


Abstract
Gene regulatory networks (GRNs) consist of different molecular interactions that closely work together to establish proper gene expression in time and space. Especially in higher eukaryotes, many questions remain on how these interactions collectively coordinate gene regulation. We study high quality GRNs consisting of undirected protein–protein, genetic and homologous interactions, and directed protein–DNA, regulatory and miRNA–mRNA interactions in the worm Caenorhabditis elegans and the plant Arabidopsis thaliana. Our data-integration framework integrates interactions in composite network motifs, clusters these in biologically relevant, higher-order topological network motif modules, overlays these with gene expression profiles and discovers novel connections between modules and regulators. Similar modules exist in the integrated GRNs of worm and plant. We show how experimental or computational methodologies underlying a certain data type impact network topology. Through phylogenetic decomposition, we found that proteins of worm and plant tend to functionally interact with proteins of a similar age, while at the regulatory level TFs favor same age, but also older target genes. Despite some influence of the duplication mode difference, we also observe at the motif and module level for both species a preference for age homogeneity for undirected and age heterogeneity for directed interactions. This leads to a model where novel genes are added together to the GRNs in a specific biological functional context, regulated by one or more TFs that also target older genes in the GRNs. Overall, we detected topological, functional and evolutionary properties of GRNs that are potentially universal in all species.
INTRODUCTION
In eukaryotic organisms, differential gene expression is a tightly controlled process that governs developmental, physiological and disease processes. At the level of transcription, specific transcription factors (TFs) bind DNA in order to activate or repress the expression of a gene. MiRNAs repress gene expression post-transcriptionally by interacting with complementary sequences located in the 3′UTR of their target mRNAs. Many molecular interactions, in which TFs and miRNAs are key players, closely work together in order to establish proper gene expression in space and time (1,2). In addition to binding DNA at specific regulatory sites in the genome, several TFs influence transcription through protein–protein interactions, either because they bind DNA as homo- or heterodimers, or because they require interaction with cofactors, chromatin modifying factors or the basal transcription machinery (3). In addition to these direct physical interactions, other molecular interactions have an indirect impact on gene regulation. Genetic interactions, in which two mutations have a combined phenotypic effect not exhibited by either mutation alone, reveal functional linkages in gene regulatory circuits (4). Together with paralogous interactions, which occur frequently between TFs and miRNAs, since duplication events significantly contributed to their evolutionary expansion (5,6), they can also pinpoint redundancy in gene regulation. Regulatory interactions between TFs and target genes are identified by expression profiling in organisms with perturbed TFs and describe both direct and indirect influences of these TFs on gene expression.
While we understand the biological consequences of single data types, we are just beginning to explore how different interaction types together influence gene regulation. For example, coexpressed genes and genes encoding interacting proteins tend to be regulated by common TFs (7,8). Synthetic genetic interactions are more likely to occur between homologous genes, although large gene families complicate the identification of digenic interactions (9). Genes encoding TFs that control miRNA expression have a higher chance to be post-transcriptionally repressed by the miRNA (10). Furthermore, genes coregulated by miRNAs are less functionally linked than genes coregulated by TFs (11). Therefore, different types of molecular interactions provide complementary insights into gene regulation and cell function, expressing the need for data integration (12).
Gene regulation can be studied in gene regulatory networks (GRNs), which map the interactions between TFs and their target genes at a systems level (13). Taking into account different types of molecular interactions that specify regulatory inputs, generates integrated GRNs. Network motifs, which are defined as patterns of interconnections occurring significantly more often than in randomized networks, have been regarded as the basic building blocks of complex networks (14). More specifically, the feed forward loop (FFL), which with positive regulations acts as a signal persistence detector, is the most prominent motif in GRNs of Escherichia coli and Saccharomyces cerevisiae (15–17), and also in higher eukaryotes like human (18,19). Similarly, integrated GRNs can be characterized by composite network motifs, which are subgraphs of which the edges can represent different interaction types, e.g. TF complexes regulating a common target gene and the transcriptional coregulation of interacting proteins (20); miRNA-TF feedback loop and miRNA-mediated FFL (10,21–23); TF-regulated kinate (kinase + substrate) motifs and interacting kinates motifs (24); and CoRePPI motifs considering coregulation of protein–protein interactions by TFs and miRNAs (25). Hence, studying composite network motifs in integrated GRNs has already revealed novel topological structures with biological implications that cannot be deduced from single interaction type networks.
The relation between motif type and biological function has been debated (26–28). Detailed information about a motif's signal integration logic, i.e. binding site affinities and molecular interactions of the regulatory TFs, is necessary for a complete understanding of the motif's function. In addition, not only network motifs, but the higher topological patterns into which they cluster, determine biological function. In GRNs of E. coli and S. cerevisiae, networks motifs such as the FFL aggregate into homogeneous motif clusters, mostly multi-output FFL generalizations, that largely overlap with known biological functions (29,30). Also, in integrated GRNs of S. cerevisiae, composite network motifs cluster together in recurring interconnecting patterns that could be tied to specific biological phenomena such as for instance in the regulonic complex theme wherein a TF regulates multiple members of a protein complex, both TF and protein complex tend to be involved in the same biological process and complexes of related function are often connected to the same TF (28,31,32). A single composite network motif can aggregate into topologically distinct motif clusters e.g. a motif composed of a transcription regulatory interaction where regulator and target both physically interact with the same protein, can cluster either into a ‘regulonic star’, where multiple targets of a TF interact with the same feedback mediator, or a ‘regulatory interacting double-star’, consisting of a regulator–target pair that share a common set of partners in the protein interaction network, which usually belong to a regulatory protein complex (32). Diverse complex networks exhibit rich higher-order organizational structures that are exposed by clustering based on higher-order connectivity patterns and hence provide biological contextualization (33).
The current GRNs are the result of evolution during millions of years. Interaction rewiring and integration of novel genes is an important step in this process. Novel genes originate through partial or full duplication of existing genes followed by divergence, incorporation of mobile elements, gene fission and fusion, and de novo gene creation from non-coding sequence (34). Through phylogenetic analysis, the age of genes can be assigned based on the oldest common species with an ortholog (35). Studies focusing only on protein–protein interaction and coexpression networks in different eukaryotic species revealed that the majority of young genes are incorporated in the periphery and slowly acquire more interactions and functions (36–39). Novel genes gain interactions and functions faster than duplicated genes (40). In addition, genes tend to interact more with proteins of the same age in protein–protein interaction networks of yeast (40,41) and human (36) and in coexpression networks of A. thaliana (39). In yeast it has been shown that proteins with the same age tend to clusters into motifs, while proteins from different age groups tend to avoid motif formation (41). Based on the observations in yeast protein–protein interaction networks, modeling approaches have tried to mimic network evolution (42,43). The best results were obtained with the network motif model where network motifs or protein clusters instead of single proteins are incorporated into pre-existing networks over evolutionary time (43). Overall, studies on GRN evolution have mainly been limited to protein–protein interaction networks in unicellular organisms.
In eukaryote organisms, the main sources of duplicates are small-scale duplication (SSD) and whole-genome duplication (WGD). In C. elegans there is a high rate of SSD, mostly single gene duplications. These SSDs are frequently partial or do not have all regulatory sequences from their original sequences (44). In plants, next to SSD, there are also WGDs. These can either be the result of interspecific or intraspecific hybridizations which lead to multiple genomic copies (polyploidy). WGDs are very abundant and an important source of duplicates in a wide range of plant species (45). In A. thaliana, there are four or five ancient WGD events described (46,47), two of which are located between the Brassicales and the Brassicaceae age groups, one between the flowering plants and the split between eudicots and monocots, and one or two between the origin of seed and flowering plants. After duplication, most of the duplicates get lost (48). However, many WGD-duplicates evolve slower than SSD-duplicates in terms of divergences of sequence (49), expression (50), protein interaction partners (51) and regulatory connections (52).
Here, we developed a data-integration framework starting from different types of interaction networks, over composite network motifs, to network motif modules, which could be dynamically investigated through integration of expression profiles and topologically interpreted in a ‘superview’ analysis (Overview pipeline in Figure 1). We studied two model organisms that are different from a structural, physiological and evolutionary point of view, i.e. the multicellular worm C. elegans and the flowering plant A. thaliana, for which many data are available. We learned that different molecular interactions interrelate in biologically relevant network motif modules to generate a coordinated response in gene regulation. Our approach enabled us to show the advantages and pitfalls of data integration of multiple data types and different experimental methodologies on the motif and motif network module level. Next, the genes were classified into evolutionary age groups by phylogenetic decomposition. Using these groups, we investigated how novel genes are incorporated in these networks. We also found that worm and plant proteins prefer to interact with proteins of a similar age. For protein–DNA interactions on the other hand, we found in both species that regulatory TFs favored to bind to older or of similar age target genes. In network motifs, undirected interactions preferentially took place between age homogeneous proteins, while directed interactions were inclined to be age heterogeneous. These preferred age patterns in the motifs were favorably incorporated in the network motif modules. Modules were mostly composed out of genes from the evolutionary age groups containing the most genes i.e. Eukaryota, Eumetazoa and Caenorhabditis in worm, and Green and Land plants in plant. Hence, especially in A. thaliana, younger genes were more inclined to attach to modules mostly composed out of older genes instead of forming modules on their own. Modules with directed interactions were only age homogeneous in the oldest evolutionary age groups or there were none. This leads to a model where novel genes are added together to the GRNs in a specific biological functional context, regulated by one or more TFs that also target older genes in the GRNs.
Figure 1.
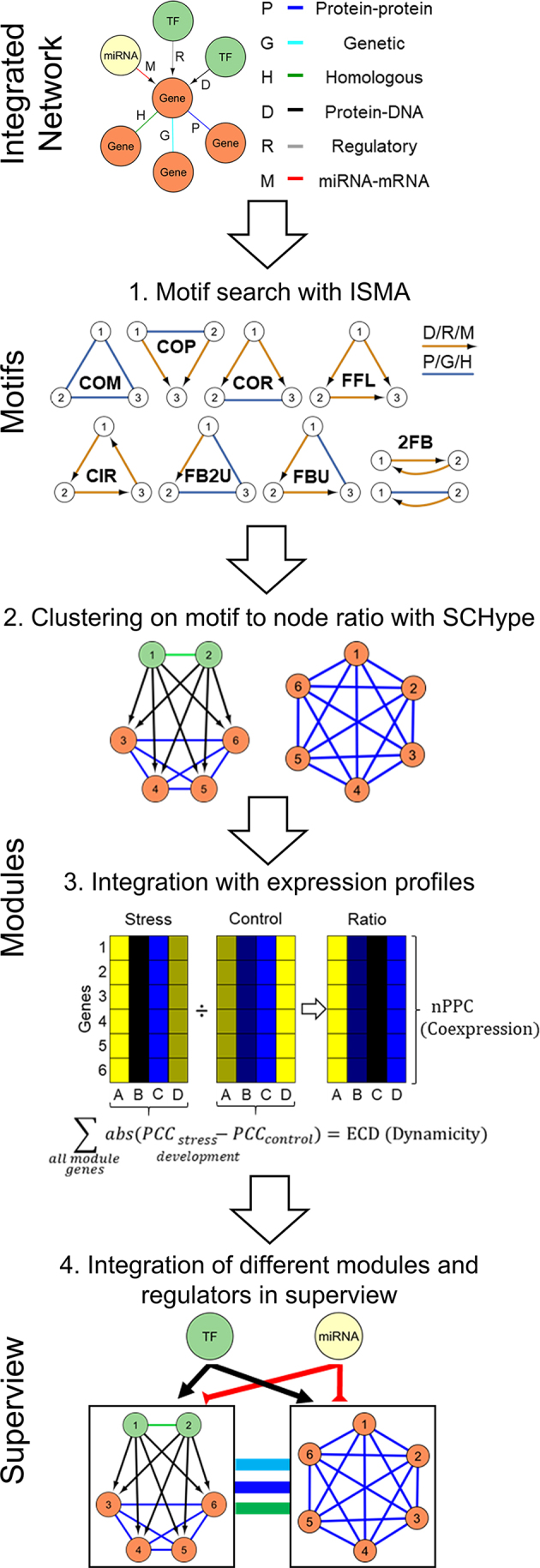
The data integration framework to study integrated GRNs. In the first step, molecular interaction data were gathered from multiple sources: protein–protein (P), genetic (G), homologous (H), protein–DNA (D), regulatory (R) and miRNA–mRNA interactions. In the motif step, all possible two-node and three-node motifs were searched with ISMA, the Index-based Subgraph Matching Algorithm that conducts a fast and efficient motif search through carefully selecting the order in which the nodes of a query motif are investigated. We grouped the motifs in eight categories (complex motif (COM), feed forward loop (FFL), co-pointing motif (COP), co-regulated motif (COR), circular feedback motif (CIR), feedback undirected motif (FBU), feedback 2 undirected motif (FB2U) and two-node feedback motif (2FB)) and named them ABC according to the interactions A between node 1 and 2, B between node 1 and 3 and C between node 2 and 3. For directed edges, if the direction is reversed e.g. interaction A between node 2 and node 1, a small case letter is used e.g. motif aBC. In the module step, motifs were clustered with SCHype, which is a spectral hyper-edge clustering algorithm maximizing the hyper-edge (i.e. motif) to node ratio. In the dynamic module step, for each module, coexpression was evaluated by the average Pearson Correlation Coefficient (nPCC) and for Arabidopsis thaliana dynamicity was assessed by the Expression Correlation Differential Score (ECD). In the superview step, modules were integrated with other modules and regulating TFs and microRNAs. This integration was based on statistical enrichment by comparing the observed versus expected interactions through comparison with random modules of the same sizes (‘Materials and Methods’ section).
MATERIALS AND METHODS
Source of interaction data
An overview of the molecular interaction data of the integrated GRNs can be found in Table 1. For C. elegans, the undirected molecular interaction data were compiled from the following resources: 9739 protein–protein interactions (P) from Wormbase WS234 (53), Worm Interactome version 8 (54), BioGRID 3.2.97 (55) and literature (56–60); 3830 genetic interactions (G) from Wormbase WS234 (53), BioGRID 3.2.97 (55) and selected publications (61–64); 6502 homologous interactions (H), which consisted on the one hand of 6348 paralogous protein-coding genes determined by an all-against-all BLASTP of the C. elegans proteome WS220 (E-value < 1e-25, percent alignment ≥ 60%) and on the other hand of 154 paralogous miRNAs with identical seed sequence identified through BLASTN. For the regulators in the directed molecular interactions, TFs were defined as in WormBook (65), while miRNAs were retrieved from miRBase (66). The 13 747 protein–DNA binding interactions (D) consist of two types of experimental data, Y1H and ChIP. The Y1H dataset contains both large and small scale datasets (10,60,67–74). The ChIP dataset was taken from modENCODE, where TF-protein-coding target gene interactions predicted from ChIP-seq data by the TIP algorithm were used with a quality score of 1 (75). The 3948 regulatory interactions (R) comprise genes with a 2-fold log2 change in gene expression upon knock-out or knock-down of the regulator (58,76–83), supplemented with regulation associated interactions from the text-mining database EVEX (84). The 6177 miRNA–mRNA interactions (M) entail experimental confirmed interactions from miRTarBase (49%) (85) and PicTar predictions conserved in five species (51%) (86,87). Gene identifiers of all protein-coding genes and miRNAs were converted to Wormbase WS245 using WormBase Converter (88) and a Perl script, only keeping the genes (and their interactions) with unchanged WS identifier or that merged/split to a new WS identifier. At last, the worm integrated GRNs contained 43 943 interactions between 845 TFs (92% of all TFs), 172 miRNAs (67% of all miRNAs) and 12 095 protein-coding genes (67% of all protein-coding genes (89)).
Table 1.
Overview of the different types of molecular interactions in the integrated GRNs of C. elegans and A. thaliana, respectively
| C. elegans | A. thaliana | |||||||
|---|---|---|---|---|---|---|---|---|
| Number of | edges | nodes | regulators | targets | edges | nodes | regulators | targets |
| P | 9739 | 4287 | / | / | 52 613 | 10 266 | / | / |
| G | 3830 | 1823 | / | / | / | / | / | / |
| H | 6502 | 4807 | / | / | 5254 | 8896 | / | / |
| D | 13 747 | 3989 | 603 | 3733 | 29 690 | 12 721 | 399 | 12 632 |
| R | 3948 | 3283 | 70 | 3235 | ||||
| M | 6177 (49% E, 51% C) | 1499 | 144 | 1355 | 2122 (5% E, 95% C) | 1623 | 171 | 1452 |
| Total | 43 943 | 13 112 | 611 | 6486 | 89 679 | 20 694 | 570 | 13 373 |
P = protein–protein, G = genetic, H = homologous, D = protein–DNA, R = transcription regulatory, M = miRNA–mRNA interactions. In the case of A. thaliana, protein–DNA and transcription regulatory interactions are combined in D, since the AtRegNet source does not specify the type of molecular interaction or experimental method i.e. protein–DNA binding or transcription regulatory interaction and several interactions from AtRegNet and literature involve both DNA binding and differential expression upon TF perturbation. Regulators indicate TFs or miRNAs. All data are experimental, except for the M data, which are a mixture of experimental (E) and computationally predicted (C) interactions.
For A. thaliana, the undirected molecular interaction data were collected from the following resources: 52 613 protein–protein interactions (P) from CORNET 3.0 (experimentally validated interactions only) (90), BioGRID 3.2.97 (55), MIND (high confidence) (91) and the Arabidopsis Interactome (51); and 5254 homologous interactions (H), which consisted on the one hand of 5226 paralogous protein-coding genes established from phylogenetic tree-based gene families (48) and on the other hand of 28 paralogous miRNAs with identical seed sequence identified through BLASTN. For the regulators in the directed molecular interactions, TFs were named by PlantTFDB 3.0 (92), while miRNAs were found in miRBase (66). The 29690 protein–DNA and transcription regulatory interactions (D) include ChIP data from a meta-analysis of publicly available data (93) and a high confidence reference set that combines ChIP binding and expression upon TF perturbation (94), Y1H data from literature (95) (96,97), protein–DNA binding and/or transcription regulatory interactions from AtRegNet (98) and differential expression analysis upon TF perturbation (94). For Arabidopsis, both protein–DNA binding and transcription regulatory interactions are combined in D, since AtRegNet does not specify the type of molecular interaction or experimental method and several interactions from AtRegNet and literature involve both DNA binding and differential expression upon TF perturbation. The 2122 miRNA-RNA interactions (M) contain experimental confirmed interactions from miRTarBase (5%) (85) and psRNATarget predictions using standard parameters on the TAIR 10 transcripts (95%) (99). In all Arabidopsis interactions, only protein coding genes and miRNAs with a TAIR 10 gene identifier were kept. Interactions involving mitochondrial and chloroplast genes were removed. As symbolic gene names, we used the primary symbol name from TAIR. At last, the plant integrated GRNs encompassed 89 679 interactions between 1519 TFs (88% of all TFs), 174 miRNAs (41% of all miRNAs) and 19001 protein-coding genes (69% of all protein-coding genes) (100). Self-interactions were removed in all the networks.
Topology of the networks
The topology of the networks was analyzed in R using the igraph package (101).
Network motif detection and enrichment
Three-node motifs were detected by ISMA (Index-based Subgraph Matching Algorithm) (102). Two-node motifs were detected by a Perl script (https://gitlab.psb.ugent.be/jofoo/NetworkMotifModules.git). To calculate motif enrichment, 1000 random networks with the same degree distributions as the real networks for each interaction type were constructed through an edge swapping algorithm in the Matlab Motif Clustering Toolbox (32). The enrichment of each detected motif compared to random networks was calculated using the Z-score 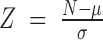 and derived P-value (significance cut-off 0.05), in which N is the number of motifs in the real networks, μ the average and σ the standard deviation of the number of motifs in the random networks. The complete search for all possible motifs in the real networks with ISMA took 3 min and 52 s on a single Linux computing node and used at maximum 250 Mb of memory. The ISMA running time per motif was between 19 and 506 ms, depending on the motif. For motif detection in the 1000 random networks, ISMA was run in parallel and gave a similar performance.
and derived P-value (significance cut-off 0.05), in which N is the number of motifs in the real networks, μ the average and σ the standard deviation of the number of motifs in the random networks. The complete search for all possible motifs in the real networks with ISMA took 3 min and 52 s on a single Linux computing node and used at maximum 250 Mb of memory. The ISMA running time per motif was between 19 and 506 ms, depending on the motif. For motif detection in the 1000 random networks, ISMA was run in parallel and gave a similar performance.
Network motif clustering
Network motif clustering was performed by the unweighted and undirected hypergraph-based spectral clustering algorithm SCHype using default settings (P = 1) (103). The different three-node motifs were clustered into seven different types. Next to these groups, all motifs were clustered together and separately. We filtered out modules smaller than five nodes and bigger than 100 nodes, and modules consisting only of homologs, because they are less informative or not interpretable.
Functional analysis of the integrated networks
All modules where visualized together with functional data in Cytoscape. For each module, GO Biological Process enrichment values (P < 0.05) were calculated by the BINGO 2.44 Cytoscape plugin using the Benjamini–Hochberg multiple testing correction (104). We used the core GO ontology release 9 January 2015 together with gene annotations files for A. thaliana GOC: 1 August 2016 and C. elegans GOC: 17 July 2016.
Integration of expression profile data
For the C. elegans microarray data, we derived expression ratios for embryonic development by dividing the expression matrix by the overall average (105) and for embryonic and post-embryonic development by dividing tissue-specific expression by its whole animal reference set at a specific developmental stage (106) (Supplementary Tables S7 and 8). The Arabidopsis abiotic stress-dedicated microarray expression profiles consisted of expression ratios in 199 experiment over control conditions (94) (Supplementary Table S9). Coexpression within modules was calculated by the average Pearson Correlation Coefficient of all the genes in a module (nPCC) and the P-value from the Z-score upon comparison to 1000 random modules of the same size picked from all clustered genes (Atha: 20 695 genes, Cele: 13 112 genes). For A. thaliana, dynamicity of the modules was analyzed by the Expression Correlation Differential score (ECD), which sums up the differences between the PCC in abiotic stress and control conditions for all the edges in the module, a measure that was originally developed for motifs (107). Therefore, stress conditions were grouped per abiotic stress type (Supplementary Table S9). Since the calculation of the PCC requires multiple replicates for a specific experimental condition, we were only able to calculate the ECD for plant, and not for worm. The PCC of every module gene was calculated in the environmental stress, as well as in the control conditions. The ECD was then calculated by the following formula: 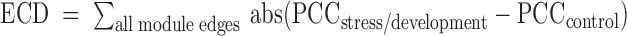 . At last, the significance of the ECD (P < 0.05) was analyzed by comparing the ECD in the real module versus the ECD of 1000 modules with the same number of genes through permutation. The used expression compendia do not contain any expression values for miRNAs and therefore no ECD analysis could be performed for miRNAs.
. At last, the significance of the ECD (P < 0.05) was analyzed by comparing the ECD in the real module versus the ECD of 1000 modules with the same number of genes through permutation. The used expression compendia do not contain any expression values for miRNAs and therefore no ECD analysis could be performed for miRNAs.
Superview
The super view representations of the networks were created using all modules of size 5–50 nodes. Modules sharing 50% or more of their genes were merged under the name of the biggest module. We counted the number of interactions going from a gene in one module to a gene in another module for each interaction type separately. This results in the total number of interactions between two modules. This observed number of interactions between modules was compared to the number of interactions between 1000 random modules with the same sizes as the original modules. The random modules were obtained by randomly selecting genes from all genes present in the modules of the integrated GRNs. A Z-score and P-value were calculated to compare the observed versus the expected value. To assign regulators to modules we integrated sets of regulatory interactions (D/R/M) with the modules. For this we counted the number of genes in a module regulated by a certain regulator. Regulators that were already in the module were not counted. We compared this count with the number of regulatory interactions going to the random modules with the same size. A Z-score and P-value were calculated to compare the observed versus the expected value.
Visualization
All network figures where made using Cytoscape. For the interactive web visualization, a custom version of CyNetShare was used (http://idekerlab.github.io/cy-net-share/). A standalone Java tool called ModuleViewer visualized the expression ratios together with other relevant biological data into customized heatmaps (94).
Phylogenetic decomposition
We applied phylostratigraphy to derive the evolutionary origin of the genes (35). Specifically, A. thaliana gene families were assigned phylogenetic ages based on the oldest lineage that still contains an ortholog of the gene family i.e. the earliest common ancestor of the gene family. As an example, if a gene family contains four genes from species in the Brassicaceae lineage and one gene from Physcomitrella patens, it is classified as Land plants/Embryophyta. Orthologous gene families were downloaded from PLAZA 4.0 dicots (http://bioinformatics.psb.ugent.be/plaza/versions/plaza_v4_dicots/) (108). They are constructed out of 55 fully sequenced species with a wide distribution over the different lineages. This resulted in 10 age groups: Green plants, Land plants, Vascular plants, Seed plants, Flowering plants, eudicots, Rosids, Brassicales, Brassicaceae, A. thaliana (Supplementary Figure S12). For C. elegans genes, phylogenetic ages were assigned according to consensus gene-age labels that are based on 13 orthology inference algorithms (109), as well as Caenorhabditis genus-specific and Caenorhabditis elegans species-specific gene labels (110). Where both methods differed, we used the oldest classification. This resulted in seven age groups: Cellular organisms, Eukaryota, Opisthokonta, Eumetazoa, Ecdysozoa, Caenorhabditis and C. elegans (Supplementary Figure S12). For both species, all genes with their phylogenetic classification are listed in Supplementary Table S20.
Interaction homogeneity and age preference
For the age homogeneity analysis, we compared the observed number of interactions between the genes in same age groups to the expected number of interactions based on 1000 randomized networks with the same age and degree distribution. Based on this comparison, a Z-score and P-value was calculated with multiple hypothesis testing correction (Benjamini–Hochberg). For the count analysis, the observed number of interactions between the genes in the age groups was compared to the expected number of interactions based on 1000 randomized networks with the same age and degree distribution. Based on this comparison, a Z-score and P-value was calculated with multiple hypothesis testing correction (Benjamini–Hochberg).
Age pattern analysis in network motifs and modules
Each motif was assigned to one of 13 motif age types (Figure 9A). Redundancy through internal symmetry within these types was removed by selecting only the motif where the nodes are in decreasing age order, e.g. for COP motifs, the motif age type OYO is the same as YOO, but OYO is chosen over YOO because there the nodes are ordered from old to young. We calculated the motif age preference by computing a Z-score and associated P-value with multiple hypothesis testing correction (Benjamini–Hochberg) of observed motif age patterns compared to expected by 1000 permutations, where nodes are shuffled for each of the three node positions separately, so the age distribution per node position is preserved. For the module age preference in each module type, we subtracted the percentage of motif age type motifs belonging to a certain motif type from the percentage of motif age type that was clustered in the corresponding module type. Formula: (relative fraction of clustered motifs) − (relative fraction of motifs in total of a certain motif age type per module type). For example, within the COM motifs of A. thaliana, 21% is age homogeneous (SSS) while in the clustered motifs in the module set COMc, 42% is age homogeneous. This results in a relative difference of +21%, which means that age homogeneous COM motifs are preferentially clustered. To test significance, a hypergeometric test was performed with multiple hypothesis testing correction (Benjamini–Hochberg).
Figure 9.

(A) The different motif age types (B) Motif age preference. Statistical significance (empirical Z-score) of the observed age patterns in motifs compared to the patterns expected by random. Due to symmetry, not every pattern is present in all motifs (blank squares). The symmetric motif age types were sorted form old to young age. Only motifs with at least one significant age type after multiple hypothesis correction (Benjamini–Hochberg, P value < 0.05) are shown in the picture, the full table can be found in Supplementary Table S16. (C) Module age preference. Preferential age patterns of the motifs clustered in network motif modules. The value represents the percentage of motifs with each age pattern that are clustered, subtracted by the percentage of a certain age pattern in all the motifs belonging to that module type. The squares are colored according to the significance of this value (hypergeometric test with multiple hypothesis correction according to Benjamini–Hochberg). Due to symmetry, not every pattern is present in all motifs (blank squares). The symmetric motif age types were sorted form old to young age.
RESULTS
High quality integrated GRNs in worm and plant feature hubs and modularity
Given that gene regulation is influenced by different physical and functional molecular interactions, we integrated high quality directed protein–DNA (D), regulatory (R) and miRNA–mRNA (M) and undirected protein–protein (P), genetic (G) and homologous (H) interactions to obtain a holistic view on gene regulation (Figure 1 and Table 1). The data contained only experimentally validated interactions, except for miRNA–mRNA interactions, where computational predictions complemented experimental interactions. Homologous relationships were also inferred through computational analysis (‘Materials and Methods’ section). The integrated GRNs of C. elegans (Cele) and A. thaliana (Atha) contained respectively 43 943 and 89 679 molecular interactions, distributed over the different molecular interaction types as depicted in Figure 2. There is limited overlap between the different types of interactions in both GRNs (Supplementary Figure S1). In the A. thaliana GRN, protein–DNA interactions and transcription regulatory interactions are merged in the same D data type due to indistinctness in experimental origin or overlap between the two types of interactions: at least 4236 interactions are both physical protein–DNA and transcription regulatory interactions. Like most biological networks, these networks are scale-free and feature hubs, highly connected proteins in the undirected networks and regulators with many targets in the directed networks (Supplementary Table S1 and Figure S2) (111,112). Many medium-degree nodes have a higher clustering coefficient than expected from the power-law fit (Supplementary Figure S3). Hence, they differ from hierarchical scale-free networks and exhibit an extra modularity than the one centered on hubs (32). The overall clustering coefficients of the protein–DNA and protein–protein interaction networks of C. elegans are 2 to 10 times higher than those of A. thaliana. Hence, the worm integrated GRNs are smaller (edge to node ratio Cele 3.4 versus Atha 4.3) and more likely to form clusters.
Figure 2.

Proportions of the different types of molecular interactions within the integrated GRNs of Caenorhabditis elegans and Arabidopsis thaliana: protein–protein (P), homologous (H), genetic (G), protein–DNA (D) (and/or transcription regulatory in the case of A. thaliana), miRNA–mRNA (M) and transcription regulatory (R) interactions. The worm integrated GRNs contained 43 943 interactions between 845 TFs (92% of all), 172 miRNAs (67% of all) and 12 095 protein-coding genes (67% of all). The plant integrated GRNs encompassed 89 679 interactions between 1519 TFs (88% of all), 174 miRNAs (41% of all) and 19 001 protein-coding genes (69% of all).
Different composite network motifs form the basic building blocks of integrated GRNs
As a first step of our data integration framework (Figure 1), we searched for possible two-node motifs using a customized Perl script and three-node composite network motifs using ISMA (Index-based Subgraph Matching Algorithm) (102). These two- and three-node motifs are the elementary building blocks of many higher-order motifs. We detected, respectively, 40 and 14 different 2- and 3-node network motifs that occurred 50 times or more in the GRNs of worm and plant (Figure 3; Supplementary Table S2 and Figure S4). The composite network motifs were grouped in 8 motif types, all of which were present in both species (Figure 3): complex motifs (COM), which represent combinations of all undirected interactions; co-pointing motifs (COP), where two interacting regulators (e.g. dimers or homologs) regulate the same gene; co-regulated motifs (COR), where one regulator controls two interacting genes; FFLs, where a regulator regulates a target gene directly and indirectly through another regulator; circular feedback motifs (CIR), where regulators act upon each other through a feedback loop; feedback undirected motifs (FBU), where two directed interactions in a cascade are connected by one undirected interaction; feedback 2 undirected motifs (FB2U), which combine two undirected interactions and one directed interaction; and two-node feedback motifs (2FB), which couple a directed edge with an undirected edge (113). The name of the motifs, e.g. RPD, determines the motif: the first letter refers to the left edge from the top node, the second letter refers to the right edge of the top node and the third letter refers to the basal edge in the motif from left to right. A lowercase letter indicates reversal of the directed edge direction (Figure 3).
Figure 3.

Overview of the different network motif types (left) and higher order modules for each motif type clustered (right). The number of specific motifs that were found at least 50 times in the GRNs of Caenorhabditis elegans and Arabidopsis thaliana is indicated in the motif. Specific examples of the clustering of motifs per motif type in modules is depicted on the right by a network figure and a Module Viewer figure of their expression profiles in developmental (C. elegans) or abiotic stress conditions (A. thaliana) (‘Materials and Methods’ section). For the abiotic stress compendium Module Viewer figure, only the top 10 conditions with most up- and downregulated expression are shown. The average Pearson Correlation Coefficient (nPCC) and if available, the abiotic stress condition with significant Expression Correlation Differential score (ECD) are shown as measures of coexpression and expression dynamicity of the modules, respectively. Complex motifs (COM) and modules (COMc): Cele COMc 104 (GGG/GPP/PPP/GGP motifs) involved in dosage compensation and sex determination and Cele COMc 70 (PPP/GPP/HPP motifs) functioning in ubiquitin-dependent protein catabolism. Co-regulated motifs (COR) and modules (CORc): Atha CORc 14 (MMP/PPP motifs) involved in leaf and flower development (149), upregulated upon cold stress and downregulated upon oxidative stress, and Cele CORc 8 (RPP/PPP motifs) involved in the endoplasmic reticulum unfolded protein response. Co-pointing motifs (COP) and modules (COPc): Cele COPc 61 (HMM motifs) involved in axon extension and Atha ALLc 70 (DDP/PDD/PPP motifs) involved in flavonoid biosynthesis, upregulated upon radiation stress and dynamic upon oxidative stress. Feed-forward motifs (FFL) and modules (FFLc): Atha FFLc 30 (DDD motifs) involved in response to water deprivation, upregulated upon cold and salt stress and dynamic upon cold stress, and Atha FFLc 48 (DDD/DMD motifs) upregulated upon cold and salt stress, downregulated upon heat stress. Circular feedback motifs (CIR) and modules (CIRc): Atha CIRc 0 (DdD/DDD/DPD/PPP/DPP motifs) involved in flower development and Cele CIRc 25 (DmD/DdD/DDD/DPP/DPD/RPD/RDD/DMM/DDG/DDH/DDP/DMD/GHH/HMM/GMM motifs) involved in the regulation of larval development. Feed-back 2 undirected motifs (FB2U) and modules (FB2Uc): Cele FB2Uc 13 (DPP motifs) involved in embryonic and larval development and Cele FB2Uc 39 (RPG /RGP/GGG/GPP motifs) involved in dauer larval development. Feed-back undirected motifs (FBU) and modules (FBUc): Atha FBUc 2 (DPD/DDD/PPP/PDD/DDP motifs) involved in the cellular response to red or far red light and upregulated upon heat and cold stress and Atha FBUc 3 (DPD motifs) functioning in flower development.
The higher presence of some motifs in one species as compared to the other can be attributed to the characteristics of the underlying data and methodologies (Figure 3 and Supplementary Figure S4). The respective fivefold and twofold higher abundance of P and D data in plant compared to worm generally resulted in higher numbers of P and D containing motifs in plant e.g. PPP (10×), DDD (2×), PDD (3×), DDP (4×), DDM (4×). HHH-motifs are only found in worm, since homologs in C. elegans are composed of direct BLAST results, while homologs in A. thaliana are based upon gene trees of gene families (‘Materials and Methods’ section). In C. elegans, extensive yeast one-hybrid (Y1H) and yeast two-hybrid (Y2H) mapping between TFs led to more widespread TF-TF interactions (60) and hence higher motif counts for the motifs DdD (30×), DmD, DD (19×) and DP (6×). The threefold higher abundance of M data in worm, as well as the large fraction of experimental data in there, compared to plant, mostly produced higher numbers of M containing motifs in worm e.g. MMD (7×), HMM (32×), MMP (3×) and DM. The higher numbers of MMH (8×), as well as HDD (5×) and DDH (3×) in plant are also possibly caused by the WGD events in A. thaliana, where upon duplication of a target gene or TF, also a novel target gene or regulator is gained. In both species, specific motifs largely overlap due to overlaying P and D, intersecting P, G and H, and bidirectional D interactions (Supplementary Figure S5): within COP and FB2U, between FBU/FFL, COP/FFL, F2BU/FBU, FBU/COR, FB2U/COR and FFL/CIR. The overlap between FFL and CIR motifs (DDD/DdD) indicate that in FFLs containing only TFs, the final targeted TF transcriptionally feedbacks on the top regulatory TF. A particular difference between plant and worm integrated GRNs is that homologous plant TFs targeting the same genes tend to physically interact more both through protein–protein and protein–DNA interactions.
Typically, the presence of network motifs is evaluated by network motif enrichment. Network motif enrichment was calculated compared to 1000 randomized networks with preserved degree distributions, as is usual done (‘Materials and Methods’ section) (22). All motif types had at least one network motif enriched in the GRNs (Supplementary Table S2 and Figure S4). We found network motif enrichment to be biased toward network topology, which is inherently connected to the experimental methodology (Supplementary Data). As a predominant example, since we integrated chromatin immunoprecipitation (ChIP) and Y1H for D type physical protein–DNA interactions, we observed an enrichment of FFL (DDD) only in the ChIP data and not in the Y1H data for both species (Figure 4 and Supplementary Table S3). Overall, the net result is a lack of enrichment of the FFL in the integrated GRNs of worm and plant (Figure 4 and Supplementary Figure S4). Accordingly, enrichment of the FFL was reported in several studies with a similar randomization methodology, most of them using ChIP data or genome-wide target gene prediction based on conserved TF binding sites for the directed edges (14,20,22,114,115). The latter are TF-centered GRN approaches, which result in genome-wide networks at the target gene level with low interconnectivity and few TF hubs. Y1H, on the contrary, is a gene-centered approach, leading to smaller networks with a higher interconnectivity distributed over more TFs and many target gene hubs (73). Therefore, these data are complementary in the construction of GRNs. This differential network motif enrichment can be mainly attributed to the randomization strategy that preserves the degree distribution, but at the same time limits the randomization in a network topology created by Y1H (Supplementary Table S4). In addition, we also observed that network motifs can be created by the integration of different experimental methodologies for a certain data type. As an example, extra CIR motifs DdD originated from the integration of ChIP and Y1H protein–DNA interaction data (Supplementary Table S3). Also, preferential interaction patterns between TF hubs in ChIP and target gene hubs in Y1H emerged in the randomized networks upon data integration, further disturbing the network motif enrichment (Supplementary Table S5).
Figure 4.

Differential FFL enrichment between ChIP and Y1H data: the FFL motif is only enriched in the ChIP data of both Caenorhabditis elegans and Arabidopsis thaliana. Number of FFL motifs (DDD) in ChIP, Y1H and the combination of ChIP and Y1H D data of C. elegans and A. thaliana. Significant over- or under-representation as compared to 1000 randomized networks (P-value = 0.05) is indicated by green or red arrow, respectively.
Hence, the experimental or computational methodology that generates a certain data type can exert an impact on the network topology, and more specifically on network motif enrichment. The presence of specific network motifs and their aggregation might therefore be a better indicator of biological functionality of a network than network motif enrichment. Moreover, network motif aggregation is independent of the over-representation of network motifs in the network (32). It is clear that the incompleteness of the data affects network topology and randomization, and this is another reason to consider network motif and module presence rather than enrichment. On top of that, the integration of different data methodologies creates network motifs that would be absent in a single data source network, further indicating that different methodologies are complementary for a given data type to obtain a systems view on gene regulation.
Network motifs aggregate into functional network motif modules
Through a general data-integration framework based on spectral clustering of hypergraphs (103), we investigated the aggregation of motifs into higher order topological structures in the integrated GRNs that represent biological and/or regulatory entities (Figure 1). The clustered structures, from now on referred to as network motif modules, can be composed of one type of motif or a combination of different motifs. We classified the modules in 7 different cluster types, depending on which three-node motifs were clustered together (‘Materials and Methods’ section) (Figure 3). In addition, we also clustered all two- and three-node motifs together. We functionally annotated the modules with GO Biological Process and investigated dynamic modules by integrating expression profiles from respectively a developmental and abiotic stress expression profile compendium for C. elegans and A. thaliana (94,105,106) (Supplementary Tables S7, S8 and S9). Here, we calculated two measures: the average nPCC as a measure of coexpression and, for A. thaliana, the ECD score, which highlights modules specific for a stress condition as compared to control conditions (‘Materials and Methods’ section). The number of different network motif modules obtained can be found in Supplementary Table S6.
The first cluster type is the complex module (COMc), generated by clustering the COM motif type (Figure 3). Complexes are built out of functionally associated genes, linked through physical protein–protein interactions or/and functional genetic or homologous interactions (cfr. protein complex theme (31)). Proteins in these modules usually show coherent coexpression patterns across conditions (Supplementary Data). In addition to clusters consisting of only one interaction type, we found clusters composed of members of a protein complex genetically interacting with the same set of proteins (Cele COMc 104 e.g.), since members of a given protein complex or biological process often have common synthetic genetic interaction partners (116). Furthermore, we observed network motif modules consisting of homologs physically interacting with the same proteins. This is in agreement with the fact that gene duplicates initially have the same interaction partners, before divergence or loss. For instance, in Cele COMc 70, functioning in ubiquitin-dependent protein catabolism, we observed a star-like configuration of protein interaction partners around the homologs cdc-48.1 and cdc-48.2. Both paralogs are also linked by genetic and protein–protein interactions and show a similar expression pattern. Their human homologs suppress the aggregate formation of a Huntington polyQ repeat (117) (See also Supplementary Data for further examples).
The second cluster type is the co-regulated module (CORc), consisting of clusters of the COR motif type and represents COR functionally associated proteins (cfr. regulonic complex theme (31)). This cluster type adds a transcriptional (e.g. Cele CORc 8) or post-transcriptional (e.g. Atha CORc 14) regulatory layer to a complex module (Figure 3). Here, we also observed star-like configured COR heterodimers (Supplementary Data).
The third type, the co-pointing module (COPc), represents interacting regulators that share a group of targets (cfr. COP theme (31)). We found homologs, heterodimers and protein complexes regulating a set of genes. In the HMM module Cele COPc 61, we observed strongly coexpressed genes involved in axon extension. We also observed interacting protein complexes that combine two groups of functionally associated proteins with regulatory interactions between them. In Atha ALLc 70, a module that combines COP, COR and COM motifs and changes dynamically upon oxidative stress, the heterodimer PIF3-HY5 targets a number of physically interacting anthocyanin biosynthetic enzymes (Figure 3). At last, we detected homologous signaling pathways like in Cele COP 193 (Supplementary Data).
The fourth type, the feed forward loop module (FFLc), consists only of regulatory links and enables universal information processing and hierarchical regulation (32). We found the feed-forward theme, where one TF regulates another one and both of them regulate a common set of target genes (31) (e.g. Atha FFLc 30), and extensions to this theme with more regulatory layers or combinations of transcriptional and post-transcriptional regulation (e.g. Atha FFLc 48) (Figure 3).
The fifth type, the circular feedback module (CIRc), has not been described before (31,32). Here, transcription and/or post-transcriptional regulatory links feed back into one another generating intrinsically clustered patterns (e.g. Cele CIRc 25, Atha CIRc 0 — see further) (Figure 3). Often, P interactions between the TFs are also present, as already indicated by the FBU/FFL/CIR motif overlap. In addition to FFLc, CIRc form the core of integrated GRNs and integrate signaling between regulators, often coordinating developmental transitions.
In C. elegans, we also observed intrinsically clustered patterns of FFLs as well as CIR, likely due to the higher clustering coefficients in the C. elegans data (Supplementary Data).
The sixth type, the feedback 2 undire cted module (FB2Uc), is formed by clusters of protein interaction-mediated transcriptional regulatory loops, a motif that mediates undirected feedback between a TF and its target, through a common partner in the protein interaction network (20). A first cluster generalization of this motif is the regulonic star, where multiple targets of a regulator interact with the same feedback protein, including the regulator itself (32). In Cele FB2Uc 13 for instance, the transcriptional co-activator EYA-1 functions as feedback mediator in the development of various tissues (118). A second cluster generalization is the regulatory interacting double-star, where one or a few regulator–target pairs share a common set of partners in the protein interaction network (32). For example, in Cele FB2Uc 39, DAF-3 is a central protein interaction partner for a number of proteins, as well as a transcriptional regulator to DAF-7 and DAF-8 that both in return genetically interact with the protein interaction partners of DAF-3 (Figure 3). This extreme example combines these two types in a protein complex, where some members transcriptionally regulate other members (e.g. Atha FB2Uc 1 (Supplementary Data), Atha CIRc 0).
The seventh type, the feedback undirected module (FBUc), is similar to the FB2Uc, but now contains clusters of motifs consisting of two coherent regulatory edges and one undirected interacting edge. Here, we also detected the regulonic star, where now the feedback protein is another regulator that targets the first regulator. In Atha FBUc 2, for instance, PIL5 and PIF4 target back to SEP3. Hence, a member of a protein complex also actively regulates its regulating TF. Due to the FBU/FFL and FBU/COR motif overlap, this module is also FFLc and CORc to some extent. Analogously, the regulatory interacting double star now consist of a regulator with a set of protein interaction partners that targets another regulator that transcriptionally regulates those protein interaction partners. In ATHA FBUc 3, MYB33 physically interacts with all the targets of LFY3 that is a direct target itself of MYB33 (Figure 3).
In the ALL modules, all motif types were clustered together. Here, we typically found integration of different motifs and combinations of different modules. As an example, in a merged module, which consists of COP, COR and FFL motifs, and functions in flower development, the homologous miRNAs miR156/157 post-transcriptionally regulate members of the squamosa–promoter binding protein-like (SPL) gene family (Figure 5A and Supplementary Figure S6). This miR156/157-SPL module is a regulatory hub important for the transition from vegetative phase into flowering. It is closely linked with environmental signals like temperature, salt and light (119). On top of that, SEPALLATA3 (SEP3) targets both miRNAs and SPLs, creating TF-mediated miRNA FFLs. From literature, it is known that SEP3 is a responsive gene of SPL3 in the ambient temperature-responsive flowering (120). Here, we observed that SEP3 also functions as an upstream regulator by binding other SPL TFs that are upregulated upon abiotic stress.
Figure 5.

ALL modules formed by clustering different motif types together. Expression profiles are depicted for the 10 abiotic stress conditions with maximal up- and downregulation. (A) Merged FFLc, COPc and CORc module from ALLc 50 largely overlapping with COPc 10 and CORc 5. Here, the homologous miRNAs miR156/157 post-transcriptionally regulate members of the squamosa–promoter binding protein-like (SPL) gene family. Both miRNAs and SPLs are targeted by SEP3. The functional diversification of the different spl-genes is illustrated by the mixture of different stresses in the conditions with maximal up- and downregulation for the abiotic stress compendium. (B) Overview of the different modules with zinc finger homeodomains: ALLc 147, COMc 14, COPc 11, COPc 47 and COPc 14. Experiments with specific tissues, root (R) and shoot (S), are marked below the expression profile matrix. Genes are sorted based on the gene family tree (121).
In the partially overlapping modules of Atha ALLc 93, ALLc 147, COMc 14, COPc 11, COPc 47 and COPc 14, which combine COP, COR, COM and FFL motifs, a gene family of eleven zinc finger homeodomains interact through P and H interactions, while several of them are targeted by the flowering regulator AGL15 and/or transcriptionally regulate genes involved in secondary cell wall and glucosinolate biosynthesis (Figure 5B). In Arabidopsis, zinc finger homeodomains are known to homo- and heterodimerize and play overlapping regulatory roles in floral development (121). We found that the HB-genes in the protein cluster COMc 14, COPc 14 and ALLc 93 are significantly coexpressed (Supplementary Figure S7). We also observed that the highly similar homologs hb30/hb34 are expressed under the same abiotic stress conditions and that they are dynamically expressed in osmotic stress conditions in roots together with zfhd1 of which the upregulation by high salinity was already reported (122) (Figure 5B and Supplementary Figure S7). Furthermore, we perceived in the abiotic stress expression compendium different expression preferences for the different zinc finger homeodomains upon osmotic, salt, heat and cold stress: most of them are preferentially expressed in root tissues, only hb23 also shows expression in shoot tissues (Figure 5B). Differential expression of zinc finger homeodomains under abiotic stress conditions has already been shown in Brassica rapa and Vitis vinifera (123,124). Together this leads to the assumption that after duplication the hb-genes have diverged to regulate development under specific abiotic stresses. Further research on the evolutionary diversification of these zinc finger homeodomains in the abiotic stress response is needed to support this hypothesis.
The advantage of our data-integration methodology, which captures different experimental methodologies and resources, is, for example, shown in the integrated complex modules of Arabidopsis SWI/SNF chromatin remodeling complexes (Supplementary Figure S8) and the C. elegans coregulated module Cele CORc 26 (Supplementary Figure S9). The SWI/SNF chromatin remodeling modules are formed by complexes that interact with each other and consist of protein–protein interactions gathered by Y2H, tandem-affinity purification (TAP), protein-fragment complementation assay and other techniques (125–127). According to their experimental methodology, TAP detected the SWI/SNF complex around the central adenosine triphosphatases BRM or SYD (127), while Y2H identified binary interactions between the SWI/SNF subunits and several TFs and cofactors (Supplementary Figure S8). In C. elegans coregulated module Cele CORc 26 with data-integration of ChIP and Y1H, Y1H has the highest incoming degree, while ChIP has the highest outgoing degree in the module, as can be expected from their experimental methodology (Supplementary Figure S9).
Overall, we found similar network motif modules in the integrated GRNs of C. elegans and A. thaliana, suggesting these topological patterns are universal in networks of gene regulation. A dynamic visualization of all modules can be found in http://bioinformatics.psb.ugent.be/supplementary_data/jofoo/networks/. This interactive visualization groups all modules per type with links to the expression matrices. Through the search bar it is possible to look for genes of interest in both species. This site also offers a downloadable file with all genes and interactions per module.
A superview analysis of network motif modules
The network motif modules are part of integrated GRNs, where they influence one another and might be active under different conditions. We developed a method to investigate modules in the network context, where we studied interactions between the modules and regulators through statistical analysis to find enrichment for functional and regulatory important edges (‘Materials and Methods’ section) (Figure 1). Linking the modules through homologous interactions and/or shared genes results in groups of modules involved in similar processes. For example, in A. thaliana we found six alternative splicing modules connected through homology edges and controlled by abiotic stress (Supplementary Figure S10). In addition, we looked for TF and miRNA regulators specifically targeting one or more modules (Supplementary Figure S11). In a first example we confirmed the regulation of the cellulose synthase complex (CSC) COMc 36 by MYB46 in A. thaliana (128) (Figure 6A).
Figure 6.

Through the superview analysis framework, we discovered previously known (A) and unknown (B) regulators for specific modules, as well as (C) novel edges for known regulators. (A) Cellulose synthase complexes (CSC) in COMc 36 are upregulated by MYB46. While MYB46 binds four module genes, the other regulators bind only one gene in the module. The module consists out of the primary cell wall CSC (CESA3, CESA1 and CESA6), the secondary cell wall CSC (CESA4, CESA7 and CESA8) and KOR1, a membrane-bound 1,4-β-D-glucanase (150,151). This module is tightly coexpressed in the abiotic stress compendium and upregulated upon brassinosteroid treatment (152) and salt stress conditions. COMc 36 has a significant ECD score under genotoxic, heat, oxidative and salt stress. In birch, overexpression mutants of MYB46 show thicker secondary cell walls and a higher tolerance to salt and osmotic stress (153). Cellulose synthases bind microtubules, hence stabilizing cellulose synthase localization at the plasma membrane and rendering plants less sensitive to salt stress (154). The relation between MYB46 and CSC is therefore important for the stress tolerance of crops. This example highlights the potential of integrating regulators with network motif modules. (B) The homeodomain TF CEH-30, which functions in neuronal cell fate and sex-specific apoptosis, was found to target a homolog group of heat shock proteins in worm. (C) CBF4 and ZML2 transcriptionally regulated the MYB/MYC module Atha COMc 48. The TFs MYB28, MYB29 and MYB76 control aliphatic glucosinolate biosynthesis (155), while MYB51 and MYB34 regulate indole glucosinolate biosynthesis (156). The JAZ-interacting TFs MYC2, MYC3 and MYC4 form together with the MYB TFs dimeric TF complexes to regulate the different glucosinolate biosynthesis pathways (95). Glucosinolates, a class of secondary metabolites mainly found in Brassicaceae, are part of a complex response to a variety of abiotic stresses. A decrease in aliphatic glucosinolates modifies the abundance of aquaporins and hence the water uptake in roots, thereby increasing drought and salt tolerance (157). Only the aliphatic glucosinolate biosynthesis TFs are directly bound by CBF4. In our abiotic stress compendium, we observed an upregulation of aliphatic glucosinolate biosynthesis (MYB26 and MYB76), indolic glucosinolate biosynthesis (MYB51), MYC2 and also of CBF4 upon salt stress; for MYB51 and CBF4 this is mostly in roots. It has been observed that CBF4 significantly alters the accumulation of at least five glucosinolates but the direct regulatory mechanism between CBF4 and glucosinolate synthesis has not been described (95). Here, we showed that the drought responsive gene cbf4 is an upstream regulator of the aliphatic glucosinolate biosynthesis which increases the tolerance to drought and salt stress. The function of zml2 in this context is still to be determined.
In a second example, we illustrated that the superview framework is able to highlight unexplored module-regulator connections. Here, the homeodomain TF CEH-30, which functions in neuronal cell fate and sex-specific apoptosis, targets a homolog group of heat shock proteins in worm (Cele COMc 35) (Figure 6B). Finally, we also found novel targets for known regulators. We link CBF4, a regulator of the ABA dependent drought response (129), and ZML2, a critical TF in the cry1-mediated photoprotective response (130), to aliphatic and indolic glucosinolate biosynthesis in Atha COMc 48 (Figure 6C). This module has a significant ECD in drought and salt stress.
These examples show how the network motif modules can be integrated into a larger context beyond individual modules and how general topological patterns can enable the study of stress related mechanisms. Through usage of different expression compendia or additional regulatory data, other processes can be explored as well.
Phylogenetic decomposition of the networks
Through phylogenetic decomposition of these integrated GRNs, we investigated how novel genes are integrated in GRNs. Therefore, genes were arranged in age groups or phylostrata based on the oldest lineage that still contained an ortholog (Supplementary Figure S12 and ‘Materials and Methods’ section). This resulted in, respectively, 7 and 10 age groups for C. elegans and A. thaliana (Supplementary Tables S10 and 11). A total of 61% of C. elegans and 99% of A. thaliana protein-coding genes in the GRNs could be given an age label. In the worm integrated GRNs, the groups Eukaryota and Caenorhabditis each contain more than 25% of all age-labeled genes, the groups Eumetazoa and Cellular organisms each have around 15% of these genes, while the other age groups each take 5% or less. In the plant integrated GRNs, nearly half of all age-labeled genes reside in the oldest age group Green plants, followed by 27% in Land plants, 7% each in Seed and Flowering plants and <5% in the other age groups. We restricted ourselves to P, G, D and R interactions in the networks: 44% and 99% of C. elegans and A. thaliana interactions respectively, have associated age labels. For worm, the age groups with most interactions were Eumetazoa (36%), Eukaryota (30%) and Caenorhabditis (15%) (Figure 7). For Arabidopsis, interactions are concentrated in Green plants (46%), Land plants (30%) and Flowering plants (10%) (Figure 7 and Supplementary Table S12). Hence, the interactions are mainly distributed over the age groups containing the most genes. Among these age groups are the oldest ones like Eukaryota in worm and Green and Land plants in plant. Another reason for the interaction distribution is the fact that older genes are better studied than young genes and therefore more represented in the networks for both species (Supplementary Tables S10 and 11, (39,40)). The average degree is mostly confirming these observations (Supplementary Table S13). For worm, the highest average undirected, incoming and outgoing degree are observed for the Eumetazoa. The further away from this age group, the lower the degrees become. For plant, the highest average undirected degrees are seen in the Land and Flowering plants, although with the exception of Rosids and A. thaliana, other age groups have only slightly lower average undirected degrees. Although average incoming degrees are similar for all plant age groups, the average outgoing degree of the Flowering plants towers.
Figure 7.

Total number of directed and undirected interactions between age groups of Arabidopsis thaliana (left) and Caenorhabditis elegans (right). The nodes are scaled according to the number of genes in the age group and colored according to age (darker = older). Red edges are within the age groups, blue edges are between the age groups. The thickness of the edge is scaled to the number of interactions (full list in Supplementary Table S12).
Protein–protein interactions preferentially occur between proteins of similar age, while for protein–DNA interactions, regulatory TFs favor older or same-age target genes
To investigate the general interaction preference of the different types of molecular interactions in our integrated GRNs, for each interaction type we analyzed whether they preferred to interact within or between age groups. In both species, we found that P interactions are preferentially age homogeneous. In A. thaliana and C. elegans, respectively, 40 and 34% of the interaction partners are of the same age. This is significantly more than expected by random (Atha P-value = 2e-16; Cele P-value = 2e-16, Z-test, 1000 random network permutations with preserved age and degree distribution). The interaction partners of protein–DNA interactions were less frequently of the same age, but still significantly more than expected by random (Atha age homogeneous only ChIP and Y1H: 31%, P-value = 0.0034; Cele age homogeneous D: 30% P-value = 0.036, Z-test). The full D set of A. thaliana, which includes the regulatory interactions, did not prefer to interact with genes of similar evolutionary age (Atha age homogeneous 25.2%, P-value = 0.50, Z-test), as well as the regulatory (Cele age homogeneous: 25.8% P-value = 0.36, Z-test) and genetic interactions (Cele age homogeneous: 33.4% P-value = 0.073, Z-test) in C. elegans. This is understandable, since the latter interactions are not necessarily direct interactions that might involve intermediate nodes in the networks.
To investigate the interaction preference in relation to evolutionary age in the integrated GRNs, for both species we compared the number of observed interactions within and pairwise between the different age groups versus the expected number of interactions based on randomized networks with the same age distribution as the real networks. Due to the differences in age homogeneity for the different interaction types (see above), we here show the results of the physical protein–protein and protein–DNA interactions, while the results of the whole set of undirected and directed interactions can be found in the Supplementary Data. This analysis indicated that some age groups attracted many more interactions than expected by random. In both C. elegans and A. thaliana protein–protein interaction networks, we observed an interaction age preference toward the own age group or to the next age groups i.e. the highest Z-scores are found on or near the main diagonal of the age group matrix (Figure 8). These results are confirmed for the undirected networks data (Supplementary Figure S13) and through the calculation of the interaction density (Supplementary Data). Hence, proteins prefer to functionally interact with proteins of similar evolutionary age. Since the overlap between the homologous interactions and the other type of interactions is small (Supplementary Figure S1), we can exclude that the interaction preference of genes in the same age group originates from interactions between homologs.
Figure 8.

Interaction age preference for physical protein–protein and protein–DNA interactions in Caenorhabditis elegans (left) and Arabidopsis thaliana (right). Enrichment (Z-score and associated P-value corrected for multiple hypothesis testing) of the comparison of the observed number of interactions within and between age groups in the real networks versus the expected number in 1000 randomized networks with the same age distribution. In the protein–DNA networks, the regulatory TFs are on the vertical and the target genes are on the horizontal axis.
Considering the protein–DNA interaction networks in worm, we noted strong preferences of Ecdysozoan TFs for target genes from Cellular Organisms and Eukaryota, of Eukaryotic TFs for Eukaryotic target genes and of Caenorhabditis TFs for Eumetazoan target genes (Figure 8). For plant physical protein–DNA data, regulatory TFs from Eudicots or older age groups preferred to bind target genes from the Green or Land plants (Figure 8). We found similar results for the directed networks (Supplementary Figure S13) and through the interaction density analysis (Supplementary Data). Hence, for the protein–DNA interaction networks in worm and plant, the highest Z-scores are found on or above the main diagonal in the age group regulatory TF-target gene matrix, indicating that regulatory TFs tend to bind target genes of similar or older evolutionary age.
Interaction age preference of motifs and modules
Since motifs are considered to be small building blocks of networks, we investigated how novel genes are incorporated in integrated GRNs at the motif level. Therefore, motifs were divided into 13 motif age types based on the age pattern of the different node positions: either all nodes are of the same phylostratum (SSS, S = Same), there are two age groups in the motif (Y = Young and O = Old) or they are all from a different phylostratum (Y = Young, M = Middle and O = Old). Motifs with internal symmetry (e.g.: PPP, DDP) were sorted according to decreasing age to remove overlap between the motif age types. We calculated motif age preference by comparison of the observed motif age patterns to the expected patterns by permuting each of the three node positions with preserved age distribution per node (‘Materials and Methods’ section and Supplementary Table S16). For the complex motifs, we observed a strong preference to be age homogeneous in both species, especially in A. thaliana (Figure 9A). Also in C. elegans, at least one edge in the complex motifs is between proteins of similar age. Similarly, the COR motifs tended to be completely age homogeneous (SSS-type) or of the OYY/YOO-type, where the targeted nodes are of similar age and interact undirectedly. In addition to the preferentially age homogeneous type (SSS-type), both the plant and worm COP motifs were composed of the YYO age type, where two younger TFs of the same age interact and target a gene of a different phylostratum. In addition, we observed a strong enrichment for the OYM age type in plant COP motifs, where a physical bound between an old and young TF regulates a middle-aged target gene. The feed-forward loops (FFL) showed the strongest preference to be age heterogeneous: OYM was the strongest age motif type enriched in both species, followed by YOO and MOY in plant, and by SSS, MOY and YYO in worm. Hence, novel genes are incorporated at every position in the FFL. CIR motifs were preferential age homogeneous in C. elegans or of the heterogeneous OMY-type in both species. The FB2U motifs followed mostly the same trends as the complex motifs while the FBU motifs were similar to the FFL motifs. This can be explained by the overlap between these motif types (Supplementary Figure S5). In C. elegans, almost all motifs displayed enrichment in the homogeneous motif age type due to the dominance of the Eumetazoa interactions, e.g. the DDD motif consists out of 23% Eumetazoa SSS type and only 0.67% other SSS-type motifs. Overall, we observed that undirected interactions in motifs tended to be age homogeneous, while directed interactions in motifs preferred to be age heterogeneous.
Several of the observed motif age types can originate from gene duplication. To investigate the contribution of duplicates to motif formation, we first looked at the number of motifs consisting out of at least one pair of homologs. We observed that homologous genes only appeared in at most 2 and 1% of motifs excluding H interactions in C. elegans and A. thaliana, respectively (Supplementary Table S17 and 18). They appeared together in up to 6% of all DdD, DDD, DDP, DPD, DPP, PDD and PPP motifs in both species. Secondly, we compared the number of genes with H interactions in the complete interaction set versus in the motifs (Supplementary Table S19). For protein–protein, regulatory and genetic interactions, we found no preferential motif formation of genes with homologous interactions in both species. For protein–DNA interactions, we found that genes with homologous interactions contribute more to motif formation in A. thaliana than expected.
Different network motifs have specific evolutionary age types associated. To investigate whether the preferred age patterns in the motifs are also preferentially incorporated into the modules upon motif clustering, we compared the set of clustered motifs to the full set of motifs (Figure 9C). In both species, we found in the COMc, COPc, CORc and FFLc modules a strong correspondence between the over-represented motif age types in their underlying network motifs and those that are clustered in their modules. In the CIRc and A. thaliana FBUc modules, we observed no real preference of clustering because almost all motifs are clustered within the modules. In FB2Uc and C. elegans FBUc modules, age heterogeneous motif types are preferentially incorporated. The age homogeneous motif types SSS in C. elegans are less clustered than expected in almost all modules. This might be explained by the over-representation of age homogeneous motif types from the Eukaryota and Eumetazoa. Overall, we observed that the over-represented age motif types clustered more than the other types in the modules i.e. we observed similar patterns in Figure 9C as compared to Figure 9B.
The evolutionary age groups contributed differently to the modules. Modules were mostly composed of genes from the evolutionary age groups containing the most genes i.e. Eukaryota, Eumetazoa and Caenorhabditis in worm, and Green and Land plants in plant (Supplementary Figure S14A and B). Hence, especially in A. thaliana, younger genes were more inclined to attach to modules mostly composed out of older genes instead of forming modules on their own. Looking at the individual module types we noted that there are COMc modules that are age homogeneous in the older groups, Green and Land plants in A. thaliana and Eukaryota and Eumetazoa in C. elegans (Supplementary Figure S14C and D). In the other modules, there is little contribution of the other younger age groups. Age homogeneous modules with directed interactions are less abundant and appeared within the oldest group of A. thaliana i.e. Green and Land plants and within the Eumetazoa in C. elegans. This is in agreement with the preferential clustering of more age heterogeneous motifs in these regulatory modules (Figure 9C).
Atha COMc 48, already discussed above, is a prime example of how innovation is introduced in GRNs (Figure 10). Indolic glucosinolate biosynthesis originated in the Land plants, and therefore the indolic glucosinolate biosynthesis TFs (MYB51 and MYB34) belong to the Land plants phylostratum. Together with the JAZ-interacting basic helix-loop-helix TFs MYC2, MYC3 and MYC4, they form heterodimer TFs that transcriptionally activate glucosinolate biosynthesis genes. From Brassicales on, not only indolic, but also aliphatic glucosinolates appeared as secondary metabolites (131). Therefore, the aliphatic glucosinolate biosynthesis TFs (MYB28, MYB29 and MYB76), which belong to the Brassicales phylostratum, were introduced in the GRNs through interactions with the MYC TFs.
Figure 10.

Atha COMc 48 colored according to evolutionary age with genes originating at the Brassicales age group in dark green, while older genes are in light green (Functional interpretation and superview of module can be found in Figure 6C). The aliphatic glucosinolate biosynthesis TFs (MYB28, MYB29 and MYB76) find their origin in the Brassicales age group (R2D3-MYB subgroup 12), while the indolic glucosinolate biosynthesis TFs (MYB51 and MYB34) and MYC TFs (MYC2, MYC3 and MYC4) have their origin in the Land plants. This is consistent with the observation that aliphatic glucosinolates are only found within the Brassicales plant lineage.
DISCUSSION
Data integration through network motif modules
Since different molecular interaction types influence gene regulation, we developed a general data integration framework to study integrated GRNs of directed protein–DNA, transcription regulatory, miRNA–mRNA interactions and undirected protein–protein, genetic and homologous interactions. Our data integration framework of composite network motif modules is unbiased, since it does not favor any interaction type or experimental methodology over the other, and preserves the identity of the interaction type as compared to other data integration methodologies that benchmark using true positive data sets, Gene Ontology or KEGG (132–134). The integration of complementary data types through two- and three-node motifs provides useful insights in the study of gene regulation and in GRN evolution. Motifs, like the well-described FFL, connect the regulatory levels (transcriptional and post-transcriptional) and integrate the directed and undirected interactions into easy interpretable patterns of gene regulation. Also, the incorporation of homologous interactions in motifs provides insights in how motifs and networks are formed by evolution. Next to the already integrated interactions, the network could still be expanded with epigenetic regulation and post-translational modifications, which are also known to affect gene regulation (24,135), or with more data alltogether.
Contrary to previous data integration studies (22), we also highlighted the effects of combining different experimental methodologies in the protein–protein interaction networks (Supplementary Figure S8) and in the protein–DNA interaction networks (Supplementary Figure S9 and Table S3). One advantage is that different methodologies are complementary for a given data type and provide a more holistic view on gene regulation. For example, the integration of Y1H and ChIP data created extra CIR motifs (DdD, DmD) in the worm networks, indicating that there is possibly condition-dependent feedback regulation at the transcriptional and post-transcriptional level. Although we barely detected the two-node miRNA-TF feedback loop in the networks, which is contrasting to other studies that used lower computational cut-offs on miRNA–mRNA interaction predictions (10), we found the three-node miRNA-TF feedback DmD in the Y1H and in the combined Y1H and ChIP networks of worm. Hence, an intermediate regulatory TF confers the feedback of a TF to the miRNA it is regulated by. The joining together of different experimental methodologies also poses some challenges, as demonstrated by the biases introduced in network randomization and hence network motif enrichment. The best-known motif in GRNs, the FFL/DDD (113), despite its abundance in both species and its important regulatory characteristics, is not found to be enriched in the integrated GRNs of both species, and only in the ChIP data of both species, as has been observed previously (22). We also noted other differences in network motif enrichment between Y1H, ChIP and the combined data (Supplementary Data). We hypothesize that this different network motif enrichment can be mainly attributed to the edge swapping randomization of the networks, which has drawn criticism before (136–138). Edge swapping randomization while preserving the degree distribution limits the randomization options for hubs and this affects the experimental methodologies differently. Since Y1H and ChIP data generate a different network topology with respectively 1–3 times more regulators than targets in Y1H and 5–20 times more targets than regulators in ChIP; more target gene versus regulator hubs, a higher clustering coefficient and a higher overall centrality for Y1H as compared to ChIP, this results in different randomized networks and therefore different network motif enrichment (see Supplementary Data). As network motif enrichment is highly sensitive to experimental methodology, network topology and randomization, we recommend to study network motif presence and aggregation into modules.
Overall, we found the same three-node motif types in both species. COM, COP, COR, FFL and FB2U motifs were already detected previously (15,20,22,31), additionally we detected the CIR motif where three regulators act upon each other through a feedback loop and the FBU motif where feedback to a linear path of directed edges is provided by an undirected interaction (Figure 3). In both species, both network motifs at the transcriptional level largely overlapped with the FFL DDD (Supplementary Figure S5), indicating that intricate regulation between TFs occurs through feedback loops consisting of both transcription regulatory and physical protein–protein interactions. As we also incorporated miRNA–mRNA interactions, we also found the miRNA-mediated FFL (MMD) and the TF-mediated miRNA FFL (DDM) at the post-transcriptional level (10,21–23).
Although network motifs are basic building blocks of GRNs, several studies have pointed out that aggregation of motifs into larger modules occurs naturally and might be more important to consider, not only from a topologically point of view, but also functionally and evolutionary (28,29,32,33). The module level is also claimed to be the most conserved one across species (139). Therefore, our data integration framework focused on network motif modules. We were able to detect topological organizations of integrated GRNS which are similar in C. elegans and A. thaliana. The network motif modules, COMc, COPc, CORc, FFLc and FB2Uc have been described in yeast and were previously detected either based on visual inspection (31), or by statistical analysis (32). Here, we confirmed these network motif modules in worm and plant and expanded them with the CIRc and FBUc modules (Figure 3). In addition, we also extended the interaction set by integrating miRNA–mRNA, regulatory and homologuous interactions. Next to this, we showed that the aggregation of different composite network motifs (ALLc) can provide useful functional insights (Figure 5). The fact that these network motif modules are detected in two unrelated species, and comparable patterns have been detected in yeast (31,32), suggests that these topological patterns might be universal throughout GRNs in all species. The network motif modules can be linked to specific functions in GRNs and by integrating gene expression data, we revealed the dynamics of these network motif modules during development or upon stress. Through the superview analysis, in which we connected the different network motif modules with one another and with regulators, we discovered novel functional and regulatory relations between modules in the integrated GRNs context. This really demonstrated the power of our data-integration framework, since genes and regulators were found to be interacting in novel, previously unstudied, biological contexts. Higher-order organization like these network motif modules has also been observed in non-molecular and non-biological networks (33). Here, we have provided a framework to study integrated GRNs in higher eukaryotes through network motif modules.
Evolution of integrated GRNs
In this study, we used phylogenetic decomposition to study the evolution of integrated GRNs and the incorporation of novel genes (35). The resolution of the age group split is dependent on the availability of genome information of the different taxa. For A. thaliana we are able to get a refined classification supported by multiple species in most age groups starting off from the Green Plants for almost all protein-coding genes in the integrated GRNs (Supplementary Figure S12) (108). However, some important taxonomic groups lack a representative with a sequenced genome e.g. ferns. For C. elegans, the availability of genomes in ‘more distant’ taxonomic groups is much sparser, which results in larger gaps between the different age groups (Supplementary Figure S12). However, for C. elegans the phylogenetic composition goes all the way down to Cellular Organisms i.e. Bacteria. Furthermore, only 61% of all protein-coding genes in the integrated GRNs of C. elegans could be classified in evolutionary age groups. Hence, the study of the evolution of the worm integrated GRNs is on only part of the networks. We found the interactions to be mainly distributed over the age groups containing the most genes, which included the oldest age groups in both species.
Several methods have been used to investigate interaction age preference in biological networks. One of the first studies characterized the age-dependent evolution of yeast protein–protein interaction networks based on the interaction density of the networks, which measures the numbers of observed over expected edges between nodes of paired age groups, normalized for the size of the network (42) (Supplementary Data). The interaction density is an intrinsic property of biological networks, but in order to infer preference patterns a comparison to randomized networks with conserved degree distribution and conserved age distribution, is needed (36). Other studies compared the observed number of interactions in the actual networks to the expected number to occur by chance in random networks that preserve the degree distribution of each age group (38,40). An intuitive view on interaction age preference is obtained by counting the edges between nodes of paired age groups and comparing these numbers to the ones obtained by permutation analysis of the gene-evolutionary age group assignments (39). In order to accurately investigate interaction age preference, we applied several of the above described computational approaches and we largely obtained similar results using different measures (count analysis in ‘Results’ section and interaction density analysis in Supplementary Data), as has been observed previously for undirected interactions. In this respect, the preferential interaction between proteins of similar age was demonstrated in protein–protein interaction networks in yeast (40) and human (36) and for coexpression networks in A. thaliana (39). However, these studies mostly used a limited number of age groups and only one type of interaction network. Using detailed phylogenetic decomposition, we showed that for undirected protein–protein interactions in C. elegans and A. thaliana, while the majority of interactions is between older and younger genes (Figure 7), genes preferentially interact with genes of a similar age (Figure 8). Similar results are obtained for all undirected interactions in C. elegans, hence including genetic interactions. Interactions between paralogs can only partially account for the age-dependency in the undirected networks. Overall, we can conclude that functional interactions tend to occur between proteins of similar evolutionary age. This indicates that introduction of a novel biological function involved the integration of a set of interacting genes in the GRNs. We expanded the interaction age preference to directed interactions (protein–DNA and regulatory) in both species. However, we have to take the distribution of TFs over the different age groups into account upon interpreting the results. In C. elegans, the TFs distribution over the age groups is shifted toward the Eumetazoa, which has more than half of the studied TFs (Supplementary Table S14). Younger TFs in A. thaliana are scarce and lack interaction data; in the Rosids and A. thaliana age group even no TFs were studied (Supplementary Table S15). With these limitations in mind, we found that regulatory TFs favored older or same age target genes. Contrary to undirected interactions, directed interactions seem to cross the age groups as is also observed on the motif and module level. We also found that interactions with experimental binding data (physical protein–protein and protein–DNA interactions) are generally age homogeneous, while interaction types that can also be indirect (genetic and regulatory) do not show any preferential age homogeneity. Our findings correspond to the observation that in the course of evolution of a GRN regulatory interactions are acquired much faster than protein–protein and genetic interactions (140).
Different mechanistic models have been introduced to explain the evolution of biological networks, especially protein–protein interaction networks. In the ‘preferential attachment’ model, new proteins preferentially attach to highly connected nodes (141). The ‘duplication and divergence’ model states that new proteins originated through duplication, initially connect to all the neighbors of the node that has been duplicated and that connections diverge over time (142). However, these models are not able to mimic the high modularity and the homogeneous age preference of protein interactions. In the ‘crystal growth’ model, the network grows by anchoring and extension, where a node increases its degree either by becoming a new module (anchoring) or by extending an existing module (42). This model incorporates the tendency of protein–protein interactions to interact within the same age group, the central aggregation of older subunits and the peripheral scattering of younger subunits and hence corresponds with our findings at the interaction level. The most recent model for protein–protein interaction evolution that mimics real protein–protein interaction networks the best, is the ‘network motif’ model, which is based upon the fact that network motifs or protein clusters are incorporated into the network instead of single proteins (43). It was confirmed in a yeast protein–protein interaction network that proteins of the same age class tend to form motifs, are densely interconnected, co-evolve, share the same biological function and tend to be within protein complexes (41). Similar to the network motif model, motifs were also used as building blocks to model transcriptional networks in bacteria (143). In accordance with these models, we looked into age patterns in the network motifs and network motif modules to get insight in the evolutionary mechanisms for GRN formation (Figure 9). Our age preference analysis at the motif and module levels indicated a strong age homogeneity preference for COM motifs and COMc modules and a strong age heterogeneity preference for FFL motifs and FFLc modules, especially in A. thaliana, which is in agreement with our results on interaction age preference of undirected and directed interactions, respectively. In C. elegans this is only partially true, since here we found COM motifs with at least one age homogeneous interaction more in COMc modules, while age homogeneous COM motifs are less clustered and we did find an over-representation of age homogeneous FFL motifs as well. This can be explained by the dominance of the Eumetazoa and Eukaryota age groups in the C. elegans interactions, motifs and modules. Overall, we found the over-represented age types in de motifs to be more incorporated in the modules. Compared to the other module types, the COMc modules were more inclined to comprise a single age group in both species (Supplementary Figure S14). However, modules were mostly composed out of genes from the evolutionary age groups containing the most genes i.e. Eukaryota, Eumetazoa and Caenorhabditis in worm, and Green and Land plants in plant. Hence, they mostly consisted of older genes and only had a smaller fraction of younger genes. This hints to the fact that the younger genes likely attach to the older core of the network during GRN evolution. Taking into account our results at interaction, motif and module level, we postulate that novel genes attach together to the GRNs in a specific biological functional context, regulated by one or more TFs that also target older genes in the GRNs. Hence, for the undirected interactions, this is in accordance with the ‘network motif’ model (43), although single genes might accompany the addition of network motifs and modules in GRN formation over evolutionary time, as low-connected genes are missed through data-integration based on network motifs or network motif modules. Despite the fact that these networks are far from complete, we hypothesize that the observations we make for the older age groups, which are already well represented in the networks, will remain. In addition, we obtained for the evolutionary analysis of the protein–protein interaction networks similar results as the coexpression networks of Ruprecht et al. (39), which are genome-wide and hence more complete.
Influence of gene duplication on network evolution
In C. elegans, SSD make up the biggest portion of the duplicates. These are frequently partial or lack the original regulatory sequences (44). In A. thaliana, WGD are the source of many duplicates, next to SSDs (45). A particular difference between integrated GRNs in A. thaliana and C. elegans is that homologous plant TFs targeting the same genes tend to physically interact more both through protein–protein and protein–DNA interactions, but homologous interactions between TFs occur more than seven times more in plant than worm. The faster divergence of genes after SSD in terms of divergences of sequence (49), expression (50), protein interaction partners (51) and regulatory connections (52) makes that homologous relations between TFs in C. elegans are no longer detected.
The differences in divergence between SSD and WGD also have an influence on the age groups classification of genes since they are categorised on the oldest occurring species in the gene family or with a shared ortholog. WGD-duplicates tend to stay within the same gene families, further expanding them, while the faster divergence of SSD-duplicates allows them to create novel gene families. This potentially explains why there is a much higher number of older genes in A. thaliana and why there are also more genes in the younger groups of C. elegans than in A. thaliana: 14% of the A. thaliana genes originated after the Brassicales split off (estimated 68 MYA ago) compared to 29%, after C. elegans diverged from other Caenorhabditis worms (estimated 60 MYA ago). This is reflected in the numbers of TFs in both species’ age groups: 14% of worm TFs belong to Caenorhabditis or younger age groups, while only 2% of plant TFs are associated to Brassicales or younger age groups (Supplementary Table S14 and 15). TFs expand through duplication, often WGD, and are retained for long periods after duplication (48,144,145). In C. elegans, the TF age distribution is diverse, which links with the fast evolution in sequence and function, and often loss probably because of dosage balance reasons, after SSD (145,146). Still despite these differences it leads to an interaction pattern with no single preferential age group but its own in the undirected networks of both species.
Motifs can originate from duplication of genes. In both species we see an overlap between HPP/PPP motifs, which hints to the contribution of duplication on complex motif formation. Since this overlap is only for a very small fraction of the total amount of PPP motifs, we exclude that duplication is a major creator of motifs but still on the cluster level this overlap gives rise to star like modules around a homolog pair (e.g. Cele COMc 70, Figure 3). The influence of duplication on network motif formation is visible within the motifs with directed interactions in A. thaliana. Similar results were obtained for genes after WGD in yeast (147). For motifs containing protein–DNA interactions, we found that homologs contribute more to motif formation in A. thaliana than expected by chance. Likewise, we found a large overlap between HDD/DDD and HDD/PDD motifs in Arabidopsis and not in C. elegans. Homologous interactions between TFs and the overlap between motifs can explain the over-representation of certain age patterns in motifs. In A. thaliana the COP motifs are preferential of the YYO/OYM-type, where two younger TFs of the same age or an old and young TF interact and target a gene of a different age group. In the context of duplication, this could be seen as a homodimer (YYO) which becomes a heterodimer after divergence (OYM). This explains the overlap between both motif types HDD/PDD. The FFL motif DDD turned out to be preferential age heterogeneous, OYM in both species, followed by YOO and MOY in plant, and by SSS, MOY and YYO in worm. This shows that novel genes are incorporated at every position in the FFL, but also that additional regulatory layers could be generated by the doubling of one of the TFs and the gain of a regulatory interaction. The gain of regulatory layers shows that evolution increases the complexity of GRNs, which allows adaptation and more specific regulation of downstream processes (140). This is in correspondence with the fact that novel TFs show a higher target binding specificity in A. thaliana as compared to TFs of ancient families (148). In C. elegans, we detected overlap between HMM/GMM, which shows that miRNAs of the same family often are genetically linked and overlap between DDH/DDP/DDG motifs, which represents interacting duplicate targets through either genetic or protein–protein interactions.
In summary, we report the presence and biological relevance of network motif and network motif modules in the integrated GRNs of C. elegans and A. thaliana. These topological patterns are potentially universal in networks of gene regulation. Depending on the interaction type being functional or regulatory, we find different interaction age preferences in GRN evolution, which are similar in both species.
DATA AVAILABILITY
A dynamic visualization of all modules and a file with all genes and edges per module can be found at (http://bioinformatics.psb.ugent.be/supplementary_data/jofoo/networks/). The source code of the computational data-integration framework can be found at https://gitlab.psb.ugent.be/jofoo/NetworkMotifModules.git.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Sofie Demeyer for technical advice on ISMA, and Tom Michoel for technical advice on SCHype and discussions on network motif clustering. We thank Thomas Van Parys for help with the design of Cytoscape figures and the accompanied website.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
European Union Seventh Framework Programme (FP7/2007-2013) under European Research Council Advanced Grant Agreement [322739-DOUBLE-UP]. Funding for open access charge: ERC Advanced Grant Agreement (322739-DOUBLE-UP).
Conflict of interest statement. None declared.
REFERENCES
- 1. Spitz F., Furlong E.E.M.. Transcription factors: from enhancer binding to developmental control. Nat. Rev. Genet. 2012; 13:613–626. [DOI] [PubMed] [Google Scholar]
- 2. Megraw M., Cumbie J.S., Ivanchenko M.G., Filichkin S.A.. Small genetic circuits and MicroRNAs: Big players in polymerase II transcriptional control in plants. Plant Cell. 2016; 28:286–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Smith N.C., Matthews J.M.. Mechanisms of DNA-binding specificity and functional gene regulation by transcription factors. Curr. Opin. Struct. Biol. 2016; 38:68–74. [DOI] [PubMed] [Google Scholar]
- 4. Dixon S.J., Costanzo M., Baryshnikova A., Andrews B., Boone C.. Systematic mapping of genetic interaction networks. Annu. Rev. Genet. 2009; 43:601–625. [DOI] [PubMed] [Google Scholar]
- 5. Schmitz J.F., Zimmer F., Bornberg-Bauer E.. Mechanisms of transcription factor evolution in Metazoa. Nucleic Acids Res. 2016; 44:6287–6297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhao M., Meyers B.C., Cai C., Xu W., Ma J.. Evolutionary patterns and coevolutionary consequences of MIRNA genes and microRNA targets triggered by multiple mechanisms of genomic duplications in soybean. Plant Cell. 2015; 27:546–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Manke T., Bringas R., Vingron M.. Correlating protein-DNA and protein-protein interaction networks. J. Mol. Biol. 2003; 333:75–85. [DOI] [PubMed] [Google Scholar]
- 8. Yu H., Luscombe N.M., Qian J., Gerstein M.. Genomic analysis of gene expression relationships in transcriptional regulatory networks. Trends Genet. 2003; 19:422–427. [DOI] [PubMed] [Google Scholar]
- 9. Costanzo M., Baryshnikova A., Bellay J., Kim Y., Spear E.D., Sevier C.S., Ding H., Koh J.L., Toufighi K., Mostafavi S. et al. . The genetic landscape of a cell. Science. 2010; 327:425–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Martinez N.J., Ow M.C., Barrasa M.I., Hammell M., Sequerra R., Doucette-Stamm L., Roth F.P., Ambros V.R., Walhout A.J.. A C. elegans genome-scale microRNA network contains composite feedback motifs with high flux capacity. Genes Dev. 2008; 22:2535–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Guo Y., Alexander K., Clark A.G., Grimson A., Yu H.. Integrated network analysis reveals distinct regulatory roles of transcription factors and microRNAs. RNA. 2016; 22:1663–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mitra K., Carvunis A.R., Ramesh S.K., Ideker T.. Integrative approaches for finding modular structure in biological networks. Nat. Rev. Genet. 2013; 14:719–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Levine M., Davidson E.H.. Gene regulatory networks for development. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:4936–4942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Milo R., Shen-Orr S., Itzkovitz S., Kashtan N., Chklovskii D., Alon U.. Network motifs: simple building blocks of complex networks. Science. 2002; 298:824–827. [DOI] [PubMed] [Google Scholar]
- 15. Shen-Orr S.S., Milo R., Mangan S., Alon U.. Network motifs in the transcriptional regulation network of Escherichia coli. Nat. Genet. 2002; 31:64–68. [DOI] [PubMed] [Google Scholar]
- 16. Mangan S., Alon U.. Structure and function of the feed-forward loop network motif. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:11980–11985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lee T.I., Rinaldi N.J., Robert F., Odom D.T., Bar-Joseph Z., Gerber G.K., Hannett N.M., Harbison C.T., Thompson C.M., Simon I. et al. . Transcriptional regulatory networks in Saccharomyces cerevisiae. Science. 2002; 298:799–804. [DOI] [PubMed] [Google Scholar]
- 18. Odom D.T., Zizlsperger N., Gordon D.B., Bell G.W., Rinaldi N.J., Murray H.L., Volkert T.L., Schreiber J., Rolfe P.A., Gifford D.K. et al. . Control of pancreas and liver gene expression by HNF transcription factors. Science. 2004; 303:1378–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Boyer L.A., Lee T.I., Cole M.F., Johnstone S.E., Levine S.S., Zucker J.P., Guenther M.G., Kumar R.M., Murray H.L., Jenner R.G. et al. . Core transcriptional regulatory circuitry in human embryonic stem cells. Cell. 2005; 122:947–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yeger-Lotem E., Sattath S., Kashtan N., Itzkovitz S., Milo R., Pinter R.Y., Alon U., Margalit H.. Network motifs in integrated cellular networks of transcription-regulation and protein-protein interaction. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:5934–5939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tsang J., Zhu J., van Oudenaarden A.. MicroRNA-mediated feedback and feedforward loops are recurrent network motifs in mammals. Mol. Cell. 2007; 26:753–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cheng C., Yan K.K., Hwang W., Qian J., Bhardwaj N., Rozowsky J., Lu Z.J., Niu W., Alves P., Kato M. et al. . Construction and analysis of an integrated regulatory network derived from high-throughput sequencing data. PLoS Comput. Biol. 2011; 7:e1002190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shalgi R., Lieber D., Oren M., Pilpel Y.. Global and local architecture of the mammalian microRNA-transcription factor regulatory network. PLoS Comput. Biol. 2007; 3:e131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ptacek J., Devgan G., Michaud G., Zhu H., Zhu X., Fasolo J., Guo H., Jona G., Breitkreutz A., Sopko R. et al. . Global analysis of protein phosphorylation in yeast. Nature. 2005; 438:679–684. [DOI] [PubMed] [Google Scholar]
- 25. Zhang J., Le T.D., Liu L., He J., Li J.. A novel framework for inferring condition-specific TF and miRNA co-regulation of protein-protein interactions. Gene. 2016; 577:55–64. [DOI] [PubMed] [Google Scholar]
- 26. Atay O., Doncic A., Skotheim J.M.. Switch-like transitions insulate network motifs to modularize biological networks. Cell Syst. 2016; 3:121–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Payne J.L., Wagner A.. Function does not follow form in gene regulatory circuits. Sci. Rep. 2015; 5:13015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mazurie A., Bottani S., Vergassola M.. An evolutionary and functional assessment of regulatory network motifs. Genome Biol. 2005; 6:R35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dobrin R., Beg Q.K., Barabasi A.L., Oltvai Z.N.. Aggregation of topological motifs in the Escherichia coli transcriptional regulatory network. BMC Bioinformatics. 2004; 5:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kashtan N., Itzkovitz S., Milo R., Alon U.. Topological generalizations of network motifs. Physi. Rev. E Stat. Nonlin. Soft Matter Phys. 2004; 70:031909. [DOI] [PubMed] [Google Scholar]
- 31. Zhang L.V., King O.D., Wong S.L., Goldberg D.S., Tong A.H., Lesage G., Andrews B., Bussey H., Boone C., Roth F.P.. Motifs, themes and thematic maps of an integrated Saccharomyces cerevisiae interaction network. J. Biol. 2005; 4:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Michoel T., Joshi A., Nachtergaele B., Van de Peer Y.. Enrichment and aggregation of topological motifs are independent organizational principles of integrated interaction networks. Mol. Biosyst. 2011; 7:2769–2778. [DOI] [PubMed] [Google Scholar]
- 33. Benson A.R., Gleich D.F., Leskovec J.. Higher-order organization of complex networks. Science. 2016; 353:163–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tautz D., Domazet-Loso T.. The evolutionary origin of orphan genes. Nat. Rev. Genet. 2011; 12:692–702. [DOI] [PubMed] [Google Scholar]
- 35. Domazet-Loso T., Brajkovic J., Tautz D.. A phylostratigraphy approach to uncover the genomic history of major adaptations in metazoan lineages. Trends Genet. 2007; 23:533–539. [DOI] [PubMed] [Google Scholar]
- 36. Chen C.Y., Ho A., Huang H.Y., Juan H.F., Huang H.C.. Dissecting the human protein-protein interaction network via phylogenetic decomposition. Sci. Rep. 2014; 4:7153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang W., Landback P., Gschwend A.R., Shen B., Long M.. New genes drive the evolution of gene interaction networks in the human and mouse genomes. Genome Biol. 2015; 16:202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wei W., Jin Y.T., Du M.Z., Wang J., Rao N., Guo F.B.. Genomic complexity places less restrictions on the evolution of young coexpression networks than protein-protein interactions. Genome Biol. Evol. 2016; 8:2624–2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ruprecht C., Proost S., Hernandez-Coronado M., Ortiz-Ramirez C., Lang D., Rensing S.A., Becker J.D., Vandepoele K., Mutwil M.. Phylogenomic analysis of gene co-expression networks reveals the evolution of functional modules. Plant J. 2017; 90:447–465. [DOI] [PubMed] [Google Scholar]
- 40. Capra J.A., Pollard K.S., Singh M.. Novel genes exhibit distinct patterns of function acquisition and network integration. Genome Biol. 2010; 11:R127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liu Z., Liu Q., Sun H., Hou L., Guo H., Zhu Y., Li D., He F.. Evidence for the additions of clustered interacting nodes during the evolution of protein interaction networks from network motifs. BMC Evol. Biol. 2011; 11:133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kim W.K., Marcotte E.M.. Age-dependent evolution of the yeast protein interaction network suggests a limited role of gene duplication and divergence. PLoS Comput. Biol. 2008; 4:e1000232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liang C., Luo J., Song D.. Network simulation reveals significant contribution of network motifs to the age-dependency of yeast protein-protein interaction networks. Mol. Biosyst. 2014; 10:2277–2288. [DOI] [PubMed] [Google Scholar]
- 44. Lipinski K.J., Farslow J.C., Fitzpatrick K.A., Lynch M., Katju V., Bergthorsson U.. High spontaneous rate of gene duplication in Caenorhabditis elegans. Curr. Biol. 2011; 21:306–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Van de Peer Y., Mizrachi E., Marchal K.. The evolutionary significance of polyploidy. Nat. Rev. Genet. 2017; 18:411–424. [DOI] [PubMed] [Google Scholar]
- 46. Bowers J.E., Chapman B.A., Rong J., Paterson A.H.. Unravelling angiosperm genome evolution by phylogenetic analysis of chromosomal duplication events. Nature. 2003; 422:433–438. [DOI] [PubMed] [Google Scholar]
- 47. Ruprecht C., Lohaus R., Vanneste K., Mutwil M., Nikoloski Z., Van de Peer Y., Persson S.. Revisiting ancestral polyploidy in plants. Sci. Adv. 2017; 3:e1603195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Li Z., Defoort J., Tasdighian S., Maere S., Van de Peer Y., De Smet R.. Gene duplicability of core genes is highly consistent across all angiosperms. Plant Cell. 2016; 28:326–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang Y., Wang X., Tang H., Tan X., Ficklin S.P., Feltus F.A., Paterson A.H.. Modes of gene duplication contribute differently to genetic novelty and redundancy, but show parallels across divergent angiosperms. PLoS One. 2011; 6:e28150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Casneuf T., De Bodt S., Raes J., Maere S., Van de Peer Y.. Nonrandom divergence of gene expression following gene and genome duplications in the flowering plant Arabidopsis thaliana. Genome Biol. 2006; 7:R13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Consortium, A.I.M. Evidence for network evolution in an Arabidopsis interactome map. Science. 2011; 333:601–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Arsovski A.A., Pradinuk J., Guo X.Q., Wang S., Adams K.L.. Evolution of cis-regulatory elements and regulatory networks in duplicated genes of arabidopsis. Plant Physiol. 2015; 169:2982–2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Harris T.W., Baran J., Bieri T., Cabunoc A., Chan J., Chen W.J., Davis P., Done J., Grove C., Howe K. et al. . WormBase 2014: new views of curated biology. Nucleic Acids Res. 2014; 42:D789–D793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Simonis N., Rual J.F., Carvunis A.R., Tasan M., Lemmens I., Hirozane-Kishikawa T., Hao T., Sahalie J.M., Venkatesan K., Gebreab F. et al. . Empirically controlled mapping of the Caenorhabditis elegans protein-protein interactome network. Nat. Methods. 2009; 6:47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chatr-Aryamontri A., Breitkreutz B.J., Oughtred R., Boucher L., Heinicke S., Chen D., Stark C., Breitkreutz A., Kolas N., O’Donnell L. et al. . The BioGRID interaction database: 2015 update. Nucleic Acids Res. 2015; 43:D470–D478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Cheeseman I.M., Chappie J.S., Wilson-Kubalek E.M., Desai A.. The conserved KMN network constitutes the core microtubule-binding site of the kinetochore. Cell. 2006; 127:983–997. [DOI] [PubMed] [Google Scholar]
- 57. Cheeseman I.M., Niessen S., Anderson S., Hyndman F., Yates J.R. 3rd, Oegema K., Desai A.. A conserved protein network controls assembly of the outer kinetochore and its ability to sustain tension. Genes Dev. 2004; 18:2255–2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Grove C.A., De Masi F., Barrasa M.I., Newburger D.E., Alkema M.J., Bulyk M.L., Walhout A.J.. A multiparameter network reveals extensive divergence between C. elegans bHLH transcription factors. Cell. 2009; 138:314–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Popovici C., Berda Y., Conchonaud F., Harbis A., Birnbaum D., Roubin R.. Direct and heterologous approaches to identify the LET-756/FGF interactome. BMC Genomics. 2006; 7:105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Reece-Hoyes J.S., Pons C., Diallo A., Mori A., Shrestha S., Kadreppa S., Nelson J., Diprima S., Dricot A., Lajoie B.R. et al. . Extensive rewiring and complex evolutionary dynamics in a C. elegans multiparameter transcription factor network. Mol. Cell. 2013; 51:116–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Byrne A.B., Weirauch M.T., Wong V., Koeva M., Dixon S.J., Stuart J.M., Roy P.J.. A global analysis of genetic interactions in Caenorhabditis elegans. J. Biol. 2007; 6:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lehner B., Crombie C., Tischler J., Fortunato A., Fraser A.G.. Systematic mapping of genetic interactions in Caenorhabditis elegans identifies common modifiers of diverse signaling pathways. Nat. Genet. 2006; 38:896–903. [DOI] [PubMed] [Google Scholar]
- 63. Tischler J., Lehner B., Chen N., Fraser A.G.. Combinatorial RNA interference in Caenorhabditis elegans reveals that redundancy between gene duplicates can be maintained for more than 80 million years of evolution. Genome Biol. 2006; 7:R69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Tischler J., Lehner B., Fraser A.G.. Evolutionary plasticity of genetic interaction networks. Nat. Genet. 2008; 40:390–391. [DOI] [PubMed] [Google Scholar]
- 65. Reinke V., Krause M., Okkema P.. Transcriptional regulation of gene expression in C. elegans. WormBook. 2013; 1–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kozomara A., Griffiths-Jones S.. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014; 42:D68–D73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Arda H.E., Taubert S., MacNeil L.T., Conine C.C., Tsuda B., Van Gilst M., Sequerra R., Doucette-Stamm L., Yamamoto K.R., Walhout A.J.. Functional modularity of nuclear hormone receptors in a Caenorhabditis elegans metabolic gene regulatory network. Mol. Syst. Biol. 2010; 6:367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Deplancke B., Dupuy D., Vidal M., Walhout A.J.. A gateway-compatible yeast one-hybrid system. Genome Res. 2004; 14:2093–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Deplancke B., Mukhopadhyay A., Ao W., Elewa A.M., Grove C.A., Martinez N.J., Sequerra R., Doucette-Stamm L., Reece-Hoyes J.S., Hope I.A. et al. . A gene-centered C. elegans protein-DNA interaction network. Cell. 2006; 125:1193–1205. [DOI] [PubMed] [Google Scholar]
- 70. Feng H., Reece-Hoyes J.S., Walhout A.J., Hope I.A.. A regulatory cascade of three transcription factors in a single specific neuron, DVC, in Caenorhabditis elegans. Gene. 2012; 494:73–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Reece-Hoyes J.S., Deplancke B., Barrasa M.I., Hatzold J., Smit R.B., Arda H.E., Pope P.A., Gaudet J., Conradt B., Walhout A.J.. The C. elegans Snail homolog CES-1 can activate gene expression in vivo and share targets with bHLH transcription factors. Nucleic Acids Res. 2009; 37:3689–3698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Reece-Hoyes J.S., Diallo A., Lajoie B., Kent A., Shrestha S., Kadreppa S., Pesyna C., Dekker J., Myers C.L., Walhout A.J.. Enhanced yeast one-hybrid assays for high-throughput gene-centered regulatory network mapping. Nat. Methods. 2011; 8:1059–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Vermeirssen V., Barrasa M.I., Hidalgo C.A., Babon J.A., Sequerra R., Doucette-Stamm L., Barabasi A.L., Walhout A.J.. Transcription factor modularity in a gene-centered C. elegans core neuronal protein-DNA interaction network. Genome Res. 2007; 17:1061–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Vermeirssen V., Deplancke B., Barrasa M.I., Reece-Hoyes J.S., Arda H.E., Grove C.A., Martinez N.J., Sequerra R., Doucette-Stamm L., Brent M.R. et al. . Matrix and Steiner-triple-system smart pooling assays for high-performance transcription regulatory network mapping. Nat. Methods. 2007; 4:659–664. [DOI] [PubMed] [Google Scholar]
- 75. Boyle A.P., Araya C.L., Brdlik C., Cayting P., Cheng C., Cheng Y., Gardner K., Hillier L.W., Janette J., Jiang L. et al. . Comparative analysis of regulatory information and circuits across distant species. Nature. 2014; 512:453–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Kirienko N.V., Fay D.S.. Transcriptome profiling of the C. elegans Rb ortholog reveals diverse developmental roles. Dev. Biol. 2007; 305:674–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Kouns N.A., Nakielna J., Behensky F., Krause M.W., Kostrouch Z., Kostrouchova M.. NHR-23 dependent collagen and hedgehog-related genes required for molting. Biochem. Biophys. Res. Commun. 2011; 413:515–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Magner D.B., Wollam J., Shen Y., Hoppe C., Li D., Latza C., Rottiers V., Hutter H., Antebi A.. The NHR-8 nuclear receptor regulates cholesterol and bile acid homeostasis in C. elegans. Cell Metab. 2013; 18:212–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Pathare P.P., Lin A., Bornfeldt K.E., Taubert S., Van Gilst M.R.. Coordinate regulation of lipid metabolism by novel nuclear receptor partnerships. PLoS Genet. 2012; 8:e1002645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Petrella L.N., Wang W., Spike C.A., Rechtsteiner A., Reinke V., Strome S.. synMuv B proteins antagonize germline fate in the intestine and ensure C. elegans survival. Development. 2011; 138:1069–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Thyagarajan B., Blaszczak A.G., Chandler K.J., Watts J.L., Johnson W.E., Graves B.J.. ETS-4 is a transcriptional regulator of life span in Caenorhabditis elegans. PLoS Genet. 2010; 6:e1001125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Troemel E.R., Chu S.W., Reinke V., Lee S.S., Ausubel F.M., Kim D.H.. p38 MAPK regulates expression of immune response genes and contributes to longevity in C. elegans. PLoS Genet. 2006; 2:e183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Van Nostrand E.L., Sanchez-Blanco A., Wu B., Nguyen A., Kim S.K.. Roles of the developmental regulator unc-62/Homothorax in limiting longevity in Caenorhabditis elegans. PLoS Genet. 2013; 9:e1003325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Van Landeghem S., Bjorne J., Wei C.H., Hakala K., Pyysalo S., Ananiadou S., Kao H.Y., Lu Z., Salakoski T., Van de Peer Y. et al. . Large-scale event extraction from literature with multi-level gene normalization. PLoS One. 2013; 8:e55814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Hsu S.D., Tseng Y.T., Shrestha S., Lin Y.L., Khaleel A., Chou C.H., Chu C.F., Huang H.Y., Lin C.M., Ho S.Y. et al. . miRTarBase update 2014: an information resource for experimentally validated miRNA-target interactions. Nucleic Acids Res. 2014; 42:D78–D85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Gerstein M.B., Lu Z.J., Van Nostrand E.L., Cheng C., Arshinoff B.I., Liu T., Yip K.Y., Robilotto R., Rechtsteiner A., Ikegami K. et al. . Integrative analysis of the Caenorhabditis elegans genome by the modENCODE project. Science. 2010; 330:1775–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Lall S., Grun D., Krek A., Chen K., Wang Y.L., Dewey C.N., Sood P., Colombo T., Bray N., Macmenamin P. et al. . A genome-wide map of conserved microRNA targets in C. elegans. Curr. Biol. 2006; 16:460–471. [DOI] [PubMed] [Google Scholar]
- 88. Engelmann I., Griffon A., Tichit L., Montanana-Sanchis F., Wang G., Reinke V., Waterston R.H., Hillier L.W., Ewbank J.J.. A comprehensive analysis of gene expression changes provoked by bacterial and fungal infection in C. elegans. PLoS One. 2011; 6:e19055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Spieth J., Lawson D., Davis P., Williams G., Howe K.. Overview of gene structure in C. elegans. WormBook. 2014; 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. De Bodt S., Hollunder J., Nelissen H., Meulemeester N., Inze D.. CORNET 2.0: integrating plant coexpression, protein-protein interactions, regulatory interactions, gene associations and functional annotations. New Phytol. 2012; 195:707–720. [DOI] [PubMed] [Google Scholar]
- 91. Jones A.M., Xuan Y., Xu M., Wang R.S., Ho C.H., Lalonde S., You C.H., Sardi M.I., Parsa S.A., Smith-Valle E. et al. . Border control–a membrane-linked interactome of Arabidopsis. Science. 2014; 344:711–716. [DOI] [PubMed] [Google Scholar]
- 92. Jin J., Zhang H., Kong L., Gao G., Luo J.. PlantTFDB 3.0: a portal for the functional and evolutionary study of plant transcription factors. Nucleic Acids Res. 2014; 42:D1182–D1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Heyndrickx K.S., Van de Velde J., Wang C., Weigel D., Vandepoele K.. A functional and evolutionary perspective on transcription factor binding in Arabidopsis thaliana. Plant Cell. 2014; 26:3894–3910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Vermeirssen V., De Clercq I., Van Parys T., Van Breusegem F., Van de Peer Y.. Arabidopsis ensemble reverse-engineered gene regulatory network discloses interconnected transcription factors in oxidative stress. Plant Cell. 2014; 26:4656–4679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Li B., Gaudinier A., Tang M., Taylor-Teeples M., Nham N.T., Ghaffari C., Benson D.S., Steinmann M., Gray J.A., Brady S.M. et al. . Promoter-based integration in plant defense regulation. Plant Physiol. 2014; 166:1803–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Brady S.M., Zhang L., Megraw M., Martinez N.J., Jiang E., Yi C.S., Liu W., Zeng A., Taylor-Teeples M., Kim D. et al. . A stele-enriched gene regulatory network in the Arabidopsis root. Mol. Syst. Biol. 2011; 7:459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Taylor-Teeples M., Lin L., de Lucas M., Turco G., Toal T.W., Gaudinier A., Young N.F., Trabucco G.M., Veling M.T., Lamothe R. et al. . An Arabidopsis gene regulatory network for secondary cell wall synthesis. Nature. 2015; 517:571–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Yilmaz A., Mejia-Guerra M.K., Kurz K., Liang X., Welch L., Grotewold E.. AGRIS: the arabidopsis gene regulatory information server, an update. Nucleic Acids Res. 2011; 39:D1118–D1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Srivastava P.K., Moturu T.R., Pandey P., Baldwin I.T., Pandey S.P.. A comparison of performance of plant miRNA target prediction tools and the characterization of features for genome-wide target prediction. BMC Genomics. 2014; 15:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Berardini T.Z., Reiser L., Li D., Mezheritsky Y., Muller R., Strait E., Huala E.. The Arabidopsis information resource: Making and mining the ‘gold standard’ annotated reference plant genome. Genesis. 2015; 53:474–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Csardi G., Nepusz T.. The igraph software package for complex network research. Inter J. Complex Syst. 2006; 1695:1–9. [Google Scholar]
- 102. Demeyer S., Michoel T., Fostier J., Audenaert P., Pickavet M., Demeester P.. The index-based subgraph matching algorithm (ISMA): fast subgraph enumeration in large networks using optimized search trees. PLoS One. 2013; 8:e61183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Michoel T., Nachtergaele B.. Alignment and integration of complex networks by hypergraph-based spectral clustering. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2012; 86:056111. [DOI] [PubMed] [Google Scholar]
- 104. Maere S., Heymans K., Kuiper M.. BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics. 2005; 21:3448–3449. [DOI] [PubMed] [Google Scholar]
- 105. Levin M., Hashimshony T., Wagner F., Yanai I.. Developmental milestones punctuate gene expression in the Caenorhabditis embryo. Dev. Cell. 2012; 22:1101–1108. [DOI] [PubMed] [Google Scholar]
- 106. Spencer W.C., Zeller G., Watson J.D., Henz S.R., Watkins K.L., McWhirter R.D., Petersen S., Sreedharan V.T., Widmer C., Jo J. et al. . A spatial and temporal map of C. elegans gene expression. Genome Res. 2011; 21:325–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Chen L., Qu X., Cao M., Zhou Y., Li W., Liang B., Li W., He W., Feng C., Jia X. et al. . Identification of breast cancer patients based on human signaling network motifs. Sci. Rep. 2013; 3:3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Van Bel M., Diels T., Vancaester E., Kreft L., Botzki A., Van de Peer Y., Coppens F., Vandepoele K.. PLAZA 4.0: an integrative resource for functional, evolutionary and comparative plant genomics. Nucleic Acids Res. 2018; 46:D1190–D1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Liebeskind B.J., McWhite C.D., Marcotte E.M.. Towards consensus gene ages. Genome Biol. Evol. 2016; 8:1812–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Zhou K., Huang B., Zou M., Lu D., He S., Wang G.. Genome-wide identification of lineage-specific genes within Caenorhabditis elegans. Genomics. 2015; 106:242–248. [DOI] [PubMed] [Google Scholar]
- 111. Barabasi A.L., Oltvai Z.N.. Network biology: understanding the cell's functional organization. Nat. Rev. Genet. 2004; 5:101–113. [DOI] [PubMed] [Google Scholar]
- 112. Newman M.E.J. The structure and function of complex networks. Soc. Ind. Appl. Math. Rev. 2003; 45:167–256. [Google Scholar]
- 113. Alon U. Network motifs: theory and experimental approaches. Nat. Rev. Genet. 2007; 8:450–461. [DOI] [PubMed] [Google Scholar]
- 114. Li J., Hua X., Haubrock M., Wang J., Wingender E.. The architecture of the gene regulatory networks of different tissues. Bioinformatics. 2012; 28:i509–i514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Gerstein M.B., Kundaje A., Hariharan M., Landt S.G., Yan K.K., Cheng C., Mu X.J., Khurana E., Rozowsky J., Alexander R. et al. . Architecture of the human regulatory network derived from ENCODE data. Nature. 2012; 489:91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Kelley R., Ideker T.. Systematic interpretation of genetic interactions using protein networks. Nat. Biotechnol. 2005; 23:561–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Nishikori S., Yamanaka K., Sakurai T., Esaki M., Ogura T.. p97 Homologs from Caenorhabditis elegans, CDC-48.1 and CDC-48.2, suppress the aggregate formation of huntingtin exon1 containing expanded polyQ repeat. Genes Cells. 2008; 13:827–838. [DOI] [PubMed] [Google Scholar]
- 118. Furuya M., Qadota H., Chisholm A.D., Sugimoto A.. The C. elegans eyes absent ortholog EYA-1 is required for tissue differentiation and plays partially redundant roles with PAX-6. Dev. Biol. 2005; 286:452–463. [DOI] [PubMed] [Google Scholar]
- 119. Wang H., Wang H.. The miR156/SPL module, a regulatory hub and versatile toolbox, gears up crops for enhanced agronomic traits. Mol. Plant. 2015; 8:677–688. [DOI] [PubMed] [Google Scholar]
- 120. Hwan Lee J., Joon Kim J., Ahn J.H.. Role of SEPALLATA3 (SEP3) as a downstream gene of miR156-SPL3-FT circuitry in ambient temperature-responsive flowering. Plant Signal. Behav. 2012; 7:1151–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Tan Q.K., Irish V.F.. The Arabidopsis zinc finger-homeodomain genes encode proteins with unique biochemical properties that are coordinately expressed during floral development. Plant Physiol. 2006; 140:1095–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Tran L.S., Nakashima K., Sakuma Y., Osakabe Y., Qin F., Simpson S.D., Maruyama K., Fujita Y., Shinozaki K., Yamaguchi-Shinozaki K.. Co-expression of the stress-inducible zinc finger homeodomain ZFHD1 and NAC transcription factors enhances expression of the ERD1 gene in Arabidopsis. Plant J. 2007; 49:46–63. [DOI] [PubMed] [Google Scholar]
- 123. Wang W., Wu P., Li Y., Hou X.. Genome-wide analysis and expression patterns of ZF-HD transcription factors under different developmental tissues and abiotic stresses in Chinese cabbage. Mol. Genet. Genomics. 2016; 291:1451–1464. [DOI] [PubMed] [Google Scholar]
- 124. Wang H., Yin X., Li X., Wang L., Zheng Y., Xu X., Zhang Y., Wang X.. Genome-wide identification, evolution and expression analysis of the grape (Vitis vinifera L.) zinc finger-homeodomain gene family. Int. J. Mol. Sci. 2014; 15:5730–5748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Saez A., Rodrigues A., Santiago J., Rubio S., Rodriguez P.L.. HAB1-SWI3B interaction reveals a link between abscisic acid signaling and putative SWI/SNF chromatin-remodeling complexes in Arabidopsis. Plant Cell. 2008; 20:2972–2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Sarnowski T.J., Rios G., Jasik J., Swiezewski S., Kaczanowski S., Li Y., Kwiatkowska A., Pawlikowska K., Kozbial M., Kozbial P. et al. . SWI3 subunits of putative SWI/SNF chromatin-remodeling complexes play distinct roles during Arabidopsis development. Plant Cell. 2005; 17:2454–2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Vercruyssen L., Verkest A., Gonzalez N., Heyndrickx K.S., Eeckhout D., Han S.K., Jegu T., Archacki R., Van Leene J., Andriankaja M. et al. . ANGUSTIFOLIA3 binds to SWI/SNF chromatin remodeling complexes to regulate transcription during Arabidopsis leaf development. Plant Cell. 2014; 26:210–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Ko J.H., Jeon H.W., Kim W.C., Kim J.Y., Han K.H.. The MYB46/MYB83-mediated transcriptional regulatory programme is a gatekeeper of secondary wall biosynthesis. Ann. Bot. 2014; 114:1099–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Haake V., Cook D., Riechmann J.L., Pineda O., Thomashow M.F., Zhang J.Z.. Transcription factor CBF4 is a regulator of drought adaptation in Arabidopsis. Plant Physiol. 2002; 130:639–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Shaikhali J., de Dios Barajas-Lopez J., Otvos K., Kremnev D., Garcia A.S., Srivastava V., Wingsle G., Bako L., Strand A.. The CRYPTOCHROME1-dependent response to excess light is mediated through the transcriptional activators ZINC FINGER PROTEIN EXPRESSED IN INFLORESCENCE MERISTEM LIKE1 and ZML2 in Arabidopsis. Plant Cell. 2012; 24:3009–3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Mithen R., Bennett R., Marquez J.. Glucosinolate biochemical diversity and innovation in the Brassicales. Phytochemistry. 2010; 71:2074–2086. [DOI] [PubMed] [Google Scholar]
- 132. Beyer A., Workman C., Hollunder J., Radke D., Moller U., Wilhelm T., Ideker T.. Integrated assessment and prediction of transcription factor binding. PLoS Comput. Biol. 2006; 2:e70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Park C.Y., Hess D.C., Huttenhower C., Troyanskaya O.G.. Simultaneous genome-wide inference of physical, genetic, regulatory, and functional pathway components. PLoS Comput. Biol. 2010; 6:e1001009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Lee I., Date S.V., Adai A.T., Marcotte E.M.. A probabilistic functional network of yeast genes. Science. 2004; 306:1555–1558. [DOI] [PubMed] [Google Scholar]
- 135. Zhao H., Zhang G., Pang L., Lan Y., Wang L., Yu F., Hu J., Li F., Zhao T., Xiao Y. et al. . ‘Traffic light rules’: Chromatin states direct miRNA-mediated network motifs running by integrating epigenome and regulatome. Biochim. Biophys. Acta. 2016; 1860:1475–1488. [DOI] [PubMed] [Google Scholar]
- 136. Beber M.E., Fretter C., Jain S., Sonnenschein N., Müller-Hannemann M., Hütt M.-T.. Artefacts in statistical analyses of network motifs: general framework and application to metabolic networks. J. R. Soc. Interface. 2012; 9:3426–3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Ginoza R., Mugler A.. Network motifs come in sets: correlations in the randomization process. Phys. Rev. E. 2010; 82:011921. [DOI] [PubMed] [Google Scholar]
- 138. Megraw M., Mukherjee S., Ohler U.. Sustained-input switches for transcription factors and microRNAs are central building blocks of eukaryotic gene circuits. Genome Biol. 2013; 14:R85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Zinman G.E., Zhong S., Bar-Joseph Z.. Biological interaction networks are conserved at the module level. BMC Syst. Biol. 2011; 5:134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Abrusan G. Integration of new genes into cellular networks, and their structural maturation. Genetics. 2013; 195:1407–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Barabasi A.L., Albert R.. Emergence of scaling in random networks. Science. 1999; 286:509–512. [DOI] [PubMed] [Google Scholar]
- 142. Ispolatov I., Krapivsky P.L., Yuryev A.. Duplication-divergence model of protein interaction network. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2005; 71:061911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Abdelzaher A.F., Al-Musawi A.F., Ghosh P., Mayo M.L., Perkins E.J.. Transcriptional network growing models using Motif-Based preferential attachment. Front. Bioeng. Biotechnol. 2015; 3:157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Lehti-Shiu M.D., Panchy N., Wang P., Uygun S., Shiu S.H.. Diversity, expansion, and evolutionary novelty of plant DNA-binding transcription factor families. Biochim. Biophys. Acta. 2017; 1860:3–20. [DOI] [PubMed] [Google Scholar]
- 145. Maere S., De Bodt S., Raes J., Casneuf T., Van Montagu M., Kuiper M., Van de Peer Y.. Modeling gene and genome duplications in eukaryotes. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:5454–5459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Haerty W., Artieri C., Khezri N., Singh R.S., Gupta B.P.. Comparative analysis of function and interaction of transcription factors in nematodes: extensive conservation of orthology coupled to rapid sequence evolution. BMC Genomics. 2008; 9:399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Ward J.J., Thornton J.M.. Evolutionary models for formation of network motifs and modularity in the Saccharomyces transcription factor network. PLoS Comput. Biol. 2007; 3:1993–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Jin J., He K., Tang X., Li Z., Lv L., Zhao Y., Luo J., Gao G.. An arabidopsis transcriptional regulatory map reveals distinct functional and evolutionary features of novel transcription factors. Mol. Biol. Evol. 2015; 32:1767–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Nag A., King S., Jack T.. miR319a targeting of TCP4 is critical for petal growth and development in Arabidopsis. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:22534–22539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. McFarlane H.E., Doring A., Persson S.. The cell biology of cellulose synthesis. Annu. Rev. Plant Biol. 2014; 65:69–94. [DOI] [PubMed] [Google Scholar]
- 151. Vain T., Crowell E.F., Timpano H., Biot E., Desprez T., Mansoori N., Trindade L.M., Pagant S., Robert S., Hofte H. et al. . The cellulase KORRIGAN is part of the cellulose synthase complex. Plant Physiol. 2014; 165:1521–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Xie L., Yang C., Wang X.. Brassinosteroids can regulate cellulose biosynthesis by controlling the expression of CESA genes in Arabidopsis. J. Exp. Bot. 2011; 62:4495–4506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Guo H., Wang Y., Wang L., Hu P., Wang Y., Jia Y., Zhang C., Zhang Y., Zhang Y., Wang C. et al. . Expression of the MYB transcription factor gene BplMYB46 affects abiotic stress tolerance and secondary cell wall deposition in Betula platyphylla. Plant Biotechnol. J. 2017; 15:107–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Endler A., Kesten C., Schneider R., Zhang Y., Ivakov A., Froehlich A., Funke N., Persson S.. A mechanism for sustained cellulose synthesis during salt stress. Cell. 2015; 162:1353–1364. [DOI] [PubMed] [Google Scholar]
- 155. Li Y., Sawada Y., Hirai A., Sato M., Kuwahara A., Yan X., Hirai M.Y.. Novel insights into the function of Arabidopsis R2R3-MYB transcription factors regulating aliphatic glucosinolate biosynthesis. Plant Cell Physiol. 2013; 54:1335–1344. [DOI] [PubMed] [Google Scholar]
- 156. Frerigmann H., Gigolashvili T.. MYB34, MYB51, and MYB122 distinctly regulate indolic glucosinolate biosynthesis in Arabidopsis thaliana. Mol. Plant. 2014; 7:814–828. [DOI] [PubMed] [Google Scholar]
- 157. Martinez-Ballesta M., Moreno-Fernandez D.A., Castejon D., Ochando C., Morandini P.A., Carvajal M.. The impact of the absence of aliphatic glucosinolates on water transport under salt stress in Arabidopsis thaliana. Front. Plant Sci. 2015; 6:524. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
A dynamic visualization of all modules and a file with all genes and edges per module can be found at (http://bioinformatics.psb.ugent.be/supplementary_data/jofoo/networks/). The source code of the computational data-integration framework can be found at https://gitlab.psb.ugent.be/jofoo/NetworkMotifModules.git.