


Abstract
Natural and lab-evolved proteins often recognize their RNA partners with exquisite affinity. Structural analysis of such complexes can offer valuable insight into sequence-selective recognition that can be exploited to alter biological function. Here, we describe the structure of a lab-evolved RNA recognition motif (RRM) bound to the HIV-1 trans-activation response (TAR) RNA element at 1.80 Å-resolution. The complex reveals a trio of arginines in an evolved β2–β3 loop penetrating deeply into the major groove to read conserved guanines while simultaneously forming cation-π and salt-bridge contacts. The observation that the evolved RRM engages TAR within a double-stranded stem is atypical compared to most RRMs. Mutagenesis, thermodynamic analysis and molecular dynamics validate the atypical binding mode and quantify molecular contributions that support the exceptionally tight binding of the TAR-protein complex (KD,App of 2.5 ± 0.1 nM). These findings led to the hypothesis that the β2–β3 loop can function as a standalone TAR-recognition module. Indeed, short constrained peptides comprising the β2–β3 loop still bind TAR (KD,App of 1.8 ± 0.5 μM) and significantly weaken TAR-dependent transcription. Our results provide a detailed understanding of TAR molecular recognition and reveal that a lab-evolved protein can be reduced to a minimal RNA-binding peptide.
INTRODUCTION
HIV/AIDS afflicts 36.7 million people worldwide, and currently there is no vaccine or cure (1,2). With the goal of eliminating latent reservoirs, it is necessary to develop new compounds that disrupt currently undrugged pathways of the viral lifecycle—particularly those targets with low levels of mutation across multiple HIV-1 clades (3). In these respects the HIV-1 TAR (trans-activation response) RNA element is attractive as a drug target because it exists in the 5′-noncoding regions of all viral mRNAs and resists mutations to maintain interactions with Tat (trans-activator of transcription) (4–6). The latter viral protein is essential to recruit the host's positive transcription elongation factor complex to TAR (7) (Figure 1A), leading to sustained proviral DNA transcription (8). TAR also serves as a pre-miRNA whose processed products block apoptosis of the infected cell, prolonging the viral lifespan in the host (9–11). For these reasons, ablation of the TAR–Tat interaction is viewed as a high-value therapeutic target since disruption would block activation of the proviral genome (12).
Figure 1.

Cartoon depicting the dependence of HIV-1 transcription upon the viral TAR–Tat interaction and overview of the ‘semi design’ approach that led to TBP6.7 (TAR-Binding Protein 6.7). (A) The viral trans-activation response (TAR) element RNA comprises lower (S1a) and upper (S1b) stems. The positive transcription elongation factor b (p-TEFb) comprising cyclin T1 (green) and CDK9 (red) is recruited to TAR by the HIV-1 protein Tat (purple), which binds the central RNA bulge allowing cyclin T1 to interact with the apical loop. The bound complex stimulates host RNA polymerase II (yellow) by phosphorylation to produce full-length viral transcripts from proviral DNA [reviewed in (12)]. (B) A yeast-display approach was used to diversify putative RNA-binding amino acids in the β2–β3 loop and C-terminus of U1A RRM1 (RNA Recognition Motif 1, depicted as a blue ribbon); selection utilized labeled TAR (star) binding under increasingly stringent conditions (21). The resulting loop consensus sequence is shown (right) along with amino acids from U1A and TBP6.7—the tightest known TAR binder (21). R47 and R52 were unaltered to exploit their innate RNA binding potential.
Designing molecules with potent and selective recognition of RNA is an unsolved and important challenge (13). Due to its key significance in the HIV-1 lifecycle, TAR has been the focus of multiple drug discovery efforts utilizing small-molecules, peptides, and proteins (14–19). While these TAR-binding reagents have taught us much about RNA recognition and assisted in the development of RNA-targeting compounds, innovative discovery techniques are needed to generate new drugs with sufficient potency and selectivity to warrant development (20). To identify such molecules, we undertook a fundamentally different ‘semi-design’ strategy that uses laboratory evolution to alter putative RNA-binding amino acids within a known protein (21). Starting from the extraordinarily tight interaction (KD,App of 32 pM) between hairpin II of the U1 snRNA and RRM1 (RNA recognition motif 1) of the U1A protein (22,23), we performed saturation mutagenesis in the RRM β2–β3 loop and C-terminus (Figure 1B). The resulting protein library was then subjected to yeast-display screening, and desired TAR-Binding Proteins (TBPs) were identified by flow cytometry (21). Variant TBP6.7 exhibits extraordinarily tight TAR affinity (KD,App of 2.5 ± 0.1 nM), impairs TAR binding to a previously reported Tat peptide, and attenuates Tat-dependent transcription. However, the chemical determinants of TAR binding by TBP6.7 remained unknown, despite their potential to yield unique insights into molecular recognition. RNA recognition possibilities included readout by the unaltered RRM binding motifs (i.e. RNP1 and RNP2) present in parental U1A (23), the evolved β2–β3 loop, the evolved C-terminus, or combinations thereof (21).
To understand the underlying molecular basis of the TAR–TBP6.7 interaction, we determined the high-resolution co-crystal structure of the complex at 1.80 Å-resolution. The results revealed that a subset of RNP residues as well as evolved amino acids of the β2–β3 loop confer an unprecedented mode of TAR recognition. In contrast, the evolved C-terminus exhibited no RNA binding. Mutagenesis, isothermal titration calorimetry (ITC), and molecular dynamics (MD) simulations support the hypothesis that a short peptide harboring the evolved β2–β3 loop is sufficient for TAR recognition. This was confirmed by the observation that fusion proteins containing the evolved loop retain TAR binding, and conformationally constrained peptides comprising the β2–β3 loop retain TAR binding and significantly weaken Tat-dependent transcription. The results have implications for HIV-1/AIDS research that seeks to suppress latent viral reservoirs by blocking proviral DNA transcription, leading to a functional cure (24). Our findings further demonstrate that our lab-evolution approach can be used to distill the RRM fold into a standalone RNA-recognition peptide. This advance is considered in the context of natural examples of molecular exaptation, whereby an existing biomolecular scaffold is co-opted for a new biological function.
MATERIALS AND METHODS
Expression and purification of TBP6.7
TBP6.7 was identified previously in our lab (21). TBP6.7 DNA was prepared as a synthetic gene (GeneScript Inc) comprising the human U1A sequence, yeast-display mutants (21), and Y31H/Q36R integrated for crystallization (25). After sub-cloning into pET28a(+) (Novagen) the thrombin site was modified by PCR to utilize TEV protease to cleave the N-terminal linker (ENLYFQ/G) (Supplementary Tables S2 and S3). Point mutations were incorporated using the Q5 kit as described by the manufacturer (NEB) with primers from IDT (Supplementary Table S3). Protein expression in Escherichia coli BL21(DE3) (NEB) was induced by 0.5 mM IPTG in LB at 20 °C. Cells were harvested after 4 h and pellets were frozen in N2(l). Cells were thawed in a cell lysis buffer (CLB): 0.05 M Na-HEPES pH 7.5, 0.5 M NaCl, 0.02 M imidazole pH 8.0, 0.0005 M EDTA, 0.005 M β-ME and 0.01% (v/v) Brij35; the cell slurry was made 2 mg ml−1 in lysozyme (VWR). After 20 min, cells were sonicated and the clarified supernatant was applied in batch to Ni-NTA resin (Pierce) equilibrated with CLB. After 2 h of nutation at 4 °C, resin was poured into a 1.5 cm × 10 cm gravity-flow column (CrystalCruz), washed with 40 column volumes of CLB, and two column volumes of wash buffer (WB): 0.05 M Na-HEPES pH 7.0, 0.3 M NaCl, 0.04 M imidazole pH 7.5, 0.005 M EDTA, 0.005 M β-ME and 0.01% (v/v) Brij35. Elution was in 3 ml fractions using elution buffer (EB): 0.15 M NaCl and 0.2 M imidazole pH 7.5. Fractions with 280 nm absorption were pooled and diluted with EB to a final imidazole concentration <0.02 M. TEV (26) was added (1:100 TEV:TBP) and the mixture was incubated at 4 °C. After 16 h, the reaction was incubated in batch with pre-equilibrated Ni-NTA, and supernatant was collected. Protein was loaded with an ÄKTA Pure (GE Lifesciences) at 0.5 ml min−1 onto a 5 ml HiTrap SP FF column (GE), followed by a linear gradient comprising: 0.15–0.85 M NaCl, 0.05 M Na-HEPES pH 7.0, 0.0025 M EDTA and 0.00025 M β-ME; TBP6.7 elutes at ∼70% as a sharp peak. The concentrated protein is polished on a HiPrep (16/60) Sephacryl S-300 HR column (GE Lifesciences). TBP6.7 (Mr of 11.5 kDa) exhibits higher retention than predicted by its Mr, eluting at or >1 CV. The yield is 2–3 mg l−1 of cells. Mutants (Supplementary Table S3) were purified similarly.
Isothermal titration calorimetry
TAR 27-mer (Figure 2C) was produced by chemical synthesis (Dharmacon) and purified by denaturing gel electrophoresis (27). Lyophilized RNA was suspended in 0.01 M Na-HEPES pH 7.5 and heated at 65 °C. After 3 min, ITC buffer (0.05 M Na-HEPES pH 7.5, 0.05 M NaCl, 0.05 M KCl, 0.002 M MgCl2 and 0.002 M 2-mercaptoethanol) at 65 °C was pipetted into the RNA, followed by 2 min at 65 °C. The sample was cooled overnight to room temperature. ITC measurements were conducted using a VP-ITC (MicroCal) (28) with protein in the syringe and RNA in the cell. Each sample was dialyzed at 4 °C overnight against 4 l of ITC buffer. RNA was diluted with dialysis buffer to 8.0–11.4 μM for R49A, 14.4–17.0 μM for R47A and R52A, and 2.0–3.9 μM for titrations with wild-type, other mutants, or 2-aminopurine (2AP)-TAR. Experiments were conducted at 20 °C unless noted. Following co-dialysis with RNA, protein samples were diluted in dialysis buffer to concentrations ∼10-fold higher than RNA. Thermograms were analyzed with Origin 7.0 (MicroCal) using a 1:1 binding model. Average thermodynamic parameters and representative curve fits are provided (Supplementary Table S1 and Supplementary Figure S4).
Figure 2.

Ribbon and schematic diagrams depicting the HIV-1 TAR–TBP6.7 complex of this investigation and parental U1hpII-U1A. (A) Global view of the co-crystal structure depicting the TBP6.7 RRM domain (blue) engaging TAR RNA (purple) in upper helical stem S1b. Arginines of the β2–β3 loop that provide the principal determinants of TAR binding are depicted as ball-and-stick models (orange); similar depictions are provided for conserved RRM amino acids known as RNP2 (Y13) and RNP1 (R52, Q54 and F56). (B) Global view of the structure in A rotated +90°, providing a view looking through the apical loop and down the helical axis. The TBP6.7 β2–β3 loop penetrates deeply into the TAR major groove. (C) Schematic diagram depicting interactions between TBP6.7 and TAR based on the co-crystal structure. Henceforth asterisks (*) indicate lab-evolved TBP6.7 residues depicted in Figure 1B. (D) Close-up of the TAR Uri23•Ade27-Uri38 major-groove base triple and the central bulge that interrupts stems S1a and S1b. Dashed lines joining ball-and-stick models represent putative hydrogen bonds unless noted otherwise. (E) Close-up view of the apical hexaloop and interface with the S1b closing base pair. (F) Global view of the U1hpII-U1A complex (23) oriented and colored as in A. U1A binds U1hpII primarily within the single-stranded region of the upper loop.
Crystallization and X-ray data collection
TAR RNA (prepared as described above) was suspended in 0.01 M Na-HEPES pH 7.5 to a concentration of 0.4 mM and heated at 65 °C. After 3 min, the RNA was diluted 10-fold with folding buffer (0.01 M Na-HEPES pH 7.5, 0.05 M NaCl and 0.002 M MgCl2) and incubated at 65 °C for 2 min. The RNA was cooled overnight to room temperature. TBP6.7 was titrated drop-wise into folded RNA at a 1.2:1 molar ratio (48 μM protein to equal volume of 40 μM RNA) with vortexing. The mixture was incubated at room temperature for 0.5 h and concentrated to 10–12 mg ml−1 based on 280 nm absorption using a Nanosep 3K Omega spin-filter (PALL); the final complex was 0.2 μm filtered (Millex, EMD). Crystals were prepared by vapor diffusion in which an equal volume of well solution (0.05 M Na-cacodylate pH 7.0, 0.1 M NaCl, 0.002 M (NH4)2SO4 and 17% (w/v) of PEG-MME 5K) was added to 1.5 μl of TAR–TBP6.7 complex with equilibration over 1 ml of well solution at 20 °C. Crystals grew within 72 h producing a half-octagon habit that reached 0.12 mm × 0.07 mm × 0.04 mm in 1 week. Cryo-protection was by serial transfer into well solution supplemented with 5–20% (v/v) glycerol followed by snap cooling in N2(l). X-ray data were recorded at the Stanford Synchrotron Radiation Lightsource (Table 1).
Table 1.
X-ray diffraction and refinement statistics
| Data collection a | |
| Space group | P43212 |
| Cell constants | |
| a = b, c (Å) | 40.4, 284.6 |
| α = β = γ (°) | 90.0 |
| Resolution (Å) | 38.90–1.80 |
| (1.83–1.80) | |
| R p.i.m. (%)b | 2.6 (45.1) |
| CC1/2 (%)c | 98.7 (69.2) |
| I/σ(I) | 19.9 (1.8) |
| Complete (%) | 99.4 (91.8) |
| Redundancy | 8.8 (7.9) |
| Refinement | |
| Resolution (Å) | 37.2–1.80 |
| No. reflections | 23 297 |
| R work /R free (%) | 18.9/22.1 |
| No. atoms | |
| Protein | 746 |
| RNA | 572 |
| Solvent | 153 |
| B -factors (Å 2 ) | |
| Protein | 39 |
| RNA | 44 |
| Waters | 47 |
| R.M.S. deviations | |
| Bonds (Å) | 0.005 |
| Angles (°) | 0.759 |
| Clash scored | 0.4 |
| Ramachandran (%) | |
| Allowed | 100.0 |
| Outliers | 0.0 |
| Coord. errore (Å) | 0.21 |
a X-ray data collection (λ = 0.9795) was conducted remotely at beamline 12-2 of the Stanford Synchrotron Radiation Lightsource (SSRL, Menlo Park, CA, USA) using Blu-Ice software and the Stanford Auto-Mounter (50).
b
R
precision-indicating merging R-value = 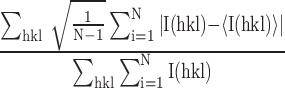 , where N is the redundancy of the data and
, where N is the redundancy of the data and 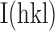 is the average intensity (51). Data were reduced with XDS and AIMLESS (52,53).
is the average intensity (51). Data were reduced with XDS and AIMLESS (52,53).
c The Pearson correlation coefficient calculated for the average intensities resulting from division of the unmerged data into two parts, each containing half of the measurements selected at random for each unique reflection (54).
d Number of unfavorable all-atom steric overlaps ≥0.4 Å per 1000 atoms (55).
e Coordinate error as implemented in PHENIX (29).
Phase determination, refinement and analysis
The structure was determined by molecular replacement in PHENIX (29,30) starting from U1A RRM1 (23) devoid of RNA. The initial TBP6.7 model was generated by Phenix.autobuild (29), although TAR required manual building in Coot (31) with intervening cycles of Phenix.refine (29). This iterative approach converged on Rcryst/Rwork/Rfree values of 19.1%/18.9%/22.1% to 1.80 Å resolution (Table 1). An unbiased electron density map envelops all TAR nucleotides and the TBP6.7 core (Supplementary Figure S1A) indicating the quality of the refined structure. Reduced-bias omit maps demonstrate atomistic features that define placement of R47, R49* and R52 side-chain rotamers and opposing bases (Supplementary Figure S1B–D). These features are representative of the high-quality model that defines the TBP6.7–TAR interface. The accompanying quality indicators (Table 1) provide confidence that the coordinates accurately describe the molecular details of protein-mediated TAR recognition. All cartoons, schematic diagrams and movies derived from coordinates were produced in PYMOL (Schrödinger, LLC). Cα superposition and Sc analysis were performed in CCP4 (32,33).
Molecular dynamics simulations
MD simulations were conducted on the TBP6.7–TAR complex, the TAR-(β2–β3-loop) peptide comprising residues L41 to F59 (Figure 4A), and isolated TAR RNA. The Amber 14 simulation package (34) was used to solvate crystallographic coordinates, or subsets thereof, in a box of OPC water (35) with 150 mM KCl. Starting coordinates were energy minimized using 500 steps each of steepest descent and conjugate gradient minimization with 25 kcal mol−1 Å−2 positional restraints on solute atoms. Then, 10 cycles of alternating between minimization with decreasing positional restraints on the solute atoms and NVT dynamics were performed. After 250 ns of NPT equilibration, production dynamics simulations were performed using the AMBER ff14SB (36–38) force field in the NPT ensemble with periodic boundary conditions, a time step of 2 fs, and a direct space cutoff of 10.0 Å for nonbonded interactions. Bond lengths for covalent bonds involving hydrogen were constrained using the RATTLE algorithm (39). Temperature was maintained at 300 K using a Langevin thermostat with a collision frequency of 1 ps−1, and pressure was maintained at 1 atm using a Monte Carlo barostat. Simulations were performed on Nvidia Tesla K20X GPU cards. Six trajectories each of TAR–TBP6.7, TAR–(β2–β3-loop) peptide, and free TAR were each run for 4 μs for an aggregate time of 72 μs. For simulations of TAR–(β2–β3-loop) peptide, the distance between the terminal carbon atoms of the β2–β3 loop was restrained using a harmonic restraint with a force constant of 250 kcal mol−1 Å−2 (roughly the strength of a covalent bond) to mimic peptide cyclization. Analysis of simulation interactions was performed using custom tools developed using the LOOS software (40).
Figure 4.

Cartoon depiction of the β2–β3-loop peptide supersecondary structure and retention of TAR binding by β2–β3-loop peptides outside the context of TBP6.7. (A) Diagram illustrating hydrogen bonds within the close-packed β-strand core (transparent surface and yellow amino acids) and loop (orange amino acids) of the TAR–TBP6.7 complex. Key TAR-binding arginines are shown, as well as wild-type and evolved amino acids that contribute peptide stability. (B) Fluorescence activated cell-sorting analysis of TAR binding. (Upper) A fusion protein expressed on the E. coli surface harbors the TBP6.7 β2–β3-loop peptide from A. (Lower) Display and TAR binding control using only E. coli. (C) ELISA analysis of TAR binding by SUMO (control) and a SUMO-β2–β3-loop fusion protein harboring the sequence from A; off target (CUG)10 RNA is used as a binding control. (D) Schematic drawing of conformationally-constrained peptide1 derived from the sequence in A, except cysteine termini were added (red) for conjugation to polyfluorinated biphenyl. (E) Titration of peptide1 into 2-aminopurine-labeled TAR produces changes in fluorescence emission at 390 nm. Filled circles represent the experimental data resulting from three independent measurements. The smooth curve shows the fit of a one-site-binding model, which gave the apparent KD with standard deviation shown. (F) Graphed densitometry data of HIV-1 TAR–Tat-dependent transcription in HeLa nuclear lysate and inhibition by peptide1s or TBP6.7. P values from t tests are: 100 μM, P = 0.040; 20 μM, P = 0.028; 2 μM, P = 0.055 and 10 μM TBP6.7, P = 0.013. *P < 0.05 is significant and P > 0.05 is not significant. (G) Representative densitometry signals for TAR–Tat-dependent transcription in HeLa nuclear extract, and suppression of transcription by peptide1s or TBP6.7. For C and F, data are plotted as the mean of three separate experiments with corresponding standard errors of the mean. An intact gel used in F and G is provided (Supplementary Figure S9).
Bacterial display and flow cytometry
The β2–β3-loop peptide sequence of TBP6.7 (Figure 4A) was cloned into the pB33eCPX construct (41) (AddGene) using restriction enzymes NdeI and XhoI (NEB), downstream of an in-frame myc tag and transformed into 5-alpha competent E. coli cells (NEB). The eCPX-β2–β3-loop plasmid DNA was purified by miniprep (Omega) and ∼200 ng were used to electroporate E. coli MC1061 F− cells (Lucigen) in 1 mm electroporation cuvettes (Fisher). Cells were grown in 50 ml LB (Fisher) containing 12.5 μg ml−1 chloramphenicol (GoldBio Technology) at 37 °C to an OD600 of 0.5 and induced overnight with 0.1% arabinose at 25 °C. ∼5 × 108 cells were pelleted (7300 × g) for 5 min at 4 °C, then washed with ice-cold CellGro PBS 1× (Corning). Cells were incubated with 100 nM Cy5-labeled TAR RNA (IDT) (annealed by heating at 95 °C for 2 min, followed by plunging into ice) and 1:1000-fold diluted FITC-conjugated anti-cMyc antibody (Abcam) with rotating at 4 °C in 1 ml PBS for 1 h. Cells were pelleted and washed once with ice-cold PBS. RNA-binding (Cy5 fluorescence) and display (FITC fluorescence) were measured using a CyAn ADP flow cytometer (Beckman-Coulter). All flow data were analyzed and plotted using FlowJo 10.3.
Preparation of TBP6.7, SUMO and SUMO-β2–β3 for ELISA or transcription
Protein sequences are provided in Supplementary Table S2. Plasmids containing appropriate DNA sequences (Supplementary Table S3) were sub-cloned into a pET plasmid and transformed into E. coli BL21(DE3) (NEB). Cells were grown in 0.5 l cultures of LB (Fisher) containing 100 μg ml−1 carbenicillin (GoldBio Technology) to an OD600 of ∼0.6 and induced with 1 mM IPTG (Thermo Scientific) at 25 °C for 4–12 h. Cells were harvested by centrifugation (5000 × g, 10 min), resuspended in Buffer 1 (10 mM HEPES pH 7.4, 50 mM KCl, 30 mM NaCl, 1 mM MgCl2 and 1 mM EDTA) prepared with cOmplete ULTRA Protease Inhibitor Tablets (Roche) and stored at −20 °C. For lysis, frozen cell suspensions were thawed and sonicated for 2 min. The lysate was cleared by centrifugation (9000 × g, 20 min) and the supernatant was mixed with 0.75 ml of Ni-NTA agarose (Fisher) for 10 min. The resin was sedimented by low-speed centrifugation for 5 min. Resin was washed with 30 ml of Buffer 1 containing 0.02 M imidazole, followed by a 10 ml wash with Buffer 1 containing 0.05 M imidazole. Proteins were eluted using 2 ml of Buffer 1 containing 0.4 M imidazole. Eluted protein was dialyzed in 10K MWCO dialysis tubing (Thermo Scientific) against 2 l of Buffer 1, and then against 2 l of PBS (20 mM phosphate pH 7.4 and 0.15 M NaCl). Purified proteins were quantified by absorbance at 280 nm using the calculated extinction coefficient. SUMO and the SUMO-β2–β3 fusion protein were prepared in an identical manner except that Buffer 1 was replaced with PBS.
ELISA
ELISA was performed using clear, 5 picomole well−1 streptavidin-coated 96-well plates (Pierce). The plate was pre-incubated for 1 h with wash buffer (20 mM phosphate pH 7.4, 150 mM NaCl, 0.05% Tween-20 and 0.1 mg ml−1 BSA). During pre-incubation 100 μl of TAR (5′-GGC AGA UCU GAG CCU GGG AGC UCU CUG CC-3′) or CUG10 (5′-CCG CUG CUG CUG CUG CUG CUG CUG CUG CUG CUG GGC-3′) RNA modified with a 5′-biotin (IDT) was incubated in 100 μl of buffer with 1 μM of either SUMO-β2–β3 loop or SUMO (Supplementary Table S2) for 1 h, rotating at 4 °C. The pre-incubation buffer was removed from the ELISA plate and the RNA–protein mixture was incubated on the plate for 2 h. Wells were then washed 3× with 200 μl of wash buffer with shaking for 5 min. Next, a 1:10 000 dilution of HRP-conjugated anti-FLAG antibody (Abcam, ab2493) was made with Odyssey Blocking Buffer (Li-Cor) and 100 μl was incubated in each well for 30 min at 25 °C; each well was then washed 4×. Colorimetry was developed for 20 min using 100 μl of TMB-One substrate (Promega). Absorbance was measured at 655 nm on a plate reader. ELISA experiments were repeated in triplicate.
General reagent information for synthesis of constrained peptide 1
1-[Bis(dimethylamino)-methylene]-1H-1,2,3-triazolo[4,5-b] pyridinium 3-oxide hexafluorophosphate (HATU) and Fmoc-l-amino acids were purchased from Chem-Impex International (Wood Dale, IL). H-Rink Amide ChemMatrix resin was obtained from PCAS BioMatrix Inc. (Quebec, Canada). Peptide synthesis-grade N,N′-dimethylformamide (DMF), dichloromethane (CH2Cl2), diethyl ether and HPLC-grade acetonitrile were obtained from VWR International (Philadelphia, PA). Decafluorobiphenyl was from Oakwood Chemicals (West Columbia, SC).
Synthesis of constrained peptide 1 and 1s
Peptide 1 (NH2-CLDILVPRQRTPRGQAFVIC-CONH2) and peptide1s (NH2-CVPRQRTPRGQAC-CONH2) containing two free cysteines (Figure 4D and Supplementary Figure S7) were each synthesized on a 0.1 mmol scale on H-Rink Amide ChemMatrix resin. Solid-phase peptide synthesis was carried out on a synthesizer for automated flow peptide synthesis (42). After completion, the resin was washed thoroughly with CH2Cl2 and dried under vacuum. The resin was transferred to a 50-ml plastic tube and the peptide was cleaved simultaneously from the resin while the side-chain was deprotected by treatment with 2.5% (v/v) water, 2.5% (v/v) 1,2-ethanedithiol and 1% (v/v) triisopropylsilane in neat trifluoroacetic acid (TFA) for 2 h at room temperature. The resulting peptide-containing solution was triturated and washed 2× with cold diethyl ether (pre-chilled at −80 °C). A gummy-like solid was dissolved in 50% H2O:50% acetonitrile containing 0.1% TFA and lyophilized to yield the crude peptide. The peptide was reacted with decafluorobiphenyl in DMF for macrocyclization (43,44). The reaction mixture in DMF was quenched by water containing 0.5% TFA for 1:10 dilution, filtered and then purified by Reverse Phase HPLC (RP-HPLC). The solvent compositions for RP-HPLC purification were water with 0.1% TFA (solvent A) and acetonitrile with 0.1% TFA (solvent B). The diluted crude mixture was injected directly into an Agilent 1260 Infinity Automated LC/MS Purification System with a semi-preparative Agilent Zorbax 300SB C3 Reverse Phase-HPLC column (21.2 mm × 250 mm, 7 μm) operated with a linear gradient of 5−65% B over 82 min at a 4 ml min−1 flow rate. Fraction purity was assessed by LC–MS. Fractions containing pure, cyclized peptide were combined and lyophilized.
LC–MS analysis of constrained peptides
LC–MS chromatograms and associated mass spectra were acquired using an Agilent 6520 ESI-Q-TOF mass spectrometer. Mobile phases used for LC–MS analysis were: solvent C (0.1% formic acid in water) and solvent D (0.1% formic acid in acetonitrile). LC utilized a Zorbax 300SB C3 column (2.1 mm × 150 mm, 5 μm) with a column temperature set at 40°C and a flow rate of 0.8 ml min−1. The gradient was: 0–2 min 5% D; 2–14 min 5–95% D; and 14–15 min 95% D. MS conditions were: positive electrospray ionization (ESI) extended dynamic mode in mass range 300–3000 m/z; temperature of drying gas equals 350°C; flow rate of drying gas equals 11 l min−1; pressure of nebulizer gas equals 60 psi; the capillary, fragmentor, and octupole rf voltages were set at 4000, 175 and 750 V. LC–MS characterization of each peptide product is shown in Supplementary Figure S7.
Fluorescence emission analysis of TAR binding to peptide 1
Fluorescence measurements were conducted at 24 °C by titrating concentrated peptide in FL buffer (0.050 M Na-HEPES pH 7.5, 0.050 M NaCl, 0.050 M KCl and 0.002 M MgCl2) into 500 μl 100 nM TAR RNA 31-mer labeled with 2-aminopurine (2AP) at position 24 [5′-CGG CAG AU(2AP) UGA GCC UGG GAG CUC UCU GCC G-3′]. The 2AP-RNA was purified by denaturing PAGE and folded as described (above). The excitation wavelength for 2AP was 320 nm and changes in emission were recorded at 390 nm as described (45) using a Fluoromax-3 fluorometer (Horiba Scientific). Data were fit to a one-site binding model, as described for TBP6.7 binding to (2AP)-TAR (Supplementary Figure S8).
Transcription assay
A previously described transcription assay (46–48) was used to evaluate peptide1s or TBP6.7 for functional suppression of TAR–Tat-dependent transcription of HIV-1 genomic DNA. A DNA fragment (-477 to +568) comprising the HIV 5′-LTR was PCR amplified from plasmid pLAI-BS (courtesy of Gerald Joyce, Scripps) (forward primer: TCTAGAACTAGTGGATCTG; reverse primer: GCTACAACCATCCCTTCAGAC). In vitro transcription was performed in a 30 μl reaction containing 18 μl of HeLa nuclear lysate (Promega) in 20 mM HEPES, 80 mM KCl, 3 mM MgCl2, 2 mM DTT, 10 μM ZnCl2, 10 mM creatine phosphate (Sigma-Aldrich), 1 μg creatine kinase (Sigma-Aldrich), 250 μM each of GTP, ATP and CTP, 50 μM UTP (Thermo), 15 U rRNasin (Promega), and 10 μCi [α-32P]UTP (PerkinElmer). Assays were conducted in the absence or presence of various peptide1s concentrations or 10 μM TBP6.7 (Figure 4F, G). Recombinant HIV-I Tat (ProSpec) was added to initiate transcription; TBP6.7 (Supplementary Table S2) was prepared as described (above). The reactions were incubated for 1 h at 37 °C, and quenched by addition of 370 μl HSCB buffer (25 mM Tris–HCl pH 7.5, 400 mM NaCl and 0.1% SDS) containing 60 μg glycogen and a 120-base radiolabeled RNA was added as a loading control. Proteins were extracted using phenol/chloroform/isoamyl alcohol, and nucleic acids were ethanol precipitated. Nucleic acids were resuspended in RNA loading dye, and separated via denaturing gel electrophoresis. Gels were exposed to a phosphor screen and visualized with a Typhoon imager (GE Lifesciences).
Statistical analysis
Unpaired, two-tailed t tests were performed with a Welch correction on data obtained from three separate transcription assays comparing untreated to inhibitor-treated conditions (Figure 4F). The analysis was performed using Prism (GraphPad Software). The t values were: 4.82 (2) for 100 μM peptide1, 5.83 (2) for 10 μM peptide1, 4.07 (2) for 2 μM peptide1; and 8.55 (2) for 10 μM TBP6.7. Parenthetical values indicate degrees of freedom.
Sequence conservation analysis of HIV-1 TAR RNA
All complete HIV-1 genome sequences available at the time of writing were retrieved from http://www.hiv.lanl.gov/ using the Web Alignments option. This produced 406 dissimilar sequences representative of common viral groups, sub-groups, and circulating recombinant forms (CRFs). These sequences were aligned in the 5′ LTR from 471 to 497 and used as input for web-logo (49). Conservation was rendered in PYMOL (Schrödinger, LLC) as a heat map on a cartoon of the TAR–TBP6.7 co-crystal structure by reassignment of the PDB temperature factors according to the web-logo probability multiplied by 100. The resulting backbone provided a reference to exactly match the web-logo graphic colors using Adobe Illustrator (CS5.1). Highest conservation is depicted in dark blue and lowest is red (Figure 5A). The analysis should be interpreted such that invariant Gua26, Gua28 and Gua36 are recognized by arginines in the β2–β3 loop. Uri23 engages in a long-range base triple interaction (Uri23•Ade27-U38) that is also invariant. Cyt24 is conserved poorly (yellow), and is deleted (Δ) in ∼5% of sequences, such as the A1 CRF. Uri25 is also poorly conserved (red) with ∼9% deletion in various CRFs (e.g. A1, AB, AE and AU).
Figure 5.

HIV-1 TAR conservation and comparison of the TAR–TBP6.7 complex to a known antiviral cyclic peptide. (A) (Upper) Sequence conservation for representative circulating forms of HIV-1 depicted as a web-logo diagram. Blue represents greatest conservation and red indicates poor conservation. (Lower) Cartoon diagram of the TAR–TBP6.7 complex with web-logo sequence conservation heat mapped onto the RNA. (B) The TAR–L-22 complex (PDB entry 2kdq) (17) reveals interactions distributed throughout the RNA-peptide interface, which yields a shape-complementarity score (Sc) of 0.60, indicating a high degree of interlocking surface. (C) The TAR–TBP6.7 complex shows a clustered trio of arginines that recognizes three highly conserved guanosine nucleotides by hydrogen bond, salt-bridge and cation-π contacts (Figure 3 and Supplementary Figure S5). Here, the Sc score is 0.79 for the co-crystal structure and 0.76 for the isolated β2–β3 loop peptide shown. These scores are comparable to the nearly ideal Sc score of 0.84 measured for the U1A–hpII interface. The Uri23•Ade27-U38 triple is intact in the presence of a canonical Cyt30–Gua34 pair located in the apical loop.
RESULTS
Structural analysis of the HIV TAR–TBP6.7 complex
To define the molecular details by which TBP6.7 recognizes TAR, we determined the co-crystal structure (Table 1, Supplementary Figure S1A-D, and Materials and Methods). In complex with TBP6.7, TAR exhibits several architectural features consistent with solution studies of small ligands bound to the RNA. Hallmarks include stems S1a and S1b interrupted by the major-groove Uri23•Ade27-Uri38 triplex, flanked by a bulge that extrudes Cyt24 and Uri25 from its core (Figure 2A–D and Supplementary Movie S1). These characteristics are consistent with NMR analyses (17,18) and persist on a μs timescale in our MD simulations (Supplementary Figure S2A and Supplementary Movie S2). Conversely, MD simulations conducted on apo-state TAR showed rapid dissolution of the triple (Supplementary Figure S2A and Supplementary Movie S3) concurring with ligand-free NMR analyses (56–60). Another hallmark of TAR is that the apical hexaloop interconverts between minor and major conformations (61). In the latter, Uri31, Gua32 and Ade35 are flexible with adenine extruded (61–63). This is again mostly consistent with our co-crystal structure wherein Ade35 projects away from the hexaloop, whereas Gua32 and Uri31 stack on Cyt30 (Figure 2A–C and E and Supplementary Movie S1). Although unrepresented in solution ensembles of TAR-peptide complexes (17,18), the TAR–TBP6.7 co-crystal structure exhibits a canonical cross-loop Cyt30-Gua34 pair (Figure 2C, E) supported by chemical modification experiments, NMR assignments, sequence conservation, and cyclin-T1 binding requirements (61,64–68). MD simulations indicate that Cyt30–Gua34 pairing is stable (Supplementary Figure S2B), although transient dissolution and spontaneous reformation are seen for the TBP6.7-bound and apo states. Nonetheless, the interaction appears to be a stable feature of the RNA conformational landscape (Supplementary Movie S2). In one trajectory, Ade35 makes an excursion into the apical loop to displace Gua34 and interact with Cyt30 (Supplementary Figure S2B, right, purple lines of trajectory four), agreeing with a low population state observed by NMR (61). A likely site of conformational variation is extruded base Gua33, which forms a crystal contact with Cyt24 of the bulged loop from a neighboring molecule (Supplementary Figure S1E, F). Neither base stacks appreciably inside the apical loop or bulged loop core on the timescale of MD simulations (Supplementary Movies S2 and S3) and this contact does not influence TBP6.7 binding.
Comparison of the TBP6.7 fold to that of U1A reveals that the evolved protein adopts the same mixed α/β architecture as parental RRM1 (Figure 2A,F). A Cα superposition produced a modest rmsd of 1.1 Å, but local conformational differences are apparent. The greatest variations include the β2–β3 loop (46–51, rmsd 3.9 Å) and the C-terminus (91–95, rmsd 3.6 Å), which were each subjected to saturation mutagenesis to achieve TAR binding (21). When oriented similarly it is evident that TBP6.7 and U1A engage their RNA targets in extraordinarily different ways (Figure 2A,F). Whereas TBP6.7 binds TAR in the S1b duplex, U1A recognizes the distinctly single-stranded loop of U1hpII between Ade66 and Cyt72 (23). Despite fundamentally different modes of engagement, TBP6.7 buries 1555 Å2 in its protein-RNA interface, which is only 278 Å2 less than the U1A–hpII complex. Importantly, the co-crystal structure reveals that numerous contacts to TAR originate in the β2–β3 loop (Figure 2A–C), which yielded a clear consensus during selection that departs from U1A (Figure 1B). Unexpectedly, the evolved C-terminus of TBP6.7 is devoid of TAR contacts, implying that the minimal lab-evolved β2–β3 loop is operative in the new mode of RNA binding.
TBP6.7 uses a single-stranded RNA recognition motif to recognize double-stranded RNA
Because TBP6.7 maintains the classical RRM fold, we asked if it uses the conserved RNP motifs to bind double-stranded S1b of TAR, since these amino acids were unaltered in our approach (21). This point is especially significant because RNP residues function classically in single-stranded RNA recognition (69). In the U1A–U1hpII complex, RNA bases stack upon aromatic RNP side chains to provide affinity and recognition (23,70–73); Y13 of RNP2 and F56 of RNP1 stack on bases Cyt70 and Ade71 (Figure 2F and Supplementary Figure S3A). In contrast, Y13 of TBP6.7 stacks on Ade35, but F56 does not engage TAR due to a lack of bulged bases flanking S1b (Figure 2A and Supplementary Figure S3B). Conversely, the Q54 amide of U1A RNP1 approaches the 2′-OH of Gua69 in U1hpII without interacting, whereas Q54 Nδ of TBP6.7 hydrogen bonds to the 2′-OH of Gua34 in TAR (Supplementary Figure S3A,B), consistent with its RNA readout role in other RRMs (69). Finally, R52 of RNP1 recognizes the Hoogsteen edge of loop-closing pair Gua76-Cyt65 in U1hpII, as well as Gua36 in TAR (Supplementary Figure S3C,D). The former interaction is the only instance of arginine-mediated base readout by U1A, although its simultaneous recognition of Ade66 N1 yields a non-optimal, inclined guanidinium-guanine interaction. A key finding is that TBP6.7 still utilizes a subset of RNP amino acids to bind TAR, but affinity and specificity appear to arise primarily from the lab-evolved β2–β3 loop, distinguishing it from U1A and other RRMs (69).
A trio of arginines in the lab-evolved β2–β3 loop reads the TAR major groove
Thermodynamic analysis of the TAR–TBP6.7 complex reveals that binding is enthalpy-driven (ΔH of −25 ± 0.2 kcal mol−1) with an unfavorable entropy (−TΔS of 13.5 ± 0.2 kcal mol−1) that yields a KD,App of 2.5 ± 0.1 nM (Supplementary Table S1 and Supplementary Figure S4A). Analysis of the co-crystal structure suggested that binding interactions can be parsed into four groups: (i) arginines in the β2–β3 loop that read guanine to impart specificity; (ii) β2–β3-loop residues that interact with phosphate or 2′-OH groups; (iii) evolved protein–protein interactions that stabilize the β2–β3 loop; and (iv) interactions outside the β2–β3 loop. To test the energetic contributions of each, we prepared TBP6.7 point mutants and evaluated them for TAR binding.
Of the arginines in the β2–β3 loop (Figure 1B), R52 makes the fewest TAR contacts, making it straightforward to evaluate its binding contributions. Its guanidinium moiety donates hydrogen bonds from NH1 and NH2 to atoms N7 and O6 of Gua36 (Figure 3A and Supplementary Movie S4), while forming a cation–π interaction with Gua34 of the apical loop (Supplementary Figure S5A). Accordingly, the R52A mutation reduced binding by a factor of 116 (ΔΔG° of +2.8 kcal mol−1) (Supplementary Table S1 and Supplementary Figure S4B). R49* is the only arginine in the β2–β3 loop that resulted from yeast display (Figure 1B). This side-chain makes an equal number of contacts to TAR compared to R52, but the modes of interaction are different. The guanidinium group not only makes a hydrogen bond that recognizes N7 of Gua28, but also forms a salt-bridge to the nucleotide's pro-Rp oxygen while engaging in a cation–π contact to Ade27 (Figure 3B, Supplementary Figure S5B and Supplementary Movie S4). Accordingly, R49A* yielded a larger ΔΔG° of +3.2 kcal mol−1, corresponding to a loss in binding by a factor of 233 (Supplementary Table S1 and Supplementary Figure S4C).
Figure 3.

Close-up views of key interactions between the evolved β2–β3 hairpin loop of TBP6.7 and HIV-1 TAR based on the co-crystal structure. ΔΔG° values from ITC analysis of R-to-A mutations are taken from Supplementary Table S1. (A) R52 forms two hydrogen bonds to the Hoogsteen edge of Gua36; for clarity, some evolved amino acids in the β2–β3 loop are omitted. (B) R49* forms a hydrogen bond with N7 of Gua28 and a salt-bridge to its non-bridging phosphate oxygen. (C) R47 forms two hydrogen bonds with the Hoogsteen edge of Gua26, as well as hydrogen bond and salt-bridge interactions to the Uri23 phosphate. Cation–π interactions and buried surface areas for each arginine are described in Supplementary Figure S5.
Although R47 is present in the U1A sequence (Figure 1B), it does not contact U1hpII RNA (23). In contrast, R47 of TBP6.7 makes the most extensive number of contacts with TAR forming an ‘arginine fork’ (74) wherein NH1 and NH2 hydrogen bond to O6 and N7 of Gua26, while Nϵ and NH2 hydrogen bond and salt-bridge to Uri23 O5′ and its pro-Rp oxygen (Figure 3C). The R47 guanidinium is sandwiched simultaneously between bases from Ade22 and Uri23 to form cation-π stacks (Supplementary Figure S5C,D). As anticipated, R47A produced a large ΔΔG° of ∼+3.8 kcal mol−1 corresponding to a loss in binding by a factor >600 (Supplementary Table S1 and Supplementary Figure S4D). The magnitude of this loss makes it tenuous to relate specific energetic contributions to the structure. An estimated 324 Å2 of buried area is ablated by this mutation—nearly double that of R52A (Supplementary Figure S5). For a more conservative change, we examined R47K, which gave a ΔΔG° of +3.4 kcal mol−1 corresponding to factor of 327 in lost binding (Supplementary Table S1 and Supplementary Figure S4E). K47 could theoretically preserve salt bridge formation between its Nϵ and the Uri23 phosphate, as well as cation-π stacking, but hydrogen bonding to Gua26 and O5′ of U23 seem unlikely. From this analysis it is clear that R47 is of paramount importance for TAR binding, and that the positive charge of lysine is insufficient to attain optimal readout.
Our collective mutagenesis results support the crystallographic observations, revealing three tiers of TAR recognition corresponding to explicit modes of arginine readout with distinct free-energy profiles. MD simulations of the TAR–TBP6.7 complex support the dynamics of the observed arginine–TAR interactions with higher maintenance of binding occupancy in more solvent-excluded regions (Supplementary Figure S6A–C). The simulations not only illustrate the feasibility of interactions to TAR in the context of full-length TBP6.7, but also in the context of the minimal β2–β3-loop peptide. An analysis of the other classes of interactions (ii) through (iv) demonstrated the roles of other evolved β2–β3 loop residues in TAR recognition, their maintenance of a loop conformation productive for RNA binding, and the dispensability of the lab-evolved C-terminus for TAR readout. The co-crystal structure also provides a strong rationale for the binding affinities of various TAR mutants that were generated previously by our lab to probe sites of TBP6.7 interaction with the RNA. These analyses are provided in the Supplementary Results.
Short β2–β3-loop peptides retain TAR binding as fusion proteins
The observation that a single, short peptide comprising a lab-evolved loop is sufficient for TAR recognition (Figure 2A–C and Supplementary Movie S4) has implications for the development of a minimal RNA binding module. To further test this possibility, we fused β2–β3-loop residues L41-F59 (Figure 4A) inside the eCPX protein. The eCPX-β2–β3-loop-cMyc tagged protein was displayed on the surface of E. coli, followed by incubation with Cy5-labeled TAR and a FITC-conjugated anti-cMyc antibody (to measure display efficiency). Upon washing to remove unbound antibody and RNA, we observed a distinct population of bacteria by cell sorting that binds both the FITC-labeled antibody (display) and Cy5-labeled RNA (TAR binding) (Figure 4B, upper); in contrast, a ‘no display’ control is devoid of this population (Figure 4B, lower). We next prepared a display protein comprising β2–β3-loop residues L41-F59 fused to the C-terminus of SUMO to facilitate overexpression in E. coli. The ability of the SUMO fusion to bind TAR or an off-target (CUG)10 (i.e. a disease-relevant, guanine-rich RNA hairpin with bulged U) was measured by ELISA. As expected, SUMO alone shows comparatively low levels of binding. In contrast, the SUMO fusion binds TAR, but has less affinity for (CUG)10 (Figure 4C). The results collectively demonstrate that the lab-evolved β2–β3 loop retains TAR binding outside the context of TBP6.7 when presented as a fusion protein.
Constrained peptides comprising the lab-evolved β2–β3 loop bind TAR and suppress Tat-dependent transcription
To test whether the β2–β3 loop serves as a standalone TAR recognition module, we synthesized a short, covalently-constrained peptide (i.e. peptide1) comprising only the lab-evolved loop sequence flanked by strands β2 and β3, as observed in the co-crystal structure (Figure 4A, D and Supplementary Figure S7A). Because ITC can require large quantities of material, we used a sensitive fluorescence emission assay (45). Our results indicate that peptide1 binds 2-aminopurine labeled (2AP)-TAR with a KD,App of 1.8 ± 0.5 μM (Figure 4E). Control experiments in which TBP6.7 binding to (2AP)-TAR was measured by fluorescence and ITC revealed close agreement of KD,App values, demonstrating methodological consistency (Supplementary Figure S8). However, a 3.2-fold loss in affinity was observed for TBP6.7 recognition of (2AP)-TAR relative to the wild-type RNA (Supplementary Table S1 and Supplementary Figures S4A and S8B, C), suggesting that peptide1 binding to TAR could be tighter than indicated by our fluorescence measurements. Our findings demonstrate that peptide1 is a standalone RNA binding motif that recognizes TAR outside the context of TBP6.7.
We then tested the ability of a shorter peptide1 variant (i.e. peptide1s of Supplementary Figure S7B) to target TAR using a known functional assay. Here efficient transcription from the HIV-1 5′-LTR requires an unfettered TAR–Tat interaction. Assays were conducted in HeLa nuclear extract to provide the endogenous transcription machinery. Exogenous Tat was required for efficacious production of the ∼500 base transcript. Reactions lacking plasmid template and Tat, or without Tat, generated low levels of product (Figure 4F, lanes I and II). In contrast, reactions containing template and exogenous Tat generated comparatively high levels of transcription product (Figure 4F, lane III). When template, exogenous Tat, and various concentrations of peptide1s (100, 20 or 2 μM) were added, we observed concentration-dependent decreases in transcript production (Figure 4F, lanes IV–VI). Statistically significant reduction occurred at 100 and 20 μM concentrations. In three separate experiments, addition of 100, 20 or 2 μM of peptide1s resulted in approximately 70%, 65% or 40% suppression of transcription product (Figure 4G, lanes 4–6 and Supplementary Figure S9). Consistent with our previous findings (21), 10 μM TBP6.7 inhibits TAR–Tat-dependent transcription (Figure 4F, lane VII and Figure 4G, lane 7). The results imply that peptide1s mimics the β2–β3 loop of TBP6.7 and serves as a minimal TAR recognition peptide capable of restricting an essential viral activity.
TAR conservation and comparison of β2–β3-loop interactions to an antiviral peptide
We next asked whether the β2–β3 loop of TBP6.7 targets conserved regions of TAR. Overall the S1b stem reveals high conservation whereas S1a and the apical loop exhibit comparatively greater variation (Figure 5A). In contrast, the central bulge is conserved poorly, consistent with the extrusion of bases 24 and 25 into solvent (Figure 5A, lower). The conservation map further reveals that each guanine base recognized by the evolved β2–β3 loop is invariant (Figure 5A, upper). As such, peptides harboring the β2–β3 loop have the potential to target a broad viral population. In this context, we then asked how TAR–TBP6.7 recognition compares to the well known L-22 antiviral peptide, which shows high affinity for TAR (KD,App ∼30 nM) and was developed using structure-based design (17,75), as compared to lab-evolution used here.
L-22 is a highly basic, cyclic peptide comprising a β-hairpin that fully traverses the TAR major groove, where it is buried partly by apical-loop bases Gua34 and Ade35 while abutting Cyt24 of the bulged loop (Figure 5B). Specificity appears to be driven by hydrogen bonding and electrostatic interactions. R3 reads N7 of Ade22, R5 reads the Gua28 Hoogsteen edge, and R8 interacts with O2 of Cyt30 and N7 of Gua34. R1, R3, K6 and R9 form salt bridges to the backbone, and the R5 and R11 guanidinium groups form cation-π stacks with Ade27 and Ade35 (Figure 5B). In these respects, L-22 runs a gamut of interactions with conserved and non-conserved nucleotides. In contrast, TBP6.7 recognizes only a single apical loop base (Ade35) while avoiding the bulged loop in favor of conserved nucleobases (Figure 5C). The finding that TBP6.7 clusters all three of its β2–β3 loop arginines (i.e. R47, R49* and R52) to recognize S1b guanines at positions 26, 28 and 36 is distinctive compared to L-22; the latter peptide displays a more distributed set of interactions (Figure 5B, C).
Interestingly, recent evidence points to a TAR–L-22 conformation that demonstrates the feasibility of ‘clustered’ arginine recognition by this cyclic peptide. Metadynamics simulations revealed R3, R5 and R8 poised to recognize Gua26, Gua28 and Gua34 in the TAR–L-22 complex (64). Remarkably, this conformer was detected in only 7% of the population wherein TAR exhibited an apo-like fold. Moreover, Cyt24 changed position with Uri25–the least conserved base–to yield ‘non-native’ backbone and R5 contacts integral to L-22 recognition (64) (Supplementary Figure S10). In this respect, the TAR–TBP6.7 complex demonstrates that major-groove peptide recognition by a close-knit cluster of arginines is compatible with formation of the central A23•A27–U38 triple, as well as a canonical Cyt30–Gua34 pair in the apical loop that heretofore was absent in known TAR–peptide complexes. This comparison also highlights how two high-affinity RNA-binding peptides employ diverse modes of readout to recognize the same target. This finding parallels natural adaptations used by diverse gene-regulatory RNAs to detect a common small-molecule effector (76,77).
DISCUSSION
Identifying sequence-selective molecules that target disease-relevant RNAs remains a daunting and significant challenge (75,78–81). HIV-1 TAR continues to be the focus of drug discovery efforts due to its high degree of sequence conservation and critical roles in the viral lifecycle. To identify and explore innovative methods to target TAR, we pursued a fundamentally different ‘semi design’ approach that relied on lab evolution of putative RNA-binding regions starting with naturally occurring RRM1 of the U1A spliceosomal protein. Although our efforts led to the discovery of potent new TAR-Binding Proteins (TBPs) (21), details of molecular recognition were unknown, thus limiting our ability to fully exploit this breakthrough and the associated methodology. Because separate sequences in the U1A β2–β3 loop and C-terminus underwent saturation mutagenesis (Figure 1B), we recognized the potential of these regions to function alone or synergistically in RNA recognition. Moreover, it was unclear if existing RNP amino acids were involved in TAR binding and whether such binding was restricted to single-stranded RNA regions—the RRM rule rather than the exception (82).
To directly assess the underlying mode of HIV-1 TAR molecular recognition by TBP6.7, we determined the co-crystal structure of the RNA-protein complex at 1.80 Å resolution, which represents the first high-resolution crystal structure of intact TAR. Prior work on apo TAR deleted the apical loop or utilized complexes that restricted the resolution to 5.9 Å (83,84). Our structure and experimental analyses complement this work and reveal three new findings: (i) a lab-evolved β2–β3-loop is sufficient to bind TAR outside the context of TBP6.7 when displayed on bacteria, fused to SUMO or as a conformationally constrained peptide; (ii) the lab-evolved RRM recognizes the TAR major groove and (iii) the mode of TAR–TBP6.7 readout differs from a well known class of antiviral cyclic peptides. These outcomes have broader ramifications that strengthen our understanding of RNA-peptide recognition, the methods used to identify such complexes, and the theoretical diversity of RNA-RRM interactions in biology.
HIV functional cure efforts are focused on eradication of latent viral reservoirs (20,24). In this respect, the TAR–TBP6.7 complex represents a positive step toward understanding the detailed molecular interactions required to target a key viral RNA with the specificity and affinity needed to suppress proviral DNA transcription. Indeed, mutation of each β2–β3-loop arginine reduced TBP6.7 affinity for TAR by two orders of magnitude confirming that the guanidinium groups are significant determinants of affinity. Moreover, peptides comprising the β2–β3-loop resist binding to off-target (CUG)10 hairpin RNA when fused to SUMO, and retain binding to TAR as fusion proteins or as constrained peptides (Figure 4B,C and E). An extraordinary finding is that peptide1 binds TAR with a KD,App of 1.8 ± 0.5 μM (Figure 4D, E), which could be a three-fold underestimate based on control experiments (Supplementary Figure S8 and Supplementary Table S1). This suggests between 200- and 700-fold loss in peptide1 binding to TAR compared to TBP6.7. This reduction is reasonable considering that the β2–β3 loop contributes slightly less than two-thirds of the total buried area in the TAR–TBP6.7 interface. Perhaps more significantly, MD simulations of the constrained TAR–(β2–β3-loop) peptide complex revealed higher flexibility and lower occupancy of amino acids at the interaction interface compared to intact TAR–TBP6.7 (Supplementary Figure S6). TBP6.7 maintained a tightly packed protein core that did not persist in MD simulations of the β2–β3-loop peptide, despite the addition of a harmonic restraint representative of the perfluoroaryl linkage. Likewise, the observation that peptide1s required 20–100 μM to significantly inhibit transcription is consistent with its truncated β-strand core and the requirement of TAR binding in a complex solution of nuclear lysate (Figure 4F and G). These observations illustrate the importance of a stable peptide core.
Future efforts to improve peptide1 binding to TAR could entail grafting the lab-evolved β2–β3 loop onto a stable β-hairpin (85). Our observations further suggest that the isolated β2–β3 loop would benefit from additional maturation by our semi-design approach (21). Another implication of our work is that next-generation semi-design methods can be simplified by focusing saturation mutagenesis solely on the β2–β3 loop of U1A RRM1, which proved to be the salient determinant of RNA binding here. With this in mind, it is useful to consider natural modes of double-stranded RNA recognition by RRMs. Such a comparison provides perspective for our current findings while potentially expanding the number of parental RRMs for use in lab-evolution experiments.
Although rare, some RRMs engage in sequence-specific recognition of the RNA major-groove. In this respect, TBP6.7 most closely resembles the RBMY protein, which uses RNP amino acids to interact with the single-stranded hairpin loop of its target while simultaneously employing an extended β2–β3 loop to recognize the major-groove (86) (Figure 6A). Like TBP6.7, RBMY uses hydrogen bonds and salt bridges to read specific bases and the phosphate backbone of its target. However, RBMY major-groove recognition is limited in scope compared to the trio of arginines and other interactions utilized by TBP6.7. Whereas RBMY makes full use of its RNP residues to bind the CA/CAA pentaloop (Figure 6A), TBP6.7 recognizes only bulged Ade35, otherwise avoiding contact with the apical loop (Figure 2A–C and Supplementary Figure S3B). Hence, TBP6.7 stands apart from RBMY due to its predominant use of double-stranded RNA recognition to achieve TAR binding.
Figure 6.

Representative atypical RRMs that recognize the RNA major groove. (A) Ribbon diagram of the human RBMY-CA/CAA pentaloop complex. Although this naturally occurring RRM uses classical single-stranded RNA recognition by RNP1 and RNP2 amino acids, its β2–β3-loop residues engage in modest double-stranded RNA readout (PDB entry 2fyi) (86). RNP residues are depicted as ball-and-stick models colored similarly to those of Figure 2A,F; idiosyncratic protein residues that recognize duplex RNA are colored lime green. (B) Ribbon diagram of the Bacillus subtilis YxiN protein in complex with 23S rRNA (PDB entry 3moj) (87). Salt bridges to the backbone and hydrophobic contacts form an array of complementary interactions between the RRM and the three-way helical junction major groove. RNP and β2–β3-loop residues are not used in RNA binding. (C) Ribbon diagram of the p65 C-terminal RRM (p65-C1ΔL2:S4) in complex with telomerase RNA stem IV (PDB entry 4erd) (88). An unusually long β2–β3 loop was truncated for structural studies but is dispensable for RNA binding. The atypical mode of double-stranded RNA binding utilizes a C-terminal α-helical extension that interacts with the major groove, along with idiosyncratic amino acids contributed from strands β2 and β3. Single- and double-stranded RNA recognition occurs without use of RNP amino acids.
A more divergent example of double-stranded RNA recognition is the bacterial YxiN protein, which belongs to the DEAD-box helicase family. YxinN uses a C-terminal RRM domain to bind the three-way helical junction of 23S rRNA (Figure 6B). Specificity is attained through shape complementarity between numerous basic residues that recognize the phosphodiester backbone (87). Gua2553 makes a pivotal interaction to the polypeptide mainchain in strand β2 but RNP residues and the β2–β3 loop do not contribute to binding (87). An even more divergent RRM is the C-terminal domain of p65 in which the signature RNP motifs and the β2–β3 loop are completely dispensable for major-groove recognition of telomerase stemloop IV RNA (88) (Figure 6C). Instead a helical extension recognizes the RNA single- and double-strand features using aromatic stacking, base-specific readout of bulged Gua121 and Ade122 by D409 and R465 in strands β2 and β3, and salt-bridge contacts to the phosphate backbone. In the three diverse instances examined, no comparable constellation of arginines reads the RNA major groove in the manner observed for the TAR–TBP6.7 complex. It remains to be seen whether other lab-evolved TAR binding proteins (e.g. TBP6.9) (21) utilize a fourth β2–β3 loop arginine to recognize another major-groove base, or if these residues are relegated to backbone interactions like many of the basic residues in the atypical RRMs (Figure 6).
An unanticipated finding of the TAR–TBP6.7 interaction is that the lab-evolved RRM was transformed into a double-stranded RNA binding module. This was unexpected especially because parental U1A uses a predominantly single-stranded mode of U1hpII recognition (23) and TAR possesses two prominent single-stranded regions (Figure 2C). RRM plasticity has been noted previously when comparing U1A to homologous U2B″. Indeed, a difference of only three amino acids and two nucleobases in the target reorganizes hydrogen-bond networks leading to altered RNA specificity (89). Here, as few as five amino acid changes to the U1A β2–β3 loop imparted high-affinity binding to TAR with no appreciable recognition of U1hpII or homologous BIV TAR (21,90). These observations prompted us to consider how many changes are needed to co-opt a biomolecular scaffold—roughly the size of an RRM domain—for a new biological function.
Indeed, exaptation plays a prominent role in evolution (91) that is relevant to the principles of semi design espoused here (Figure 1B). For example, endogenous retrovirus (ERV) envelope (Env) proteins have been exapted for placental morphogenesis in mammals (92). One of these ‘enslaved’ ERV proteins, human syncytin-1, maintains cell-cell fusion function but lacks the immunosuppressive activity characteristic of ERV Env (93). Using a structure-guided approach based on Mason-Pfizer Monkey Virus Env, only two amino acid mutations were needed to restore syncytin-1 immunosuppression while maintaining fusogenicity. Such preservation seems extraordinary given millions of years since the original env gene capture event (92). Eye crystallins represent a different case in which housekeeping genes were co-opted to promote transparency and optical clarity of the lens. Remarkably, only four point mutations were required to restore the glutathione S-transferase activity of cephalopod S-crystallin, which was exapted from an ancestral GST (94). Minute numbers of base changes are also effective in reassignment of non-coding RNA functions. The glmS riboswitch requires only three mutations to supplant its glucosamine-6-phosphate cleavage-dependence with divalent ions (95). Eight additional changes co-opt a non-specific ion-binding site for Ca2+-dependent cleavage (96). More surprisingly, a change in effector specificity from cyclic-di-GMP to 3′,3′-cyclic-GMP-AMP can be achieved by a single base change in the Vc2 cyclic-di-GMP riboswitch aptamer. This result demonstrates the facility by which some riboswitches can acquire new ligand-binding properties in structurally homologous scaffolds (97). Collectively, these observations reinforce our findings that lab-based exaptation of U1A for TAR binding required only modest changes to the β2–β3 loop without altering the fundamental RRM fold (Figure 1B and Figure 2A,F).
CONCLUSIONS
Overall, our results provide a detailed molecular-level understanding of HIV-1 TAR recognition and energetics used by the lab-evolved protein TBP6.7. Molecular dynamics simulations and biochemical experiments indicate that the β2–β3-loop motif of TBP6.7 is sufficient for TAR binding, and short conformationally-constrained peptides thereof can bind TAR and suppress Tat-mediated transcription in a dose-dependent manner. Comparing the mode of TAR–TBP6.7 recognition to a known TAR-binding antiviral peptide revealed that different specificity determinants are used to target the same RNA. Double-stranded RNA recognition by TBP6.7 has parallels to a least one naturally occurring RRM that recognizes the major groove, although the majority of its contacts occur in the flanking single-stranded RNA loop. The juxtaposition of canonical single-stranded RNA recognition with diverse modes of duplex RNA binding suggests that the RRM motif could be of broad utility in lab-based evolution experiments. Our results further imply that new modes of RRM-mediated major-groove recognition exist in nature but have yet to be discovered.
DATA AVAILABILITY
Coordinates and structure factor amplitudes have been deposited into the Protein Data Bank as entry 6cmn.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr J.L. Jenkins of the Structural Biology & Biophysics Facility for helpful discussions. We thank Prof. B.L. Miller for helpful advice on peptide binding. We also thank Profs. S.E. Butcher and F.H. Allain for helpful RRM suggestions. Computer time was provided by the University of Rochester Center for Integrated Research Computing.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
HIV RNA Biology SWG CFAR Pilot Award (to J.E.W.) from the University of Rochester Center for AIDS Research funded by National Institutes of Health [P30AI078498]; National Institutes of Health research awards [GM107520 to B.R.M., GM076485 to D.H.M, RR026501 and GM063162 to J.E.W., and GM123864 to B.R.M. and J.E.W]; National Institutes of Health training grants [T32GM068411 and T32GM118283 to C.E.C.]; Stanford Synchrotron Radiation Lightsource, which is funded by the National Institutes of Health [GM103393 and RR001209]; Department of Energy; Sontag Foundation (to B.L.P.). Funding for open access charge: NIH [GM123864].
Conflict of interest statement. None declared.
References
- 1. Fauci A., Dieffenbach C.. Thirty Years of HIV and AIDS: future challenges and opportunities. Ann. Intern. Med. 2011; 154:766–771. [DOI] [PubMed] [Google Scholar]
- 2. HIV/AIDS, J.U.N.P.o. 2016; Geneva: www.unaids.org/sites/default/files/media_asset/global-AIDS-update-2016_en.pdf. [Google Scholar]
- 3. Cary D.C., Peterlin B.M.. Targeting the latent reservoir to achieve functional HIV cure. F1000Res. 2016; 5:F1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Harrich D., Ulich C., Gaynor R.B.. A critical role for the TAR element in promoting efficient human immunodeficiency virus type 1 reverse transcription. J. Virol. 1996; 70:4017–4027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Huthoff H., Berkhout B.. Mutations in the TAR hairpin affect the equilibrium between alternative conformations of the HIV-1 leader RNA. Nucleic Acids Res. 2001; 29:2594–2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Feng S., Holland E.C.. HIV-1 Tat trans-activation requires the loop sequence within TAR. Nature. 1988; 334:165–167. [DOI] [PubMed] [Google Scholar]
- 7. Peterlin B.M., Price D.H.. Controlling the elongation phase of transcription with P-TEFb. Mol. Cell. 2006; 23:297–305. [DOI] [PubMed] [Google Scholar]
- 8. Karn J., Stoltzfus C.M.. Transcriptional and posttranscriptional regulation of HIV-1 gene expression. Cold Spring Harb. Perspect. Med. 2012; 2:a006916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Klase Z., Kale P., Winograd R., Gupta M.V., Heydarian M., Berro R., McCaffrey T., Kashanchi F.. HIV-1 TAR element is processed by Dicer to yield a viral micro-RNA involved in chromatin remodeling of the viral LTR. BMC Mol. Biol. 2007; 8:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Klase Z., Winograd R., Davis J., Carpio L., Hildreth R., Heydarian M., Fu S., McCaffrey T., Meiri E., Ayash-Rashkovsky M. et al. HIV-1 TAR miRNA protects against apoptosis by altering cellular gene expression. Retrovirology. 2009; 6:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ouellet D.L., Plante I., Landry P., Barat C., Janelle M.E., Flamand L., Tremblay M.J., Provost P.. Identification of functional microRNAs released through asymmetrical processing of HIV-1 TAR element. Nucleic Acids Res. 2008; 36:2353–2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mousseau G., Valente S.. Strategies to block HIV transcription: focus on small molecule tat inhibitors. Biology. 2012; 1:668–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Guan L., Disney M.D.. Recent advances in developing small molecules targeting RNA. ACS Chem. Biol. 2012; 7:73–86. [DOI] [PubMed] [Google Scholar]
- 14. Patwardhan N.N., Ganser L.R., Kapral G.J., Eubanks C.S., Lee J., Sathyamoorthy B., Al-Hashimi H.M., Hargrove A.E.. Amiloride as a new RNA-binding scaffold with activity against HIV-1 TAR. Medchemcomm. 2017; 8:1022–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Richter S., Parolin C., Gatto B., Del Vecchio C., Brocca-Cofano E., Fravolini A., Palu G., Palumbo M.. Inhibition of human immunodeficiency virus type 1 tat-trans-activation-responsive region interaction by an antiviral quinolone derivative. Antimicrob. Agents Chemother. 2004; 48:1895–1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Stelzer A.C., Frank A.T., Kratz J.D., Swanson M.D., Gonzalez-Hernandez M.J., Lee J., Andricioaei I., Markovitz D.M., Al-Hashimi H.M.. Discovery of selective bioactive small molecules by targeting an RNA dynamic ensemble. Nat. Chem. Biol. 2011; 7:553–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Davidson A., Leeper T.C., Athanassiou Z., Patora-Komisarska K., Karn J., Robinson J.A., Varani G.. Simultaneous recognition of HIV-1 TAR RNA bulge and loop sequences by cyclic peptide mimics of Tat protein. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:11931–11936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Davidson A., Patora-Komisarska K., Robinson J.A., Varani G.. Essential structural requirements for specific recognition of HIV TAR RNA by peptide mimetics of Tat protein. Nucleic Acids Res. 2011; 39:248–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mei H.Y., Mack D.P., Galan A.A., Halim N.S., Heldsinger A., Loo J.A., Moreland D.W., Sannes-Lowery K.A., Sharmeen L., Truong H.N. et al. Discovery of selective, small-molecule inhibitors of RNA complexes–I. The Tat protein/TAR RNA complexes required for HIV-1 transcription. Bioorg. Med. Chem. 1997; 5:1173–1184. [DOI] [PubMed] [Google Scholar]
- 20. Zhan P., Pannecouque C., De Clercq E., Liu X.. Anti-HIV drug discovery and development: current innovations and future trends. J. Med. Chem. 2016; 59:2849–2878. [DOI] [PubMed] [Google Scholar]
- 21. Crawford D.W., Blakeley B.D., Chen P.H., Sherpa C., Le Grice S.F., Laird-Offringa I.A., McNaughton B.R.. An evolved RNA recognition motif that suppresses HIV-1 Tat/TAR-dependent transcription. ACS Chem. Biol. 2016; 11:2206–2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Katsamba P.S., Myszka D.G., Laird-Offringa I.A.. Two functionally distinct steps mediate high affinity binding of U1A protein to U1 hairpin II RNA. J. Biol. Chem. 2001; 276:21476–21481. [DOI] [PubMed] [Google Scholar]
- 23. Oubridge C., Ito N., Evans P.R., Teo C.H., Nagai K.. Crystal structure at 1.92 Å resolution of the RNA-binding domain of the U1A spliceosomal protein complexed with an RNA hairpin. Nature. 1994; 372:432–438. [DOI] [PubMed] [Google Scholar]
- 24. Mousseau G., Mediouni S., Valente S.T.. Targeting HIV transcription: the quest for a functional cure. Curr. Top. Microbiol. Immunol. 2015; 389:121–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Oubridge C., Ito N., Teo C.H., Fearnley I., Nagai K.. Crystallisation of RNA-protein complexes. II. The application of protein engineering for crystallisation of the U1A protein-RNA complex. J. Mol. Biol. 1995; 249:409–423. [DOI] [PubMed] [Google Scholar]
- 26. Kapust R.B., Tozser J., Fox J.D., Anderson D.E., Cherry S., Copeland T.D., Waugh D.S.. Tobacco etch virus protease: mechanism of autolysis and rational design of stable mutants with wild-type catalytic proficiency. Protein Eng. 2001; 14:993–1000. [DOI] [PubMed] [Google Scholar]
- 27. Lippa G.M., Liberman J.A., Jenkins J.L., Krucinska J., Salim M., Wedekind J.E.. Crystallographic analysis of small ribozymes and riboswitches. Methods Mol. Biol. 2012; 848:159–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liberman J.A., Bogue J.T., Jenkins J.L., Salim M., Wedekind J.E.. ITC analysis of ligand binding to preQ(1) riboswitches. Methods Enzymol. 2014; 549:435–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Adams P.D., Afonine P.V., Bunkoczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010; 66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. McCoy A.J., Grosse-Kunstleve R.W., Adams P.D., Winn M.D., Storoni L.C., Read R.J.. Phaser crystallographic software. J. Appl. Crystallogr. 2007; 40:658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Emsley P., Lohkamp B., Scott W.G., Cowtan K.. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010; 66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Winn M.D., Ballard C.C., Cowtan K.D., Dodson E.J., Emsley P., Evans P.R., Keegan R.M., Krissinel E.B., Leslie A.G., McCoy A. et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 2011; 67:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lawrence M.C., Colman P.M.. Shape complementarity at protein/protein interfaces. J. Mol. Biol. 1993; 234:946–950. [DOI] [PubMed] [Google Scholar]
- 34. Case D.A., Cerutti D.S., Cheatham I.T.E., Darden T.A., Duke R.E., Giese T.J., Gohlke H., Goetz A.W., Greene D., Homeyer N. et al. AMBER. 2014; San Francisco: University of California. [Google Scholar]
- 35. Izadi S., Anandakrishnan R., Onufriev A.V.. Building water models: a different approach. J. Phys. Chem. Lett. 2014; 5:3863–3871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Maier J.A., Martinez C., Kasavajhala K., Wickstrom L., Hauser K.E., Simmerling C.. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015; 11:3696–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Perez A., Marchan I., Svozil D., Sponer J., Cheatham T.E. 3rd, Laughton C.A., Orozco M.. Refinement of the AMBER force field for nucleic acids: improving the description of alpha/gamma conformers. Biophys. J. 2007; 92:3817–3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zgarbova M., Otyepka M., Sponer J., Mladek A., Banas P., Cheatham T.E., Jurecka P.. Refinement of the Cornell et al. nucleic acids force field based on reference quantum chemical calculations of glycosidic torsion profiles. J. Chem. Theory Comput. 2011; 7:2886–2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Andersen H.C. Rattle - a velocity version of the shake algorithm for molecular-dynamics calculations. J. Comput. Phys. 1983; 52:24–34. [Google Scholar]
- 40. Romo T.D., Leioatts N., Grossfield A.. Lightweight object oriented structure analysis: tools for building tools to analyze molecular dynamics simulations. J. Comput. Chem. 2014; 35:2305–2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rice J.J., Daugherty P.S.. Directed evolution of a biterminal bacterial display scaffold enhances the display of diverse peptides. Protein Eng. Des. Sel. 2008; 21:435–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mijalis A.J., Thomas D.A. 3rd, Simon M.D., Adamo A., Jensen K.F., Pentelute B.L.. A fully automated flow-based approach for accelerated peptide synthesis. Nat. Chem. Biol. 2017; 13:464–466. [DOI] [PubMed] [Google Scholar]
- 43. Zou Y., Spokoyny A.M., Zhang C., Simon M.D., Yu H., Lin Y.S., Pentelute B.L.. Convergent diversity-oriented side-chain macrocyclization scan for unprotected polypeptides. Org. Biomol. Chem. 2014; 12:566–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Spokoyny A.M., Zou Y., Ling J.J., Yu H., Lin Y.S., Pentelute B.L.. A perfluoroaryl-cysteine S(N)Ar chemistry approach to unprotected peptide stapling. J. Am. Chem. Soc. 2013; 135:5946–5949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bradrick T.D., Marino J.P.. Ligand-induced changes in 2-aminopurine fluorescence as a probe for small molecule binding to HIV-1 TAR RNA. RNA. 2004; 10:1459–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Marciniak R.A., Calnan B.J., Frankel A.D., Sharp P.A.. HIV-1 Tat protein trans-activates transcription in vitro. Cell. 1990; 63:791–802. [DOI] [PubMed] [Google Scholar]
- 47. Arzumanov A., Walsh A.P., Liu X., Rajwanshi V.K., Wengel J., Gait M.J.. Oligonucleotide analogue interference with the HIV-1 Tat protein-TAR RNA interaction. Nucleosides Nucleotides Nucleic Acids. 2001; 20:471–480. [DOI] [PubMed] [Google Scholar]
- 48. Sczepanski J.T., Joyce G.F.. Binding of a structured D-RNA molecule by an L-RNA aptamer. J. Am. Chem. Soc. 2013; 135:13290–13293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Crooks G.E., Hon G., Chandonia J.M., Brenner S.E.. WebLogo: a sequence logo generator. Genome Res. 2004; 14:1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Soltis S.M., Cohen A.E., Deacon A., Eriksson T., Gonzalez A., McPhillips S., Chui H., Dunten P., Hollenbeck M., Mathews I. et al. New paradigm for macromolecular crystallography experiments at SSRL: automated crystal screening and remote data collection. Acta Crystallogr. D Biol. Crystallogr. 2008; 64:1210–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Weiss M.S. Global indicators of X-ray data quality. J. Appl. Crystallogr. 2001; 34:130–135. [Google Scholar]
- 52. Kabsch W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010; 66:125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Evans P.R., Murshudov G.N.. How good are my data and what is the resolution. Acta Crystallogr. D Biol. Crystallogr. 2013; 69:1204–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Karplus P.A., Diederichs K.. Linking crystallographic model and data quality. Science. 2012; 336:1030–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Word J.M., Lovell S.C., LaBean T.H., Taylor H.C., Zalis M.E., Presley B.K., Richardson J.S., Richardson D.C.. Visualizing and quantifying molecular goodness-of-fit: small-probe contact dots with explicit hydrogen atoms. J. Mol. Biol. 1999; 285:1711–1733. [DOI] [PubMed] [Google Scholar]
- 56. Puglisi J.D., Tan R., Calnan B.J., Frankel A.D., Williamson J.R.. Conformation of the TAR RNA-arginine complex by NMR spectroscopy. Science. 1992; 257:76–80. [DOI] [PubMed] [Google Scholar]
- 57. Aboul-ela F., Karn J., Varani G.. The structure of the human immunodeficiency virus type-1 TAR RNA reveals principles of RNA recognition by Tat protein. J. Mol. Biol. 1995; 253:313–332. [DOI] [PubMed] [Google Scholar]
- 58. Aboul-ela F., Karn J., Varani G.. Structure of HIV-1 TAR RNA in the absence of ligands reveals a novel conformation of the trinucleotide bulge. Nucleic Acids Res. 1996; 24:3974–3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Tao J., Chen L., Frankel A.D.. Dissection of the proposed base triple in human immunodeficiency virus TAR RNA indicates the importance of the Hoogsteen interaction. Biochemistry. 1997; 36:3491–3495. [DOI] [PubMed] [Google Scholar]
- 60. Long K.S., Crothers D.M.. Characterization of the solution conformations of unbound and Tat peptide-bound forms of HIV-1 TAR RNA. Biochemistry. 1999; 38:10059–10069. [DOI] [PubMed] [Google Scholar]
- 61. Dethoff E.A., Petzold K., Chugh J., Casiano-Negroni A., Al-Hashimi H.M.. Visualizing transient low-populated structures of RNA. Nature. 2012; 491:724–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Michnicka M.J., Harper J.W., King G.C.. Selective isotopic enrichment of synthetic RNA: application to the HIV-1 TAR element. Biochemistry. 1993; 32:395–400. [DOI] [PubMed] [Google Scholar]
- 63. Jaeger J.A., Tinoco I. Jr. An NMR study of the HIV-1 TAR element hairpin. Biochemistry. 1993; 32:12522–12530. [DOI] [PubMed] [Google Scholar]
- 64. Borkar A.N., Bardaro M.F. Jr., Camilloni C., Aprile F.A., Varani G., Vendruscolo M.. Structure of a low-population binding intermediate in protein-RNA recognition. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:7171–7176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Foley B., Leitner T., Apetrei C., Hahn B., Mizrachi I., Mullins J., Rambaut A., Wolinsky S., Korber B.. HIV Sequence Compendium. 2016; Los Alamos National Laboratory; LA-UR-16–25625. [Google Scholar]
- 66. Kulinski T., Olejniczak M., Huthoff H., Bielecki L., Pachulska-Wieczorek K., Das A.T., Berkhout B., Adamiak R.W.. The apical loop of the HIV-1 TAR RNA hairpin is stabilized by a cross-loop base pair. J. Biol. Chem. 2003; 278:38892–38901. [DOI] [PubMed] [Google Scholar]
- 67. Richter S., Cao H., Rana T.M.. Specific HIV-1 TAR RNA loop sequence and functional groups are required for human cyclin T1-Tat-TAR ternary complex formation. Biochemistry. 2002; 41:6391–6397. [DOI] [PubMed] [Google Scholar]
- 68. Dethoff E.A., Hansen A.L., Musselman C., Watt E.D., Andricioaei I., Al-Hashimi H.M.. Characterizing complex dynamics in the transactivation response element apical loop and motional correlations with the bulge by NMR, molecular dynamics, and mutagenesis. Biophys. J. 2008; 95:3906–3915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Martin-Tumasz S., Richie A.C., Clos L.J. 2nd, Brow D.A., Butcher S.E.. A novel occluded RNA recognition motif in Prp24 unwinds the U6 RNA internal stem loop. Nucleic Acids Res. 2011; 39:7837–7847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Allain F.H., Gubser C.C., Howe P.W., Nagai K., Neuhaus D., Varani G.. Specificity of ribonucleoprotein interaction determined by RNA folding during complex formulation. Nature. 1996; 380:646–650. [DOI] [PubMed] [Google Scholar]
- 71. Allain F.H., Howe P.W., Neuhaus D., Varani G.. Structural basis of the RNA-binding specificity of human U1A protein. EMBO J. 1997; 16:5764–5772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Jessen T.H., Oubridge C., Teo C.H., Pritchard C., Nagai K.. Identification of molecular contacts between the U1A small nuclear ribonucleoprotein and U1 RNA. EMBO J. 1991; 10:3447–3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Nolan S.J., Shiels J.C., Tuite J.B., Cecere K.L., Baranger A.M.. Recognition of an essential adenine at a protein-RNA interface: comparison of the contributions of hydrogen bonds and a stacking interaction. J. Am. Chem. Soc. 1999; 121:8951–8952. [Google Scholar]
- 74. Calnan B.J., Tidor B., Biancalana S., Hudson D., Frankel A.D.. Arginine-mediated RNA recognition: the arginine fork. Science. 1991; 252:1167–1171. [DOI] [PubMed] [Google Scholar]
- 75. Lalonde M.S., Lobritz M.A., Ratcliff A., Chamanian M., Athanassiou Z., Tyagi M., Wong J., Robinson J.A., Karn J., Varani G. et al. Inhibition of both HIV-1 reverse transcription and gene expression by a cyclic peptide that binds the Tat-transactivating response element (TAR) RNA. PLoS Pathog. 2011; 7:e1002038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Liberman J.A., Salim M., Krucinska J., Wedekind J.E.. Structure of a class II preQ1 riboswitch reveals ligand recognition by a new fold. Nat. Chem. Biol. 2013; 9:353–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Liberman J.A., Suddala K.C., Aytenfisu A., Chan D., Belashov I.A., Salim M., Mathews D.H., Spitale R.C., Walter N.G., Wedekind J.E.. Structural analysis of a class III preQ1 riboswitch reveals an aptamer distant from a ribosome-binding site regulated by fast dynamics. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:E3485–E3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Howe J.A., Wang H., Fischmann T.O., Balibar C.J., Xiao L., Galgoci A.M., Malinverni J.C., Mayhood T., Villafania A., Nahvi A. et al. Selective small-molecule inhibition of an RNA structural element. Nature. 2015; 526:672–677. [DOI] [PubMed] [Google Scholar]
- 79. Hilimire T.A., Chamberlain J.M., Anokhina V., Bennett R.P., Swart O., Myers J.R., Ashton J.M., Stewart R.A., Featherston A.L., Gates K. et al. HIV-1 frameshift RNA-targeted triazoles inhibit propagation of replication-competent and multi-drug-resistant HIV in human cells. ACS Chem. Biol. 2017; 12:1674–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Warf M.B., Nakamori M., Matthys C.M., Thornton C.A., Berglund J.A.. Pentamidine reverses the splicing defects associated with myotonic dystrophy. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:18551–18556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Wang H., Mann P.A., Xiao L., Gill C., Galgoci A.M., Howe J.A., Villafania A., Barbieri C.M., Malinverni J.C., Sher X. et al. Dual-targeting small-molecule inhibitors of the Staphylococcus aureus FMN riboswitch disrupt riboflavin homeostasis in an infectious setting. Cell Chem. Biol. 2017; 24:576–588. [DOI] [PubMed] [Google Scholar]
- 82. Clery A., Blatter M., Allain F.H.. RNA recognition motifs: boring? Not quite. Curr. Opin. Struct. Biol. 2008; 18:290–298. [DOI] [PubMed] [Google Scholar]
- 83. Ippolito J.A., Steitz T.A.. A 1.3-Å resolution crystal structure of the HIV-1 trans-activation response region RNA stem reveals a metal ion-dependent bulge conformation. Proc. Natl. Acad. Sci. U.S.A. 1998; 95:9819–9824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Schulze-Gahmen U., Echeverria I., Stjepanovic G., Bai Y., Lu H., Schneidman-Duhovny D., Doudna J.A., Zhou Q., Sali A., Hurley J.H.. Insights into HIV-1 proviral transcription from integrative structure and dynamics of the Tat:AFF4:P-TEFb:TAR complex. Elife. 2016; 5:e15910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Hughes R.M., Waters M.L.. Influence of N-methylation on a cation-pi interaction produces a remarkably stable beta-hairpin peptide. J. Am. Chem. Soc. 2005; 127:6518–6519. [DOI] [PubMed] [Google Scholar]
- 86. Skrisovska L., Bourgeois C.F., Stefl R., Grellscheid S.N., Kister L., Wenter P., Elliott D.J., Stevenin J., Allain F.H.. The testis-specific human protein RBMY recognizes RNA through a novel mode of interaction. EMBO Rep. 2007; 8:372–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Hardin J.W., Hu Y.X., McKay D.B.. Structure of the RNA binding domain of a DEAD-box helicase bound to its ribosomal RNA target reveals a novel mode of recognition by an RNA recognition motif. J. Mol. Biol. 2010; 402:412–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Singh M., Wang Z., Koo B.K., Patel A., Cascio D., Collins K., Feigon J.. Structural basis for telomerase RNA recognition and RNP assembly by the holoenzyme La family protein p65. Mol. Cell. 2012; 47:16–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Price S.R., Evans P.R., Nagai K.. Crystal structure of the spliceosomal U2B"-U2A' protein complex bound to a fragment of U2 small nuclear RNA. Nature. 1998; 394:645–650. [DOI] [PubMed] [Google Scholar]
- 90. Blakeley B.D., McNaughton B.R.. Synthetic RNA recognition motifs that selectively recognize HIV-1 trans-activation response element hairpin RNA. ACS Chem. Biol. 2014; 9:1320–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Gould S.J., Vrba E.S.. Exaptation - a missing term in the science of form. Paleobiology. 1982; 8:4–15. [Google Scholar]
- 92. Lavialle C., Cornelis G., Dupressoir A., Esnault C., Heidmann O., Vernochet C., Heidmann T.. Paleovirology of ‘syncytins’, retroviral env genes exapted for a role in placentation. Philos. Trans. R Soc. Lond. B Biol. Sci. 2013; 368:20120507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Mangeney M., Renard M., Schlecht-Louf G., Bouallaga I., Heidmann O., Letzelter C., Richaud A., Ducos B., Heidmann T.. Placental syncytins: Genetic disjunction between the fusogenic and immunosuppressive activity of retroviral envelope proteins. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:20534–20539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Tan W.H., Cheng S.C., Liu Y.T., Wu C.G., Lin M.H., Chen C.C., Lin C.H., Chou C.Y.. Structure of a highly active cephalopod S-crystallin mutant: new molecular evidence for evolution from an active enzyme into lens-refractive protein. Sci. Rep. 2016; 6:31176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Lau M.W., Ferre-D’Amare A.R.. An in vitro evolved glmS ribozyme has the wild-type fold but loses coenzyme dependence. Nat. Chem. Biol. 2013; 9:805–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Lau M.W., Trachman R.J. 3rd, Ferre-D’Amare A.R.. A divalent cation-dependent variant of the glmS ribozyme with stringent Ca(2+) selectivity co-opts a preexisting nonspecific metal ion-binding site. RNA. 2017; 23:355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Ren A., Wang X.C., Kellenberger C.A., Rajashankar K.R., Jones R.A., Hammond M.C., Patel D.J.. Structural basis for molecular discrimination by a 3′,3′-cGAMP sensing riboswitch. Cell Rep. 2015; 11:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Coordinates and structure factor amplitudes have been deposited into the Protein Data Bank as entry 6cmn.