


Abstract
DNA polymerase fidelity is affected by both intrinsic properties and environmental conditions. Current strategies for measuring DNA polymerase error rate in vitro are constrained by low error subtype sensitivity, poor scalability, and lack of flexibility in types of sequence contexts that can be tested. We have developed the Magnification via Nucleotide Imbalance Fidelity (MagNIFi) assay, a scalable next-generation sequencing assay that uses a biased deoxynucleotide pool to quantitatively shift error rates into a range where errors are frequent and hence measurement is robust, while still allowing for accurate mapping to error rates under typical conditions. This assay is compatible with a wide range of fidelity-modulating conditions, and enables high-throughput analysis of sequence context effects on base substitution and single nucleotide deletion fidelity using a built-in template library. We validate this assay by comparing to previously established fidelity metrics, and use it to investigate neighboring sequence-mediated effects on fidelity for several DNA polymerases. Through these demonstrations, we establish the MagNIFi assay for robust, high-throughput analysis of DNA polymerase fidelity.
INTRODUCTION
DNA polymerase fidelity is critical to maintaining faithful replication of the genome (1). Despite their overall low frequency, DNA replication errors drive important biological phenomena like evolution and heritable disease genesis (2–5). During replication, DNA polymerases rely on built-in fidelity checkpoints, such as nucleotide selectivity and proofreading, to ensure faithful replication of genomic DNA (1,6,7). Beyond mechanisms intrinsic to the DNA polymerase, external factors such as mismatch repair, nucleotide supply, template sequence context and other environmental modulators also influence fidelity outcomes (2,8–11). In vivo, these factors influence a wide population of different DNA polymerases, each with their own fidelity characteristics (12,13). The interplay between these different types of DNA polymerases and their corresponding in vivo replication environments can result in unique error signatures that have been difficult to pinpoint (14–17). Since the complexity of in vivo systems can obscure mechanistic insight into DNA polymerase fidelity, it is important to have robust methods for fidelity characterization that allow for dissection of key modulators in specified contexts.
Traditionally, in vitro forward mutation assays that link replication errors with phenotype upon introducing copied DNA into bacterial cells have been used for quantifying error rates of DNA polymerases. These commonly-used lacZ-based assays suffer from drawbacks such as (a) lack of base-specific observations because only mutations that inactivate lacZ are reported, (b) low throughput as each assay requires significant effort and is not easily scaled, (c) limited capacity to interrogate sequence context effects on fidelity due to copying a defined reporter sequence (e.g., lacZ), and (d) additional sequencing steps to identify error subtypes (18–20). Alternatively, gel-based assays, such as denaturing gradient gel electrophoresis (DGGE), can be used to measure DNA polymerase fidelity. This method is optimal for resolving products with fewer, dominant mutation types as opposed to a highly diverse mix of error-containing products, which requires repeated rounds of separation, purification, and sequencing (21–23). Ultimately, the low-throughput nature of both lacZ and DGGE mutation assays render these techniques suboptimal for assaying the impact of a multitude of conditions on fidelity.
Recently, high-throughput assays based on next-generation sequencing (NGS) have been successfully employed for direct detection of DNA polymerase errors (23–28). These approaches substantially improve throughput and data quality, and allow for fine-grained testing and analysis of fidelity in different sequence contexts. Even inherent limitations such as errors introduced during sample preparation and sequencing can be circumvented using different barcoding strategies (23,25–27). However, NGS-based approaches require extensive sequencing (at least as many reads as the inverse of the error rate that is being measured) to identify naturally rare error events, limiting sample scaling capacity within a fixed sequencing lane. Thus, current approaches do not scale economically when investigating the impact of a large set of conditions on DNA polymerase fidelity, which may be useful for directed evolution or compound library screens.
Available techniques for measuring DNA polymerase error rate require trade-offs between scalability, error subtype sensitivity, and flexibility in types of error-modulating conditions that can be tested. To overcome these limitations, we have developed a novel assay that combines high-throughput NGS with an error rate amplification strategy that dramatically reduces the amount of sequencing reads required. This technique, we term the Magnification via Nucleotide Imbalance Fidelity (MagNIFi) assay, takes advantage of the observation that error rates increase proportionally to imbalances in nucleotide concentrations (29–31). This allows the MagNIFi assay to amplify naturally low DNA polymerase error rates well above noise levels. Forced misincorporation through either limiting or completely withholding one or more nucleotides during replication has been employed previously for analysis of DNA polymerase fidelity and as a strategy for random and site-specific mutagenesis (32–37). The MagNIFi assay employs similar principles by titrating the concentration of a designated ‘rare’ base (dRTP) during synthesis until either base substitutions or single nucleotide deletions are induced (Figure 1A). Custom creation of extension templates by DNA synthesis allows us to (a) design the exact site where errors will be made (by synthesizing a template that only contains the complement to the dRTP at a specific location), and (b) test the full combinatorial space of neighboring nucleotides to determine the effect of sequence context on the type and frequency of errors. By coupling the MagNIFi assay with a NGS readout, we enable a standardized platform for obtaining reproducible, high-resolution DNA polymerase fidelity profiles. We measure five distinct DNA polymerases spanning families A, B, Y and reverse transcriptase (RT) and show strong agreement between our fidelity outputs and literature error rate values. We also establish the MagNIFi assay as a powerful tool for exploring the impact of local sequence context on error propensity and type. Through these results, we demonstrate the MagNIFi assay as a robust method for high-throughput interrogation of DNA polymerase fidelity.
Figure 1.

MagNIFi assay concept. (A) The MagNIFi assay relies on imbalanced dNTP pools (e.g., [dATP]<<<[dTTP, dCTP, dGTP]) to magnify native DNA polymerase error rates through forced misincorporation at a specified Error-Enriched Site (EES, e.g., T) on a template consisting of three bases (e.g., A, G, C). Rare base concentration can be tuned to a threshold where a low fidelity DNA polymerase (LF DNAP) preferentially creates replication errors but a high fidelity DNA polymerase (HF DNAP) still correctly copies. Therefore, rare base concentration can be used to resolve differences in DNA polymerase fidelity. (B) Each extension template (yellow) consists of a primer-binding site (dark blue) and a 6-base combinatorial sequence context library (gray) flanking the EES (pink).
MATERIALS AND METHODS
DNA polymerases
All enzymes and corresponding reaction buffers were commercially obtained (Supplementary Table S1). Purified Taq polymerase, Avian Myeloblastosis Virus (AMV) RT, Phi29 and Sulfolobus islandicus Dpo4 were purchased through New England Biolabs. Purified Sequenase 2.0 was purchased through Affymetrix.
Extension template design
Extension templates (TT, TA, TC and TG) were designed for all four rare base contexts (Supplementary Table S2). Templates were 100 bp in length and contained a single T, A, C or G, or Error-Enriched Site (EES), near the middle of the template. Extension templates were designed to contain only three bases, with the exception of the EES (fourth base) and the extension primer-binding site. For each template type, the EES was flanked by three degenerate bases before the EES and three degenerate bases after the EES in order to create the following sequence context libraries: VVVTVVV, BBBABBB, DDDCDDD and HHHGHHH (using IUPAC ambiguity codes). Because each of the six degenerate sites can be composed of three possible bases, each template library contained 729 (36) unique sequence contexts surrounding the EES. With the exception of the 6 bases flanking the EES, template sequences within a given library were identical. All templates contained a 3′ dideoxy-C modification to prevent extension from the template strand during a final PCR amplification step. Extension templates were purified via PAGE (Integrated DNA Technologies).
Primer design
A universal extension primer (PEXT) was designed to bind to all assay templates (TT, TA, TC and TG) and was used for all DNA polymerase extension reactions. From 5′-3′, the primer contained a 22-base universal tag called common sequence 1 (CS1) of the Fluidigm Access Array Barcode Library for Illumina Sequencers (Fluidigm), a 12-base DNA barcode, and 20 bases of homology with the template (Supplementary Table S2). To enable assay scalability, a DNA barcode was built into PEXT as a placeholder to allow dual-barcoding of reactions for experimental set ups requiring multiplexing beyond 384 reaction conditions. A library of 2168 barcodes has been published by Caporaso et al. (38) and is available here: http://www.nature.com/ismej/journal/v6/n8/extref/ismej20128x2.txt. PEXT was purified via standard desalting (Integrated DNA Technologies).
MagNIFi assay
Individual annealing reactions were performed for each template type (TT, TA, TC and TG). Primer/template DNA was prepared by mixing PEXT with each template library in a 1:1.5 molar ratio (70 nM primer: 105 nM template) in a 1X reaction buffer specific to each DNA polymerase being tested (Supplementary Table S1). The extension primer was annealed to the template DNA by incubation at 95°C for 2 min, followed by a −0.1°C/s ramp until reaching 4°C.
Primer extension reactions were set up in duplicate for each condition being tested. All biological replicate reactions were performed in parallel, with the same annealed primer/template sample used for both replicates. For each DNA polymerase and rare base context of interest, nine rare base (dRTP) concentrations were tested (log-fold dilutions from 10 μM to 0.1 pM) while the concentration of the three non-rare bases was held constant at 10 μM. Stocks of dNTPs were prepared using commercially obtained individual solutions of dATP, dCTP, dGTP, and dTTP (Bioline). Due to the nature of commercial dNTP manufacturing, a zero [dRTP] condition revealed contaminating trace levels of dRTP in non-rare base stocks. Although trace dRTP contamination could impact the true concentration of dRTP propagated in each dilution series, we concluded that since the same dNTP stocks were used for each reaction, potential contaminating effects were systematic and did not affect the FC50 estimate.
Primer extension reactions consisted of 1 μl of annealing reaction, 1X dNTP stocks (10 μM non-rare bases + variable [dRTP]), variable DNA polymerase units, and 1X DNA polymerase reaction buffer in a 10 μl reaction. Extension reaction conditions for all DNA polymerases tested are described in Supplementary Table S1. All extension reactions were incubated for 1 h and stored at −20°C until purification. The Fluent Automated Liquid Handling Platform (Tecan) was used to set up all primer extension reactions.
Illumina library preparation and sequencing
Our sample preparation pipeline for NGS was adapted from a previous protocol (28). Products from individual rare base extension reactions were column purified in 96-well plate format using the ZR-96 DNA Clean & Concentrator-5 (Deep well format) kit (Zymo Research). Purified DNA products were eluted in 10 μL of water and stored at −20°C until ligation. Next, a 22 bp universal tag, common sequence 2 (CS2) of the Fluidigm Access Array Barcode Library for Illumina Sequencers (Fluidigm), synthesized as duplex DNA with a 5′ phosphate modification and PAGE purified (Integrated DNA Technologies, Supplementary Table S2), was blunt-end ligated to the 3′ end of extended products. Ligation reactions were carried out in 10 μl volumes and consisted of 6 μL of purified product, 30 nM CS2 duplex DNA, 1X T4 DNA Ligase Reaction Buffer (New England Biolabs), and 2000 units of T4 DNA Ligase (New England Biolabs). Ligation reactions were incubated at 16°C for 16 h. Ligated products were stored at −20°C until PCR.
PCR was performed with barcoded primer sets from the Access Array Barcode Library for Illumina Sequencers (Fluidigm) to label extension products from up to 384 individual reactions. Each PCR primer set contained a unique barcode in the reverse primer. From 5′-3′, the forward PCR primer (PE1_CS1) contained a 25-base paired-end Illumina adapter 1 sequence followed by CS1. The binding target of the forward PCR primer was the reverse complement of the CS1 tag built into PEXT. From 5′-3′, the reverse PCR primer (PE2_BC_CS2) consisted of a 24-base paired-end Illumina adapter 2 sequence, a 10-base Fluidigm barcode, and the reverse complement of CS2. CS2 DNA that had been ligated onto the 3′ end of extended products served as the reverse PCR primer-binding site.
Each PCR reaction consisted of 2 μl of ligation product, 1X Phusion High-Fidelity PCR Master Mix with HF Buffer (New England Biolabs), and 400 nM forward and reverse Fluidigm PCR primers in a 20 μl reaction volume. Products were initially denatured for 30 s at 98°C, followed by 20 cycles of 10 s at 98°C (denaturation), 30 s at 60°C (annealing) and 30 s at 72°C (extension). Final extensions were performed at 72°C for 10 min. Amplified products were stored at −20°C until clean up and pooling. All ligation and PCR reactions were performed in 96-well plate format. The Fluent Automated Liquid Handling Platform (Tecan) and Mosquito Crystal (TTP Labtech) were used to set up all reactions.
The SequalPrep Normalization 96-well Plate Kit (ThermoFisher Scientific) was used to clean up and normalize the recovery of PCR reaction products up to 25 ng per reaction. Normalized, barcoded products were pooled together to form a library. AMPure XP beads (Beckman Coulter) were used to concentrate each product library 10-fold. Concentrated libraries were analyzed using a 2200 TapeStation (Agilent) to determine size and quality. Concentration of each library was measured using a Qubit 2.0 Fluorometer (Life Technologies). Sequencing was performed using a MiSeq v2 500 cycle kit on a MiSeq Benchtop Sequencer (Illumina). A 15% phiX DNA control was spiked in alongside product libraries during sequencing. Fluidigm sequencing primers, targeting the CS1 and CS2 linker regions, were used to initiate sequencing. De-multiplexing of reads was performed on the instrument based on Fluidigm barcodes. Library concentration, quality analysis, and quantification were performed at the DNA services (DNAS) facility, Research Resources Center (RRC), University of Illinois at Chicago (UIC). Sequencing was performed at the W. M. Keck Center for Comparative and Functional Genomics at the University of Illinois at Urbana-Champaign (UIUC).
Error rate analysis
Forward and paired-end sequences were obtained in FASTQ format. Forward sequences were filtered for exact matches to the extension primer and CS2 sequences, as well as for the presence of a corresponding paired-end read. The sequence from the start of the read to the beginning of the Fluidigm reverse PCR primer was isolated, leaving only the sequence corresponding to the extension product. Reads in which the paired-end read did not contain the exact reverse complement of this extension sequence were discarded. Next, reads where any base call in this sequence had a quality score less than 20 were discarded. This sequence was then aligned to the expected sequence using the Needleman–Wunsch algorithm and sequences that had an alignment score outside of a specified set of cutoffs (using the EDNAFULL scoring matrix, a gap-opening penalty of 10, and a gap-extension penalty of 0.5) were filtered for alignments with scores between 300 and 1000 (39,40). Extension sequences that were shorter than 70 bp or longer than 150 bp were discarded. The filtering steps described were inspired by those used in a previous study (28).
Extension sequences were indexed based on their alignments to the expected template sequence. To determine error rates at EESs, occurrences of the correct incorporation or error of interest at the given EES were counted and divided by the total number of reads that passed the filtering procedure. Calculated errors included nucleotide substitutions and single nucleotide deletions at the EES. Descriptive statistics for experimental error rates were calculated over the results of two biological replicates.
DNA polymerase error rate data was collected in biological duplicate at nine concentrations of the rare base (log-fold [dRTP] dilutions from 10 μM to 0.1 pM) for each template type tested. To obtain a rare base titration curve, log[dRTP] was plotted against mean error rates (n = 2) and nonlinear regression was performed. Sampling error between replicates was plotted using standard deviation values. Curves were fit to a dose response equation accounting for variable slope, four parameters and a least squares (ordinary) fit. From each nonlinear fit we obtained the concentration of rare base that yields the half maximal error rate, the Fidelity Concentration-50 (FC50), as well as 95% confidence intervals for the FC50, and R-squared values.
Error rate measurement simulations
To estimate the coefficient of variation (CV), 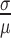 , for a given error rate estimate, we assumed the number of errors present in a given sample, X, was distributed as
, for a given error rate estimate, we assumed the number of errors present in a given sample, X, was distributed as 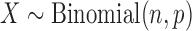 , where n is the number of sequencing reads in the sample and p is the underlying DNA polymerase error rate. The error rate estimator
, where n is the number of sequencing reads in the sample and p is the underlying DNA polymerase error rate. The error rate estimator 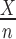 has variance
has variance 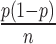 ; thus, the CV for the error rate estimator is
; thus, the CV for the error rate estimator is 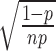 . We calculated the CV for error rates and read counts representative of potential NGS-based experiments analyzing various natural DNA polymerases.
. We calculated the CV for error rates and read counts representative of potential NGS-based experiments analyzing various natural DNA polymerases.
FC50 simulations
For FC50 sensitivity analysis, 1000 rare base titration curve experiments were simulated for both Dpo4 and Phi29 copying in a ‘T’ template context. For each rare base condition, sequencing results based on 50 sequencing reads were simulated by drawing 50 samples from a Bernoulli process with a true error rate equivalent to the experimentally derived value. A FC50 value was then determined for each simulated experiment using the fitting procedure described previously.
Mapping FC50 onto literature error rates
For each DNA polymerase, an average FC50 value was plotted against several literature error rates to enable calibration of FC50 with error rate. Nonlinear regression was performed on a log–log plot using a least squares (ordinary) fit and the following equation:  . Nonlinear fitting between literature error rates and average FC50 values revealed the following equation:
. Nonlinear fitting between literature error rates and average FC50 values revealed the following equation:  with a RMSE of 0.0008998 errors/bp.
with a RMSE of 0.0008998 errors/bp.
Sequence context analysis
The three bases before (−3, −2, −1) the EES and the three bases after (+1, +2, +3) the EES were analyzed for their fidelity impact at the EES. Reads were identified based on the composition of the −3, −2, −1, +1, +2 and +3 bases flanking the EES and the counts of each error (or correct incorporation) at the EES were determined for each possible 6-base identity. Counts over all possible 6-base sequences were then aggregated by base identity and position surrounding the EES and error rates for each base identity and neighboring position were calculated.
RESULTS
MagNIFi assay description
Our error rate amplification strategy for characterizing fidelity (Figure 1A) uses a DNA polymerase extension assay with a uniquely designed template and carefully controlled levels of dNTPs. Our extension template consists of primarily three nucleotides (e.g., A, G, C) with a fourth base (e.g., T) reserved as the Error-Enriched Site (EES) positioned near the middle of the template (Figure 1B). During primer extension, we supply asymmetric levels of dNTPs, where three out of the four dNTPs (e.g., dTTP, dCTP, dGTP) are fixed at an equimolar concentration but the concentration of the dRTP, the base complementary to the EES (e.g., dATP in Figure 1A), is limited. The concentration of the dRTP is reduced until a concentration is found where a DNA polymerase is more likely to misincorporate one of the more abundant dNTPs or make a deletion at the EES than incorporate the dRTP correctly. For a given DNA polymerase, a critical concentration of dRTP exists (the FC50) for which a DNA polymerase will create replication errors as frequently as correct incorporations. We hypothesized that this FC50 metric, while not a direct measurement of error rate, would correlate strongly with a DNA polymerase's underlying error rate.
The MagNIFi assay procedure is displayed in Supplementary Figure S1. First, a universal extension primer is annealed to one of four possible templates. The DNA polymerase of interest is allowed to extend in several parallel reactions where the dRTP for a given template is diluted log-fold across those parallel reactions. After extension, several sample preparation steps are performed before deep sequencing to ensure that sequence-specific PCR amplification bias is suppressed and only the extended strand is sequenced (28). This procedure allows us to robustly examine the behavior of DNA polymerases over a wide number of conditions in a high-throughput manner.
MagNIFi assay is scalable, robust and sensitive
Before using the MagNIFi assay for DNA polymerase fidelity characterization, we considered how scalable our assay would be for high-throughput screening of different fidelity conditions, and how this scaling compared to other methods. In order to maintain satisfactory precision in our apparent error rate measurements, a minimum number of NGS reads would be required. To determine this number, we simulated a DNA polymerase as a Bernoulli process at various error rates, and determined the variance of the measured error rate based on the number of NGS reads we observed (Figure 2A). We found that in order to capture error rates between 4% and 30% with a coefficient of variation (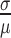 ) < 15%, only 1000 sequencing reads were required, and to capture error rates greater than 30% only 100 reads were needed. These simulations demonstrated that we could accurately determine amplified error rates (induced by low dRTP concentration) with substantially fewer reads than those required for extremely low error rates under normal dNTP conditions. Given that the FC50 (our previously defined fidelity metric) would correspond to error rate measurements near 50%, we concluded that as few as 100 sequencing reads per rare base condition would enable reliable fitting of the FC50 value. Thus, assuming that a correlation could be established between magnified error rates and true error rates, these findings suggested that the MagNIFi assay would allow us to analyze DNA polymerase fidelity while using substantially less sequencing resources.
) < 15%, only 1000 sequencing reads were required, and to capture error rates greater than 30% only 100 reads were needed. These simulations demonstrated that we could accurately determine amplified error rates (induced by low dRTP concentration) with substantially fewer reads than those required for extremely low error rates under normal dNTP conditions. Given that the FC50 (our previously defined fidelity metric) would correspond to error rate measurements near 50%, we concluded that as few as 100 sequencing reads per rare base condition would enable reliable fitting of the FC50 value. Thus, assuming that a correlation could be established between magnified error rates and true error rates, these findings suggested that the MagNIFi assay would allow us to analyze DNA polymerase fidelity while using substantially less sequencing resources.
Figure 2.

Assessing MagNIFi assay potential. (A) Simulation of the minimum number of sequencing reads required to accurately measure different error rates. Black line denotes a CV of 15%. (B) Sensitivity of FC50 values to sampling error and DNA polymerase stochasticity. Distribution of calculated FC50 values for 1000 simulated rare base titration experiments, with each rare base condition receiving 50 simulated reads. Each condition was simulated by drawing 50 samples from a Bernoulli process with an underlying error rate equal to the experimentally derived error rate. FC50 values were determined using the fitting procedure described in Methods. Histograms are shown for simulations based on Dpo4 (blue) and Phi29 (pink) error rates in a ‘T’ template context. (C) Determination of MagNIFi assay sample variability across different read counts. Error rate data collected for Dpo4 copying in all four template contexts reveal that error rate differences between 36 sets of biological replicates (n = 2) do not vary with read count differences between the same set of biological replicates. (D) Resolving power of the MagNIFi assay based on FC50 sensitivity. Minimum detectable fold change in FC50 was determined based on the 95% confidence interval of the fitted FC50 (see Supplementary Table S3 for values). The ratio of the Upper Bound 95% CI: Fitted FC50 was determined for Dpo4 copying in ‘T’, ‘A’, ‘C’ and ‘G’ template contexts.
To obtain a better sense of how the apparent error rate uncertainty would affect the FC50, we performed an initial MagNIFi assay trial with a low fidelity DNA polymerase, S. islandicus Dpo4 (41–43) and a high fidelity DNA polymerase, Phi29 (44–46), copying in a ‘T’ template context. Using these data, we simulated many parallel error rate readouts based on only 50 sequencing reads per rare base condition, allowing us to obtain a distribution of fitted FC50 values for each DNA polymerase (Figure 2B). We found our system to be well calibrated, since random variance resulted in distributions around our measured FC50 values that enabled clear separation of two different polymerases.
Next, we set out to determine assay robustness and sensitivity by performing additional MagNIFi assay experiments with Dpo4 copying in the remaining three rare base contexts. We first determined whether variation in the number of sequencing reads between biological replicates had any significant impact on error rate readout. We examined 36 sets of biological replicates (n = 2) of Dpo4 copying four different template types and found that error rate values did not vary substantially with read counts (Figure 2C). Finally, we determined whether the MagNIFi assay could resolve small differences in fidelity. From our Dpo4 data set, we calculated that 10% changes in FC50 (and even smaller) could be resolved with 95% confidence (Figure 2D). In all, the MagNIFi assay appeared to have robust properties for large-scale characterization of DNA polymerase fidelity. Our assay required significantly fewer sequencing reads than other NGS-based methods, was robust against random variance, had low sample variability, and showed promise as a highly sensitive reporter of fidelity changes.
FC50 effectively captures native error rates of DNA polymerases
After demonstrating our methodology was robust, we next validated that the MagNIFi assay could recapitulate previously reported error rates for a range of DNA polymerases. We performed rare base extension assays for five DNA polymerases: Sequenase 2.0, AMV RT, Phi29, Taq and Dpo4, and compared dose response fidelity curves of all five DNA polymerases copying in ‘T’ template contexts (Figure 3A). The lowest fidelity DNA polymerase tested, Dpo4, began making replication errors more than half the time between 0.01 and 0.1 μM dATP, while higher fidelity polymerases maintained lower error rates. Further log dilutions in [dATP] ultimately resulted in error saturation, suggesting that the ideal range for resolving fidelity differences was between 0.0001 and 0.1 μM dATP. Rare base titration curves revealed FC50 values of these DNA polymerases (Supplementary Table S3) that generally agreed with documented error rates from previous studies (Supplementary Table S4).
Figure 3.

MagNIFi assay validation. (A) Fidelity dose response curves of Sequenase 2.0, AMV RT, Phi29, Dpo4 and Taq copying in ‘T’ template contexts. Values are the average of two experiments. Standard deviation error bars (n = 2) are smaller than data points. Curves show qualitative agreement between DNA polymerase FC50 values (indicated by black dotted line) and the expected rank order of natural DNA polymerase error rates (Supplementary Table S4). In general, fidelity increases from right to left. (B) Calibration between error rate and [FC50]. We show a calibration curve relating multiple reported error rates per DNA polymerase (Supplementary Table S4) and the average FC50 value of each DNA polymerase (Supplementary Table S3). Nonlinear fitting on a log–log plot (line of best fit in grey) revealed the following equation: 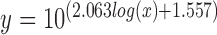 , RMSE = 0.0008998 errors/bp.
, RMSE = 0.0008998 errors/bp.
To validate how well our approach could represent DNA polymerase error rates, we fit dose response data from all four rare base contexts to obtain an average FC50 for each DNA polymerase studied (Supplementary Table S3). We next performed nonlinear calibration between average FC50 values and previously reported literature error rates to determine how well the FC50 metric would map onto traditional measures of fidelity (Figure 3B). Nonlinear fitting showed good agreement with the spread of literature values reported for each DNA polymerase. For Phi29, the estimated error rate fell 2.7-fold from the mean of the two reported literature error rates.
Correlation between error rates that were measured using a variety of fidelity assays and our FC50 outputs allowed us to establish MagNIFi assay fidelity readouts as biologically relevant. Analyses of FC50 sensitivity (Supplementary Figure S2) and error rate variability between replicates (Supplementary Figure S3) for all five DNA polymerases copying in all four template contexts further established MagNIFi assay robustness and sensitivity.
MagNIFi assay resolves DNA polymerase base substitution and single nucleotide deletion preferences
Sequencing data from each set of rare base conditions revealed high-resolution information on DNA polymerase fidelity preferences. In Figure 4, we calculated in-depth fidelity profiles of Dpo4 copying in all four rare base contexts: ‘T’ template (Figure 4A), ‘A’ template (Figure 4B), ‘C’ template (Figure 4C), and ‘G’ template (Figure 4D). Each profile serves as a fidelity fingerprint, revealing DNA polymerase mutation preferences by displaying the fraction of base substitutions and single nucleotide deletions that were created. When copying a ‘T’ template, Dpo4 favored T:dGTP substitutions over other error types. Similarly, in a ‘G’ template context, Dpo4 preferentially created G:dTTP mismatches. A:dATP and C:dATP substitutions were only marginally preferred in ‘A’ template and ‘C’ template contexts, respectively. A previous study that measured all 12 base substitution rates for Sulfolobus solfataricus Dpo4, a close homolog of S. islandicus Dpo4, reported the following error preferences: T:dGTP > T:dCTP ∼ T:dTTP at T sites, A:dATP > A:dCTP > A:dGTP at A sites, G:dTTP > G:dGTP > G:dATP at G sites, and C:dCTP > C:dATP = C:dTTP at C sites (41). We observed similar trends with S. islandicus Dpo4, with the exception of a slight preference for C:dATP over nearly equivalent C:dCTP and C:dTTP mispairs. Another report that measured S. solfataricus Dpo4 error preferences further corroborated our findings (28). High-resolution profiles for remaining DNA polymerases in this study are shown in Supplementary Figure S4A–D.
Figure 4.

High-resolution Dpo4 fidelity profiles. Rare base titration curves are shown for Dpo4 copying in a (A) ‘T’ template context, (B) ‘A’ template context, (C) ‘C’ template context and (D) ‘G’ template context. Preferred error type (nucleotide substitutions and single nucleotide deletions) is quantified for Dpo4 synthesizing in all four template contexts. Total error represents the sum of all error types. Values are the average of two experiments. Standard deviation error bars (n = 2) are smaller than data points.
For all DNA polymerases, we quantified error preference as the fraction of total errors that resulted in a particular error type (i.e., a specific base substitution or single nucleotide deletion) at the lowest rare base concentration tested (since the lowest [dRTP] produced the largest error response) (Figure 5A–E). Consistent with past reports, we found Dpo4 to have a higher average deletion error rate than DNA polymerases from other families (i.e., A, B, RT) (41–43). Dpo4 also displayed the highest C:dCTP substitution rate, a typically rare mutation that almost never occurred with the other DNA polymerases tested (10,41). In general, Dpo4 errors were more evenly distributed across all possible error subtypes compared to the higher fidelity DNA polymerases measured. As expected, the breakdown of error type at different template sites varied with the DNA polymerase. At ‘T’ and ‘G’ template sites, all DNA polymerases made dominant T:dGTP and G:dTTP substitutions, respectively. At ‘A’ template sites, all DNA polymerases preferentially misincorporated A:dATP with the exception of AMV RT, which preferred to make A:dCTP substitutions. At ‘C’ template sites, all DNA polymerases except for Phi29 created preferential C:dATP mispairs. Phi29 displayed a marginal preference for C:dTTP mismatches. Overall, we found that MagNIFi assay data could comprehensively reveal DNA polymerase error preferences at all possible bases, recapitulating past findings and uncovering new insights into DNA polymerase fidelity tendencies.
Figure 5.

DNA polymerase error preferences. Heat maps reflect nucleotide substitution and single nucleotide deletion preferences of (A) Dpo4, (B) Taq, (C) Sequenase 2.0, (D) AMV RT and (E) Phi29 copying in ‘T’, ‘A’, ‘C’ and ‘G’ template contexts. Error fraction was determined by normalizing individual error subtype frequencies to the total error rate measured at the lowest [dRTP] tested (10−7 μM) for each template context. Values are the average of two experiments.
MagNIFi assay reveals sequence context effects on fidelity
It is well established that template sequence context can impact DNA polymerase fidelity (9,10,15–17). Here, we sought to examine the effect of neighboring bases on DNA polymerase error rates and error preferences. To examine the effects of sequence context on error rate decisions, we designed each template type to contain six degenerate base positions (−3, −2, −1, +1, +2, +3) flanking the EES (Figure 1B, Supplementary Table S2). Each resulting template library consisted of 729 unique 6-base combinations surrounding the EES and allowed us to thoroughly investigate the positional effect of base identity on DNA polymerase error tendencies. For a given rare base context, we calculated position-dependent error rates by grouping sequencing readouts that shared the same base identity at the −3, −2, −1, +1, +2 or +3 positions. Sequence context-fixed error rates were then fit to obtain FC50 values as described previously.
To determine whether sequence context could substantially impact the FC50 readout, we calculated the extent to which sequence context-specific FC50 values deviated from the average FC50 of a given template library. For each DNA polymerase and template library, we calculated the change in FC50 (logFC50_Average – logFC50_Fixed Template Base) for a given template base at each position surrounding the EES (Supplementary Figures S5-S9). Results showed that the −3, −2, −1, +1, +2 and +3 base identities and positions could modulate changes in FC50 that indicated either increased or decreased fidelity. For example, when Phi29 replicated a VVVTVVV template library (Figure 6A), we observed that G and C template bases could increase or decrease fidelity depending on their proximity to the EES, whereas A template bases consistently increased fidelity regardless of position. Further, we observed template-mediated fold-changes in FC50 as large as 2-fold in both directions. For Dpo4, a +1 G in a DDDCDDD context led to a 2-fold increase in FC50, signifying lower fidelity, whereas a +1 A in the same context yielded a ∼2-fold decrease in FC50, indicating higher fidelity (Supplementary Figure S9). These data support error rate modulation by sequence context and furthermore demonstrate the sensitivity of the FC50 metric to sequence-driven changes in DNA polymerase fidelity.
Figure 6.

Local sequence context effects on DNA polymerase fidelity. (A) The identity and position of template bases neighboring a ‘T’ EES impact the measured FC50 of Phi29 copying in a VVVTVVV template context. Change in logFC50 (logFC50_Average – logFC50_Fixed Template Base) was calculated for each base identity/position. Positive ΔlogFC50 values pertain to an increase in fidelity whereas negative values signify a decrease in fidelity. (B) The identity and position of template bases flanking ‘C’ and ‘G’ EESs impact the % total error Phi29 creates at 10−7 μM dRTP when copying in DDDCDDD and HHHGHHH template contexts, respectively. A gray dotted line represents the average % total error made by Phi29 in a given context. (C) Base identity at the +1 template position in DDDCDDD, VVVTVVV, and BBBABBB contexts impacts the distribution of Dpo4 error preferences (nucleotide substitutions and single nucleotide deletions). Error preference was determined by normalizing error subtype frequency to the total error rate measured at 10−7 μM dRTP. For all graphs in (A)–(C), values represent the average of two experiments. Error bars signify standard deviation (n = 2).
Apart from modulating FC50, sequence context in certain cases also had an effect on the total error response (defined as the error rate measured at the lowest [dRTP] tested) that a DNA polymerase could create within a given rare base context (Supplementary Figures S10-S14). For instance, Phi29’s total error depended substantially on sequence context when replicating DDDCDDD and HHHGHHH template libraries (Figure 6B). With a couple of exceptions, total error tended to increase with surrounding C and G template bases and decrease with surrounding A and T template bases. Such observations corroborate past reports that A+T-richness at the primer terminus helps to improve strand separation and therefore increase proofreading efficiency (9,47–50), enabling higher fidelity outcomes for DNA polymerases such as Phi29 bearing 3′-5′ exonuclease activity.
Lastly, we investigated whether DNA polymerase error preference could be modulated by the identity and position of neighboring template bases. Similar to before, we determined error preference by normalizing error subtype frequencies to total error rate at the lowest rare base concentration tested. For most DNA polymerases studied, we found that base identity at the −1 template position tended to affect the preferred error distribution at the EES (Supplementary Figures S15–S18). For instance, a −1 G in a DDDCDDD context led to Phi29 preferentially producing C:dATP errors, whereas a −1 T in the same context yielded dominant C:dTTP errors (Supplementary Figure S17). In contrast, we found that Dpo4 error preferences were predominantly modulated by the +1 template base position (Supplementary Figure S19). For instance, in a DDDCDDD context, when the +1 base was A or T, Dpo4 predominantly misincorporated C:dATP, however, when the +1 base was G, Dpo4 error preference shifted to C:dCTP and single nucleotide deletions also increased (Figure 6C). This unique phenomenon, attributed to active site misalignment of Dpo4, has been previously reported to explain the unusually high rate of C:dCTP and deletion mutations in this particular sequence context (41,43). Interestingly, we also observed +1 G-mediated increases in T:dCTP and A:dCTP rates when Dpo4 replicated in VVVTVVV and BBBABBB contexts, respectively (Figure 6C), further supporting a misalignment mechanism (41). Overall, our sequence context data collectively point to the strong influence template base position and identity can exert on DNA polymerase fidelity decisions.
DISCUSSION
Most DNA polymerases in nature rarely make mistakes (51), which makes accurate measurement of their fidelity dependent on many observations. To overcome this technical barrier, we created an assay that substantially magnifies DNA polymerase error rates using imbalanced dNTP pools during extension, allowing for robust measurement of otherwise difficult-to-obtain values simply by tracking the concentration of the dRTP. We coupled this error rate amplification strategy with a NGS readout, measuring DNA polymerase fidelity under varying levels of dNTP pool asymmetry. Through the MagNIFi assay, we measured a robust metric, DNA polymerase FC50, which strongly correlates with DNA polymerase error rate while requiring far fewer sequencing reads for estimation, allowing for high-throughput determination of DNA polymerase fidelity.
Using the MagNIFi assay, we interrogated the fidelity properties of five DNA polymerases and recapitulated known fidelity trends for these polymerases based on the FC50 metric. The DNA polymerases we tested included two widely used commercial polymerases (Taq and Sequenase 2.0, a modified T7 polymerase without 3′-5′ exonuclease activity), a reverse transcriptase (AMV RT) (52), a high fidelity polymerase with proofreading ability (Phi29) (44–46), and a low fidelity translesional polymerase (S. islandicus Dpo4) (41–43). Agreement with the literature suggested that the MagNIFi assay is a valid approach for rapidly assessing DNA polymerase fidelity. Beyond capturing general error rates, the MagNIFi assay also recapitulated known sequence context-dependent fidelity effects for a couple of the DNA polymerases that we examined using a simple, generalizable template library approach. These results suggest a role for the MagNIFi assay as a high-throughput tool in the DNA polymerase toolkit, alongside established measures of DNA polymerase fidelity (18–28,36).
Assay properties
An advantage of using error rate magnification in combination with NGS is the technique's inherent scalability. Elevating DNA polymerase error rates means that far fewer observations are required for accurate estimates of polymerase error behavior. On top of this, it pushes error rates substantially above the baseline imposed by phosphoramidite synthesis (∼0.05–0.09%) (26,28) and NGS (∼0.1%) (53), removing the need for more intricate error-correction methods (23,25–27). In addition, NGS allows for substantially multiplexed samples using DNA barcoding. This makes the MagNIFi assay suitable for medium- to high-throughput investigations of DNA polymerase fidelity properties.
Compared to standard NGS approaches for measuring error rates, we determined that to obtain an estimated, FC50-based error rate of a moderate fidelity DNA polymerase (error rate of 10−5 errors/bp) using the MagNIFi assay, the required number of sequenced bases would be reduced by 250-fold. The MagNIFi assay would require sequencing of 4 × 104 bases compared to a required ∼107 bases using other approaches (Supplementary Discussion). Additionally, if the objective were to simply analyze how DNA polymerase error preference changed across conditions, FC50 calculation would not be necessary, and a single rare base condition where error rate is maximal (i.e., 10−7 μM dRTP) would suffice for determining error fraction. In this case, the required number of sequenced bases using the MagNIFi assay would be reduced to 2000 bases per template type. Overall, the MagNIFi assay requires substantially less sequencing coverage compared to standard NGS-based methods that rely on balanced dNTP levels.
Another advantage of amplifying errors is that we free up sequencing reads that can be used to gain other types of fidelity information, such as how unique sequence contexts may change DNA polymerase error rate or preferred error type under any set of conditions. By embedding this information capacity in every rare base condition tested, we enable a powerful tool for rapidly dissecting the effect of a particular sequence context on a given fidelity outcome. This is particularly useful since commonly used fidelity assays lack the flexibility to systematically evaluate the role of a particular sequence context in dictating error frequency and type. At the same time, by encoding a library of many different sequence contexts into each reaction, we are able to circumvent potential sequence bias (which is inherent when a fixed extension template is used, like lacZ) by considering the average effect of sequence composition on DNA polymerase error rate. Therefore, even without exploiting the built-in capacity to parse sequence effects, we can still reduce sequence bias, enabling the detection of errors that may be rare or even non-existent in commonly used template sequences. In all, the MagNIFi assay platform is well positioned to help further substantiate proposed template-driven DNA polymerase fidelity mechanisms and also facilitate discovery of novel sequence-based modulators of fidelity.
Assay limitations
Although the MagNIFi assay is well-suited for a variety of DNA polymerase investigations, it is important to note inherent limitations of using our approach. For one, the MagNIFi assay does not measure natural DNA polymerase error rates. Despite simplifying error measurement and being a robust indicator of fidelity, our assay is not a direct error rate measurement technique. For many applications, such as screening and relative measurements, direct measurement of natural error rates is not necessary. However, even in scenarios where such measures of fidelity are required, it is useful to recall that MagNIFi assay outputs map reasonably well onto traditional measures of error rate.
Furthermore, it is worth noting that previous error rate measurements are only moderately consistent between themselves. Although our nonlinear calibration model for mapping FC50 values to literature error rates reveals a slight divergence for Phi29 (2.7-fold from the average literature value), we note a ∼3-fold discrepancy in the reported error rates used for the calibration. This suggests the inherent difficulty in measuring highly infrequent errors, regardless of the methodology employed.
Finally, it should be mentioned that due to the nature of the MagNIFi assay design, we are only able to characterize a subset of replication error types: base substitutions and single nucleotide deletions occurring at non-iterated sequences. Therefore, alternative approaches should be considered for measuring other classes of common DNA polymerase errors, such as single nucleotide insertions and multiple insertions/deletions at either iterated or non-iterated sequences.
Assay validation and new insights
Using the MagNIFi assay, we were able to capture a substantial number of DNA polymerase fidelity trends that were consistent with the known literature. This allowed us to establish rare base dose response curves as valid measurements of DNA polymerase fidelity. Further, we also observed a number of sequence context- and polymerase-dependent phenomena that suggested that the error rate magnification of the MagNIFi assay was done in a relatively unbiased manner. We observed amplification of DNA polymerase errors in the correct proportion to their natural error rates, revealing magnified error preferences that matched known DNA polymerase error preferences. For instance, we correctly captured the general DNA polymerase preference for dGTP and dTTP misincorporations at T and G bases, respectively (20,22,28,37,41,54–57). We also captured polymerase-specific preferences such as AMV RT’s unique tendency to misincorporate dCTP at A bases (56,58).
At the same time, we were able to characterize a number of DNA polymerase fidelity characteristics that, to our knowledge, have not been interrogated. For instance, although base substitution preferences for exonuclease-deficient Phi29 have been previously measured (46), amplification of errors using the MagNIFi assay enabled detection of Phi29 error preferences without having to disable 3′-5′ proofreading. As a consequence, we were able to more naturally characterize Phi29 fidelity, and even detect sequence context-dependent fidelity phenomena that supported previously cited sequence effects on 3′-5′ exonuclease activity (9,47–50).
Another advantage of amplifying DNA polymerase errors through the MagNIFi assay is that we enable observations of rare error subtypes that previously may not have been detectable. For instance, traditional fidelity assays report T:dGTP mismatches as the dominant error preference of Taq polymerase (20,22,54), but are unable to report higher resolution of error preferences beyond that particular mismatch. Our assay enabled further detection of preferred mispairs at the three remaining types of template bases: A:dATP, C:dATP and G:dTTP.
Interestingly, we noted that reported DNA polymerase error preferences could be heavily biased by the sequence context used to measure them. For instance, we observed initial discrepancies in our observed error preferences for Dpo4 at ‘C’ template sites (preference for C:dATP) and previous measurements that used a lacZ template (preference for C:dCTP) (41). However, further investigation of sequence context effects revealed the template-driven nature of that preference. Although, on average, Dpo4 preferred misincorporating dATP at ‘C’ template sites, Dpo4 distinctly preferred C:dCTP in a context where +1 G flanked the EES. This +1 G-driven error preference, confirmed by the literature (41,43), emphasizes just how important it is to consider the bias introduced by the template used to measure the fidelity of a DNA polymerase.
High-throughput screening application
Beyond using the MagNIFi assay platform to study DNA polymerase fidelity, we envision adapting assay principles for rapidly screening DNA polymerase variants. By supplying only one rare base concentration (near the FC50) during extension, many DNA polymerases could be rapidly characterized as having a FC50 that is higher or lower than the rare base concentration. Since even relatively small changes in DNA polymerase fidelity would result in a large change in error frequency near the FC50, this strategy would enable reliable fidelity screening while requiring only a small number of sequencing reads for error rate measurement. DNA polymerases with specified fidelity responses could be developed for applications including but not limited to DNA data storage (59), molecular recording (28), random mutagenesis (60) and DNA/RNA sequencing (53,61).
Altogether, we present a reliable, sensitive, and standardized platform for measurement of all 12 possible mispairs and single nucleotide deletions for DNA polymerases spanning all families. Our error rate amplification approach coupled with NGS helps to overcome previous challenges with measuring DNA polymerase fidelity. The MagNIFi assay allows us to collect high-resolution data on DNA polymerase error preferences, including highly rare events. Our assay also considers the prominent role of sequence context on fidelity outcomes, and simultaneously enables the removal and further study of sequence bias under the same platform. Thus, the MagNIFi assay could prove a useful tool for the discovery of mutational hotspots, DNA sequence motifs where error probability is high (62). In addition, the MagNIFi assay may be readily adapted for high-throughput screening of a wide range of fidelity-modulating conditions due to its high scalability and resolving power. Looking forward, our strategy may also enable development of DNA polymerases with tailored fidelity responses.
DATA AVAILABILITY
Sequencing data can be accessed from the Sequence Read Archive (SRA) with accession numbers SAMN08053154–SAMN08053333. Python scripts for FASTQ processing and analysis are available on GitHub: https://github.com/tyo-nu/nextgen4b.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Dr. Stefan Green, Kevin Kunstman and Weihua Wang of the University of Illinois at Chicago DNA Services Facility, who assisted with the design and implementation of next-generation sequencing. The authors would also like to thank Sara Fernandez Dunne and Mariam George of the High Throughput Analysis Laboratory at Northwestern University, who helped with liquid handling robots and assay automation. Finally, we thank Dr. Doug Bishop, Dr. Lucia Rothman-Denes and Joseph Muldoon for helpful conversations.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health (NIH) Common Fund [5R01MH103910-02]; Chicago Biomedical Consortium with support from the Searle Funds at The Chicago Community Trust; National Science Foundation Graduate Research Fellowship [DGE-1324585]. Funding for open access charge: National Institutes of Health (NIH) Common Fund [5R01MH103910-02].
Conflict of interest statement. The authors have filed a provisional patent for the methods described in this manuscript.
REFERENCES
- 1. Kunkel T.A., Bebenek K.. DNA replication fidelity. Annu. Rev. Biochem. 2000; 69:497–529. [DOI] [PubMed] [Google Scholar]
- 2. Ganai R.A., Johansson E.. DNA replication—a matter of fidelity. Mol. Cell. 2016; 62:745–755. [DOI] [PubMed] [Google Scholar]
- 3. Loh E., Salk J.J., Loeb L.A.. Optimization of DNA polymerase mutation rates during bacterial evolution. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:1154–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tomasetti C., Li L., Vogelstein B.. Stem cell divisions, somatic mutations, cancer etiology, and cancer prevention. Science. 2017; 355:1330–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lange S.S., Takata K., Wood R.D.. DNA polymerases and cancer. Nat. Rev. Cancer. 2011; 11:96–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Freudenthal B.D., Beard W.A., Shock D.D., Wilson S.H.. Observing a DNA polymerase choose right from wrong. Cell. 2013; 154:157–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hohlbein J., Aigrain L., Craggs T.D., Bermek O., Potapova O., Shoolizadeh P., Grindley N.D., Joyce C.M., Kapanidis A.N.. Conformational landscapes of DNA polymerase I and mutator derivatives establish fidelity checkpoints for nucleotide insertion. Nat. Commun. 2013; 4:2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zakour R.A., Kunkel T.A., Loeb L.A.. Metal-induced infidelity of DNA synthesis. Environ. Health Perspect. 1981; 40:197–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Petruska J., Goodman M.F.. Influence of neighboring bases on DNA polymerase insertion and proofreading fidelity. J. Biol. Chem. 1985; 260:7533–7539. [PubMed] [Google Scholar]
- 10. Mendelman L.V., Boosalis M.S., Petruska J., Goodman M.F.. Nearest neighbor influences on DNA polymerase insertion fidelity. J. Biol. Chem. 1989; 264:14415–14423. [PubMed] [Google Scholar]
- 11. Mathews C.K. DNA precursor metabolism and genomic stability. FASEB J. 2006; 20:1300–1314. [DOI] [PubMed] [Google Scholar]
- 12. McCulloch S.D., Kunkel T.A.. The fidelity of DNA synthesis by eukaryotic replicative and translesion synthesis polymerases. Cell Res. 2008; 18:148–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kunkel T.A. Evolving views of DNA replication (in)fidelity. Cold Spring Harb. Symp. Quant. Biol. 2009; 74:91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jack B.R., Leonard S.P., Mishler D.M., Renda B.A., Leon D., Suarez G.A., Barrick J.E.. Predicting the genetic stability of engineered DNA Sequences with the EFM Calculator. ACS Synth. Biol. 2015; 4:939–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schroeder J.W., Hirst W.G., Szewczyk G.A., Simmons L.A.. The effect of local sequence context on mutational bias of genes encoded on the leading and lagging strands. Curr. Biol. 2016; 26:692–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lujan S.A., Clausen A.R., Clark A.B., MacAlpine H.K., MacAlpine D.M., Malc E.P., Mieczkowski P.A., Burkholder A.B., Fargo D.C., Gordenin D.A. et al. . Heterogeneous polymerase fidelity and mismatch repair bias genome variation and composition. Genome Res. 2014; 24:1751–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sung W., Ackerman M.S., Gout J.F., Miller S.F., Williams E., Foster P.L., Lynch M.. Asymmetric Context-Dependent mutation patterns revealed through mutation-accumulation experiments. Mol. Biol. Evol. 2015; 32:1672–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Keith B.J., Jozwiakowski S.K., Connolly B.A.. A plasmid-based lacZalpha gene assay for DNA polymerase fidelity measurement. Anal. Biochem. 2013; 433:153–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bebenek K., Kunkel T.A.. Analyzing fidelity of DNA polymerases. Methods Enzymol. 1995; 262:217–232. [DOI] [PubMed] [Google Scholar]
- 20. Tindall K.R., Kunkel T.A.. Fidelity of DNA synthesis by the Thermus aquaticus DNA polymerase. Biochemistry. 1988; 27:6008–6013. [DOI] [PubMed] [Google Scholar]
- 21. Cariello N.F., Swenberg J.A., Skopek T.R.. Fidelity of Thermococcus litoralis DNA polymerase (Vent) in PCR determined by denaturing gradient gel electrophoresis. Nucleic Acids Res. 1991; 19:4193–4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Keohavong P., Thilly W.G.. Fidelity of DNA polymerases in DNA amplification. Proc. Natl. Acad. Sci. U.S.A. 1989; 86:9253–9257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lee D.F., Lu J., Chang S., Loparo J.J., Xie X.S.. Mapping DNA polymerase errors by single-molecule sequencing. Nucleic Acids Res. 2016; 44:e118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hestand M.S., Van Houdt J., Cristofoli F., Vermeesch J.R.. Polymerase specific error rates and profiles identified by single molecule sequencing. Mutat. Res. 2016; 784–785:39–45. [DOI] [PubMed] [Google Scholar]
- 25. Gregory M.T., Bertout J.A., Ericson N.G., Taylor S.D., Mukherjee R., Robins H.S., Drescher C.W., Bielas J.H.. Targeted single molecule mutation detection with massively parallel sequencing. Nucleic Acids Res. 2016; 44:e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kinde I., Wu J., Papadopoulos N., Kinzler K.W., Vogelstein B.. Detection and quantification of rare mutations with massively parallel sequencing. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:9530–9535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schmitt M.W., Kennedy S.R., Salk J.J., Fox E.J., Hiatt J.B., Loeb L.A.. Detection of ultra-rare mutations by next-generation sequencing. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:14508–14513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zamft B.M., Marblestone A.H., Kording K., Schmidt D., Martin-Alarcon D., Tyo K., Boyden E.S., Church G.. Measuring cation dependent DNA polymerase fidelity landscapes by deep sequencing. PLoS One. 2012; 7:e43876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kunz B.A., Kohalmi S.E., Kunkel T.A., Mathews C.K., McIntosh E.M., Reidy J.A.. International commission for protection against environmental mutagens and carcinogens. deoxyribonucleoside triphosphate levels: a critical factor in the maintenance of genetic stability. Mutat. Res. 1994; 318:1–64. [DOI] [PubMed] [Google Scholar]
- 30. Bebenek K., Roberts J.D., Kunkel T.A.. The effects of dNTP pool imbalances on frameshift fidelity during DNA replication. J. Biol. Chem. 1992; 267:3589–3596. [PubMed] [Google Scholar]
- 31. Echols H., Goodman M.F.. Fidelity mechanisms in DNA replication. Annu. Rev. Biochem. 1991; 60:477–511. [DOI] [PubMed] [Google Scholar]
- 32. Baldwin J.E., Martin S.L., Sutherland J.D.. Site-specific forced misincorporation mutagenesis using modified T7 DNA polymerase. Protein Eng. 1991; 4:579–584. [DOI] [PubMed] [Google Scholar]
- 33. Spee J.H., de Vos W.M., Kuipers O.P.. Efficient random mutagenesis method with adjustable mutation frequency by use of PCR and dITP. Nucleic Acids Res. 1993; 21:777–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liao X.B., Wise J.A.. A simple high-efficiency method for random mutagenesis of cloned genes using forced nucleotide misincorporation. Gene. 1990; 88:107–111. [DOI] [PubMed] [Google Scholar]
- 35. Bertram J.G., Oertell K., Petruska J., Goodman M.F.. DNA polymerase fidelity: comparing direct competition of right and wrong dNTP substrates with steady state and pre-steady state kinetics. Biochemistry. 2010; 49:20–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Creighton S., Bloom L.B., Goodman M.F.. Gel fidelity assay measuring nucleotide misinsertion, exonucleolytic proofreading, and lesion bypass efficiencies. Methods Enzymol. 1995; 262:232–256. [DOI] [PubMed] [Google Scholar]
- 37. Sinha N.K., Haimes M.D.. Molecular mechanisms of substitution mutagenesis. An experimental test of the Watson-Crick and topal-fresco models of base mispairings. J. Biol. Chem. 1981; 256:10671–10683. [PubMed] [Google Scholar]
- 38. Caporaso J.G., Lauber C.L., Walters W.A., Berg-Lyons D., Huntley J., Fierer N., Owens S.M., Betley J., Fraser L., Bauer M. et al. . Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012; 6:1621–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Needleman S.B., Wunsch C.D.. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 1970; 48:443–453. [DOI] [PubMed] [Google Scholar]
- 40. Rice P., Longden I., Bleasby A.. EMBOSS: the european molecular biology open software suite. Trends Genet. 2000; 16:276–277. [DOI] [PubMed] [Google Scholar]
- 41. Kokoska R.J., Bebenek K., Boudsocq F., Woodgate R., Kunkel T.A.. Low fidelity DNA synthesis by a y family DNA polymerase due to misalignment in the active site. J. Biol. Chem. 2002; 277:19633–19638. [DOI] [PubMed] [Google Scholar]
- 42. Pata J.D. Structural diversity of the Y-family DNA polymerases. Biochim. Biophys. Acta. 2010; 1804:1124–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wu Y., Wilson R.C., Pata J.D.. The Y-family DNA polymerase Dpo4 uses a template slippage mechanism to create single-base deletions. J. Bacteriol. 2011; 193:2630–2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nelson J.R., Cai Y.C., Giesler T.L., Farchaus J.W., Sundaram S.T., Ortiz-Rivera M., Hosta L.P., Hewitt P.L., Mamone J.A., Palaniappan C. et al. . TempliPhi, phi29 DNA polymerase based rolling circle amplification of templates for DNA sequencing. Biotechniques. 2002; Suppl:44–47. [PubMed] [Google Scholar]
- 45. Paez J.G., Lin M., Beroukhim R., Lee J.C., Zhao X., Richter D.J., Gabriel S., Herman P., Sasaki H., Altshuler D. et al. . Genome coverage and sequence fidelity of phi29 polymerase-based multiple strand displacement whole genome amplification. Nucleic Acids Res. 2004; 32:e71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Esteban J.A., Salas M., Blanco L.. Fidelity of phi 29 DNA polymerase. Comparison between protein-primed initiation and DNA polymerization. J. Biol. Chem. 1993; 268:2719–2726. [PubMed] [Google Scholar]
- 47. Reha-Krantz L.J., Woodgate S., Goodman M.F.. Engineering processive DNA polymerases with maximum benefit at minimum cost. Front. Microbiol. 2014; 5:380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bloom L.B., Otto M.R., Eritja R., Reha-Krantz L.J., Goodman M.F., Beechem J.M.. Pre-steady-state kinetic analysis of sequence-dependent nucleotide excision by the 3′-exonuclease activity of bacteriophage T4 DNA polymerase. Biochemistry. 1994; 33:7576–7586. [DOI] [PubMed] [Google Scholar]
- 49. Reha-Krantz L.J. DNA polymerase proofreading: Multiple roles maintain genome stability. Biochim. Biophys. Acta. 2010; 1804:1049–1063. [DOI] [PubMed] [Google Scholar]
- 50. Bessman M.J., Reha-Krantz L.J.. Studies on the biochemical basis of spontaneous mutation. V. Effect of temperature on mutation frequency. J. Mol. Biol. 1977; 116:115–123. [DOI] [PubMed] [Google Scholar]
- 51. Garcia-Diaz M., Bebenek K.. Multiple functions of DNA polymerases. CRC Crit. Rev. Plant Sci. 2007; 26:105–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rittie L., Perbal B.. Enzymes used in molecular biology: a useful guide. J. Cell Commun. Signal. 2008; 2:25–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Goodwin S., McPherson J.D., McCombie W.R.. Coming of age: ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016; 17:333–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Eckert K.A., Kunkel T.A.. DNA polymerase fidelity and the polymerase chain reaction. Genome Res. 1991; 1:17–24. [DOI] [PubMed] [Google Scholar]
- 55. Koag M.C., Nam K., Lee S.. The spontaneous replication error and the mismatch discrimination mechanisms of human DNA polymerase beta. Nucleic Acids Res. 2014; 42:11233–11245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Boutabout M., Wilhelm M., Wilhelm F.X.. DNA synthesis fidelity by the reverse transcriptase of the yeast retrotransposon Ty1. Nucleic Acids Res. 2001; 29:2217–2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yu H., Goodman M.F.. Comparison of HIV-1 and avian myeloblastosis virus reverse transcriptase fidelity on RNA and DNA templates. J. Biol. Chem. 1992; 267:10888–10896. [PubMed] [Google Scholar]
- 58. Taube R., Avidan O., Bakhanashvili M., Hizi A.. DNA synthesis exhibited by the reverse transcriptase of mouse mammary tumor virus: processivity and fidelity of misinsertion and mispair extension. Eur. J. Biochem. 1998; 258:1032–1039. [DOI] [PubMed] [Google Scholar]
- 59. Erlich Y., Zielinski D.. DNA Fountain enables a robust and efficient storage architecture. Science. 2017; 355:950–954. [DOI] [PubMed] [Google Scholar]
- 60. Camps M., Naukkarinen J., Johnson B.P., Loeb L.A.. Targeted gene evolution in Escherichia coli using a highly error-prone DNA polymerase I. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:9727–9732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ellefson J.W., Gollihar J., Shroff R., Shivram H., Iyer V.R., Ellington A.D.. Synthetic evolutionary origin of a proofreading reverse transcriptase. Science. 2016; 352:1590–1593. [DOI] [PubMed] [Google Scholar]
- 62. Rogozin I.B., Pavlov Y.I.. Theoretical analysis of mutation hotspots and their DNA sequence context specificity. Mutat. Res. 2003; 544:65–85. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing data can be accessed from the Sequence Read Archive (SRA) with accession numbers SAMN08053154–SAMN08053333. Python scripts for FASTQ processing and analysis are available on GitHub: https://github.com/tyo-nu/nextgen4b.