


Abstract
Deep mutational scanning has been used to create high-resolution DNA sequence maps that illustrate the functional consequences of large numbers of point mutations. However, this approach has not yet been applied to libraries of genes created by random circular permutation, an engineering strategy that is used to create open reading frames that express proteins with altered contact order. We describe a new method, termed circular permutation profiling with DNA sequencing (CPP-seq), which combines a one-step transposon mutagenesis protocol for creating libraries with a functional selection, deep sequencing and computational analysis to obtain unbiased insight into a protein’s tolerance to circular permutation. Application of this method to an adenylate kinase revealed that CPP-seq creates two types of vectors encoding each circularly permuted gene, which differ in their ability to express proteins. Functional selection of this library revealed that >65% of the sampled vectors that express proteins are enriched relative to those that cannot translate proteins. Mapping enriched sequences onto structure revealed that the mobile AMP binding and rigid core domains display greater tolerance to backbone fragmentation than the mobile lid domain, illustrating how CPP-seq can be used to relate a protein's biophysical characteristics to the retention of activity upon permutation.
INTRODUCTION
Deep mutational scanning can be used to analyze the functional consequences of many mutations on a protein in parallel (1–4). By quantifying library sequence diversity before and after selecting (or screening) a library for proteins with specific functional characteristics, this approach is increasingly used to determine how a selection alters the relative abundances of many mutants sampled in a single experiment. Changes in sequence abundances can be used to calculate a fitness score, which estimates the relative biological activities of each protein (5–7). The generation of such large-scale mutational data has yielded sequence-function maps that assist with understanding residue-level contributions to protein solubility, protein–protein interactions and enzymatic function (8–13). By performing parallel deep mutational scanning experiments on proteins differing by a single mutation, these studies have also begun to provide insight into mutational epistasis (14–19). Additionally, experiments analyzing how sequence enrichment changes with selection pressure have revealed how environmental conditions influence the relative impact of mutations on fitness (20). Recent studies have begun to apply this knowledge to protein engineering (21–24), although generalizable design rules have not yet been established.
During evolution, genomic rearrangements can alter protein structure and function by altering length and contact order (25–27). The functional consequences of these types of topological mutational lesions can be studied in vitro by applying deep mutational scanning to combinatorial libraries created in the lab (28). A recent study employed deep sequencing to analyze the functional consequences of randomly inserting a ligand-binding domain into the RNA-guided DNA endonuclease Cas9 (29). Backbone insertion sites were identified in Cas9 that yielded allosteric protein switches whose gene editing activities depend upon ligand binding to the inserted domain (29). This domain-insertion profiling has also been applied to a green fluorescent protein to accelerate the creation of fluorescent protein biosensors for applications in metabolite sensing (30). These studies have demonstrated the power of using deep mutational scanning to identify hot spots that are most likely to yield folded proteins when engineering new biomolecules. However, deep mutational scanning has not been applied to libraries encoding circularly permuted proteins (31,32), i.e. proteins of similar length that differ in their arrangements of primary structure. Libraries encoding circularly permuted proteins are frequently used in the design of protein switches through domain insertion (33–36), so a better understanding of permutation tolerance would be synergistic to information gleaned from domain-insertion profiling.
Bioinformatic studies have provided evidence that genomic rearrangements that alter protein length, e.g. gene duplication followed by gene fission, underlie the evolution of circularly permuted proteins in nature (37). Unfortunately, the approaches used to discover circularly permuted proteins have not provided a complete understanding of protein tolerance to permutation because they utilize sequences and structures after natural selection has occurred (38,39). An alternative way to study this type of mutational lesion is to create combinatorial libraries of vectors that express different circularly permuted variants of a protein and to analyze the functions of those variants using biochemical and cellular methods. Biochemical approaches provide detailed insight into the folding, stability and activity of individual permuted proteins (40). However, these methods are slow and arduous to perform, and they cannot be easily implemented to provide insight into all permuted variants of a protein in parallel. In contrast, when cellular approaches are coupled to screens and selections (31,32), data can be rapidly generated about multiple variants in parallel. While high-throughput selections have been used for permuted protein discovery, they have not yet been coupled to deep sequencing to generate comprehensive profiles quantifying the enrichment of individual variants in a combinatorial library.
Here we develop a method to profile a protein’s tolerance to circular permutation, using an adenylate kinase (AK) as a model system. We first show that a library of circularly permuted genes can be created in a single step using a one-step transposition protocol (Figure 1), which is simplified compared with previous approaches (41–44). We then characterize the sequence diversity in the library before and after selection for variants that retain biological activity, and we apply a simple computational approach to show how this sequence information can be used to generate a map of protein tolerance to circular permutation.
Figure 1.

A one-step method for constructing libraries. With e-PERMUTE, libraries are created by mixing a circular gene, a permuteposon and MuA. The transposase inserts the permuteposon into the circular gene in two orientations. In the orientation that is designated as parallel, the regulatory elements, i.e. promoter (Pc) and RBS, and the permuted genes are in the same orientation. When an ORF is parallel and in frame, a circularly permuted protein is expressed with an 18-residue peptide amended to the new N-terminus. In the antiparallel orientation, the regulatory elements and the permuted AK genes are in different orientations such that the antisense strand of each permuted gene is transcribed. In this case, the permuted protein cannot be translated.
MATERIALS AND METHODS
Library construction
A previously described transposase method was modified to create our combinatorial library (41). The pMT2 vector (880 ng) was linearized using BglII, the product was run on a 0.8% agarose gel and the 1.8 kb permuteposon P1 was excised and gel purified using a Zymoclean Gel DNA Recovery Kit (Zymo Research). The gene for Thermotoga neapolitana AK was polymerase chain reaction (PCR) amplified to create an amplicon flanked by NotI sites and lacks a stop codon. The gene was cloned into a pET26b vector using Golden Gate assembly and sequence verified (45). To generate a circularized AK gene, 4000 ng of this plasmid was digested with NotI overnight at 37°C, agarose gel electrophoresis was used to separate AK gene from the vector backbone, the AK gene was purified, the gene (400 ng) was circularized through incubation with T4 DNA ligase for 16 h at 16°C, and purified using a DNA Clean & Concentrator Kit (Zymo Research).
A mixture containing circular AK gene (350 ng) and linearized pMT2 (50 ng) was incubated with 1 unit of MuA transposase (Thermo Fisher Scientific) at 37°C for 16 h. Following incubation at 75°C for 10 min, DNA was purified using a DNA Clean & Concentrator kit and transformed into library grade MegaX DH10B Ultracompetent cells (Thermo Fisher Scientific) using electroporation. Cells were allowed to recover for 45 min at 37°C while shaking before plating cells onto five LB-agar plates (150 × 15 mm) containing 25 μg/ml of kanamycin. After incubation at 37°C for 24 h, cfu were quantified visually. LB medium (3 ml) was added to each plate, colonies were scraped with a sterile cell spreader, the cell slurries were pooled, and the naïve library was purified using a QIAprep Spin Miniprep Kit (Qiagen).
AK selection
The naïve library (300 ng) was transformed into Escherichia coli CV2 using electroporation (46), cells were allowed to recover in SOC medium for 45 min at a non-selective temperature (30°C) and cells were spread across five LB-agar plates (150 × 15 mm) containing 25 μg/ml kanamycin. Plates were incubated at a selective temperature (42°C) for 48 h, colonies were counted, cells were harvested and selected library DNA was purified. As a control, a circularized transposon lacking a phosphotransferase was transformed into E. coli CV2, spread on LB-agar plates containing 25 μg/ml kanamycin and incubated at 30°C and 42°C for 48 h. With this control, growth was observed at 30°C but not at 42°C. In our permuteposon, the ribosomal binding site (RBS) used to initiate permuted protein translation is located adjacent to the transposon binding site, such that the genetic context remains constant across all variants in our combinatorial library. When analyzed using a thermodynamic model for translation initiation (47), considering a 60 bp window encompassing the RBS site using version 1.0 of the software, this analysis yielded a calculate translation initiation rate value (40264 au) that corresponds to a strong RBS site. Based on the dilution of AP variants following selection, we estimate that three rounds of our selection protocol would be sufficient to create a selected library that lacks vectors containing non-functional AK variants.
Deep sequencing
Our unselected and selected libraries (≥700 ng) were digested with ClaI and PciI at 37°C for 15 h to excise the circularly permuted AK genes. ClaI cuts ∼750 bp upstream of the permuted genes, while PciI cuts ∼150 bp downstream of the permuted gene. The permuted AK genes were separated from the vector backbone using 0.8% agarose gels, and the band encoding the permuted AK genes (∼1.6 kb) was excised and purified. All subsequent processing and sequencing was performed by the Baylor College of Medicine Genomic and RNA Profiling Core. The Nextera XT kit was used to fragment the DNA and attach sequencing adapters to the ends, and an Illumina MiSeq System was then used to collect short sequencing reads (150 bp).
Sequence analysis
Sequencing data were analyzed with a custom Python pipeline (Supplementary Figure S1), which can be found at github.com/SilbergLab/CPP-seq and carries out the following operations. Total reads containing a permuteposon were first identified using a pair of 9 bp sequences (GCCGCTCAA and TTGAGCGGC), which represent inverted repeat motifs found on the sense and antisense strands at the end of each permuteposon (Figure 2). To determine whether reads occurred at the beginning or end of the permuteposon, we included additional sequence directly adjacent to the 9 bp motifs. By adding 45 bp to our search so that the total permuteposon search motif was 54 bp, the sequences could be further resolved into reads of sequence at the different ends of the permuteposon. Reads including the start codon were designated ‘start motif’ reads, while reads including the stop codon were designated the ‘stop motif’ reads. Each of these motifs could also be further divided into sense and antisense reads.
Figure 2.

Sequence motifs used to identify the orientation of each AK gene. MiSeq data contained four types of sequence reads for the P variants, including (A) two different types of sense-strand reads and (B) two different types of antisense-strand reads. Reads that occurred at the different ends of permuteposon either contained the start codon (green) or stop codon (red). These were designated Start and Stop motifs. (C) Unique 54 bp sequences were used to differentiate each type of sequence read in our analysis. After identifying whether a sequence read corresponded to the sense versus antisense strand (and Start or Stop motifs), the adjacent 11 bp sequence was compared with all possible 11 bp sequences within both the sense and antisense strands of the circular AK gene. This analysis allowed us to identify the orientation and sequence of the circularly permuted gene in the different sequence reads obtained from MiSeq analysis. The 5 bp sequence directly adjacent to the Start and Stop motifs was used to determine the AK residue at the beginning of each polypeptide.
To establish the permuted AK genes encoded by each variant, we next evaluated the first 11 bp of AK-derived sequence adjacent the 54 bp start/stop motifs and determined how they related to all possible 11 bp sequences within the AK gene. For the sequence reads corresponding to the permuteposon sense strand, every possible 11 bp sequence within the AK gene was counted adjacent to the start and stop motif sequences. Those 11 bp sequences that corresponded to the AK sense strand were designated as P variants, while those from the antisense strand were designed AP. In the case of the permuteposon antisense strand reads, those 11 bp sequences that corresponded to the sense strand of the AK gene were designated as AP variants, while those from the antisense strand were designed P.
When MuA inserts the permuteposon, it duplicates 5 bp of the AK gene sequence such that the first 5 bp of each circularly permuted AK gene is also found at the end of the gene (48). This duplication was used to simplify the identification of permuted proteins expressed by the vectors identified from our four types of P sequence reads, which included: (i) sense start motif, (ii) antisense start motif, (iii) sense stop motif and (iv) antisense stop motif reads. In the case of the start motif reads encoding P variants, the AK-derived sequence adjacent to the permuteposon motif contains the codon that encodes the N-terminus of each circularly permuted protein. In the case of the stop motif reads encoding P variants, the 5 bp sequence adjacent to the permuteposon also represents the sequence at the beginning of each permuted gene, and the first AK codon in this motif encodes the residue at the beginning of those permuted proteins. A similar strategy was used to consider the reads corresponding to AP variants.
Statistical analysis
Two methods were used to analyze whether vectors were significantly enriched by the selection. Within our sequencing datasets, each circularly permuted gene can have four values: P-unselected, AP-unselected, P-selected and AP-selected. Using a two by two contingency table containing this data for each variant, we tested whether the ratio of selected to unselected P variants is equal to the AP variants using Fisher’s Exact Test. This test can only establish a statistic value provided that cognate P and AP variants are observed in the unselected library, which was not the case for all of our variants.
To obtain statistics on P variant activity in cases where cognate AP variants were absent from the unselected library, we took a similar approach to the DESeq method (49). DEseq was developed to analyze if there are significant changes in gene expression with RNA-seq and ChIP-seq datasets, using no change as the frame of reference and the null hypothesis. With our dataset, we used the ratio of selected to unselected AP variant counts as a frame of reference for the expected effect of selective pressure as the null hypothesis instead of the no change null hypothesis that DESeq assumes. We modeled the AP variant counts (i = 1 to 223 for each unique variant) using a negative binomial model where we defined 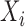 and
and 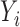 as the unselected and selected counts, respectively. In our model, the AP counts in the unselected library are defined as mi and the AP counts in the selected library are mi multiplied by a scalar dilution factor (β), such that
as the unselected and selected counts, respectively. In our model, the AP counts in the unselected library are defined as mi and the AP counts in the selected library are mi multiplied by a scalar dilution factor (β), such that 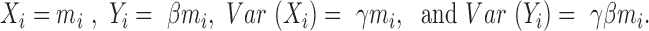 There are two global parameters shared by all the loci,
There are two global parameters shared by all the loci, 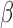 and
and 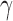 . The former reflects the average selection effect on all the AP variants, and the latter reflects the overdispersion compared with Poisson distribution.
. The former reflects the average selection effect on all the AP variants, and the latter reflects the overdispersion compared with Poisson distribution. 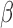 was estimated using sample median, and
was estimated using sample median, and 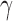 was estimated by assuming a negative binomial distributions for
was estimated by assuming a negative binomial distributions for 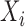 and
and 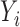 and then maximizing the likelihood. Using the estimated negative binomial distribution, the P-value of each P variant pair (
and then maximizing the likelihood. Using the estimated negative binomial distribution, the P-value of each P variant pair (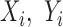 ) was computed by conditioning on the sum of
) was computed by conditioning on the sum of 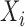 and
and 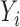 as outlined in equation eleven of DESeq (49). Finally, P-values were adjusted for multiple testing using the Benjamini–Hochberg procedure.
as outlined in equation eleven of DESeq (49). Finally, P-values were adjusted for multiple testing using the Benjamini–Hochberg procedure.
Testing individual variants
In a previous study (52), we created vectors for assessing the cellular activity of individual circularly permuted AK that were generated in our e-PERMUTE library. These vectors were transformed into E. coli CV2 and plated on LB-agar plates containing kanamycin (25 μg/ml) at 30°C. Individual colonies arising from each transformation (n = 3) were used to inoculate LB liquid cultures containing kanamycin (25 μg/ml), cultures were grown at 30°C for 28 h and each overnight culture (1 μl) was used to inoculate fresh 200 μl cultures in a Costar 3595 flat bottom 96-well plate. Cell growth was then monitored at 42°C using a Tecan M1000 plate reader every 10 min for 15 h using double orbital shaking.
RESULTS
One-step library construction
Previous studies have shown that the transposase MuA can be used to create libraries of vectors that express circularly permuted variants of a native protein by inserting synthetic transposons into a gene encoding the protein of interest (41–44). One advantage of these approaches is that they yield vectors that express proteins lacking deletions and duplications that can arise with other methods (50). However, all of the transposase methods that have been reported to date require multiple DNA manipulation steps, which limits their sampling efficiencies. For example, Permutation Using Transposase Engineering (PERMUTE) uses MuA to randomly insert a synthetic transposon containing all of the attributes of a vector (called a permuteposon) into a temperature-sensitive vector containing the gene being targeted for permutation (41,43). Only a fraction of the reaction products ultimately yield a desired product because the permuteposon must be inserted within the gene of interest to yield the desired DNA products. Many of the transposition events lead to insertion of the permuteposon within the vector backbone, rather than the gene of interest. Furthermore, the temperature-sensitive vector must be excised after performing the transposition reaction, and the resulting DNA must be circularized through ligation to yield the final library. An additional limitation with these libraries is the sequence bias associated with the MuA transposition reaction (48).
We sought to improve upon the PERMUTE method by decreasing the number of steps required for library construction. As shown in Figure 1, we hypothesized that a transposase reaction could be used to create libraries in a single step that sample the vast majority of possible circularly permuted variants without requiring a temperature-sensitive vector. In this enhanced-PERMUTE (e-PERMUTE) approach, we used MuA to randomly insert a permuteposon directly into a circularized gene encoding the target protein. This library construction approach is expected to experience the same sequence biases arising from the MuA transposition reaction. However, it avoids undesired off-pathway transposition reactions in the vector backbone, and thus, samples only the desired transposition reactions.
To evaluate e-PERMUTE, we built a library of vectors that express different circularly permuted variants of T. neapolitana AK (51), a thermostable phosphotransferase. The circular AK gene was synthesized by digesting a previously described vector (pMM1) with NotI (41), purifying the linear AK gene, and circularizing the gene through ligation. The circular gene lacked a stop codon and had the first and last codons connected by a NotI restriction site followed by an adenine. This sequence encodes a tripeptide (Ala-Ala-Ala) that ultimately connects the original N- and C-termini of AK within each circularly permuted protein. To generate the vector library, the circular AK gene was incubated with a permuteposon and MuA. In the permuteposon used for this study, named P1 (52), the RBS that drives translation initiation is adjacent to the transposase-binding site (TBS), such that protein translation adds an 18-amino acid polypeptide, encoded by the TBS, to the N-terminus of every circularly permuted protein variant (41). A smaller two amino acid scar is amended to the C-terminus of every variant, whose identity depends upon the location of insertion since these extra residues arise from a 5 bp duplication generated during the MuA insertion reaction (48). P1 was chosen instead of permuteposons that add smaller peptides to the N-terminus, because it is thought to maintain more consistent translation initiation across all variants in the library (52).
To assess the diversity of expression vectors generated by our protocol, the product of the transposase reaction was transformed into E. coli, cells were spread onto agar plates containing vector-selective medium, and colony forming units (cfu) were quantified following an overnight incubation. This protocol yielded ∼164 000 cfu. Before plating, the cells were allowed to double, so the number of unique colonies was 82 000. We next harvested these colonies, purified the vectors from pooled cells and analyzed the fraction of vectors containing permuted genes using agarose gel electrophoresis. Two distinct vectors were observed in the library (Supplementary Figure S2). One vector has a size consistent with the desired AK expression vectors, while the other had a size consistent with that of a permuteposon lacking an AK gene. These vectors presented similar intensities, indicating that only half of the unique colonies from the transformation (41 000 cfu) represent the desired vector library.
Sequence analysis of the unselected library
To isolate DNA for deep mutational scanning, we separated the vectors containing permuted AK genes from those lacking AK genes, excised only those DNA fragments containing the permuted AK genes and adjacent permuteposon sequence (Supplementary Figure S3), barcoded the DNA and analyzed the DNA sequences using MiSeq (53). This analysis yielded 3 477 712 total reads. Table 1 shows the abundance of reads containing permuteposon and AK-derived sequences. Among these reads, only ∼11.6% (402 805) contained the 9 bp inverted repeat sequences (GCCGCTCAA or TTGAGCGGC) that are found at both ends of every permuteposon and adjacent to every permuted AK gene (Supplementary Figure S2). We chose this 9 bp sequence for our analysis because it represents the shortest motif that is unique within all of the variants created by e-PERMUTE.
Table 1.
Abundance of sequence reads within each library
| Unselected | Selected | S/US ratio | |
|---|---|---|---|
| Total reads | 3 477 712 | 3 934 541 | 1.13 |
| +9 bp repeat | 402 805 | 528 526 | 1.31 |
| +start/stop motif | 298 707 | 405 857 | 1.36 |
| +start motif | 171 922 | 216 079 | 1.26 |
| +stop motif | 126 785 | 189 778 | 1.50 |
| +11 bp AK gene | 236 324 | 327 908 | 1.42 |
| all in frame AK gene | 114 754 | 327 908 | 2.86 |
| in frame P variants | 59 494 | 326 503 | 5.49 |
| in frame AP variants | 55 260 | 1405 | 0.03 |
| all +1 frame AK gene | 82 085 | 4373 | 0.05 |
| +1 frame P variants | 42 719 | 3581 | 0.08 |
| +1 frame AP variants | 39 366 | 792 | 0.02 |
| all −1 frame AK gene | 39 485 | 2727 | 0.07 |
| −1 frame P variants | 21 909 | 2398 | 0.11 |
| −1 frame AP variants | 17 576 | 329 | 0.02 |
The data generated by MiSeq analysis, including the total sequence reads having the bar code corresponding to the unselected and selected libraries, the number of reads that contain a 9 bp motif in the permuteposon (GCCGCTCAA and TTGAGCGGC), either the 54 bp start or stop motifs shown in Figure 2, and a start or stop motif and at least 11 bp of adjacent AK gene sequence. Among the variants containing a start (or stop) motif and 11 bp of adjacent AK sequence, we list the number of P and AP that are in frame, −1 frame, and +1 frame.
With e-PERMUTE, two types of circularly permuted AK genes are generated. In one case, the coding sequence is found on the same strand as the promoter, which we designate the parallel (P) orientation (Figure 1). In the other case, the coding sequence is on the complementary antisense strand, which we designate the antiparallel (AP) orientation. Only those vectors having the P orientation are able to transcribe and translate an AK variant. One challenge with analyzing our sequence data is identifying the orientation of each permuted gene relative to the permuteposon. Because the R1R2 and R2R1 transposase binding sites found at each end of the permuteposon are inverted repeats sharing the same sequences, the short 9 bp sequence flanking the permuted genes was not sufficient to determine the orientation of transposon within the AK gene. To identify whether genes were P or AP, larger unique 54 bp sequence motifs (Figure 2) were used to determine if sequence reads initiated within the end of the permuteposon contain the start codon (start motifs) or stop codon (stop motifs).
Among the 402 805 reads containing a unique 9 bp permuteposon sequence, 42.6% contained the start motif and 31.4% contained the stop motif. To identify the circularly permuted AK genes found in these sequence reads, we compared the gene sequence adjacent to the start and stop motifs with all possible 11 bp sequences in the circular AK gene (sense and antisense). Among the 298 707 reads that contained either the start or stop motif, 86% contained 11 bp of adjacent sequence that were identical to some region of the AK gene.
We next compared the relative abundances of each possible permuted gene in the P and AP orientation (Figure 3A). We found that the genes with the highest prevalence in both orientations arose from permuteposon insertion at the same locations within the AK gene. These findings are consistent with previous studies, which found that MuA insertion efficiency depends upon DNA sequence and is independent of orientation (48). We also analyzed the relative abundance of each in frame sequence in our library, since in frame P variants are able to translate a circularly permuted AK. In total, 114 754 in frame sequences were observed among the reads that contained a start or stop motif. This number represents 45% of the total sequence reads that contained either the start or stop motif and adjacent AK-derived sequence. We found that the number of occurrences of each unique variant varied by over three orders of magnitude (Figure 3B). The observed enrichment of the in frame variants (45%) over the value expected for an unbiased library (33%) is thought to occur because this frame had the most abundant individual variants in the library. In this frame, the permuted AK gene that started with the codon for Ala209 occurred 7 906 times in the P orientation and 8 377 times in the AP orientation, with the sum of these two variants representing 7% of the total sequence reads having a start (or stop) motif adjacent to 11 bp of AK gene sequence.
Figure 3.

Permuted gene abundances are independent of orientation. (A) The relative abundances of every possible cognate P (purple) and AP (green) variant mapped adjacent to one another on a circle as a function of the distance from the start codon, which is shown as a closed black symbol. Within the unselected library, the relative abundance of identical genes in P and AP orientations is similar. (B) For each unique P (purple) and AP (green) sequence, we evaluated the number of degenerate in frame sequences observed for each variant and plotted these as stacked bars. In the unselected library, 159 of the P variants and 148 of the AP variants were observed in one or more reads of the deep mutational scanning data. (C) Following selection, the relative abundances of every possible gene in the P and AP orientation differed as well as (D) the number of degenerate sequences. Among the selected sequence reads, 144 of the P variants and 85 of the AP variants were observed. Among both the naïve and selected libraries, a total of 171 unique P variants were observed out of 223.
Among the in frame permuted gene sequence reads in our naïve library, 51.8% were in the P orientation, while 48.2% encoded permuted AK genes in the AP orientation. In addition, a subset of the possible in frame variants were not observed in our sequences. Approximately 29% (n = 64) of the possible 223 P variants were absent from the naïve sequencing data, while 34% (n = 75) of the possible AP variants were not observed. This sampling of possible variants is similar to that previously achieved with domain insertion profiling (29,30), a method that also uses transposon mutagenesis to create combinatorial libraries.
Effect of selection on sequence diversity
To identify the subset of vectors in our library that encode active circularly permuted AK, we transformed our library into E. coli CV2, a strain with a temperature-sensitive AK that displays growth defects above 40°C (46) and selected for vectors that complemented growth at 42°C. The chromosomally encoded AK in this strain has a Pro87Ala mutation (46), whose activity at 42°C is not sufficient to maintain cellular energy homeostasis. After incubation at the selective temperature for two days, >105 colonies were observed and harvested, vector DNA was purified from pooled cells, and this DNA was subjected to deep sequencing using MiSeq. MiSeq analysis yielded 3 934 541 total reads, ∼1.1× more than the unselected library. The prevalence of different types of reads are compared with the unselected library in Table 1. Among these reads, 528 526 contained one of the 9 bp inverted repeat sequences (GCCGCTCAA or TTGAGCGGC) in the permuteposon that is adjacent to every permuted AK gene. In addition, 66% of these reads contained 11 bp of adjacent sequence that were identical to some region of the AK gene, either the sense or antisense gene sequence.
We next identified those sequence reads that contained in frame AK genes. Among the reads from the selected library, in frame AK genes were observed in 94% of the reads, compared with 45% in the unselected library. In contrast to the unselected library, which presented similar abundances of P and AP variants, the vast majority of the selected reads (99.5%) encoded circularly permuted genes in the P orientation (Figure 3C). The relative abundance of the different P variants spanned four orders of magnitude (Figure 3D). Among the P variants, one circularly permuted AK gene was observed 92 915 times. This permuted gene encoded the permuted AK variant whose sequence started with Ala209. In total, 79 of the possible in frame P variants were absent from this sequencing data. In the case of the AP variants, the most abundant selected sequence was only observed in 224 of the reads, which corresponded to the most abundant permuted gene in the P orientation. In total, 138 of the possible in frame AP variants were absent from the selected sequencing data.
A comparison of the P sequences in the naïve and selected libraries revealed differences in the identities of the variants observed. Twelve of the variants observed following selection were among the 64 P variants that were absent from the naïve library. This finding indicates that these twelve variants were present in our original library but not sufficiently abundant to be detected in our deep sequencing analysis. Taken together with the detection of 159 unique P variants in the unselected library, the detection of 12 additional variants post selection indicates that at least 171 vectors that express permuted AK were sampled in our original library out of 223 total possible in frame and P variants (77%).
As shown in Figure 3, a unique feature transposon-mediated library construction methods like e-PERMUTE is the generation of equal proportions of cognate P and AP variants that represent paired data across the broad range of initial sequence frequencies found in the naive library. In these libraries, the AP variants provide an internal frame of reference for evaluating the effect of the selection on their cognate P vectors. These AP vectors cannot express a circularly permuted protein and should not be enriched by the selection. Instead, the AP variants are expected to be uniformly diluted by the selection such that their relative abundances remain similar. The AP representation in the total population (AP + P variant counts) becomes lower as cells expressing active AK grow and take over the population during the selection. To test this idea, we quantified the abundance of every AP variant in the selected library and compared these values with those observed in our library before selection (Figure 4). We found that the abundance of these AP variants uniformly decreased. In contrast, the abundances of the P variants were differentially affected by the selection. These differences were also visualized by plotting the abundances of each gene in the P and AP orientations before and after selection (Figure 5A). With the unselected variants, the P and AP abundances are proportional, yielding a strong linear correlation (R2 = 0.97). In contrast, the abundances of the selected P variants deviate from this trend. These findings demonstrate that the dilution of the AP variants upon selection can be used as a frame of reference to identify P vectors that are significantly enriched by the selection.
Figure 4.

Relationship between abundance and the AK codon at the beginning of each permuted genes. A comparison of the number of (A) P and (B) AP sequences before (top) and after selecting (bottom) for biological activity. The residue position represents the AK residue found at the beginning of each ORF regardless of orientation. Only those ORFs encoding in frame variants are shown. In cases where a P or AP variant was absent, black bars are shown below the x-axis.
Figure 5.

Effect of selection on P and AP sequence abundance. (A) A comparison of the P and AP sequence abundances for each variant in the unselected library (left panel) reveals a linear correlation (y = 0.979x + 30.829; R2 = 0.97), which is shown in blue. Following selection (right panel), the relative abundance of P to AP counts diverged from this trend. The solid black line represents the expectation when cognate P and AP variants occur with identical frequencies. (B) The abundance of each P and AP sequence before (left) and after (right) selection. The AP variants display a linear correlation (y = 0.026x − 0.358; R2 = 0.95), which is shown in blue. Selected P variants are colored as a function of the P-value obtained from Fisher’s Exact Test (PVF), with variants presenting P-values > 0.01 in red, those presenting P-values ≤ 10−300 in black, and those displaying intermediate values shaded as indicated in the bar.
To visualize how the selection influenced the relative abundances of P and AP variants, we compared the number of reads observed for every variant before and after selection (Figure 5B). In the case of the AP variants, a strong linear correlation was observed between the abundances of each variant before and after selection (R2 = 0.95). This line is shifted from the trend expected (1:1) if the selection had no effect on AP abundance, illustrating how the negative selective pressure uniformly diluted the vectors harboring AP variants. This analysis also suggested that a large fraction of the P variants are enriched relative to the AP variants, with some of the P variants being enriched three orders of magnitude over that observed with the AP variants following the selection.
One challenge with our deep mutational scanning data is establishing which of the P variants are biologically active. To identify active AK variants, we sought to determine which of the P variants present changes in abundance that are significantly different from the AP variants. Because the relative abundances of each unique permuted gene was similar in the P and AP orientation before selection, we generated a two by two contingency table containing abundance information about each circularly permuted gene before and after selection in the P and AP orientations. Using the data for each circularly permuted gene, we analyzed whether the ratio of the selected to unselected abundances is equal in the P and AP orientations using Fisher’s Exact Test. We initially chose this approach because vectors containing an AK gene in the AP orientation presented uniform dilution upon selection (Figure 5), consistent with these vectors being unable to transcribe and translate a circularly permuted AK. With this statistical test, we obtained evidence that 56% (n = 96) of the total variants sampled (n = 171) present ratios of selected to unselected sequence reads that are higher in the P orientation compared with the AP orientation (P-values < 0.01).
Fisher’s Exact Test cannot assess biological activity of P variants whose sequences were observed in the selected library in cases when their cognate AP variants are absent from the unselected and selected libraries (n = 15 for the in frame variants). Additionally, this test cannot assess biological activity when the P and cognate AP variants are both absent from the selected library (n = 27 for the in frame variants), even if they are present in the unselected library. Because of this limitation, we used a negative binomial distribution to model the mean and variance of the ratio of AP variant abundances before and after the selection, i.e. log2(fold change). We used the mean and variance from this model to investigate which of our P variants were significantly enriched by the selection (two tailed t-test, P < 0.01). With this analysis (Figure 6), 65.5% of the P variants (n = 112) sampled in our library (n = 171) presented log2(fold change) ratios of selected to unselected sequence reads that were higher in the P orientation compared with that modeled for the AP orientation (P-values < 0.01). Using this global error fitting approach, we were able to assess the significance of an additional ∼19% of the possible variants (n = 42) that weren’t accessible using Fisher’s Test alone. Among these 42 variants, <25% (n = 10) were significantly enriched. These ten variants all presented increases in P counts following the selection but had no AP observed before and after selection. Additionally, the p values generated by Fisher’s test and the negative binomial model presented a strong linear correlation (R2 = 0.99, Supplementary Figure S4).
Figure 6.

Enrichment of parallel sequences following selection. The log2(fold change) in sequence abundances of the AP (open symbols) and P variants (closed symbols). The significance of P variant enrichment obtained using the negative binomial model (PVNB) is colored as a function of the P-value obtained with the variants presenting values >0.01 in red, variants having values ≤10−300 in black and those variants displaying intermediate values shaded as indicated in the bar. The black line represents the mean dilution for AP variants relative to their initial abundance in the unselected library, while the dashed line represents two standard deviations greater than the mean. Variants not observed in the selected library (infinitely diluted) are plotted in the shaded region.
To determine the robustness of our method for identifying permuted proteins that are biologically active, we compared the log2(fold change) values of thirty one variants with the ability of those variants to complement the growth of E. coli CV2 in liquid growth. With this analysis, we observed a linear correlation between log2(fold change) in our deep sequencing data and growth analysis of individual variants (Figure 7A) using vectors that were previously reported (52). This analysis also shows that the P-values obtained from circular permutation profiling with DNA sequencing (CPP-seq) correlate with growth complementation of the individual variants. Permuted AK yielding the strongest complementation had the smallest P-values, while those presenting the weakest complementation presented the largest P-values.
Figure 7.

Relationship between AK structure and retention of biological activity. (A) For thirty one variants, we compared the log2(fold change) values with growth complementation of Escherichia coli CV2 transformed with vectors that constitutively express each variant. This data displays a linear correlation (y = 0.066x + 0.533; R2 = 0.783). P-values obtained from the negative binomial model (PNB) are color coded and analyzed as described in Figure 6. (B) For each P variant, the log2(fold change) is shown as a function of the AK residue found at the N-terminus of the circularly permuted protein. The AK domain structure is shown as a frame of reference. Variants no longer observed in the selected library (infinitely diluted) are shown as bars that reach the line at the bottom of the graph. Red variants above the shaded region were observed in the selected library but were not significantly enriched (P-values > 0.01). Those cognate P and AP variant pairs absent from both the unselected and selected datasets (n = 52) are indicated as black lines shown below the x-axis.
To generate a profile that relates circular permutation tolerance to primary structure, we designated in frame permuted AK variants as active if we observed a significant enrichment upon selection, defined as variants presenting a P < 0.01. Mapping the first residue in biologically active variants onto AK structure, as well as their log2(fold change) upon selection, reveals that new protein termini were functionally tolerated at distal locations within the overall structure (Figure 7B). In addition, new protein termini were tolerated in all three domains, including the AMP binding, core and lid domains. The regions of the native AK structure that appeared most tolerant to harboring new protein termini following circular permutation included: (i) the residues adjacent to the original N- and C-termini, (ii) residues within the mobile AMP binding domain and (iii) residues found within the portion of the core domain that is located between the two mobile domains. The log2(fold change) upon selection for the AP variants is shown as a frame of reference (Supplementary Figure S5).
Analyzing out of frame variants
In our library, only one third of the possible P variants have the permuted AK gene in the same frame as the intended start codon, which is encoded by the permuteposon. This trend occurs because the permuteposon is inserted in all three frames of the circular gene when constructing the library. Because previous studies have shown that proteins can be translated even when they are out of frame of an intended start codon (54), we investigated if any of the out of frame P variants were enriched by our selection compared with the AP variants.
The abundances of out of frame variants before and after selection is shown in Supplementary Figure S6. Prior to the selection, the abundances of the P sequence reads in the +1 (42 719) and −1 (21 909) frames were similar to the in frame reads (59 494). In each alternative frame, the fraction of P variants was also similar to that observed with the in frame variants, which presented 52% P before selection. Among the variants in the +1 frame, 52% were in the P orientation, while 55% of the variants in the −1 frame were P. A plot analyzing the relative abundances of the out of frame variants in the P and AP orientations revealed linear trends before and after selection (Supplementary Figure S7), although some variants displayed a small enrichment by the selection. This enrichment of the out of frame variants was also visualized by comparing the counts of the selected and unselected P and AP variants (Supplementary Figure S8). This analysis revealed that some P variants were significantly enriched by the selection, i.e. presented P-values < 0.01. While a small enrichment was observed for some out of frame P variants (Supplementary Figures S9 and 10), the magnitude of this enrichment was much lower on average than the in frame variants. Furthermore, the dispersion of enriched variants appears more randomly dispersed throughout the primary structure.
DISCUSSION
Our results provide evidence that libraries encoding circularly permuted variants of a gene can be made through a simplified one-step enzymatic method called e-PERMUTE. This new approach is similar to PERMUTE, but it requires fewer manipulations to generate a library. In addition, a larger fraction of the transposase reaction products from e-PERMUTE contain permuteposons inserted within the gene of interest. Alternative methods have been described for creating libraries of vectors that express circularly permuted variants of a protein, including nuclease- and PCR-based approaches. In contrast to nuclease methods, which create permuted genes encoding proteins with random deletions of primary sequence proximal to their new termini (31,32), e-PERMUTE generates libraries encoding full-length permuted genes that lack deletions. Like e-PERMUTE, PCR-based methods can avoid these deletions by amplifying a portion of a gene fusion (55). However, these methods increase the likelihood of creating point mutations within permuted genes in a library because they utilize DNA polymerases that can introduce errors, and they do not generate the AP frame of reference for each gene like e-PERMUTE.
Deep mutational scanning of an e-PERMUTE library revealed that 77% of the possible vectors that are capable of expressing full-length circularly permuted AK variants (i.e. P genes that are in frame) were present in our library. Our library additionally sampled a large fraction of the out of frame P variants, which could express truncated permuted proteins through alternative start sites and ribosomal frameshifting (56). This idea is supported by our finding that a subset of the out of frame P variants are significantly enriched by the selection. The protein that we targeted is similar to the average size of many prokaryotic proteins (57), so these findings suggest that CPP-seq can be applied to diverse proteins in cases where high-throughput selections or screens are available for enriching active variants (58). Our results also suggest that CPP-seq could be further improved by creating unselected libraries that have larger numbers of degenerate sequences. Three approaches can be used to accomplish improved sampling in future studies. First, the transposon insertion reaction could be scaled up to obtain a larger number of colonies from the transformation, and a larger number of unique colonies can be sampled during the bacterial transformation step used to generate the naïve library. Second, a larger number of sequence reads can be obtained when analyzing the library using MiSeq, and multiple MiSeq experiments can be performed to improve the sampling of low abundance variants before and after selection. Third, the target gene sequence could be optimized to avoid sequence motifs that represent MuA hot spots (48).
Our results demonstrate that MuA inserts permuteposons at diverse locations within DNA, yielding variants with abundances spanning over three orders of magnitude, similar to previous observations with distinct artificial transposons (48). Even though our naïve library contained a large sequence bias, we were able to demonstrate that >65% of the unique P vectors that were sampled express circularly permuted AK that are biologically active even though they have a large eighteen amino acid peptide amended to their N-terminus. This value indicates that 50% of all possible permuted AK retain biological activity. As with other deep mutational scanning studies (1–4), this analysis was accomplished by quantifying the ratio of abundances for every possible variant before and after the selection. Because every circularly permuted gene exhibited similar abundance in the P and AP orientation, we initially used Fisher’s Exact Test to evaluate whether the ratio of their abundances were altered by the selection. This approach can be applied to other libraries created using transposon mutagenesis, such as domain insertion and split protein libraries (29,30,59–62). However, this statistical approach is limited by the requirement that at least one AP sequence be observed within the naïve library. Because our naïve library did not present exhaustive sampling of each unique P and AP variant, the mean dilution of AP vectors and their variance was modeled using a negative binomial distribution (49), and the values obtained from this model were used to determine which P variants were biologically active.
A comparison of AK structure with the in frame P sequences significantly enriched by the selection revealed that circularly permuted variants with the greatest enrichment initiate translation using native AK residues that cluster within different regions of the primary structure. Not surprisingly, highly enriched clusters of sequences were observed with N-terminal residues that cluster near the termini of native AK, with the largest cluster observed within the last two dozen residues within the core domain. This result is similar to previous studies (32,63), which have demonstrated that proteins often display a high tolerance to permutation when backbone fission occurs near the native protein termini. Additionally, a large cluster of highly enriched variants was observed within the mobile AMP binding domain, and smaller clusters occurred within the middle of the core domain region that connects the two different mobile domains. The underlying cause of these trends is not known, although they are not expected to arise from changes in translation initiation. The RBS used to initiate protein synthesis is adjacent to the large R1R2 sequence, which is unchanged in all of our expression vectors, and calculations of RBS strength using a thermodynamic model yields identical values corresponding to a strong RBS for every P and in frame variant in our library (47).
Structural and enzymatic studies have provided evidence that the AMP binding and lid domains undergo large conformational motions as AK cycles between substrate-bound and substrate-free conformational states (64–66), and the opening of these domains is thought to be rate-limiting for AK catalysis (67). The paucity of active, circularly permuted AK with new protein termini within the lid domain suggests that this mobile domain is particularly sensitive to mutations that increase conformational fluctuations. Consistent with this trend, a previous biochemical study found that point mutations (valine to glycine) that increase conformational fluctuations have deleterious effects on AK and substrate binding even though they do not alter the ground state structure (68). Why the mobile AMP binding domain differs from the mobile lid domain in its tolerance to these types of mutations is not clear and will require future biochemical studies that compare the structure and stability of circularly permuted variants with new protein termini in each of these domains.
In future studies, it will be interesting to apply CPP-seq to protein orthologs with divergent sequences, stabilities, and activities. By comparing the circular permutation tolerance of structurally related proteins using more comprehensive sampling of sequences, CPP-seq can be used to examine how the profiles generated by this approach relate to the protein stability as well as the linkers used to connect the original protein termini. Furthermore, by evaluating how circular permutation tolerance changes with temperature or other environmental conditions, combinatorial experiments can be used to establish how selection pressure influences the impact of circular permutation on protein structure and function.
DATA AVAILABILITY
The CPP-seq processing code can be found online at: github.com/SilbergLab/CPP-seq.
Illumina sequencing data has been submitted to the NCBI Sequence Read Archive (SRA) under accession numbers SRR6327683 (unselected) and SRR6327684 (selected).
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to express their appreciation to Gerda Saxer, Kathryn Beabout and Philip Ernst for helpful discussions related to acquisition of deep sequencing data and statistical analysis.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Science Foundation (NSG) [1150138]; National Science Foundation Graduate Research Fellowship Program [R3E821 to A.M.J., J.T.A.]; Department of Energy Office of Science Graduate Student Research Program (to J.T.A.). Funding for open access charge: NSF Grant [1150138].
Conflict of interest statement. None declared.
REFERENCES
- 1. Fowler D.M., Araya C.L., Fleishman S.J., Kellogg E.H., Stephany J.J., Baker D., Fields S.. High-resolution mapping of protein sequence-function relationships. Nat. Methods. 2010; 7:741–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hietpas R.T., Jensen J.D., Bolon D.N.A.. Experimental illumination of a fitness landscape. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:7896–7901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fowler D.M., Fields S.. Deep mutational scanning: a new style of protein science. Nat. Methods. 2014; 11:801–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Starita L.M., Fields S.. Deep mutational Scanning: a highly parallel method to measure the effects of mutation on protein function. Cold Spring Harb. Protoc. 2015; 2015:711–714. [DOI] [PubMed] [Google Scholar]
- 5. Fowler D.M., Araya C.L., Gerard W., Fields S.. Enrich: software for analysis of protein function by enrichment and depletion of variants. Bioinformatics. 2011; 27:3430–3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bloom J.D. Software for the analysis and visualization of deep mutational scanning data. BMC Bioinformatics. 2015; 16:168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Starita L.M., Fields S.. Deep mutational scanning: calculating enrichment scores for protein variants from DNA sequencing output files. Cold Spring Harb. Protoc. 2015; 2015:781–783. [DOI] [PubMed] [Google Scholar]
- 8. Klesmith J.R., Bacik J.-P., Wrenbeck E.E., Michalczyk R., Whitehead T.A.. Trade-offs between enzyme fitness and solubility illuminated by deep mutational scanning. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:2265–2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. McLaughlin R.N., Poelwijk F.J., Raman A., Gosal W.S., Ranganathan R.. The spatial architecture of protein function and adaptation. Nature. 2012; 491:138–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Doolan K.M., Colby D.W.. Conformation-dependent epitopes recognized by prion protein antibodies probed using mutational scanning and deep sequencing. J. Mol. Biol. 2015; 427:328–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wagenaar T.R., Ma L., Roscoe B., Park S.M., Bolon D.N., Green M.R.. Resistance to vemurafenib resulting from a novel mutation in the BRAFV600E kinase domain. Pigment Cell Melanoma Res. 2014; 27:124–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shin H., Cho Y., Choe D.-H., Jeong Y., Cho S., Kim S.C., Cho B.-K.. Exploring the functional residues in a flavin-binding fluorescent protein using deep mutational scanning. PLoS One. 2014; 9:e97817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Romero P.A., Tran T.M., Abate A.R.. Dissecting enzyme function with microfluidic-based deep mutational scanning. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:7159–7164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Araya C.L., Fowler D.M., Chen W., Muniez I., Kelly J.W., Fields S.. A fundamental protein property, thermodynamic stability, revealed solely from large-scale measurements of protein function. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:16858–16863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wu N.C., Young A.P., Dandekar S., Wijersuriya H., Al-Mawsawi L.Q., Wu T.-T., Sun R.. Systematic identification of H274Y compensatory mutations in influenza A virus neuraminidase by high-throughput screening. J. Virol. 2013; 87:1193–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jacquier H., Birgy A., Le Nagard H., Mechulam Y., Schmitt E., Glodt J., Bercot B., Petit E., Poulain J., Barnaud G. et al. Capturing the mutational landscape of the beta-lactamase TEM-1. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:13067–13072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Melamed D., Young D.L., Gamble C.E., Miller C.R., Fields S.. Deep mutational scanning of an RRM domain of the Saccharomyces cerevisiae poly(A)-binding protein. RNA. 2013; 19:1537–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Olson C.A., Wu N.C., Sun R.. A comprehensive biophysical description of pairwise epistasis throughout an entire protein domain. Curr. Biol. 2014; 24:2643–2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bank C., Hietpas R.T., Jensen J.D., Bolon D.N.A.. A systematic survey of an intragenic epistatic landscape. Mol. Biol. Evol. 2015; 32:229–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wrenbeck E.E., Azouz L.R., Whitehead T.A.. Single-mutation fitness landscapes for an enzyme on multiple substrates reveal specificity is globally encoded. Nat. Commun. 2017; 8:15695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Whitehead T.A., Chevalier A., Song Y., Dreyfus C., Fleishman S.J., De Mattos C., Myers C.A., Kamisetty H., Blair P., Wilson I.A. et al. Optimization of affinity, specificity and function of designed influenza inhibitors using deep sequencing. Nat. Biotechnol. 2012; 30:543–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Melnikov A., Rogov P., Wang L., Gnirke A., Mikkelsen T.S.. Comprehensive mutational scanning of a kinase in vivo reveals substrate-dependent fitness landscapes. Nucleic Acids Res. 2014; 42:e112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Koenig P., Lee C.V., Sanowar S., Wu P., Stinson J., Harris S.F., Fuh G.. Deep Sequencing-guided design of a high affinity dual specificity antibody to target two angiogenic factors in neovascular Age-related macular degeneration. J. Biol. Chem. 2015; 290:21773–21786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Harris D.T., Wang N., Riley T.P., Anderson S.D., Singh N.K., Procko E., Baker B.M., Kranz D.M.. Deep mutational scans as a guide to engineering high affinity t cell receptor interactions with Peptide-bound major histocompatibility complex. J. Biol. Chem. 2016; 291:24566–24578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Peisajovich S.G., Rockah L., Tawfik D.S.. Evolution of new protein topologies through multistep gene rearrangements. Nat. Genet. 2006; 38:168–174. [DOI] [PubMed] [Google Scholar]
- 26. Weiner J., Bornberg-Bauer E.. Evolution of circular permutations in multidomain proteins. Mol. Biol. Evol. 2006; 23:734–743. [DOI] [PubMed] [Google Scholar]
- 27. Vogel C., Morea V.. Duplication, divergence and formation of novel protein topologies. Bioessays. 2006; 28:973–978. [DOI] [PubMed] [Google Scholar]
- 28. Higgins S.A., Savage D.F.. Protein science by DNA Sequencing: How advances in molecular biology are accelerating biochemistry. Biochemistry. 2018; 57:38–46. [DOI] [PubMed] [Google Scholar]
- 29. Oakes B.L., Nadler D.C., Flamholz A., Fellmann C., Staahl B.T., Doudna J.A., Savage D.F.. Profiling of engineering hotspots identifies an allosteric CRISPR-Cas9 switch. Nat. Biotechnol. 2016; 34:646–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nadler D.C., Morgan S.-A., Flamholz A., Kortright K.E., Savage D.F.. Rapid construction of metabolite biosensors using domain-insertion profiling. Nat. Commun. 2016; 7:12266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hennecke J., Sebbel P., Glockshuber R.. Random circular permutation of DsbA reveals segments that are essential for protein folding and stability. J. Mol. Biol. 1999; 286:1197–1215. [DOI] [PubMed] [Google Scholar]
- 32. Graf R., Schachman H.K.. Random circular permutation of genes and expressed polypeptide chains: application of the method to the catalytic chains of aspartate transcarbamoylase. Proc. Natl. Acad. Sci. U.S.A. 1996; 93:11591–11596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Baird G.S., Zacharias D.A., Tsien R.Y.. Circular permutation and receptor insertion within green fluorescent proteins. Proc. Natl. Acad. Sci. U.S.A. 1999; 96:11241–11246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Guntas G., Mansell T.J., Kim J.R., Ostermeier M.. Directed evolution of protein switches and their application to the creation of ligand-binding proteins. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:11224–11229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ostermeier M. Engineering allosteric protein switches by domain insertion. Protein Eng. Des. Sel. 2005; 18:359–364. [DOI] [PubMed] [Google Scholar]
- 36. Tullman J., Guntas G., Dumont M., Ostermeier M.. Protein switches identified from diverse insertion libraries created using S1 nuclease digestion of supercoiled-form plasmid DNA. Biotechnol. Bioeng. 2011; 108:2535–2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lindqvist Y., Schneider G.. Circular permutations of natural protein sequences: structural evidence. Curr. Opin. Struct. Biol. 1997; 7:422–427. [DOI] [PubMed] [Google Scholar]
- 38. Lo W.-C., Lyu P.-C.. CPSARST: an efficient circular permutation search tool applied to the detection of novel protein structural relationships. Genome Biol. 2008; 9:R11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bliven S.E., Bourne P.E., Prlić A.. Detection of circular permutations within protein structures using CE-CP. Bioinformatics. 2015; 31:1316–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yu Y., Lutz S.. Circular permutation: a different way to engineer enzyme structure and function. Trends Biotechnol. 2011; 29:18–25. [DOI] [PubMed] [Google Scholar]
- 41. Mehta M.M., Liu S., Silberg J.J.. A transposase strategy for creating libraries of circularly permuted proteins. Nucleic Acids Res. 2012; 40:e71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pierre B., Shah V., Xiao J., Kim J.R.. Construction of a random circular permutation library using an engineered transposon. Anal. Biochem. 2015; 474:16–24. [DOI] [PubMed] [Google Scholar]
- 43. Jones A.M., Atkinson J.T., Silberg J.J.. PERMutation Using Transposase Engineering (PERMUTE): a simple approach for constructing circularly permuted protein libraries. Methods Mol. Biol. 2017; 1498:295–308. [DOI] [PubMed] [Google Scholar]
- 44. Shah V., Kim J.R.. Transposon for protein engineering. Mob. Genet. Elements. 2016; 6:e1239601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Engler C., Gruetzner R., Kandzia R., Marillonnet S.. Golden gate shuffling: a one-pot DNA shuffling method based on type IIs restriction enzymes. PLoS One. 2009; 4:e5553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Haase G.H., Brune M., Reinstein J., Pai E.F., Pingoud A., Wittinghofer A.. Adenylate kinases from thermosensitive Escherichia coli strains. J. Mol. Biol. 1989; 207:151–162. [DOI] [PubMed] [Google Scholar]
- 47. Salis H.M., Mirsky E.A., Voigt C.A.. Automated design of synthetic ribosome binding sites to control protein expression. Nat. Biotechnol. 2009; 27:946–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Haapa-Paananen S., Rita H., Savilahti H.. DNA transposition of bacteriophage Mu. A quantitative analysis of target site selection in vitro. J. Biol. Chem. 2002; 277:2843–2851. [DOI] [PubMed] [Google Scholar]
- 49. Anders S., Huber W.. Differential expression analysis for sequence count data. Genome Biol. 2010; 11:R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Judd J., Wei F., Nguyen P.Q., Tartaglia L.J., Agbandje-McKenna M., Silberg J.J., Suh J.. Random insertion of mCherry into VP3 domain of Adeno-associated virus yields fluorescent capsids with no loss of infectivity. Mol. Ther. Nucleic Acids. 2012; 1:e54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Vieille C., Krishnamurthy H., Hyun H.-H., Savchenko A., Yan H., Zeikus J.G.. Thermotoga neapolitana adenylate kinase is highly active at 30 degrees C. Biochem. J. 2003; 372:577–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Jones A.M., Mehta M.M., Thomas E.E., Atkinson J.T., Segall-Shapiro T.H., Liu S., Silberg J.J.. The structure of a thermophilic kinase shapes fitness upon random circular permutation. ACS Synth. Biol. 2016; 5:415–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Caporaso J.G., Lauber C.L., Walters W.A., Berg-Lyons D., Huntley J., Fierer N., Owens S.M., Betley J., Fraser L., Bauer M. et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012; 6:1621–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Whitaker W.R., Lee H., Arkin A.P., Dueber J.E.. Avoidance of truncated proteins from unintended ribosome binding sites within heterologous protein coding sequences. ACS Synth. Biol. 2015; 4:249–257. [DOI] [PubMed] [Google Scholar]
- 55. Kanwar M., Wright R.C., Date A., Tullman J., Ostermeier M.. Protein switch engineering by domain insertion. Meth. Enzymol. 2013; 523:369–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Seligmann H. Cost minimization of ribosomal frameshifts. J. Theor. Biol. 2007; 249:162–167. [DOI] [PubMed] [Google Scholar]
- 57. Netzer W.J., Hartl F.U.. Recombination of protein domains facilitated by co-translational folding in eukaryotes. Nature. 1997; 388:343–349. [DOI] [PubMed] [Google Scholar]
- 58. Aharoni A., Griffiths A.D., Tawfik D.S.. High-throughput screens and selections of enzyme-encoding genes. Curr. Opin. Chem. Biol. 2005; 9:210–216. [DOI] [PubMed] [Google Scholar]
- 59. Ostermeier M., Nixon A.E., Shim J.H., Benkovic S.J.. Combinatorial protein engineering by incremental truncation. Proc. Natl. Acad. Sci. U.S.A. 1999; 96:3562–3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Segall-Shapiro T.H., Nguyen P.Q., Santos Dos E.D., Subedi S., Judd J., Suh J., Silberg J.J.. Mesophilic and hyperthermophilic adenylate kinases differ in their tolerance to random fragmentation. J. Mol. Biol. 2011; 406:135–148. [DOI] [PubMed] [Google Scholar]
- 61. Mahdavi A., Segall-Shapiro T.H., Kou S., Jindal G.A., Hoff K.G., Liu S., Chitsaz M., Ismagilov R.F., Silberg J.J., Tirrell D.A.. A genetically encoded and gate for cell-targeted metabolic labeling of proteins. J. Am. Chem. Soc. 2013; 135:2979–2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Segall-Shapiro T.H., Meyer A.J., Ellington A.D., Sontag E.D., Voigt C.A.. A ‘resource allocator’ for transcription based on a highly fragmented T7 RNA polymerase. Mol. Syst. Biol. 2014; 10:742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Qian Z., Lutz S.. Improving the catalytic activity of Candida antarctica lipase B by circular permutation. J. Am. Chem. Soc. 2005; 127:13466–13467. [DOI] [PubMed] [Google Scholar]
- 64. Müller C.W., Schlauderer G.J., Reinstein J., Schulz G.E.. Adenylate kinase motions during catalysis: an energetic counterweight balancing substrate binding. Structure. 1996; 4:147–156. [DOI] [PubMed] [Google Scholar]
- 65. Müller C.W., Schulz G.E.. Structure of the complex between adenylate kinase from Escherichia coli and the inhibitor Ap5A refined at 1.9 A resolution. A model for a catalytic transition state. J. Mol. Biol. 1992; 224:159–177. [DOI] [PubMed] [Google Scholar]
- 66. Henzler-Wildman K.A., Thai V., Lei M., Ott M., Wolf-Watz M., Fenn T., Pozharski E., Wilson M.A., Petsko G.A., Karplus M. et al. Intrinsic motions along an enzymatic reaction trajectory. Nature. 2007; 450:838–844. [DOI] [PubMed] [Google Scholar]
- 67. Wolf-Watz M., Thai V., Henzler-Wildman K., Hadjipavlou G., Eisenmesser E.Z., Kern D.. Linkage between dynamics and catalysis in a thermophilic-mesophilic enzyme pair. Nat. Struct. Mol. Biol. 2004; 11:945–949. [DOI] [PubMed] [Google Scholar]
- 68. Schrank T.P., Bolen D.W., Hilser V.J.. Rational modulation of conformational fluctuations in adenylate kinase reveals a local unfolding mechanism for allostery and functional adaptation in proteins. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:16984–16989. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The CPP-seq processing code can be found online at: github.com/SilbergLab/CPP-seq.
Illumina sequencing data has been submitted to the NCBI Sequence Read Archive (SRA) under accession numbers SRR6327683 (unselected) and SRR6327684 (selected).