Kaneshiro T., Arakaki J., Nishimaki T., Yoshimi N. J. Exp. Clin. Cancer Res. 2006;25:89–95. [PubMed] [Google Scholar]
- Muthu R., Selvaraj N., Vaiyapuri M. Hum. Exp. Toxicol. 2016;35:1–14. doi: 10.1177/0960327115618245. [DOI] [PubMed] [Google Scholar]
- Bird R. P. Cancer Lett. 1987;37:147–151. doi: 10.1016/0304-3835(87)90157-1. [DOI] [PubMed] [Google Scholar]
- Rudolph R. E., Dominitz J. A., Lampe J. W., Levy L., Qu P., Li S. S., Lampe P. D., Bronner M. P., Potter J. D. Cancer Epidemiol. Biomarkers Prev. 2005;14:605–608. doi: 10.1158/1055-9965.EPI-04-0058. [DOI] [PubMed] [Google Scholar]
- Zhu Q. C., Gao R. Y., Wu W., Guo B. M., Peng J. Y., Qin H. L. World J. Gastroenterol. 2014;20:8119–8129. doi: 10.3748/wjg.v20.i25.8119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalini N., Manju V., Menon V. P. Clin. Chim. Acta. 2004;342:203–210. doi: 10.1016/j.cccn.2004.01.001. [DOI] [PubMed] [Google Scholar]
- Davie J. R. J. Nutr. 2003;133:2485–2493. doi: 10.1093/jn/133.7.2485S. [DOI] [PubMed] [Google Scholar]
- Reddy B. S., Weisburger J. H., Wynder E. L. Science. 1974;183:416–417. doi: 10.1126/science.183.4123.416. [DOI] [PubMed] [Google Scholar]
- Robertson W., Ropes M. W., Bauer W. J. Biol. Chem. 1940;133:261–276. [Google Scholar]
- Hecht H. J., Erdmann H., Park H. J., Sprinzl M., Schmid R. D. Nat. Struct. Biol. 1995;2:1109–1114. doi: 10.1038/nsb1295-1109. [DOI] [PubMed] [Google Scholar]
- Johnson J., Maher P., Hanneken A. Invest. Ophthalmol. Visual Sci. 2009;50:2398–2406. doi: 10.1167/iovs.08-2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C. Z., Jin H. H., Sun H. X., Zhang Z. Z., Zheng J. X., Li S. H., Han S. H. Eur. J. Pharmacol. 2016;772:124–130. doi: 10.1016/j.ejphar.2015.12.042. [DOI] [PubMed] [Google Scholar]
- Imen M. B., Chaabane F., Nadia M., Soumaya K. J., Kamel G., Leila C. G. Life Sci. 2015;135:173–178. doi: 10.1016/j.lfs.2015.06.022. [DOI] [PubMed] [Google Scholar]
- Havsteen B. Biochem. Pharmacol. 1983;32:1141–1148. doi: 10.1016/0006-2952(83)90262-9. [DOI] [PubMed] [Google Scholar]
- Clavin M., Gorzalczany S., Macho A., Muñoz E., Ferraro G., Acevedo C., Martino V. J. Ethnopharmacol. 2007;112:585–589. doi: 10.1016/j.jep.2007.04.007. [DOI] [PubMed] [Google Scholar]
- Wang F., Wang Yu.-H., Wang J.-J., Xu He.-L., Wang C.-M. Bangladesh J. Pharmacol. 2016;11:285–291. [Google Scholar]
- Nalini N., Manju V., Menon V. P. Clin. Chim. Acta. 2004;342:203–210. doi: 10.1016/j.cccn.2004.01.001. [DOI] [PubMed] [Google Scholar]
- Sivakumar A. S., Viswanathan P., Anuradha C. V. Redox Rep. 2010;15:224–232. doi: 10.1179/135100010X12826446921545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossato M. F., Trevisan G., Walker C. I., Klafke J. Z., de Oliveira A. P., Villarinho J. G., Zanon R. B., Royes L. F., Athayde M. L., Gomez M. V., Ferreira J. Biochem. Pharmacol. 2011;81:544–551. doi: 10.1016/j.bcp.2010.11.004. [DOI] [PubMed] [Google Scholar]
- Nalini N., Manju V., Menon V. P. Clin. Chim. Acta. 2004;342:203–210. doi: 10.1016/j.cccn.2004.01.001. [DOI] [PubMed] [Google Scholar]
- Ohkawa H., Ohishi N., Yagi K. Anal. Biochem. 1979;95:35l–358. doi: 10.1016/0003-2697(79)90738-3. [DOI] [PubMed] [Google Scholar]
- Rao K. S., Recknagel R. O. Exp. Mol. Pathol. 1968;9:271–278. doi: 10.1016/0014-4800(68)90041-5. [DOI] [PubMed] [Google Scholar]
- Jiang Z. Y., Woollard A. C., Wolff S. P. Lipids. 1991;26:856–856. doi: 10.1007/BF02536169. [DOI] [PubMed] [Google Scholar]
- Freeman H. J. Cancer Res. 1986;46:5529–5532. [PubMed] [Google Scholar]
- Shiau S. Y., Chang G. W. J. Nutr. 1983;113:138–144. doi: 10.1093/jn/113.1.138. [DOI] [PubMed] [Google Scholar]
- Rowland I. R., Mallett A. K., Wise A. Toxicol. Appl. Pharmacol. 1983;69:l43–l48. doi: 10.1016/0041-008x(83)90130-8. [DOI] [PubMed] [Google Scholar]
- Bratton A. C., Marshall E. K. J. Biol. Chem. 1939;128:537–550. [Google Scholar]
- Ellman G. L., Fiches F. T. Biochem. Biophys. 1959;82:70–72. doi: 10.1016/0003-9861(59)90090-6. [DOI] [PubMed] [Google Scholar]
- Rotruck J. T., Pope A. L., Ganther H. E., Swanson A. B., Hafeman D. G., Hoekstra W. G. Science. 1973;179:588–590. doi: 10.1126/science.179.4073.588. [DOI] [PubMed] [Google Scholar]
- Habig W. H., Pabst M. J., Jakoby W. B. J. Biol. Chem. 1974;249:7130–7139. [PubMed] [Google Scholar]
- Carlberg I., Mannervik B. J. Biol. Chem. 1975;250:5475–5480. [PubMed] [Google Scholar]
- Kakkar P., Das B., Viswanathan P. N. Indian J. Biochem. Biophys. 1984;21:130–132. [PubMed] [Google Scholar]
- Sinha A. K. Anal. Biochem. 1972;47:389–394. doi: 10.1016/0003-2697(72)90132-7. [DOI] [PubMed] [Google Scholar]
- Muthu R., Selvaraj N., Vaiyapuri M. Hum. Exp. Toxicol. 2016;35:1–14. doi: 10.1177/0960327115618245. [DOI] [PubMed] [Google Scholar]
- Trere D., Pession A., Derenzini M. Exp. Cell Res. 1989;184:131–137. doi: 10.1016/0014-4827(89)90371-6. [DOI] [PubMed] [Google Scholar]
- Duncan B. D. Biometrics. 1957;13:164–176. [Google Scholar]
- Fiala E. S. Cancer. 1977;40:2436–2445. doi: 10.1002/1097-0142(197711)40:5+<2436::aid-cncr2820400908>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- Traverso G., Shuber A., Olsson L., Levin B., Johnson C., Hamilton S. R., Boynton K., Kinzler K. W., Vogelstein B. Lancet. 2002;359:403–404. doi: 10.1016/S0140-6736(02)07591-8. [DOI] [PubMed] [Google Scholar]
- Takada H., Hirooka T., Hiramatsu Y., Yamamoto M. Cancer Res. 1982;42:33l–334. [PubMed] [Google Scholar]
- Karthik Kumar V., Vennila S., Nalini N. Food Chem. Toxicol. 2009;47:309–315. doi: 10.1016/j.fct.2008.11.017. [DOI] [PubMed] [Google Scholar]
- Youssef K. M., Ezzo A. M., El-Sayed M. I., Hazzaa A. A., El-Medany A. H., Arafa M. Future J. Pharmaceut Sci. 2015;1:57–72. [Google Scholar]
- Peluso M., Munnia A., Risso G. G., Catarzi S., Piro S., Ceppi M., Giese R. W., Brancato B. Free Radical Res. 2011;45:477–482. doi: 10.3109/10715762.2010.549485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma R., Ellis B., Sharma A. Phytother. Res. 2010;25:563–568. doi: 10.1002/ptr.3297. [DOI] [PubMed] [Google Scholar]
- Ashokkumar P., Sudhandiran G. Biomed. Pharmacother. 2008;9:590–597. doi: 10.1016/j.biopha.2008.06.031. [DOI] [PubMed] [Google Scholar]
- Manju V., Nalini N. J. Biochem. Technol. 2010;2:161–167. [Google Scholar]
- Hanene J. H., Sonia B. H. K., Aya M., Rabeb E. G., Touhami M. J. Funct. Foods. 2013;5:1310–1316. [Google Scholar]
- Ray G., Batra S., Shukla N. K., Deo S., Raina V., Ashok S., Husain S. A. Breast Cancer Res. Treat. 2000;59:163–170. doi: 10.1023/a:1006357330486. [DOI] [PubMed] [Google Scholar]
- Arigesavan K., Sudhandiran G. Biochem. Biophys. Res. Commun. 2015;461:314–320. doi: 10.1016/j.bbrc.2015.04.030. [DOI] [PubMed] [Google Scholar]
- Byung Pal Y. U. Physiol. Rev. 1994;14:139–162. [Google Scholar]
- Hayes J. D., McLellan L. I. Free Radical Res. 1999;31:273–300. doi: 10.1080/10715769900300851. [DOI] [PubMed] [Google Scholar]
- Gopalan P., Jensen D. E., Lotlikar P. D. Cancer Lett. 1992;64:225–233. doi: 10.1016/0304-3835(92)90047-y. [DOI] [PubMed] [Google Scholar]
- Navarro J., Obrador E., Carretero J., Petschen I., Avino J., Perez P., Estrela J. M. Free Radical Biol. Med. 1999;26:410–418. doi: 10.1016/s0891-5849(98)00213-5. [DOI] [PubMed] [Google Scholar]
- Manju V., Nalini N. Clin. Chim. Acta. 2005;358:60–67. doi: 10.1016/j.cccn.2005.02.018. [DOI] [PubMed] [Google Scholar]
- Vinoth Kumar R., Karthikkumar V., Viswanathan P., Kabalimoorthy J., Nalini N. Environ. Toxicol. Pharmacol. 2014;37:174–184. doi: 10.1016/j.etap.2013.11.022. [DOI] [PubMed] [Google Scholar]
- Manoj G. R., Thampi B. S. H., Menon V. P., Leelamma S. J. Nutr. Biochem. 1999;10:555–560. doi: 10.1016/s0955-2863(99)00035-2. [DOI] [PubMed] [Google Scholar]
- Lokesh Kumar B., Sathishkumar V., Nandakumar N., Rengarajan T., Madankumar A., Balasubramanian M. P. J. Food Drug Anal. 2016;24:206–213. [Google Scholar]
- Reddy B. S., Engle A., Simi B., Goldman M. Gastroenterology. 1992;102:1475–1482. doi: 10.1016/0016-5085(92)91704-8. [DOI] [PubMed] [Google Scholar]
- Reddy B. S., Narisawa T., Weisburger J. H. Cancer Res. 1976;36:2874–2876. [PubMed] [Google Scholar]
- de Graaf M., van Veen I. C., van der Meulen-Muileman I. H., Gerritsen W. R., Pinedo H. M., Haisma H. J. Biochem. J. 2001;356:907–910. doi: 10.1042/0264-6021:3560907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karthik Kumar V., Vennila S., Nalini N. Food Chem. Toxicol. 2009;47:309–315. doi: 10.1016/j.fct.2008.11.017. [DOI] [PubMed] [Google Scholar]
- Ram K., Melinda D. C., Priya D., Shiladitya D. BMC Biotechnol. 2013;13:3. [Google Scholar]
- Shiau S. Y., Ong Y. O. J. Nutr. Sci. Vitaminol. 1992;38:49–55. doi: 10.3177/jnsv.38.49. [DOI] [PubMed] [Google Scholar]
- Goldin B. R., Gorbach S. L. J. Natl. Cancer Inst. 1984;73:689–695. [PubMed] [Google Scholar]
- Goldin B. R., Gorbach S. L. J. Natl. Cancer Inst. 1980;64:263–265. doi: 10.1093/jnci/64.2.263. [DOI] [PubMed] [Google Scholar]
- Hanene J. H., Sonia B. H. K., Wassim Y. A., Aya M., Zohra H., Touhami M. Eur. J. Cancer. 2013;49:1127–1135. [Google Scholar]
- Ragunath M., Prabu T., Nadanasabapathy S., Revathi R., Manju V. Biomed. Prevent Nutr. 2013;3:74–82. [Google Scholar]
- Roussel P., Hernandez-Verden D. Exp. Cell Res. 1994;214:465–472. doi: 10.1006/excr.1994.1283. [DOI] [PubMed] [Google Scholar]
- Jaafari-Ashkavandi Z., Fatemi F. S. J. Craniofac. Surg. 2013;24:788–791. doi: 10.1097/SCS.0b013e31828b6e0e. [DOI] [PubMed] [Google Scholar]