


Abstract
Vitally important for controlling gene expression in eukaryotes and prokaryotes, the deprotection of mRNA 5′ termini is governed by enzymes whose activity is modulated by interactions with ancillary factors. In Escherichia coli, 5′-end-dependent mRNA degradation begins with the generation of monophosphorylated 5′ termini by the RNA pyrophosphohydrolase RppH, which can be stimulated by DapF, a diaminopimelate epimerase involved in amino acid and cell wall biosynthesis. We have determined crystal structures of RppH–DapF complexes and measured rates of RNA deprotection. These studies show that DapF potentiates RppH activity in two ways, depending on the nature of the substrate. Its stimulatory effect on the reactivity of diphosphorylated RNAs, the predominant natural substrates of RppH, requires a substrate long enough to reach DapF in the complex, while the enhanced reactivity of triphosphorylated RNAs appears to involve DapF-induced changes in RppH itself and likewise increases with substrate length. This study provides a basis for understanding the intricate relationship between cellular metabolism and mRNA decay and reveals striking parallels with the stimulation of decapping activity in eukaryotes.
INTRODUCTION
Critical to the survival of all living organisms is the ability to precisely regulate the production of proteins needed to respond to an ever-changing environment. Because the rate of protein biosynthesis depends on the availability of mRNA for translation, the modulation of mRNA lifetimes represents a commonly used strategy to influence protein production in both eukaryotes and prokaryotes (1).
In eukaryotes, a key event that enables mRNA degradation in the 5′-to-3′ direction is the deprotection of the capped 5′ end by the decapping enzyme Dcp2 (2). The resulting monophosphorylated intermediate is rapidly degraded by the 5′ exonuclease XRN1. The activity of Dcp2 is modulated by several protein factors with which it associates, such as Dcp1 and Edc proteins, which stimulate or inhibit decapping in either a generic or a pathway-specific manner and play central roles in a variety of cellular processes (Figure 1) (3).
Figure 1.

Parallels between 5′-end-dependent mRNA degradation pathways in eukaryotes and E. coli. In eukaryotes, cap removal by mRNA-decapping enzyme subunit 2 (Dcp2) bound to Dcp1 triggers mRNA decay (1). The activity of Dcp2 is modulated by several protein factors, including Enhancer of mRNA Decapping (Edc1) (3). The decapped RNA is then degraded by 5′-3′ exoribonuclease 1 (XRN1). In E. coli, the corresponding pathway begins with stepwise removal of the γ and β phosphates of a triphosphorylated transcript by the consecutive action of an unidentified enzyme and RppH (6). RppH activity is stimulated by its interaction with the metabolic enzyme DapF. The resulting monophosphorylated RNA 5′ end potentiates internal cleavage by the endonuclease RNase E.
In E. coli, accumulated evidence suggests that triphosphates or diphosphates on the 5′ end of mRNA play a protective role similar to that of caps on eukaryotic mRNA (4–7). Conversion of such ends to 5′-monophosphates creates decay intermediates vulnerable to rapid internal cleavage by the endoribonuclease RNase E, which is able to bind monophosphorylated but not di- or triphosphorylated 5′ ends and to initiate a cascade of endo- and exoribonucleolytic cleavages leading to full RNA degradation (Figure 1) (6,8). A key mediator of the 5′-end-dependent RNA decay pathway is the RNA pyrophosphohydrolase RppH (5). Although RppH can cleave between the α and β phosphates at the 5′ end of triphosphorylated primary transcripts and release pyrophosphate, the enzyme is much more efficient at severing orthophosphate off of diphosphorylated RNAs, which are abundant decay intermediates in E. coli (6). Therefore, the conversion of triphosphorylated RNA to monophosphorylated RNA appears to occur in E. coli by the sequential removal of the γ phosphate by an unidentified enzyme and the β phosphate by RppH (6). The function of RppH seems to parallel that of the eukaryotic decapping enzyme Dcp2, as these two enzymes both belong to the family of Nudix hydrolases and their catalytic mechanisms share significant commonalities.
RppH is widespread in diverse bacterial species. In E. coli, it initiates the 5′-end-dependent degradation of many but not all mRNAs for reasons that include its 5′-terminal substrate specificity (9–12). In particular, E. coli RppH requires two and prefers three or more unpaired 5′ nucleotides and favors a purine at the second position of its substrates (11). The enzyme binds the nucleobase of the second nucleotide in a discrete cleft and interacts with the 5′ phosphates of the RNA and 2-3 Mg2+ cations in a separate catalytic site (13). The Mg2+ cations mediate interactions between RppH and the RNA 5′ end and promote nucleophilic attack by water on the β phosphate (13).
A recent study identified DapF as a modulator of the bacterial 5′-end-dependent RNA degradation pathway (14). In E. coli, DapF forms a tight complex with RppH and stimulates its activity, thereby accelerating mRNA degradation (14). DapF is not only a regulator of RppH activity but also a metabolic enzyme (diaminopimelate (DAP) epimerase) that catalyzes the stereoconversion of L,L-DAP to meso-DAP (15). The product of this reaction is both an intermediate in lysine biosynthesis and a peptidoglycan precursor (16). Given the contribution of RppH to diverse cellular processes (17) ranging from virulence (18–20) to ribosome biogenesis (21) and resistance to osmotic lysis (14), the connection between a metabolic enzyme and RNA degradation is an intriguing phenomenon, especially since the catalytic activity of DapF is not required for this protein to stimulate RNA decay (14).
To understand the mechanism by which DapF potentiates RppH activity, we have determined crystal structures of several complexes that include RppH, DapF and RNA, and examined the effect of DapF on the reactivity of RppH with RNA substrates bearing different numbers of 5′-terminal phosphates. Our studies have revealed distinct, length-dependent modes of RppH stimulation by DapF for diphosphorylated and triphosphorylated substrates. These unexpected observations demonstrate the versatility of RppH, which is capable of hydrolyzing a variety of substrates and of employing multiple activation strategies in response to binding its protein modulator.
MATERIALS AND METHODS
Protein preparation
Full-length E. coli RppH and its truncated variant EcRppH(1–158)AA (here called RppHt) were purified as previously described (13). Escherichia coli DapF bearing six additional residues (SGSGSG) at the amino terminus was generated from a protein fusion in which hexahistidine and SUMO tags were joined in tandem to the amino terminus of DapF. The protein fusion was produced from a plasmid bearing a T7 promoter in E. coli BL21(DE3) after IPTG induction at 25°C for 16 h. Cells were lysed in a buffer containing 20 mM Tris–HCl, pH 8.0, 250 mM NaCl, 20 mM imidazole, and 0.05% (v/v) β-mercaptoethanol by EmulsiFlex C-5 (Avestin) homogenization, and the protein lysate clarified by centrifugation was loaded onto a 5-ml HisTrap FF column (GE Healthcare). The recombinant protein was eluted with lysis buffer supplemented with 0.3 M imidazole. Peak fractions were combined, and the tag was cleaved off by treatment with His-tagged ULP1 protease at 4°C for 14–16 h. The products of digestion were dialyzed against lysis buffer lacking imidazole, and the His-tagged ULP1 and His-SUMO tag were removed by retention on a HisTrap FF column. Finally, DapF in the flow-through from a second HisTrap column (no imidazole) was concentrated and further fractionated by gel filtration on Superdex 200 16/60 (GE Healthcare) in 20 mM Tris–HCl, pH 8.0, 250 mM NaCl, 1 mM dithiothreitol (DTT) and stored at 4°C. Mutagenesis was conducted by inverse PCR, and the mutated proteins were purified by the same procedure as their wild type counterparts.
RNA preparation
RNAs were prepared by run-off transcription with T7 RNA polymerase and DNA templates that comprised a double-stranded T7 promoter and a single-stranded RNA-encoding extension, as described (22). 3–9 nt RNAs resembled the 5′-end of yeiP mRNA except that they contained a U-to-G substitution at the second nucleotide to facilitate transcription by T7 RNA polymerase and an A-to-U substitution at the third nucleotide (6). Full-length yeiP mRNA for in vitro studies contained only the U2G substitution. DNA templates for oligoribonucleotide synthesis were prepared by annealing top and bottom oligodeoxynucleotides: TAATACGACTCACTATT (top strand); AGACTAATAGTGAGTCGTATTA (3-mer bottom); AGAAACTAATAGTGAGTCGTATTA (5-mer bottom); AGAAAACTAATAGTGAGTCGTATTA (6-mer bottom); AGAAAAACTAATAGTGAGT CGTATTA (7-mer bottom); AGAAAAAACTAATAGTGAGTCGTATTA (8-mer bottom); AGCAAAAAACTAATAGTGAGTCGTATTA (9-mer bottom). Transcription was performed in 5 mL at 37°C for 4 h. The total concentration of NTPs, ADP and modified NTPs was kept at 15–16 mM while the concentration of each nucleotide was adjusted according to the RNA sequence. For example, to prepare ppcpAGU, we used 5 mM each of ppcpA, GTP, and UTP; to prepare ppAGUUUU, we used 2.6 mM ADP, 2.6 mM GTP, and 10.7 mM UTP. All nucleotides were purchased from Sigma/Aldrich and ppcpA was obtained from TriLink Biotechnologies. For purification, oligoribonucleotides were loaded onto a 5-ml HiTrap Q column (GE Healthcare) in 20 mM Tris–HCl, pH 8.0, eluted with a 15–25% gradient of 1 M NaCl in the loading buffer, precipitated with ethanol, washed with 80% (v/v) ethanol, dried, and dissolved in water. They were then verified by gel electrophoresis, chromatography, and mass spectrometry. For MALDI-ToF mass spectrometry, the RNAs were desalted on a ZipTipC18 and eluted in 2,4,6-trihydroxyacetophenone-containing matrix for direct spotting, according to Technical Note 225 (Millipore Corporation).
Crosslinking
1 μM RppH or RppHt and 1 μM His6-SUMO-DapF, alone or mixed, were crosslinked with 2.5 mM of the amine-specific crosslinking reagent bis(sulfosuccinimidyl)suberate (BS3) (Thermo Fisher Scientific) in a buffer containing 20 mM HEPES–NaOH, pH 7.5, 150 mM NaCl, 5 mM MgCl2 and 0.1% (v/v) Tween 20. 100-μl reactions were incubated at room temperature for 30 min and quenched with 5 μl of 1 M Tris–HCl, pH 7.5, followed by a 15-min incubation at room temperature. Proteins were precipitated with 10% (w/v) trichloroacetic acid, redissolved, and analyzed by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis.
Size-exclusion chromatography
RppH was added in ∼20–50% molar excess to DapF, and complex formation was analyzed by size-exclusion chromatography (SEC). SEC was conducted at a flow rate of 1 ml/min on Superdex 200 16/600 in a buffer containing 20 mM Tris–HCl, pH 8.0, 500 mM NaCl and 1 mM DTT and monitored at 280 nm.
Crystallization
RppH–DapF complexes were formed by mixing DapF and RppH at a molar ratio of 1:1.2 in a buffer containing 20 mM Tris–HCl pH 8.0, 500 mM NaCl, and 1 mM DTT, and purified by gel-filtration on Superdex 200 16/600. Crystals of the RppH–DapF complex were prepared by mixing 0.5 μl of the complex (16 mg/ml) with 0.5 μl of reservoir solution containing 15% (v/v) PEG4,000, 0.1 M Tris–HCl, pH 8.0, and 0.2 M KI. Crystals of the RppHt-DapFm complex were prepared by mixing 0.5 μl of the complex (16 mg/ml) with 0.5 μl of reservoir solution containing 30% (v/v) PEG400 and 0.1 M CHES, pH 9.2. Crystals were grown against 80 μl of the reservoir solution in 96-well plates at 18°C for 5–7 days. To soak RNA into the crystals of the RppHt–DapFm complex, the crystals were transferred from the growth solution into a 1.0 μl drop containing 0.1 mM RNA, 30% (v/v) PEG400, 0.1 M CHES, pH 9.2 and 10 mM MgCl2 for 1–4 h. Crystals were cryoprotected in reservoir solution supplemented with 16% (v/v) glycerol.
Data collection and structure determination
Diffraction data were collected at 100 K on beamlines 24ID-C of the Advanced Photon Source (Argonne National Laboratory) and FMX of the National Synchrotron Light Source-II (Brookhaven National Laboratory). Data were processed by using the XDS suite (23). The crystal structures were solved by molecular replacement using E. coli DapF (PDB code: 4IJZ) and RppH (PDB code: 4S2X) as search models and PHENIX (24). The models were adjusted manually in COOT (25) and refined in PHENIX. RNA and ions were added at the late stages of refinement on the basis of the Fo – Fc, 2Fo – Fc and anomalous electron density maps. The γ phosphate was refined at 60% occupancy on the basis of the residual density map.
Bio-layer interferometry of protein-protein interactions
An Octet Red96 system (PALL FortéBio) was used to measure the binding affinity of RppH for DapF. His6-SUMO-tagged RppH or DapF was immobilized by applying 1 μg/ml protein solution in a buffer containing 20 mM HEPES–NaOH, pH 7.5, 500 mM NaCl, 1 mM DTT, 10 mM imidazole, 5 mM MgCl2 and 0.05% (v/v) Tween to Dip and Read™ Ni-NTA Biosensors (PALL FortéBio) hydrated in the sample buffer. An 8.3-min baseline recording was obtained prior to the binding measurements. Association and dissociation of the dissolved partner protein present at various concentrations (5 nM–22.5 μM) were monitored for 5 min each. Nonspecific protein binding was monitored by using a sensor bearing no immobilized protein. The contribution of the buffer to the signal was measured with a protein-coated sensor. Both controls were subtracted from the binding signal prior to calculating KD values. Binding affinities were verified by reciprocal measurements in which each interacting protein was tested both in solution and after immobilization. Kinetic parameters were determined by global fitting using an integrated software package (PALL FortéBio) and the appropriate stoichiometry.
Kinetics of RppH reactivity monitored by chromatography
DapF stimulation of the reactivity of RppH with diphosphorylated ppAGU and triphosphorylated pppAGU RNA was compared by kinetic analysis, as described in (13). Each reaction mixture contained 100 μM RNA and 0.1 μM RppH, with or without 0.4 μM DapF, in a 350-μl solution containing 50 mM HEPES–NaOH, pH 7.5, 100 mM NaCl, 10 mM MgCl2, 0.1% Triton X-100, and 0.005% (v/v) β-mercaptoethanol at 37°C. 50-μL aliquots taken at time intervals were quenched with 500 μl of 50 mM sodium acetate, pH 5. The quenched reaction samples were analyzed by anion-exchange chromatography on a 5 × 50 mm MonoQ column (GE Healthcare) eluted with a 0–500 mM NaCl gradient in 20 mM Tris–HCl, pH 7.5. Reaction substrates and products were detected at 254 nm, and the amount of each was calculated by using the integration function in UNICORN software. The data for triphosphorylated RNA were fit to an exponential decay equation by using QtiPlot software. Each assay was repeated twice.
Kinetics of orthophosphate removal monitored by phosphomolybdate assay
Rates of phosphate removal from diphosphorylated oligoribonucleotides were determined for RppH in the presence and absence of DapF by a colorimetric phosphomolybdate assay (26). Each reaction mixture contained 100 μM RNA and 0.1 μM RppH, with or without 0.4 μM DapF, in a 175-μl solution containing 50 mM HEPES–NaOH, pH 7.5, 100 mM NaCl and 10 mM MgCl2 at 37°C. 25-μl aliquots taken at time intervals were quenched with 2.5 μl of 0.5 M EDTA. The samples were then mixed with 27.5 μl of reagent ‘C’ and incubated at 37°C for 1.5 h to develop the color. Spectroscopic absorbance measurements were conducted at 820 nm. For comparison, a standard curve was generated with 5–160 μM potassium phosphate. Reagent ‘C’ was prepared fresh by mixing 1 volume of 6N sulfuric acid, 2 volumes of water and 1 volume of 2.5% (w/v) ammonium molybdate and then adding 1 volume of 10% (w/v) ascorbic acid. Each assay was repeated at least twice. The reaction rate was calculated from the linear part of the reaction time course by using QtiPlot software.
Kinetics of pyrophosphate removal monitored by phosphomolybdate assay
Rates of pyrophosphate removal from triphosphorylated oligoribonucleotides were measured as described earlier (13). Briefly, the reactions were conducted in a 20 μl solution containing 50 mM HEPES–Na, pH 7.5, 10 mM MgCl2, 0.1% (v/v) Triton X-100, 150 mM NaCl, 100 μM RNA and 0.1 μM RppH in presence or absence of 0.4 μM DapF. Reactions were incubated at 37°C for 0–20 min and quenched by 5-fold dilution with 1% SDS. Inorganic pyrophosphate from the quenched reactions was precipitated in the presence of 1 mM CaCl2 and 100 mM NaF. The precipitate was collected by centrifugation at 20 000 × g for 10 min, washed with acetone, dried, and re-dissolved in a solution containing 1.25N H2SO4, 10 mM ammonium heptamolybdate, and 40 mM β-mercaptoethanol. The intensity of the green color developed after 1 h of incubation at room temperature was measured at 700 nm, and the amount of the pyrophosphate liberated during the reaction was calculated by comparison to a calibration curve generated with standard solutions of sodium pyrophosphate. The data for triphosphorylated RNA were fit to an exponential decay equation by using QtiPlot software.
Effect of DapF on the reactivity of RppH with yeiP RNA in vitro
Diphosphorylated and triphosphorylated yeiP-U2G RNAs were synthesized by in vitro transcription and gel-purified as previously described (6). Each transcript (1 pmol) was dissolved in 50 μl of a buffer containing 20 mM HEPES, pH 7.5, 10 mM MgCl2, 1 mM DTT, 1% (v/v) glycerol and 2 units/μl rRNasin (Promega). A 5-μl sample was removed to represent the unreacted starting material, and another 5-μl sample was rendered fully monophosphorylated by exhaustive treatment with excess RppH (200 nM at 37°C for 2 h). The remainder (40 μl) was prewarmed to 37°C and combined with prewarmed RppH ± DapF (40 μl) in the same buffer to initiate the reaction. The final reaction mixture (80 μl) contained yeiP-U2G RNA (10 nM), RppH (5 nM), and DapF (0 or 10 nM). After 30 s, a reaction sample (10 μl) was quenched with excess EDTA, phenol extracted, and ethanol precipitated. To detect monophosphorylated yeiP-U2G RNA, each sample was then examined by PABLO (27) in conjunction with site-specific cleavage of the RNA and its ligation product by the 10–23 deoxyribozyme DZyeiP69 (GTAATTCAGTAGGCTAGCTACAACGACATACCTTTT), as described (6). The extent of conversion of diphosphates or triphosphates to monophosphates was calculated by normalizing the measured ligation yield to that of the fully monophosphorylated ligation control.
Decay rate of yeiP mRNA in E. coli
Measurements of the half-life of yeiP mRNA were performed in E. coli strain BW25113 (28) and an isogenic derivative thereof bearing a mutated dapF allele (dapF-V19S-F58S-L89S), which was constructed by allelic exchange as previously described (9,29) and verified by DNA sequencing. To improve detection of yeiP mRNA, each strain also contained a multicopy plasmid (pYeiP1) bearing the yeiP gene (11,12).
To measure rates of yeiP mRNA decay, E. coli cells were grown to mid-log phase at 37°C in LB medium, transcription was inhibited with rifampicin (0.2 mg/ml), and total cellular RNA was extracted at time intervals (6). Equal amounts of each RNA sample (10 μg) were then subjected to gel electrophoresis on 4.5% polyacrylamide containing 8 M urea. RNA was transferred to an Immobilon-NY+ membrane (Millipore) by electroblotting, and the yeiP transcript was detected by probing with a complementary 5′-end-labeled oligodeoxynucleotide (TTCGTTCGCTCTTGGCATCG). Radioactive bands were visualized with a Typhoon Trio imager (GE Healthcare) and quantified by using ImageQuant TL software. RNA half-lives were calculated by linear regression analysis.
RESULTS
The RppH–DapF complex forms a dimer of heterodimers
A previous study reported that E. coli RppH and DapF interact tightly with a KD of ∼5 nM and form a complex with 2:2 stoichiometry (14). These results are consistent with dimerization of an RppH–DapF heterodimer through DapF-DapF interactions, as earlier studies reported that DapF forms a stable dimer with a dimer-monomer KD of ∼22 nM (30). By size-exclusion chromatography (SEC) and cross-linking, we confirmed the 2:2 stoichiometry of the complex and showed that formation of this complex was not affected by adding a His6-SUMO tag at the N-terminus of DapF or by removing 18 structurally disordered amino acids from the C-terminus of RppH (RppHt construct) (Supplementary Figure S1A).
To determine the structure of the RppH–DapF complex, we purified full-length E. coli RppH and DapF, formed a complex in vitro, purified the complex by gel-filtration, and obtained crystals of the complex diffracting at 3.1 Å resolution (Supplementary Table S1). The structure contained a single RppH–DapF heterodimer in the asymmetric unit. Examination of symmetry-related molecules in the crystal revealed extensive interactions between the N-terminal domains of adjacent two-domain DapF protomers, as previously observed in the structure of the E. coli DapF homodimer (30). Therefore, the biological assembly comprises two RppH–DapF heterodimers related by two-fold symmetry and connected through a DapF-DapF interface, according to the observed 2:2 stoichiometry of the complex (Figure 2, Supplementary Figure S2A, B).
Figure 2.

Crystal structure of the E. coli RppH–DapF complex in the apo form with 2:2 stoichiometry. Cartoon representation of the structure as viewed from the front (A) and side (B). The structure comprises two 1:1 RppH–DapF complexes related by 2-fold symmetry in the crystal.
In the structure, each monomer of RppH interacts with the apical region of the N-terminal domain of a DapF protomer. The collinear alignment of RppH with the two domains of DapF results in an RppH–DapF heterotetramer that resembles a butterfly whose ‘wings’ are bridged by a ‘body’ where the DapF subunits contact one another (Figure 2A). A side view of the complex reveals that the two ‘wings’ are not parallel and instead form an ‘X’ shape with a ∼40° angle between the long axes of the RppH–DapF heterodimers (Figure 2B).
Improved quality of the RppH–DapF structural model upon disrupting the DapF-DapF interface
Comparison of the heterotetrameric RppH–DapF structure with the structures of RppH (13), DapF (30), and an RppH-RNA complex (13) did not reveal pronounced conformational changes, which might have an impact on RppH activity. To trap RppH in the active conformation, we soaked the crystals containing the RppH–DapF complex with the substrate-like RNA trinucleotide ppcpAGU and Mg2+ cations, as we did earlier to obtain the RppH-RNA structure (13). Replacement of the bridging oxygen between the α and β phosphates by a methylene moiety (‘c’) prevented cleavage by RppH. The addition of RNA did not affect the structure globally; however, the resolution (3.1 Å) and quality of the electron density map for the RNA ligand and the RNA-binding regions of RppH were insufficient to visualize the entire RNA molecule or to identify DapF-induced changes in RppH compared to previously determined high-resolution structures (1.5–2.0 Å).
To improve the resolution and quality of the RppH electron density map, we removed the disordered C-terminal region of RppH, which is not important for catalysis (13). This truncated form of RppH (RppHt) has a similar affinity for DapF (KD = 4.7 nM) as full-length RppH (KD = 6.7 nM) (Supplementary Figure S1C, D). Since truncation did not significantly improve the quality of the electron density map, we created Y268A and R36A mutations in DapF (DapFm variant) to disrupt the DapF-DapF dimerization interface (30) with the goal of reducing the mobility of RppH in the crystals by changing crystal-packing interactions. These alterations resulted in a 1:1 RppH–DapFm complex (Supplementary Figure S1B) in which the binding affinities of full-length RppH and truncated RppHt are similar (KD values of 7.5 and 6.0 nM, respectively) (Supplementary Figure S1C,E). The RppHt-DapFm complex yielded a new crystal form that diffracted to 1.8 Å and produced an electron density map of excellent quality for the entire RppH protomer, both in the apo state and when bound to ppcpAGU and Mg2+ cations (Figure 3A, B).
Figure 3.

Crystal structure of an RppHt-DapFm heterodimer bound to ppcpAGU RNA. (A) Overall view. The proteins are in cartoon representation, and the RNA ligand is in stick representation. RNA is colored violet, with nitrogen, oxygen and phosphorus atoms shown in their ‘atomic’ colors (blue, red and yellow, respectively). Mg2+ cations are depicted by light pink spheres. (B) Surface representation of the proteins, highlighting the shape complementarity of the RppH–DapF interface. (C) All-atom superposition of the RppHt-DapFm-RNA complex with isolated E. coli DapF and RppH-RNA structures (shown in grey color).
As observed for the RppH–DapF structure, the RppHt-DapFm and RppHt-ppcpAGU-DapFm structures did not reveal significant conformational changes when compared to the structures of the individual enzymes (Figure 3C). Neither structure of the RppH–DapF complex provided evidence of bound DAP when it was soaked into the crystals. Furthermore, both structures of the complex showed all enzyme catalytic sites to be positioned far from the RppH–DapF interface (Figure 3A). The catalytic site of DapF is located in a cleft formed by the two domains of the protein and is positioned on the side of the N-terminal domain opposite to the interface. Likewise, the active site of RppH, where the 5′ phosphates of the RNA substrate bind, is positioned almost on the opposite side of the protein from the DapF interface. A cleft that accommodates the guanine nucleobase of the second nucleotide of the RNA ligand is positioned closer to this interface but is still far away (>16 Å). Thus, any effect of the RppH–DapF interface on the catalytic and nucleobase-binding sites of RppH would have to be propagated over a long distance.
The binding interface of the RppH–DapF complex contains several key residues
The ∼709 Å2 interface between RppH and DapF, calculated by Proteins, Interfaces, Structures and Assemblies (PISA) (31), involves a two-helix bundle on RppH and a platform on DapF composed of three loop regions (Figure 4A). The binding interface of RppH comprises amino acid residues located at the end of the β7 strand (aa 125-126), in the loop connecting the β7 strand and the α4 helix (aa 127–129), in the α4 helix (aa 130–135), and in part of the α5 helix (aa 145–156). The binding interface of DapF comprises amino acids located in the apical part of the β2–α1 loop (aa 18–22), the β3–β4 loop (aa 49–58), and the α2–β5 loop (aa 89–91).
Figure 4.

The RppH–DapF interface. (A) Interface between RppHt (green) and DapFm (cyan). The locations of several amino acids defining the boundaries of the interacting regions are indicated. (B) Contacts between RppH and DapF. Amino acids located ≤3.5 Å from atoms of the partner protein are shown in sticks. Potential hydrogen bonds and cation-π interactions are represented by dashed black and red lines, respectively. (C) View highlighting intermolecular interactions that involve W130, R134 and R145 of RppH. (D–F) Conservation of the amino acids shown in panel (B). (D) and (F) show the interacting surfaces of RppH and DapF, respectively. (E) depicts a side view of the RppH–DapF complex. Identical and similar residues conserved in >70% (orange), 50–69% (yellow), or <50% (green for RppH and cyan for DapF) of a representative set of protein sequences from γ- and β-proteobacteria (43) are indicated. Residues colored grey are >3.5 Å away from the partner protein. Panel (F) highlights the position of W130, R134 and R145 (in sticks) of RppH near conserved interface residues of DapF.
The interacting surfaces are held together by multiple direct and water-mediated hydrogen bonds, hydrophobic contacts and other interactions, and they contain several protruding amino acids in RppH that interlock the complex through excellent shape complementarity (Figures 3B and 4B, C). Despite the large number of RppH residues potentially contributing to interactions with DapF, only seven of them are located ≤3.5 Å from DapF; those seven appear to make the most significant contribution to the binding affinity by interacting directly with 13 DapF residues (Figure 4D–F). Interestingly, among these DapF residues, the majority are evolutionarily conserved, seven of them being present in >70% of orthologs in a representative set of sequences from γ- and β-proteobacterial species (Figure 4F and Supplementary Figure S3). The RppH residues at the interface are less well conserved, such that only two are present in >70% of RppH orthologs from the same set of species (Figure 4D). These two conserved RppH residues, W130 and R145, are involved in multiple interactions with DapF (Figure 4C, F). Another RppH residue forming multiple contacts with DapF is the non-conserved amino acid R134.
W130 of RppH is positioned near the center of the binding interface. Its side chain contacts a hydrophobic cavity formed by V19, T20, F58, and L89 of DapF (Figure 4B, F) and makes a hydrogen bond with the main chain of DapF L89 (Figure 4C). W130 is located on the surface of RppH and appears not to be critical for the stability or folding of this protein, as it adopts two alternative conformations in the RppH–RNA complex when DapF is absent (13). In the RppH–DapF complex, W130 adopts only one, inward-oriented conformation because P51 of DapF occupies the space needed for W130 to adopt the other, outward-oriented conformation. W130 is likely a key residue of the binding interface because it is highly conserved in γ- and β-proteobacteria. The high evolutionary conservation of DapF F58 and L89 and moderate conservation of DapF V19 and T20, where substitutions of similar amino acids are sometimes observed, support the importance of W130 recognition for RppH–DapF binding.
R134 and R145 of RppH are located more peripherally and participate in both stitching the complex together and re-orientating its components (Figure 4B, C). R145 is highly conserved, and its conformation changes only slightly upon DapF binding to maximize intermolecular interactions. The side chain of R145 is at a suitable distance for hydrogen bonding to the main chain carbonyl oxygens of A18, V19 and Q21 (Figure 4B), and it appears to lock in place the DapF residues involved in the recognition of W130. The significance of R134 for complex formation is less obvious. In the apo RppH and RppH-RNA structures, the side chain of R134 is not well ordered (13), but it becomes ordered upon DapF binding. The side chain of R134 is situated in a shallow DapF cavity formed by the side chains of P50 and Y52 and the main chain of P51. The guanidinium moiety of R134 makes cation-π interactions with Y52 and hydrogen bonds with E49, P50 and P51 (Figure 4C). Despite these extensive interactions, R134 is present only in some RppH orthologs and is probably responsible for tuning RppH–DapF interactions in a species-specific manner, as it typically covaries with Y52 of DapF, its partner for cation–π interactions.
Mutational studies confirmed the importance of the conserved interface residues for RppH–DapF binding. As shown by SEC (Figure 5A, B) and BLI (Figure 5C–E), a double W130A-R145A mutation in RppH disrupted complex formation by increasing the KD to 1.9 μM, which corresponds to a ∼284-fold decrease in affinity for DapF. Altering the interaction site on DapF by the double mutations V19S-F58S, V19S-L89S and F58S-L89S also impaired complex formation by reducing the binding affinity 59-, 77- and 290-fold, respectively (Figure 5C). Simultaneously mutating all three of these hydrophobic residues (V19S–F58S–L89S) decreased binding by ∼5800 fold (Figure 5C, F). Mutating the less conserved R134 to alanine (R134A) in RppH reduced the binding affinity only 2-fold (Figure 5C, G).
Figure 5.

Heteromeric binding affinity of RppH and DapF. (A) Size exclusion chromatography (SEC) of the RppH–DapF complex (black line), RppH (red line) and wild type DapF (blue line). RppH was added in excess to DapF during complex formation. The RppH–DapF peak at 70 ml corresponds to a complex with 2:2 stoichiometry. (B) SEC of a mixture of RppHW130A,R145A and DapF. W, R correspond to W130A and R145A mutations in RppH. (C) Binding affinity of RppH–DapF complexes, as determined by bio-layer interferometry (BLI). KD values are averages of two experiments in which the concentration of the protein in solution was varied. SE, standard error. ‘Surface’ and ‘solution’ identify the immobilized protein and the protein in solution, respectively. V, F and L correspond to V19S, F58S and L89S mutations in DapF, W and R to W130A and R145A mutations in RppH, and R* to R134A in RppH, respectively. (D–G) Association and dissociation curves for representative BLI experiments with RppH and DapF. (D), RppH and DapF; (E), DapF and RppHW130A,R145A; (F), RppH and DapF V19S,F58S,L89S; (G), RppHR134A and DapF. The first protein named in each panel title was immobilized. Concentrations of the protein in solution were 5 nM (violet), 10 nM (blue), 20 nM (cyan), 39 nM (green), 79 nM (yellow), 156 nM (orange) and 312.5 nM (red) in (D), (F) and (G). In (E), the concentrations were 0.35 μM (violet), 0.7 μM (blue), 1.4 μM (cyan), 2.8 μM (green), 5.6 μM (yellow), 11.25 μM (orange) and 22.5 μM (red).
DapF binding potentiates RppH activity on small substrates bearing at least three 5′ phosphates
Previous in vitro studies showed that DapF doubles the rate at which RppH hydrolyzes diadenosine pentaphosphate (Ap5A) and the short triphosphorylated RNAs pppGCA and pppACG (14). We used a chromatographic assay (13) to monitor the reaction of RppH with pppAGU RNA and observed a similar 2-fold acceleration of pyrophosphate removal by RppH in the presence of DapF (Figure 6A). However, in light of recent evidence that diphosphorylated, not triphosphorylated, RNA is the principal substrate of RppH in E. coli (6), we also examined the influence of DapF on the reactivity of ppAGU RNA. Unexpectedly, DapF did not increase the rate at which RppH removed orthophosphate from this short diphosphorylated substrate (Figure 6A).
Figure 6.

Identification of potential allosteric interactions of DapF with RppH. (A) Effect of DapF on RppH reactivity with ppAGU and pppAGU RNA substrates, as analyzed by chromatography. pppAGU substrate: RppH alone, black lines and circles; RppH with DapF, red lines and triangles. ppAGU substrate: RppH alone, blue lines and inverted triangles; RppH with DapF, orange lines and diamonds. Error bars represent standard deviations. Inset: the straight lines obtained in semilogarithmic plots show that the reaction of the triphosphorylated substrate proceeded with first-order kinetics. The rate constants with pppAGU were 0.080 ± 0.001 min−1 for RppH and 0.165 ± 0.014 min−1 for the RppH and DapF mixture (average ± SD, n = 2). (B) All-atom superposition of the RppHt–ppcpAGU–DapFm (protein in green, RNA and Mg2+ cations in violet) and RppHt–2Mg–ppcpAGU (orange) structures. Several RNA- and Mg2+-binding residues are shown in sticks. Blue arrows show conformational shifts in the upper part of the catalytic and Gua-2 binding sites. (C) All-atom superposition of the RppH–DapF (green) and RppH (orange) structures. RNA- and Mg2+-binding residues that adopt different conformations in the two structures are shown in sticks. (D and E) Zoomed-in views of the structure superposition (panel B), centered on the Gua2 binding cleft (D) and the active site (E). All residues are shown in lines except residues participating in RNA and Mg2+ recognition, which are in sticks. (F) Zoomed-in view of the structure superposition (panel C), centered on the Gua2 binding cleft and the upper part of the active site. (G) Schematics of RNA 5′-end recognition in different RppH complexes. The left and central panels are for structures from (13), and the right panel is from the current study. Magenta circles, magnesium ions; green circles, phosphates; dashed white circle, structurally disordered γ phosphate; arrow, location of a water molecule poised for nucleophilic attack.
DapF does not induce pronounced allosteric transitions in RppH
To understand the mechanism by which DapF stimulates RppH activity on short triphosphorylated RNAs, we compared the high-resolution structures of the RppHt–DapFm complex in the apo and RNA-bound states with the corresponding structures of RppH without DapF from our previous work (Figure 6B–F) (13).
The RppHt–ppcpAGU–DapFm structure shows that the DapF interface and RNA 5′-end binding site of RppH are far from one another. Nevertheless, the effects of DapF binding could, in principle, be transmitted through structural elements of RppH to both the catalytic and nucleobase-recognition sites of RppH and influence RNA and/or magnesium binding. Indeed, V137, F139 and K140, which recognize the Gua2 nucleobase, are located on the tips of the α4 and α5 helices involved in DapF binding (Figure 6B, D). In addition, the Gua2-contacting residue R27 is situated at the end of a beta strand (β2) that contacts the DapF-bound β7 strand, which itself is adjacent to a loop containing a fourth residue (E120) that participates in Mg2+-binding. Therefore, a conformational change affecting these amino acid residues in the DapF-bound state could in theory influence substrate binding and/or catalysis.
Comparison of the RppHt-ppcpAGU-DapFm structure with the structures of the RppH-RNA complexes (13) revealed a high degree of resemblance to the RppHt–2Mg–ppcpAGU structure (Figure 6G, center and right panels). The RppHt–2Mg–ppcpAGU structure contains similarly positioned 5′ phosphates bound to two Mg2+ cations and aligned for cleavage between the β and γ phosphates (Figure 6B, G, central panel) to release diphosphorylated RNA, an RppH reaction product not observed under normal conditions. Despite also having three Mg2+ cations in the active site, the RppHt–ppcpAGU–DapFm structure was less similar to the RppHt–3Mg–ppcpAGU structure, in which the 5′ phosphates were positioned for cleavage between the α and β phosphates to release monophosphorylated RNA and pyrophosphate as the reaction products (Figure 6G, left panel).
The protein and RNA conformations in the RppHt–ppcpAGU–DapFm and RppHt–2Mg–ppcpAGU structures are very similar, resulting in a low all-atom RMSD of 0.293 Å between them, excluding flexible regions (M1-D5 and V84-K96) (Figure 6B). However, the structures have a few notable differences. First, relative to the bound RNA, there are 0.3–1.1 Å conformational shifts at the top of the Nudix helix and adjacent regions, which comprise the upper part of the catalytic and Gua2-binding sites and contain R27, E57 and E120 (Figure 6D, E). Second, in the RppHt–2Mg–ppcpAGU structure, the γ phosphate was almost entirely hydrolyzed during soaking and therefore was not modeled (13) (Figure 6B, G, center), while in the RppHt–ppcpAGU–DapFm structure the γ phosphate is only partially hydrolyzed (Figure 6G, right). Third, in the RppHt–ppcpAGU–DapFm structure, we observe three Mg2+ cations, while in the RppHt–2Mg–ppcpAGU structure one of the three Mg2+ cations was replaced by a sulfate ion from the crystallization solution. Removal of sulfate allowed binding of the third Mg2+ cation in the RppHt–3Mg–ppcpAGU structure (13). Fourth, the four glutamates involved in Mg2+ coordination (E53, E56, E57 and E120) adopt a single Mg2+-bound conformation in the RppHt–ppcpAGU–DapFm structure, while in the RppHt–2Mg–ppcpAGU structure, these amino acids adopted both Mg2+-bound and -unbound conformations (Figure 6E).
While the small differences between the DapF-free and DapF-bound structures of RppH bearing an RNA ligand might help to explain the ability of DapF to potentiate RppH activity, the structural data alone are not sufficient to ascribe a major role to any of the observed changes. To explore the alternative possibility that DapF binding potentiates RppH activity by making the apo state of RppH more amenable to RNA and Mg2+ binding, we compared the apo-RppHt and apo-RppHt–DapFm structures. The conformation of RppH was very similar in each of them (all-atom RMSD of 0.414 Å, excluding the flexible regions), with minor changes in the conformation of the long side chains of R8, R27, E53 and E120 (Figure 6C, F), residues involved in RNA and Mg2+ binding. However, the presence of DapF causes only R8 and E53 to adopt conformations potentially more favorable for binding Mg2+ and RNA, and these two residues are located far from DapF. Thus, DapF does not appear to induce significant conformational changes in the ground state of RppH that would accelerate the catalytic reaction.
The effect of DapF on the reactivity of RNAs depends on their length
Since RppH is thought to preferentially target diphosphorylated mRNA decay intermediates in cells, the accelerated reaction of triphosphorylated RNAs in vitro is unlikely to explain the effect of DapF on RppH-dependent mRNA degradation in E. coli. The inability of DapF to enhance the reactivity of diphosphorylated trinucleotides (Figure 6A) prompted us to look for an alternative mechanism by which DapF might increase the reactivity of diphosphorylated mRNA. One possible mechanism could involve an effect of DapF on the binding or release of longer RNAs.
ppAGU is too short to span the distance from the catalytic site of RppH to DapF in the ternary complex. To test whether DapF stimulates phosphate removal from substrates long enough to reach this protein, we compared the reactivity of a series of diphosphorylated RNAs ranging from three to nine nucleotides in length. These in vitro-transcribed RNAs were treated with RppH in the presence or absence of DapF, and the release of inorganic phosphate was monitored as a function of time by a colorimetric assay involving phosphomolybdate reduction (26).
These rate measurements showed that DapF is unable to stimulate the reaction of RppH with diphosphorylated RNA substrates ranging in length from three to seven nucleotides (Figure 7A, B). However, it enhanced the reactivity of RppH 1.4-fold with an 8-nt RNA and 1.9-fold with a 9-nt RNA. As expected, the DapF V19S,F58S,L89S mutant, incapable of RppH binding, had no such effect. Since the 8- and 9-nt RNAs were long enough to span the distance from the RppH catalytic site to DapF in the RppH–DapF complex, these results suggest that the effect of DapF on the reactivity of diphosphorylated substrates results directly from the presence of DapF on the surface of RppH and not from any pronounced allosteric change that DapF binding may induce. The influence of DapF is unrelated to its ability to homodimerize, as monomeric DapFm is as effective as dimeric DapF at enhancing the reactivity of the 8-nt diphosphorylated substrate (Figure 7B, C).
Figure 7.

DapF stimulation of RppH reactivity with diphosphorylated RNA substrates. (A) Variable effect of DapF on RppH reactivity with diphosphorylated RNA substrates of different lengths. Phosphate release was quantified by a colorimetric molybdate assay. RppH alone, black lines and circles; RppH complexed with DapF, red lines and triangles; mixture of RppH and DapF V19S,F58S,L89S, blue lines and inverted triangles. The RNA substrates were: ppAGU (3 nt), ppAGUUU (5 nt), ppAGUUUU (6 nt), ppAGUUUUU (7 nt), ppAGUUUUUU (8 nt), and ppAGUUUUUUG (9 nt). Error bars represent standard deviations. (B) Rate constants of the experiments in (A). Each value is the average of at least two experiments (n = 2). SD, standard deviations. (C) Effect of monomeric DapFm on the reactivity of RppH with the 8-nt diphosphorylated substrate (in purple). A control reaction with wild-type DapF is in red.
A closer examination of the data reveals that, for diphosphorylated RNAs 3–7 nt in length, the reaction rate decreased with increasing substrate length whether or not DapF was present (Figure 7B). In the absence of DapF, the reaction rate continued its decline for a substrate >8 nt long, but it was faster for longer substrates when DapF was present. Thus, DapF appears to counteract the impaired turnover of longer diphosphorylated RNAs without affecting the turnover of shorter diphosphorylated RNAs.
The linearity of the plots depicting the reaction of diphosphorylated RNA substrates with RppH and the RppH–DapF complex (Figures 6A and 7A) indicates that, in every case, the reaction rate was unaffected by the diminished concentration of the substrate that remained at later times. Such kinetics indicate an enzyme operating under saturating conditions (S >> Km). Under these conditions, it would be possible to observe the effect of DapF only on kcat of the RppH-catalyzed reaction; any potential effect on Km would escape detection.
By contrast, the curvature of the plots depicting the reaction of triphosphorylated AGU with RppH and RppH–DapF and their linearity when graphed semilogarithmically (Figure 6A) are indicative of first-order kinetics. Such kinetics suggest that, with this substrate, the enzyme either was operating under subsaturating conditions (S < Km), implying a significantly higher Km than for its diphosphorylated counterpart, or was susceptible to product inhibition. Therefore, the influence of DapF on the reactivity of this trinucleotide could reflect an increase in kcat and/or a decrease in Km and, in view of the short length of this particular substrate, likely results from an indirect, allosteric effect on RppH.
To determine whether DapF stimulation of the reactivity of RppH with triphosphorylated substrates is length-dependent, we compared the reaction rates of 3-, 6-, 7- and 9-nt long triphosphorylated RNAs in the presence and absence of DapF (Figure 8). Since the chromatographic assay used in the previous experiment with the 3-nt RNA (Figure 5A) lacks sufficient resolution for monitoring the reaction of longer RNAs, we employed a molybdate-based colorimetric assay to detect the release of inorganic pyrophosphate and plotted the data on linear and semilogarithmic scales (Figure 8A, B). The excellent fit of the data to a straight line in the semilogarithmic graphs indicates that, in contrast to the reaction of their diphosphorylated counterparts, the reaction of triphosphorylated RNAs follows first-order kinetics (Figure 8). As observed for the diphosphorylated RNAs, pppAGU was significantly more reactive than triphosphorylated substrates ≥6 nt in length in the absence of DapF. Furthermore, as judged by colorimetry, pyrophosphate release from pppAGU was 2.4-fold faster in the presence of DapF than in its absence (Figure 8C), a finding consistent with the results of the chromatographic assay (Figure 6A). The DapF-induced rate acceleration increased 6-fold for the 6-nt RNA and was further enhanced by a factor of 1.7–1.9 for the 7- and 9-nt RNAs. As in the case of the diphosphorylated RNAs, this length-dependent rate acceleration resulted primarily from counteracting the diminished reactivity of the longer triphosphorylated substrates, although some of the acceleration in the presence of DapF (∼2-3-fold) was due to the greater reactivity of triphosphorylated RNAs ≥7 nt in length. The magnitude of the stimulatory effect of DapF for the longer triphosphorylated substrates (14–26-fold) was significantly greater than observed for the longest of the diphosphorylated substrates (∼1.9-fold).
Figure 8.

DapF stimulation of RppH reactivity with triphosphorylated RNA substrates. (A) Variable effect of DapF on RppH reactivity with triphosphorylated RNA substrates of different lengths. Pyrophosphate release was quantified by a colorimetric assay. RppH alone, black lines and circles; RppH complexed with DapF, red lines and circles. The RNA substrates were: pppAGU (3 nt), pppAGUUUU (6 nt), pppAGUUUUU (7 nt), and pppAGUUUUUUG (9 nt). Error bars represent standard deviations. Insets: the same data plotted semilogarithmically. (B) Comparison of the reaction time courses for RppH in the presence of DapF, as shown in (A). 3-, 6-, 7- and 9-nt RNAs are shown in black, red, blue and purple colors, respectively. (C) Rate constants of the reactions in (A). SD, standard deviations, n = 2.
DapF binding enhances the reactivity of RppH with a natural mRNA target
The 0.65-kb transcript of the yeiP gene is known to be degraded by an RppH-dependent mechanism in E. coli (5,12). Therefore, to examine the effect of DapF on the reactivity of RppH with a natural mRNA substrate, we compared rates of orthophosphate or pyrophosphate removal from diphosphorylated and triphosphorylated yeiP RNA, respectively, in vitro (Figure 9A). In each case, the generation of the monophosphorylated reaction product was monitored by PABLO, a splinted ligation assay in which monophosphorylated RNAs are selectively joined to a DNA oligonucleotide, thereby retarding their electrophoretic mobility (27). These experiments showed that DapF increases the reactivity of purified RppH with both forms of yeiP by more than a factor of 10 while maintaining the preference of RppH for diphosphorylated RNA. The basis for the greater effect of DapF on the reactivity of diphosphorylated yeiP RNA versus the diphosphorylated oligonucleotide substrates is not known but could be related to the difference in substrate length or the much lower substrate concentration used in the yeiP experiments.
Figure 9.
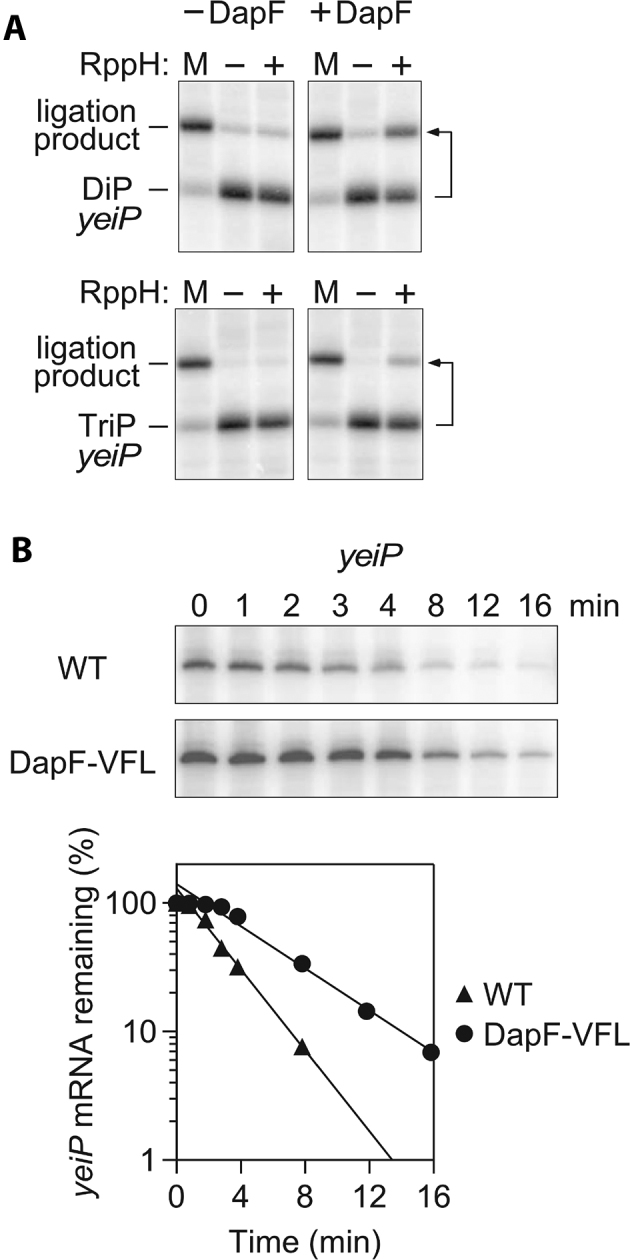
Effect of DapF on the reactivity of RppH with yeiP mRNA. (A) Influence of DapF on the reaction of RppH with yeiP-U2G mRNA in vitro. Diphosphorylated (DiP) or triphosphorylated (TriP) mRNA that had been synthesized by in vitro transcription was treated for 30 s with purified RppH in the absence or presence of DapF, and the production of monophosphorylated yeiP-U2G was subsequently detected by PABLO. M, PABLO ligation yield of fully monophosphorylated mRNA. Almost all of the monophosphorylated reaction product underwent ligation. The small amount of ligation product observed before treatment with RppH was due to trace ligation of the diphosphorylated starting material (6). Images for the reactions in the presence of DapF are reproduced from portions of Fig. 1 in (6). The second nucleotide of the yeiP transcript was changed from U to G to enable synthesis by T7 RNA polymerase. (B) Decay of yeiP mRNA in E. coli containing wild-type DapF or the DapFV19S,F58S,L89S (VFL) mutant. (Top) Northern blots. Log-phase cultures of isogenic E. coli strains bearing either a wild-type (WT) or mutant (VFL) dapF allele were treated with rifampicin to arrest transcription, and equal amounts of total RNA isolated at time intervals thereafter were analyzed by gel electrophoresis and blotting to detect yeiP mRNA. (Bottom) Semilogarithmic plots of the intensity of the yeiP band as a function of time. Data from a representative experiment are shown. The half-life of yeiP mRNA increased from 1.9 ± 0.1 min in wild-type DapF cells (WT) to 3.7 ± 0.2 min in isogenic DapFV19S,F58S,L89S cells (VFL).
We next investigated the effect of DapF association on RppH-dependent mRNA turnover in vivo. This was accomplished by constructing an E. coli strain bearing a mutant dapF allele that encodes a form of the protein (DapFV19S,F58S,L89S) shown in Figure 5C, F to be greatly impaired for RppH binding and by then comparing the decay rate of yeiP mRNA in this strain and in its isogenic counterpart bearing a wild-type dapF allele (Figure 9B). The half-life of yeiP mRNA doubled from 1.9 to 3.7 min in the mutant strain, indicating the importance of DapF binding for stimulating the reactivity of RppH in E. coli.
DISCUSSION
A small subset of metabolic enzymes not only fulfill their primary responsibility of providing energy or consumables to cells but also moonlight by participating in an additional cellular process uncharacteristic of their principal physiological role (32). In some cases, the moonlighting functions are tangentially related to the primary function of the enzymes, as when they enable a dissimilar cellular process to sense the metabolic state of the cell (33). In other cases, the physiological connection between a metabolic enzyme's two roles is not readily apparent. An intriguing relationship of the latter kind is the stimulation of the bacterial RNA pyrophosphohydrolase RppH by DapF, an enzyme involved in amino acid and peptidoglycan biosynthesis (14). Through its direct association with RppH, DapF accelerates the conversion of the 5′ termini of primary transcripts to monophosphates, thereby hastening mRNA degradation via the 5′-end-dependent pathway (14). Interestingly, another important enzyme in this pathway, the 5′-monophosphate-assisted endonuclease RNase E, itself associates with the glycolytic enzyme enolase (34–36); however, little is understood about the mechanistic basis for any effect that enolase may have on RNA cleavage by RNase E or about any interplay between glycolysis and mRNA degradation (37–39).
The crystal structure of the RppH–DapF complex, determined in the current study, has revealed that the proteins associate to form a rotationally symmetrical 2:2 complex comprising two RppH–DapF heterodimers held together by DapF–DapF interactions. In the resulting heterotetramer, each RppH subunit interacts exclusively with one DapF subunit and does not contact the other RppH–DapF heterodimer. The RppH–DapF interface is compact and stabilized by a variety of interactions involving a set of RppH residues that are fewer in number and less conserved than the DapF residues at the interface. The conserved RppH residues there are essential for DapF binding, and mutating two of them (W130A and R145A) or the DapF residues with which they interact (V19S, F58S and L89S) prevents complex formation in vitro. Disrupting formation of the RppH–DapF complex in vivo by introducing these three point mutations into the E. coli dapF gene prolongs the lifetime of the yeiP mRNA. This finding demonstrates the importance of DapF binding for RppH-dependent mRNA degradation in E. coli without the interpretive ambiguity inherent to alternative approaches in which the dapF gene is deleted and the biosynthesis of lysine and peptidoglycan is thereby impaired. The dapF point mutant will be invaluable in future studies to assess the impact of DapF binding on the degradation of other transcripts.
Remarkably, neither protein changes significantly in conformation upon binding. The lack of pronounced structural changes in the RppH–DapF complex is in stark contrast to the large conformational rearrangement that occurs when the functionally analogous yeast decapping enzyme Dcp2 forms a complex with its obligatory activator Dcp1 and an intrinsically disordered region of the coactivator Edc1 (40). The two complexes also differ in the spatial arrangement of the catalytic Nudix domain and the activating proteins (Figure 10A); while DapF binds to the top of RppH, the activators of Dcp2 bind to the side of its Nudix domain.
Figure 10.

Parallels between 5′-terminal RNA deprotection complexes in bacteria and eukaryotes. (A) Superposition of E. coli RppHt–ppcpAGU–DapFm (green, violet and cyan, respectively) and yeast Dcp2–Dcp1–Edc1 (40) (orange, pink, and hot pink, respectively) structures on the Nudix domains. The Box B helix of Dcp2 is indicated. (B, C) Views of protein surfaces in electrostatic potential representation for RppH–RNA–DapF (B) and Dcp2–Dcp1–Edc1 (C) complexes, highlighting RNA-binding regions and putative paths (violet lines) of a long RNA ligand. RNA from the RppH–RNA–DapF complex is projected into the Dcp2–Dcp1–Edc1 structure for orientation. Two 5′-terminal RNA nucleotides bound to the catalytic center and nucleobase-binding site of RppH are shown in violet sticks, and the positively charged surface patches mentioned in the text (A, B and C) are encircled by yellow dashed lines. (D) Formation of the RppH–DapF complex. The proteins are separated in orientations favorable for binding. (E) RppH–DapF interface. DapF, whose interface residues are depicted in cyan sticks, partially blocks positively-charged patch C on RppH, which is depicted in surface representation.
Unexpectedly, our study has identified two distinct modes by which DapF potentiates RppH activity. The original report of the association of DapF with RppH showed that the formation of this complex stimulates the reaction of RppH with triphosphorylated trinucleotides in vitro and full-length mRNAs in vitro and in vivo. These results implied that the same mechanism of RppH activation was operative in both cases (14). However, we recently reported that the principal role of RppH in E. coli appears to be the removal of orthophosphate from diphosphorylated decay intermediates rather than pyrophosphate from triphosphorylated primary transcripts (6). Our present findings confirm that DapF accelerates RppH-catalyzed pyrophosphate removal from short triphosphorylated RNAs, perhaps through an allosteric mechanism, but reveal no effect on orthophosphate removal from equally short diphosphorylated RNAs. The latter observation is seemingly at odds with the ability of DapF to accelerate mRNA degradation in E. coli (14), where RppH appears to act primarily on intermediates bearing a 5′-terminal diphosphate (6). The ability of DapF to hasten the degradation of long mRNAs in cells appears more likely to result from a second mode of stimulation in which it selectively accelerates orthophosphate removal from diphosphorylated RNA substrates ≥8 nt in length. Because these RNAs are long enough to reach from the RppH catalytic site to the RppH–DapF interface, this mode of stimulation likely involves a direct effect of DapF on RNA substrate binding and/or RNA product release rather than an allosteric transition in the structure of RppH.
Interestingly, the reactivity of diphosphorylated RNA with RppH alone is inversely related to its length, declining in two steps from 3 to 6 nt and again from 8 to 9 nt. The diminished reaction rate (kcat) of longer diphosphorylated substrates under the saturating conditions used in this study suggests either rate-limiting product release impeded by more extensive contacts of the enzyme with longer RNAs or suboptimal positioning of the diphosphorylated 5′ end in the RppH active site as a consequence of structural constraints imposed by those additional contacts. For diphosphorylated RNAs ≥8 nt long, DapF appears to counteract the deleterious effect of substrate length on the magnitude of kcat. Although the potential of long substrates to interact more extensively with RppH might also increase their binding affinity and while interactions with DapF may influence this binding, any such effect on Km would not have been observable under the saturating conditions that we employed.
Our experiments also demonstrate that the effect of DapF on the reactivity of RppH with triphosphorylated substrates likewise increases with RNA length. Therefore, the DapF-mediated enhancement of the reaction rate of triphosphorylated RNAs appears to involve two distinct mechanisms, one allosteric and the other dependent on substrate length. Differences in the magnitude of the effect of DapF on the reactivity of the diphosphorylated and triphosphorylated substrates and in the minimum RNA length at which this effect becomes apparent may well be related to the fact that one set of rate measurements was made under saturating conditions (influence of DapF on kcat) while the other appears to have been made under subsaturating conditions (influence on kcat / Km).
The structures of the RppH–DapF complex show that long diphosphorylated and triphosphorylated RNA ligands could follow a variety of paths to reach DapF (Figure 10B). The first possibility (path 1, magenta color) would be to span the ∼35-Å distance from the RppH catalytic site to a positively charged region of DapF (patch A, yellow color) near R96 and R103. Assuming a spacing of ∼6 Å between the phosphates of adjacent nucleotides in unstructured RNA, a 6–8-nucleotide RNA ligand would be sufficiently long to span that distance. A second possibility (path 2) would be for the RNA to bend towards a DapF cavity beneath the β3-α1 loop, ∼35 Å from the catalytic site. A third possibility (path 3) would be for the RNA to reach ∼25 Å from the catalytic site to a hydrophobic cavity of DapF potentially suitable for nucleobase binding. The last of these options (Figure 10B) is particularly appealing because (i) nucleotides starting from approximately position 6–8 could enter the hydrophobic cavity in DapF; (ii) this mode of substrate binding would allow the RNA to interact electrostatically with two successive positively charged regions of RppH (patches B and C, yellow color) between the nucleobase-binding site and DapF; and (iii) by sterically hindering access to part of the positively charged region near patch C of RppH (Figure 10D, E), DapF binding could facilitate the release of long monophosphorylated RNA products by reducing their affinity for RppH. Interaction with patches B and C may explain the biphasic nature of the dependence of the reaction rate on RNA length and the stimulatory effect of DapF if product release is rate-determining and is slowed by reaching each patch and if RNA access to the patch C is hindered when DapF is present.
Because short substrates cannot reach DapF in the complex, any stimulation of their hydrolysis by RppH must involve changes in the catalytic and/or nucleobase-binding sites induced by DapF. Their reaction with RppH may be relevant to the breakdown of alarmones like diadenosine tetraphosphate, which is thought to be targeted by RppH (41). Although a comparison of published RppH-RNA structures (13) to the structure that we now have obtained for the RppH–DapF complex bound to a triphosphorylated RNA mimetic does not entirely explain the stimulatory effect of DapF on RppH, it has limited the potential mechanisms to possibilities that include changes in the dynamics or rigidity of the catalytic and nucleobase-binding sites, improved recruitment of a catalytically relevant Mg2+ cation, and a reduction in the conformational heterogeneity of the amino acid side chains that bind RNA or Mg2+. The first of these possibilities seems the most probable explanation because of the shortcomings of the other two. For a couple of reasons, the inconsistent crystallographic detection of a third Mg2+ cation in the active site is not likely to explain the catalytic acceleration caused by DapF. First, the differing number of Mg2+ cations depended on the crystallization conditions and may therefore be unrepresentative of Mg2+ binding in solution. Furthermore, because the third Mg2+ cation helps to activate the attacking water molecule (13), it should be crucial for the reactivity of both diphosphorylated and triphosphorylated RNA, contrary to our rate measurements for ppAGU. Likewise, the conformational heterogeneity and differences observed for the side chains of the Mg2+-coordinating and RNA-binding residues in the RppHt–ppcpAGU–DapFm and RppHt–2Mg–ppcpAGU structures are unlikely to adequately explain the DapF-induced rate acceleration observed for pppAGU because the RppHt–3Mg–ppcpAGU structure (13) contains many side chains in the conformations observed in the DapF-bound structure.
Our study also suggests a plausible basis for the significant difference in the reactivity of RppH with substrates bearing two versus three 5′-terminal phosphates. The reaction rate of RppH with diphosphorylated RNA (ppAGU and longer derivatives thereof) did not decline as the substrate was consumed, a finding indicative of an initial substrate concentration well in excess of Km (i.e. saturating conditions). By contrast, the reaction of RppH with triphosphorylated RNAs proceeded with first-order kinetics, suggesting either subsaturation or product inhibition. In view of the resistance of the diphosphorylated RNAs to detectable product inhibition despite the identical RNA reaction products of the two sets of substrates, it seems likely that the Km of the diphosphorylated substrates is lower than that of their triphosphorylated counterparts.
The preference of RppH–DapF for long RNA substrates is reminiscent of a similar preference exhibited by the activated Dcp2–Dcp1–Edc1 decapping complex (40,42). In the latter complex (Figure 10C), the bound activators are thought to extend the RNA-binding surface by creating a cleft lined with positively charged residues, thereby increasing the binding affinity of capped mRNA (40). Some of the key Dcp2 residues in this cleft are located in the Box B helix of the Nudix domain of Dcp2 (40). This helix is equivalent to helix α5 of RppH (Figure 10A), which also contains positively charged residues and contributes to DapF binding. Thus, despite the limited structural similarities of the bacterial and eukaryotic complexes that deprotect RNA 5′ ends, there are interesting parallels in the mechanisms by which they are activated.
DATA AVAILABILITY
Coordinates of the structures were deposited in the Protein Data Bank with PDB ID codes: 6D13, RppH–DapF complex; 6D1Q, RppHt-DapFm complex; 6D1V, RppHt-ppcpAGU-DapFm complex. All relevant data and materials are available upon request.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to the personnel of beamlines 24-ID (APS) and 17-ID-2 (NSLS-II) for their help with data collection.
Authors Contributions: A.G., N.V., N.T., J.G.B. and A.S. designed the experiments, A.G., N.V., D.J.L., R.L.P, J.R. and B.M. conducted the experiments, A.G., N.V., N.T., J.G.B. and A.S. interpreted the data, and J.G.B. and A.S. wrote the paper.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
This work used funds from National Institutes of Health (NIH) [R01GM112940 to A.S., R01GM035769 to J.G.B., R01AI108889 to N.J.T., F99CA212474 to W.M.M.]; the Northeastern Collaborative Access Team beamlines are funded by NIH [P41 GM103403, S10 RR029205] and resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory [DE-AC02-06CH11357]; beamline 17-ID-2 is supported in part by the DOE Office of Biological and Environmental Research [KP1605010, KC0401040] and NIH [P41GM111244]; NSLS-II is supported in part by the DOE, the Office of Science, and the Office of Basic Energy Sciences Program [DE-SC0012704]. Funding for open access charge: NIH [R01GM112940].
Conflict of interest statement. None declared.
REFERENCES
- 1. Belasco J.G. All things must pass: contrasts and commonalities in eukaryotic and bacterial mRNA decay. Nat. Rev. Mol. Cell Biol. 2010; 11:467–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jonas S., Izaurralde E.. The role of disordered protein regions in the assembly of decapping complexes and RNP granules. Genes Dev. 2013; 27:2628–2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Arribas-Layton M., Wu D., Lykke-Andersen J., Song H.. Structural and functional control of the eukaryotic mRNA decapping machinery. Biochim. Biophys. Acta. 2013; 1829:580–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Celesnik H., Deana A., Belasco J.G.. Initiation of RNA decay in Escherichia coli by 5′ pyrophosphate removal. Mol. Cell. 2007; 27:79–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Deana A., Celesnik H., Belasco J.G.. The bacterial enzyme RppH triggers messenger RNA degradation by 5′ pyrophosphate removal. Nature. 2008; 451:355–358. [DOI] [PubMed] [Google Scholar]
- 6. Luciano D.J., Vasilyev N., Richards J., Serganov A., Belasco J.G.. A novel RNA phosphorylation state enables 5′ end-dependent degradation in Escherichia coli. Mol. Cell. 2017; 67:44–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Richards J., Liu Q., Pellegrini O., Celesnik H., Yao S., Bechhofer D.H., Condon C., Belasco J.G.. An RNA pyrophosphohydrolase triggers 5′-exonucleolytic degradation of mRNA in Bacillus subtilis. Mol. Cell. 2011; 43:940–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hui M.P., Foley P.L., Belasco J.G.. Messenger RNA degradation in bacterial cells. Annu. Rev. Genet. 2014; 48:537–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Luciano D.J., Hui M.P., Deana A., Foley P.L., Belasco K.J., Belasco J.G.. Differential control of the rate of 5′-end-dependent mRNA degradation in Escherichia coli. J. Bacteriol. 2012; 194:6233–6239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hsieh P.K., Richards J., Liu Q., Belasco J.G.. Specificity of RppH-dependent RNA degradation in Bacillus subtilis. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:8864–8869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Foley P.L., Hsieh P.K., Luciano D.J., Belasco J.G.. Specificity and evolutionary conservation of the Escherichia coli RNA pyrophosphohydrolase RppH. J. Biol. Chem. 2015; 290:9478–9486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Richards J., Luciano D.J., Belasco J.G.. Influence of translation on RppH-dependent mRNA degradation in Escherichia coli. Mol. Microbiol. 2012; 86:1063–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vasilyev N., Serganov A.. Structures of RNA complexes with the Escherichia coli RNA pyrophosphohydrolase RppH unveil the basis for specific 5′-end-dependent mRNA decay. J. Biol. Chem. 2015; 290:9487–9499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lee C.R., Kim M., Park Y.H., Kim Y.R., Seok Y.J.. RppH-dependent pyrophosphohydrolysis of mRNAs is regulated by direct interaction with DapF in Escherichia coli. Nucleic Acids Res. 2014; 42:12746–12757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Richaud C., Higgins W., Mengin-Lecreulx D., Stragier P.. Molecular cloning, characterization, and chromosomal localization of dapF, the Escherichia coli gene for diaminopimelate epimerase. J. Bacteriol. 1987; 169:1454–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mengin-Lecreulx D., Michaud C., Richaud C., Blanot D., van Heijenoort J.. Incorporation of LL-diaminopimelic acid into peptidoglycan of Escherichia coli mutants lacking diaminopimelate epimerase encoded by dapF. J. Bacteriol. 1988; 170:2031–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Al Mamun A.A., Lombardo M.J., Shee C., Lisewski A.M., Gonzalez C., Lin D., Nehring R.B., Saint-Ruf C., Gibson J.L., Frisch R.L. et al. Identity and function of a large gene network underlying mutagenic repair of DNA breaks. Science. 2012; 338:1344–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Badger J.L., Wass C.A., Kim K.S.. Identification of Escherichia coli K1 genes contributing to human brain microvascular endothelial cell invasion by differential fluorescence induction. Mol. Microbiol. 2000; 36:174–182. [DOI] [PubMed] [Google Scholar]
- 19. Gaywee J., Radulovic S., Higgins J.A., Azad A.F.. Transcriptional analysis of Rickettsia prowazekii invasion gene homolog (invA) during host cell infection. Infect. Immun. 2002; 70:6346–6354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lundin A., Nilsson C., Gerhard M., Andersson D.I., Krabbe M., Engstrand L.. The NudA protein in the gastric pathogen Helicobacter pylori is an ubiquitous and constitutively expressed dinucleoside polyphosphate hydrolase. J. Biol. Chem. 2003; 278:12574–12578. [DOI] [PubMed] [Google Scholar]
- 21. Jiang M., Sullivan S.M., Walker A.K., Strahler J.R., Andrews P.C., Maddock J.R.. Identification of novel Escherichia coli ribosome-associated proteins using isobaric tags and multidimensional protein identification techniques. J. Bacteriol. 2007; 189:3434–3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vasilyev N., Serganov A.. Preparation of short 5′-triphosphorylated oligoribonucleotides for crystallographic and biochemical studies. Methods Mol. Biol. 2016; 1320:11–20. [DOI] [PubMed] [Google Scholar]
- 23. Kabsch W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010; 66:125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Adams P.D., Afonine P.V., Bunkoczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010; 66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Emsley P., Lohkamp B., Scott W.G., Cowtan K.. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010; 66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen P.S., Toribara T.Y., Warner H.. Microdetermination of phosphorus. Anal. Chem. 1956; 28:1756–1758. [Google Scholar]
- 27. Celesnik H., Deana A., Belasco J.G.. PABLO analysis of RNA: 5′-phosphorylation state and 5′-end mapping. Methods Enzymol. 2008; 447:83–98. [DOI] [PubMed] [Google Scholar]
- 28. Datsenko K.A., Wanner B.L.. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A. 2000; 97:6640–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Edwards R.A., Keller L.H., Schifferli D.M.. Improved allelic exchange vectors and their use to analyze 987P fimbria gene expression. Gene. 1998; 207:149–157. [DOI] [PubMed] [Google Scholar]
- 30. Hor L., Dobson R.C., Downton M.T., Wagner J., Hutton C.A., Perugini M.A.. Dimerization of bacterial diaminopimelate epimerase is essential for catalysis. J. Biol. Chem. 2013; 288:9238–9248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Krissinel E., Henrick K.. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007; 372:774–797. [DOI] [PubMed] [Google Scholar]
- 32. Jeffery C.J. An introduction to protein moonlighting. Biochem. Soc. Trans. 2014; 42:1679–1683. [DOI] [PubMed] [Google Scholar]
- 33. Kaptain S., Downey W.E., Tang C., Philpott C., Haile D., Orloff D.G., Harford J.B., Rouault T.A., Klausner R.D.. A regulated RNA binding protein also possesses aconitase activity. Proc. Natl. Acad. Sci. U.S.A. 1991; 88:10109–10113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Miczak A., Kaberdin V.R., Wei C.L., Lin-Chao S.. Proteins associated with RNase E in a multicomponent ribonucleolytic complex. Proc. Natl. Acad. Sci. U.S.A. 1996; 93:3865–3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chandran V., Luisi B.F.. Recognition of enolase in the Escherichia coli RNA degradosome. J. Mol. Biol. 2006; 358:8–15. [DOI] [PubMed] [Google Scholar]
- 36. Nurmohamed S., McKay A.R., Robinson C.V., Luisi B.F.. Molecular recognition between Escherichia coli enolase and ribonuclease E. Acta Crystallogr. D Biol. Crystallogr. 2010; 66:1036–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bernstein J.A., Lin P.H., Cohen S.N., Lin-Chao S.. Global analysis of Escherichia coli RNA degradosome function using DNA microarrays. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:2758–2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Morita T., Kawamoto H., Mizota T., Inada T., Aiba H.. Enolase in the RNA degradosome plays a crucial role in the rapid decay of glucose transporter mRNA in the response to phosphosugar stress in Escherichia coli. Mol. Microbiol. 2004; 54:1063–1075. [DOI] [PubMed] [Google Scholar]
- 39. Mackie G.A. RNase E: at the interface of bacterial RNA processing and decay. Nat. Rev. Microbiol. 2013; 11:45–57. [DOI] [PubMed] [Google Scholar]
- 40. Valkov E., Muthukumar S., Chang C.T., Jonas S., Weichenrieder O., Izaurralde E.. Structure of the Dcp2-Dcp1 mRNA-decapping complex in the activated conformation. Nat. Struct. Mol. Biol. 2016; 23:574–579. [DOI] [PubMed] [Google Scholar]
- 41. Hand N.J., Silhavy T.J.. Null mutations in a Nudix gene, ygdP, implicate an alarmone response in a novel suppression of hybrid jamming. J. Bacteriol. 2003; 185:6530–6539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Steiger M., Carr-Schmid A., Schwartz D.C., Kiledjian M., Parker R.. Analysis of recombinant yeast decapping enzyme. RNA. 2003; 9:231–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Drozdetskiy A., Cole C., Procter J., Barton G.J.. JPred4: a protein secondary structure prediction server. Nucleic Acids Res. 2015; 43:W389–W394. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Coordinates of the structures were deposited in the Protein Data Bank with PDB ID codes: 6D13, RppH–DapF complex; 6D1Q, RppHt-DapFm complex; 6D1V, RppHt-ppcpAGU-DapFm complex. All relevant data and materials are available upon request.