


Abstract
The clustered regularly interspersed short palindromic repeat (CRISPR) gene-editing system has been repurposed for live-cell genomic imaging, but existing approaches rely on fluorescent protein reporters, making sensitive and continuous imaging difficult. Here, we present a fluorophore-based live-cell genomic imaging system that consists of a nuclease-deactivated mutant of the Cas9 protein (dCas9), a molecular beacon (MB), and an engineered single-guide RNA (sgRNA) harboring a unique MB target sequence (sgRNA-MTS), termed CRISPR/MB. Specifically, dCas9 and sgRNA-MTS are first co-expressed to target a specific locus in cells, followed by delivery of MBs that can then hybridize to MTS to illuminate the target locus. We demonstrated the feasibility of this approach for quantifying genomic loci, for monitoring chromatin dynamics, and for dual-color imaging when using two orthogonal MB/MTS pairs. With flexibility in selecting different combinations of fluorophore/quencher pairs and MB/MTS sequences, our CRISPR/MB hybrid system could be a promising platform for investigating chromatin activities.
INTRODUCTION
Since its discovery, the clustered regularly interspersed short palindromic repeat (CRISPR) gene-editing system has garnered significant attention and widespread use in the scientific community, owing to its versatility and simplicity for targeting gene sequences of interest. The CRISPR system consists of CRISPR-associated (Cas9) endonuclease and a single-guide RNA (sgRNA), which contains a specialized motif for Cas9 association and a spacer sequence complementary to the target DNA sequence of interest (1). Binding of Cas9 to the specific locus occurs in an sgRNA-guided fashion, destroying the sequence of the target by introducing mutagenic double-strand breaks. Based on this RNA-guided nuclease activity, recent work has demonstrated the capacity to repurpose CRISPR for non-gene-editing applications, including live-cell imaging of genomic loci (2–11). In this context, Cas9 is replaced with a nuclease-deactivated mutant (dCas9), which still retains the ability to associate with sgRNA and to bind to target loci (2–11).
Currently, CRISPR/dCas9-based studies of chromatin dynamics have been achieved with the use of fluorescent protein (FP) reporters (2–11). By using dCas9 fused to FP or sgRNA engineered to harbor a unique RNA sequence (an aptamer) that can bind specifically to a cognate protein fused to FP, valuable information has been revealed with unprecedented detail. Despite these advances, sensitive measurement of chromatin activities using FP-based approaches still remains elusive, owing to low signal-to-background due to the low photon output of commonly-used FPs and the high background fluorescence contributed by unbound probes. To improve signal detection, a common strategy has been to modify dCas9 or sgRNA to carry multiple copies of FPs, but extensive modification risks influencing expression or stability of each component, resulting in decreased genomic labeling (12). One tool that can achieve high signal-to-background is the molecular beacon (MB), a class of small, single-stranded, hybridization-activated oligonucleotide probe with a fluorophore and a quencher at the two termini (13). In the absence of target, the short arm sequences flanking the loop domain hybridize to each other to form a stable duplex stem. In this configuration, the probe emits a low fluorescence signal because the fluorophore is significantly quenched by the quencher situated in close spatial proximity. Hybridization of the loop domain to a complementary nucleic acid sequence disrupts the stem, leading to separation of the fluorophore from the quencher to restore fluorescence. With careful selection of fluorophore/quencher pair, it has been demonstrated that MB fluorescence can increase up to 100-fold upon hybridization to target RNA (14). This unique ability of MBs to convert target recognition into a measurable fluorescence signal without the need to wash away unbound probes has made MBs a probe of choice for analyzing various types of RNA species in living cells (15–35).
The genomic DNA double helix is highly stable and inaccessible to MBs and potentially other oligonucleotide-based probes in the native cellular context. Recognizing the capacity of dCas9–sgRNA complexes to bind specific genomic loci, the ease and cost-effectiveness of modifying sgRNAs for genomic labeling, and the sensitivity of MBs for RNA labeling, in this study we aimed to develop a CRISPR/MB hybrid genomic labeling strategy by combining dCas9, an MB, and an sgRNA scaffold modified to harbor a unique MB target sequence (MTS) (Figure 1). We created three different sgRNA scaffolds by inserting the MTS at the tetraloop (TL-sgRNA-MTS) (Figure 1 upper panel), the stem–loop 2 (SL2-sgRNA-MTS) (Figure 1 middle panel) and the 3′-tail region (3′-sgRNA-MTS) (Figure 1 bottom panel). Since these regions were previously shown to be amendable to modification with aptamer sequence (2,4–6), we anticipated that MTS insertion should not interfere with formation of the dCas9–sgRNA complex and its binding to a target DNA sequence, enabling the target sequence to fluoresce upon hybridization of the MB to the MTS.
Figure 1.

Schematic illustration of the CRISPR/MB labeling strategy. A CRISPR/MB system consists of dCas9, an MB, and an sgRNA scaffold harboring a unique MB target sequence (MTS) (green). Three different sgRNA scaffolds were created by inserting an MTS at the tetraloop (TL-sgRNA-MTS) (upper panel), the stem–loop 2 (SL2-sgRNA-MTS) (middle panel), and the 3′-tail region (3′-sgRNA-MTS) (bottom panel), respectively. The spacer sequence (purple) specifies genomic binding. To target a genomic locus, dCas9 and sgRNA-MTS were first co-expressed in cells, followed by delivery of MBs. It was hypothesized that hybridization of the MB to the MTS should not interfere with dCas9 binding, leading to illumination of a specific genomic locus.
MATERIALS AND METHODS
Cloning
pSLQ1658-dCas9-EGFP, which encodes the nuclease-deactivated Streptococcus pyogenes Cas9 (dCas9) protein, fused to EGFP, was a gift from Bo Huang & Stanley Qi (Addgene plasmid # 51023) (8). The derivative construct, pdCas9-C1, was generated by inserting the PCR product of dCas9 from pSLQ1658-dCas9-EGFP into pEGFP-C1 (Clontech) digested with AgeI and BamHI to excise EGFP (Forward primer: GCTACCGGTCGCCACCATGGTGCCCAAAAAGAAGAGGAAAGTGGACAAGA; reverse primer: ACTGCTGGATCCGTGGAACTACCTACCTTGCGCTTTTTCTTGGGA). The backbone plasmids containing genomic-targeting sgRNAs harboring two types of MTSs, named MTSa and MTSb, were custom-made by Beijing Genomics Institute (Beijing, China) (see Supplementary Table S1 for the sgRNA sequences). The backbone plasmid containing the telomere-targeting sgRNA lacking MTS (sgTelo-control) was a gift from Bo Huang & Stanley Qi (Addgene plasmid # 51024) (8).
For each sgRNA, we generated a mammalian expression vector sgRNA/EGFP/pdCas9-C1 in which the sgRNA, EGFP and dCas9 were expressed under the control of separate promoters. The U6-sgRNA cassette and the CMV-EGFP cassette were PCR amplified from each sgRNA backbone plasmid and pEGFP-C1, respectively. The PCR products were then cloned into AseI-digested pdCas9-C1 using Gibson Assembly. For SL2-sgTelo-MTSa and SL2-sgSat-MTSb, sgRNA/BFP/pdCas9-C1 was similarly generated, with the CMV-BFP cassette derived from the pmTagBFP2-C1 plasmid (a gift from Prabuddha Sengupta, Janelia Research Campus, US) (see Supplementary Table S2 for primer information). The pEGFP-TRF1 construct was created by inserting the PCR product of TRF1 from cDNA of HEK293 cells into the EcoRI- and BamHI-digested pEGFP-C1 vector (forward primer: AGCTTCGAATTCTATGGCGGAGGATGTTTCCTCAGC; reverse primer: TCCGGTGGATCCACTTAACTGTGTCCTTTCATCAAGC).
Synthesis of MBs
The anti-MTSa MB was labeled with an ATTO647N fluorophore at the 5′ end and an Iowa Black RQ quencher at the 3′ end or an ATTO488 fluorophore at the 5′ end and an Iowa Black FQ quencher at the 3′ end, and has the sequence: 5′-mCmUmUmCmG*mU*mC*mC*mA*mC*mA*mA*mA*mC*mA*mC*mA*mA*mC*mU*mC*mC*mU*mGmAmAmG-3′ (m represents 2′-O-methyl RNA modification; * represents phosphorothioate linkage modification). The anti-MTSb MB was labeled with an Iowa Black FQ quencher at the 5′ end and an ATTO488 fluorophore at the 3′ end and has the sequence: 5′-mCmUmCmAmG*mC*mG*mU*mA*mA*mG*mU*mG*mA*mU*mG*mU*mC*mG*mU*mG*mA*mCmUmGmAmG-3′. The MB sequences are designed to avoid hybridization with endogenous RNAs in mammalian cells. All MBs were synthesized by Integrated DNA Technologies (Coralville, IA, USA).
Antibodies
Human anti-CREST antibodies (Cat #15-235-0001) were purchased from Antibodies Incorporated. Alexa647-labeled anti-human secondary antibodies (A21445) were purchased from Life Technologies.
Cell culture and transfection
HEK293 and HeLa cells (American Type Culture Collection) were cultured in Dulbecco's Modified Eagle's Medium (DMEM, Mediatech), supplemented with 10% (vol/vol) FBS (PAN™ Biotech) and 1× GlutaMAX™ (Thermo Fisher) at 37°C, 5% (vol/vol) CO2, and 90% relative humidity. Plasmid transfection was performed with FuGENE® 6 (Promega) according to the manufacturer's protocols when cells reached 50%–70% confluency. All experiments were performed with cells at passage numbers between 5 and 25.
Delivery of MBs
MBs were delivered into cells by microporation according to previously described methods (15,16). In brief, cells grown to 70% confluency were trypsinized, washed with 1× PBS and pelleted, followed by resuspension in 11 μl of resuspension buffer R (Thermo Fisher) at a final cell concentration of 5000 cells/μl and a final MB concentration of 1 μM. Thereafter, 10 μl of the cell mixture (∼50 000 cells) were microporated using the Neon® Transfection System with the parameters set at 1150 V with a 20 ms pulse width and 2 pulses total for HEK293 cells and 1005 V with a 35 ms pulse width and 2 pulses total for HeLa cells. Following microporation and three washes in culture medium to remove free MBs, the cells were seeded on 8-well Lab-Tek Chambered Coverglass previously coated with fibronectin.
In addition to microporation, we anticipate that other cellular delivery methods previously used in MB research, including nanoparticles, TAT-peptide, lipofectamine and streptolysin-O (20,28,30–35), should allow for efficient delivery of MBs into cells.
Fluorescence in situ hybridization
To verify that CRISPR/MB could label single telomere loci, cells containing dCas9, SL2-sgTelo-MTSa and MBs were subjected to fluorescence in situ hybridization experiments as previously described (36), with modifications. In brief, cells were fixed with 4% paraformaldehyde (PFA) in 1× PBS for 30 min at room temperature, followed by two washes with 1× PBS. Next, the cells were permeabilized with 0.5% (vol/vol) NP-40 in 1× PBS for 10 min. After washing the cells once with 1× PBS for 5 min, hybridization was carried out in hybridization buffer (1% Tween® 20, 10% dextran sulfate, 50% (vol/vol) formamide, 500 ng/ml Salmon sperm DNA in 2× SSC buffer) containing 100 nM telomere leading strand-targeting FISH probes (TAMRA-CCCTAACCCTAACCCTAA) or nonsense FISH probes (TAMRA-CTGCAGACATGGGTGATCCTCATGTTTTCTAG) for 24 h at 37°C in a humidified chamber. The cells were washed in 2× SSC, 10% (vol/vol) formamide followed by 2× SSC, 1× SSC and 0.2× SSC to remove unhybridized probes and then incubated in 1× PBS prior to imaging. It should be noted that as dCas9 and sgRNA interaction leads to separation of the DNA duplex to allow hybridization of the sgRNA spacer sequence to the lagging strand, this makes the leading strand accessible to hybridization with FISH probes without the need for high-temperature heating.
Fluorescence microscopy
Fluorescence microscopy experiments were performed on an Olympus IX 83 motorized inverted fluorescence microscope equipped with a 100× UPlanSApo 1.4NA objective lens, a back-illuminated EMCCD camera (Andor), Sutter excitation and emission filter wheels and X-Cite Series 120 light source housing a Mercury Lamp (EXFO) under the control of the CellSens Dimension software. Images were acquired using the Olympus MT20 filter set for DAPI, EGFP and TAMRA and a Chroma filter set for Cy5 (ET620/60x, ET700/75m, T660lpxr, Chroma) or a DV2-cube (ET525/50m, 585dcxr, ET655lp, Photometrics). Three-dimensional image stacks were acquired with 0.25 μm increments in the z-direction. All images were analyzed using Fiji (37), the AutoQuant deconvolution software (MediaCybernetics) or custom-written MATLAB (Version R2014b 64-bit, Mathworks) programs.
Identification of single telomere loci
Single telomere loci in HEK293 and HeLa cells were identified using methods described previously (15,16). In brief, rolling-ball background subtraction (background = 2) was first applied on all 3D images to enhance particulate objects. This was followed by identification of particles using the 3D Laplacian of Gaussian plug-in available for Fiji (37). After manually setting the threshold to remove low-intensity spots, a region of interest (ROI) was drawn around each nucleus and applied to the filtered stack. The Find Stack Maxima macro plug-in (Exclude Edge Maxima; Noise Tolerance = 0) was then used to identify all local maxima in each slice of the z-stack. To identify which 2D local maxima were 3D local maxima and to quantify the total number of 3D local maxima, a custom MATLAB program compared the intensity of each local maximum in each slice with the intensity of the neighboring pixels in the current slice and the two adjacent slices (nine pixels in the slice above, eight surrounding pixels in the same slice and nine pixels in the slice below). Each 3D maximum was considered a single telomere locus and the total number of telomere loci per nucleus was computed.
Colocalization analysis
After determining the 3D coordinates of telomere loci in FISH and CRISPR/MB images using the methods described above, a custom MATLAB program was employed to identify the colocalization level in 3D as previously described (15,16). In brief, an MB 3D local maximum was considered to be an MB colocalization event if a FISH 3D local maximum was found within a 5 × 5 × 5 voxel cube centered around the MB maximum. The percentage of MB signals that were colocalized with FISH signals (%MB colocalizing with FISH) was calculated by dividing the number of MB colocalization events by the total number of MB local maxima. A FISH 3D local maximum was considered to be a FISH colocalization event if an MB 3D local maximum was found within a 5 × 5 × 5 voxel cube centered around the FISH maximum. The percentage of FISH signals that were colocalized with MB signals was calculated by dividing the number of FISH colocalization events by the total number of FISH local maxima (%FISH colocalizing with MB).
Single-particle tracking analysis
Single genomic loci were identified and localized in time-lapse images acquired at 100 ms per frame using the TrackMate plugin of Fiji, as shown previously (15,16). In brief, individual peaks and their coordinates were determined by using Laplacian of Gaussian followed by Differences of Gaussian filters. Thereafter, peaks that belong to the same track were determined by simple Linear Assignment Problem tracker (parameters used: linking max distance = 0.5 μm; gap-closing max distance = 1 μm for telomeres and = 0.7 μm for α-satellite (centromeres); gap-closing max frame gap = 4). The assigned tracks were imported into @msdanalyzer written in MATLAB (38) and tracks containing at least 15 time lags (Δτ) were selected for calculating the Mean Square Displacement (MSD). For simplicity, the two-dimensional diffusion coefficient Deff and the diffusive exponent α of all trajectories were obtained from log-log fit of the following formula:
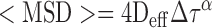 |
using the first 25% of total time lags, with a minimum fitting threshold of R2 > 0.8. Tracks with α < 1.2 represented diffusion while tracks with α > 1.2 represented directed transport.
Data analysis
Statistics were performed using one-way ANOVA with post hoc testing of pairwise comparisons using Tukey's Honestly Significant Difference test unless otherwise stated.
RESULTS AND DISCUSSION
Optimizing CRISPR/MB for genomic labeling
We designed three sgRNA scaffolds each with a spacer sequence complementary to the highly repetitive element within the human telomere locus (Telo) and a previously validated MTS sequence not found in the human genome (MTSa) (15,16,25) and named the resulting sgRNAs TL-sgTelo-MTSa, SL2-sgTelo-MTSa, and 3′-sgTelo-MTSa (Figure 1). Each sgRNA was co-expressed with dCas9 and EGFP (as a transfection control) in HEK293 cells (see Materials and Methods for plasmid construction and expression). For comparison, cells were also transfected with sgTelo lacking MTS (sgTelo-control). This was followed by delivery of MBs targeting MTSa (anti-MTSa MBs), synthesized with an oligonucleotide backbone composed of 2′-O-methyl RNA with a fully phosphorothioate-modified loop domain (2Me/PSLOOP MBs), an architecture previously shown to be highly resistant to nonspecific opening in living cells (15,16). We hypothesized that if the inserted MTS does not interfere with sgTelo-guided association of dCas9 with telomere repetitive elements, hybridization of anti-MTSa MBs to the MTSa sequence should illuminate the target loci as discrete bright spots when imaged by conventional widefield fluorescence microscopy. Fluorescence microscopy imaging showed that, 24 h following MB delivery, the majority of the MBs were localized in the nucleus, as shown previously (15,16). However, the subnuclear localization of MBs in cells with the three sgTelo-MTSa designs and the sgTelo-control were quite different. Specifically, distinct bright spots were observed in 73%, 90% and 72% of EGFP-positive cells transfected with TL-, SL2- and 3′-sgTelo-MTSa, respectively (Figure 2A). In contrast, very few spots were detected in cells transfected with the sgTelo-control, with the majority of the MB signal exhibiting a diffuse distribution. Since bright spots were observed in cells with the sgTelo-MTSa designs but not with the sgTelo-control, we concluded that these spots arose due to specific MB opening upon hybridization to the MTS, rather than nonspecific MB opening or imperfect quenching of the fluorophore. The lack of punctate signal in cells with the sgTelo-control is also consistent with our previous finding that 2Me/PSLOOP MBs, when unhybridized, remain largely quenched in the cellular environment (15,16).
Figure 2.

Specific labeling of telomeres using CRISPR/MB. Following transfection of HEK293 cells with dCas9 and either TL-, SL2-, 3′-sgTelo-MTSa or sgTelo-control, cells were microporated with 1 μM anti-MTSa MBs (ATTO647N-labeled) and imaged at 24 h post-microporation. (A) Representative maximum intensity projection images of MB signals in fixed cells. Scale bar = 10 μm. (B) Quantification of MB fluorescence spot number per cell. Data represent mean ± S.D. of at least 60 cells. (C) The distribution of spot number per cell in cells transfected with SL2-sgTelo-MTSa. n = 100 cells. (D) Detection of telomere loci by MBs and DNA FISH. Cells transfected with SL2-sgTelo-MTSa were fixed and permeabilized and then processed by FISH using telomere-targeting or nonsense FISH probes (TAMRA-labeled). Representative maximum intensity projection images of anti-MTSa MBs and telomere-targeting FISH probes are shown. DAPI stains the nucleus. Scale bar = 10 μm. (E) The percentage of MB signals that were colocalized with FISH signals (%MB colocalizing with FISH) and the percentage of FISH signals that were colocalized with MB signals (%FISH colocalizing with MB) on a cell-by-cell basis were calculated using a custom MATLAB program (see Materials and Methods: Colocalization analysis). Data represent mean ± S.D. of at least 13 cells. For (B) and (E), asterisks indicate P-values (* P < 0.05, ** P < 0.01).
To determine whether the observed punctate bright spots accurately identified telomeres rather than background signals emitted by MB hybridization to free non-targeting sgTelo-MTSa or dCas9-sgTelo-MTSa complexes, total numbers of spots in cells transfected with each sgTelo-MTSa construct were quantified in three dimensions (see Materials and Methods). We found that, despite the qualitative similarity in punctate staining appearances, cells expressing the three different sgTelo-MTSa designs showed a quantitative difference in spot formation. Specifically, the mean (± S.D.) spot number per cell was 69 ± 35 for cells with TL-sgTelo-MTSa, 123 ± 28 for cells with SL2-sgTelo-MTSa and 67 ± 31 for cells with 3′-sgTelo-MTSa (Figure 2B). This difference suggests that the stem–loop 2 region is more amendable to modifications than the other two regions, as shown in previous findings (12,39). In contrast, cells with the sgTelo-control contained an average of 0.5 ± 0.9 spots per cell, which is not significantly different from 0 (P = 0.60, two-tailed t-test assuming equal variance). The distribution of spot number in cells with SL2-sgTelo-MTSa ranged from 80 to 219 (Figure 2C), with a mode of 121 and nearly 30% of cells having between 120 and 140 spots, which is in good agreement with reported values of HEK293 cells (https://www.atcc.org/products/all/CRL-1573.aspx#characteristics) (expected mode = 128). These findings suggest that the SL2-sgTelo-MTSa design can associate with both dCas9 and MBs to enable accurate quantification of genomic loci. Supporting this finding, analogous experiments performed in HeLa cells with SL2-sgTelo-MTSa showed that 85% of the cells have spot numbers between 140 and 328, with a mode of 163 (Supplementary Figure S1), which is in good agreement with reported values of HeLa cells (https://www.atcc.org/products/all/CCL-2.aspx#characteristics) (expected mode = 164). Additional evidence that the observed MB spots resembled telomere loci came from single-molecule DNA fluorescence in situ hybridization (FISH) experiments showing colocalization between MB signals and FISH signals acquired when using telomere-targeting FISH probes but not when using nonsense FISH probes (Figure 2D, E and Supplementary Figure S2). Finally, reducing the stem sequence flanking the MTS of SL2-sgTelo-MTSa (-10_SL2-sgTelo-MTSa) resulted in a reduction in the number of spots observed, while extending it by adding a 10 bp stem (+10_SL2-sgTelo-MTSa), a 20 bp stem (+20_SL2-sgTelo-MTSa), or a 30 bp stem (+30_SL2-sgTelo-MTSa) led to no further increase in spot number (Supplementary Figure S3). This suggests that SL2-sgTelo-MTSa is an efficient design for genomic labeling and that sufficient stem length is essential for effective imaging with the CRISPR/MB system. Together, these findings indicate that a design utilizing dCas9, an MB, and an sgRNA scaffold harboring an MTS in the stem–loop 2 region can be used to image telomere loci in living cells with high specificity and accuracy.
Evaluation of CRISPR/MB for dynamic and continuous imaging
Having successfully combined CRISPR and MB technologies to label individual genomic loci, we next examined whether this strategy can be used to monitor chromatin dynamics, which can provide information regarding chromatin configurations in the native cellular context not easily attainable by biochemistry-based techniques. To investigate this, we compared the movements of telomere loci in HEK293 cells visualized by CRISPR/MB (SL2-sgTelo-MTSa) or EGFP-tagged telomere-repeat binding factor 1 (EGFP-TRF1), a fusion-protein commonly used to monitor telomere dynamics in mammalian cells (40–42). Single-particle tracking analysis revealed that diffusion properties of telomere loci reported by both methods were very similar (Figure 3A, B and Supplementary Movie S1 and S2). Specifically, all of the detected loci exhibited diffusive motion (α < 1.2, see Materials and Methods), with loci labeled by CRISPR/MB and EGFP-TRF1 moving with mean diffusion coefficients (Deff) of 0.00124 ± 0.00005 and 0.00125 ± 0.00008 μm2/s, respectively. Thus, CRISPR/MB and EGFP-TRF1 can detect similar telomere dynamics. Furthermore, CRISPR/MB was found to exhibit greater detection efficiency than EGFP-TRF1, as the number of telomere loci detected by CRISPR/MB was closer to the expected value in the HEK293 cell line than the number visualizable by EGFP-TRF1 (Figure 3C and Supplementary Figure S4). The improved detection offered by CRISPR/MB is likely due to the use of MBs, which can remain quenched when unbound to sgRNAs and thus emit a low background signal, a feature not attainable by EGFP-TRF1 that relies on FP reporters. Furthermore, photostability analysis showed that telomere loci labeled by CRISPR/MB are much more resistant to photobleaching than those labeled by EGFP-TRF1 (Figure 3D), as expected since fluorophores are in general more photostable than FPs. Hence, our CRISPR/MB technology has the potential for sensitive analysis of chromatin dynamics with high temporal resolution in living cells.
Figure 3.

Comparison of CRISPR/MB and EGFP-TRF1 for detection and dynamic imaging of telomeres. HEK293 cells expressing dCas9 and SL2-sgTelo-MTSa were microporated with anti-MTSa MBs (ATTO488-labeled). Fluorescence microscopy images were taken at 24 h post-microporation and results were compared with cells transfected with EGFP-TRF1. (A) Representative full-track movements of single telomere loci as determined by CRISPR/MB and EGFP-TRF1. (B) The distribution of diffusion coefficients of single telomere loci, as determined by CRISPR/MB (n = 1499 tracks) and EGFP-TRF1 (n = 384 tracks), analyzed from at least 40 cells. Inset shows the mean ± S.E. diffusion coefficients. (C) Measurement of total telomere loci per cell, as determined by CRISPR/MB and EGFP-TRF1. Data represent mean ± S.D. of at least 60 cells. Asterisks indicate P < 0.01. (D) Representative photostability analysis. Cells with telomeres labeled by CRISPR/MB or EGFP-TRF1 were fixed and then subjected to fluorescence imaging at 10 frames per second. The fluorescence intensity of a detected spot was determined for each time point and then normalized to the initial fluorescence intensity acquired at t = 0 for the respective probe. Normalized fluorescence intensity was plotted against time.
Dual-color imaging of chromatin dynamics
Another crucial attribute of a genomic imaging system is the ability to illuminate more than one locus at a time. In our CRISPR/MB system, the sgRNA-MTS scaffold specifies both genomic binding, via its spacer sequence, and fluorophore recruitment, via MB/MTS hybridization. This raises the possibility of imaging multiple genomic loci simultaneously in living cells by using different sgRNA-MTS scaffolds and MB sequences. To test this, we created a second sgRNA-MTS scaffold by inserting a different previously-validated MTS sequence (MTSb) not expressed in human cells (15,16,23) at the stem–loop 2 region and an sgRNA spacer targeting a unique element of the human α satellite sequence that is present at high frequencies in centromeres (SL2-sgSat-MTSb) (see Materials and Methods for plasmid construction). We then synthesized a corresponding MB specific for MTSb (anti-MTSb MB). Following co-delivery of anti-MTSa MBs (ATTO647N-labeled) and anti-MTSb MBs (ATTO488-labeled) into HEK293 cells expressing both SL2-sgTelo-MTSa and SL2-sgSat-MTSb along with dCas9, fluorescence microscopy imaging showed distinct subnuclear localization patterns of the two MBs (Figure 4A and Supplementary Movie S3). Specifically, telomere-targeting anti-MTSa MBs exhibited a punctate staining pattern characteristic of telomeres, whereas centromere-targeting anti-MTSb MBs exhibited a patchy staining pattern that resembled centromere loci as revealed by immunofluorescence staining using centromere-specific antibodies (anti-CREST) (Supplementary Figure S5). Both MB signals are specific, as anti-MTSa MBs in cells transfected with only SL2-sgSat-MTSb did not exhibit a detectable signal and vice versa for anti-MTSb MBs in cells transfected with only SL2-sgTelo-MTSa (Supplementary Figure S6). Finally, single-particle tracking analysis showed that telomere and centromere loci exhibited similar diffusion characteristics (Figure 4B and C), as reported in previous studies using FP-fused CRISPR imaging approaches (4). Together, these findings indicate that the MTS of the sgRNA-MTS scaffold can be modified to enable simultaneous, dual-color imaging of different genomic loci in living cells.
Figure 4.

Dual-color imaging of telomeres and centromeres using CRISPR/MB. HEK293 cells were dual-labeled by expressing dCas9, SL2-sgTelo-MTSa, and SL2-sgSat-MTSb, microporated with a mixture of anti-MTSa MBs (ATTO647N-labeled) and anti-MTSb MBs (ATTO488-labeled) and then imaged at 24 h post-microporation. (A) Representative maximum intensity projection images of anti-MTSa MB (labeling telomere loci) and anti-MTSb MB (labeling centromere loci) signals in fixed cells. Scale bar = 10 μm. (B) Representative full-track movements of single telomere loci and single centromere loci in living cells. (C) The distribution of diffusion coefficients of single telomere loci (n = 670 tracks) and single centromere loci (n = 243 tracks) analyzed from at least 18 cells. Inset shows the mean ± S.E. diffusion coefficients.
CONCLUSION
In summary, we have developed a novel live-cell genomic imaging platform, termed CRISPR/MB, which comprises dCas9, an MB, and an sgRNA engineered with an MB target sequence in the stem–loop 2 region, and demonstrated its capacity for quantitative, dynamic and dual-color analysis of genomic loci in human cells. To our knowledge, CRISPR/MB is the first fluorophore-based CRISPR system for studying the dynamics of individual chromosomal loci in living cells, which carries several potential advantages over existing genomic-labeling approaches, including those that rely on tagging dCas9 or sgRNA with FP reporters. For example, CRISPR/MB can incorporate a wide variety of organic fluorophores that are brighter and more photostable than FPs. Additionally, MB fluorophores are quenched when not bound to the sgRNA scaffold, which significantly reduces background signals as compared with unquenchable FPs. Furthermore, simultaneous multiplexed imaging of different genomic loci is likely to be more feasible with CRISPR/MB, as many orthogonal combinations of unique MB/MTS pairs could in principle be synthesized, and less feasible when using FP-based approaches as there are a limited number of well-characterized, orthogonal Cas9/sgRNA species or RNA aptamers. Last but not least, in CRISPR/MB, dCas9 is not fluorescently-labeled, which may avoid generation of high background signals in the nucleolus as seen in dCas9-FP based approaches, due to the tendency of dCas9 to localize in the nucleolus (8,43).
We should also emphasize that this study is the first work that reports the use of MBs for visualizing genomic loci. CRISPR/MB facilitates live-cell visualization of dynamics of repetitive genomic sequences. We envision CRISPR/MB could be further adapted for visualization of non-repetitive sequences, or in conjunction with super-resolution microscopy for visualization of genomic dynamics at nanoscopic resolution. Furthermore, because of the relative ease of modification of sgRNA, the CRISPR/MB system could be modified to use other oligonucleotide-based probes.
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Key R&D Program of China [2016YFA0100702, 2016YFA0501603]; National Natural Science Foundation of China [31771583, 81371613]; Beijing Natural Science Foundation [7162114]; China’s 1000 Young Talent Award program. Funding for open access charge: National Key R&D Program of China [2016YFA0100702].
Conflict of interest statement. None declared.
REFERENCES
- 1. Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E.. A programmable Dual-RNA-Guided DNA endonuclease in adaptive bacterial immunity. Science. 2012; 337:816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Qin P.W., Parlak M., Kuscu C., Bandaria J., Mir M., Szlachta K., Singh R., Darzacq X., Yildiz A., Adli M.. Live cell imaging of low- and non-repetitive chromosome loci using CRISPR-Cas9. Nat. Commun. 2017; 8:14725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Knight S.C., Tjian R., Doudna J.A.. Genomes in focus: development and applications of CRISPR-Cas9 imaging technologies. Angew. Chem. Int. Ed. Engl. 2018; 57:4329–4337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shao S., Zhang W., Hu H., Xue B., Qin J., Sun C., Sun Y., Wei W.. Long-term dual-color tracking of genomic loci by modified sgRNAs of the CRISPR/Cas9 system. Nucleic Acids Res. 2016; 44:e86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ma H., Tu L.C., Naseri A., Huisman M., Zhang S., Grunwald D., Pederson T.. Multiplexed labeling of genomic loci with dCas9 and engineered sgRNAs using CRISPRainbow. Nat. Biotechnol. 2016; 34:528–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fu Y., Rocha P.P., Luo V.M., Raviram R., Deng Y., Mazzoni E.O., Skok J.A.. CRISPR-dCas9 and sgRNA scaffolds enable dual-colour live imaging of satellite sequences and repeat-enriched individual loci. Nat. Commun. 2016; 7:11707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ma H., Naseri A., Reyes-Gutierrez P., Wolfe S.A., Zhang S., Pederson T.. Multicolor CRISPR labeling of chromosomal loci in human cells. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:3002–3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen B.H., Gilbert L.A., Cimini B.A., Schnitzbauer J., Zhang W., Li G.W., Park J., Blackburn E.H., Weissman J.S., Qi L.S. et al. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell. 2013; 155:1479–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cheng A.W., Jillette N., Lee P., Plaskon D., Fujiwara Y., Wang W.B., Taghbalout A., Wang H.Y.. Casilio: a versatile CRISPR-Cas9-Pumilio hybrid for gene regulation and genomic labeling. Cell Res. 2016; 26:254–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gu B., Swigut T., Spencley A., Bauer M.R., Chung M., Meyer T., Wysocka J.. Transcription-coupled changes in nuclear mobility of mammalian cis-regulatory elements. Science. 2018; 359:1050–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ochiai H., Sugawara T., Yamamoto T.. Simultaneous live imaging of the transcription and nuclear position of specific genes. Nucleic Acids Res. 2015; 43:e127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zalatan J.G., Lee M.E., Almeida R., Gilbert L.A., Whitehead E.H., La Russa M., Tsai J.C., Weissman J.S., Dueber J.E., Qi L.S. et al. Engineering complex synthetic transcriptional programs with CRISPR RNA scaffolds. Cell. 2015; 160:339–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tyagi S., Kramer F.R.. Molecular beacons: Probes that fluoresce upon hybridization. Nat. Biotechnol. 1996; 14:303–308. [DOI] [PubMed] [Google Scholar]
- 14. Marras S.A.E., Kramer F.R., Tyagi S.. Efficiencies of fluorescence resonance energy transfer and contact-mediated quenching in oligonucleotide probes. Nucleic Acids Res. 2002; 30:e122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chen M.M., Ma Z., Wu X.T., Mao S.Q., Yang Y.T., Tan J., Krueger C.J., Chen A.K.. A molecular beacon-based approach for live-cell imaging of RNA transcripts with minimal target engineering at the single-molecule level. Sci. Rep.-UK. 2017; 7:1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhao D., Yang Y.T., Qu N., Chen M.M., Ma Z., Krueger C.J., Behlke M.A., Chen A.K.. Single-molecule detection and tracking of RNA transcripts in living cells using phosphorothioate-optimized 2′-O-methyl RNA molecular beacons. Biomaterials. 2016; 100:172–183. [DOI] [PubMed] [Google Scholar]
- 17. Zhang X.M., Song Y., Shah A.Y., Lekova V., Raj A., Huang L., Behlke M.A., Tsourkas A.. Quantitative assessment of ratiometric bimolecular beacons as a tool for imaging single engineered RNA transcripts and measuring gene expression in living cells. Nucleic Acids Res. 2013; 41:e152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chen A.K., Davydenko O., Behlke M.A., Tsourkas A.. Ratiometric bimolecular beacons for the sensitive detection of RNA in single living cells. Nucleic Acids Res. 2010; 38:e148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rhee W.J., Bao G.. Simultaneous detection of mRNA and protein stem cell markers in live cells. BMC Biotechnol. 2009; 9:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yeh H.Y., Yates M.V., Mulchandani A., Chen W.. Visualizing the dynamics of viral replication in living cells via Tat peptide delivery of nuclease-resistant molecular beacons. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:17522–17525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wu Y.R., Yang C.J., Moroz L.L., Tan W.H.. Nucleic acid beacons for long-term real-time intracellular monitoring. Anal. Chem. 2008; 80:3025–3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang W., Cui Z.Q., Han H., Zhang Z.P., Wei H.P., Zhou Y.F., Chen Z., Zhang X.E.. Imaging and characterizing influenza A virus mRNA transport in living cells. Nucleic Acids Res. 2008; 36:4913–4928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chen A.K., Behlke M.A., Tsourkas A.. Avoiding false-positive signals with nuclease-vulnerable molecular beacons in single living cells. Nucleic Acids Res. 2007; 35:e105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Santangelo P., Nitin N., LaConte L., Woolums A., Bao G.. Live-cell characterization and analysis of a clinical isolate of bovine respiratory syncytial virus, using molecular beacons. J. Virol. 2006; 80:682–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vargas D.Y., Raj A., Marras S.A.E., Kramer F.R., Tyagi S.. Mechanism of mRNA transport in the nucleus. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:17008–17013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Drake T.J., Medley C.D., Sen A., Rogers R.J., Tan W.H.. Stochasticity of manganese superoxide dismutase mRNA expression in breast carcinoma cells by molecular beacon imaging. Chembiochem. 2005; 6:2041–2047. [DOI] [PubMed] [Google Scholar]
- 27. Tyagi S., Alsmadi O.. Imaging native beta-actin mRNA in motile fibroblasts. Biophys. J. 2004; 87:4153–4162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Santangelo P.J., Nix B., Tsourkas A., Bao G.. Dual FRET molecular beacons for mRNA detection in living cells. Nucleic Acids Res. 2004; 32:e57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bratu D.P., Cha B.J., Mhlanga M.M., Kramer F.R., Tyagi S.. Visualizing the distribution and transport of mRNAs in living cells. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:13308–13313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wiraja C., Yeo D.C., Chong M.S.K., Xu C.J.. Nanosensors for continuous and noninvasive monitoring of mesenchymal stem cell osteogenic differentiation. Small. 2016; 12:1342–1350. [DOI] [PubMed] [Google Scholar]
- 31. Kim J.K., Choi K.J., Lee M., Jo M.H., Kim S.. Molecular imaging of a cancer-targeting theragnostics probe using a nucleolin aptamer- and microRNA-221 molecular beacon-conjugated nanoparticle. Biomaterials. 2012; 33:207–217. [DOI] [PubMed] [Google Scholar]
- 32. Kang W.J., Cho Y.L., Chae J.R., Lee J.D., Choi K.J., Kim S.. Molecular beacon-based bioimaging of multiple microRNAs during myogenesis. Biomaterials. 2011; 32:1915–1922. [DOI] [PubMed] [Google Scholar]
- 33. Dong H.F., Ding L., Yan F., Ji H.X., Ju H.X.. The use of polyethylenimine-grafted graphene nanoribbon for cellular delivery of locked nucleic acid modified molecular beacon for recognition of microRNA. Biomaterials. 2011; 32:3875–3882. [DOI] [PubMed] [Google Scholar]
- 34. Yeh H.Y., Yates M.V., Mulchandania A., Chen W.. Molecular beacon-quantum dot-Au nanoparticle hybrid nanoprobes for visualizing virus replication in living cells. Chem. Commun. 2010; 46:3914–3916. [DOI] [PubMed] [Google Scholar]
- 35. Nitin N., Santangelo P.J., Kim G., Nie S.M., Bao G.. Peptide-linked molecular beacons for efficient delivery and rapid mRNA detection in living cells. Nucleic Acids Res. 2004; 32:e58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chen B.H., Huang B.. Imaging genomic elements in living cells using CRISPR/Cas9. Method Enzymol. 2014; 546:337–354. [DOI] [PubMed] [Google Scholar]
- 37. Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012; 9:676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tarantino N., Tinevez J.Y., Crowell E.F., Boisson B., Henriques R., Mhlanga M., Agou F., Israel A., Laplantine E.. TNF and IL-1 exhibit distinct ubiquitin requirements for inducing NEMO-IKK supramolecular structures. J. Cell Biol. 2014; 204:231–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Konermann S., Brigham M.D., Trevino A.E., Joung J., Abudayyeh O.O., Barcena C., Hsu P.D., Habib N., Gootenberg J.S., Nishimasu H. et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature. 2015; 517:583–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bronshtein I., Kepten E., Kanter I., Berezin S., Lindner M., Redwood A.B., Mai S., Gonzalo S., Foisner R., Shav-Tal Y. et al. Loss of lamin A function increases chromatin dynamics in the nuclear interior. Nat. Commun. 2015; 6:8044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Arora R., Brun C.M., Azzalin C.M.. Transcription regulates telomere dynamics in human cancer cells. RNA. 2012; 18:684–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang X., Kam Z., Carlton P.M., Xu L., Sedat J.W., Blackburn E.H.. Rapid telomere motions in live human cells analyzed by highly time-resolved microscopy. Epigenet. Chromatin. 2008; 1:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fujii W., Kawasaki K., Sugiura K., Naito K.. Efficient generation of large-scale genome-modified mice using gRNA and CAS9 endonuclease. Nucleic Acids Res. 2013; 41:e187. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.