

 Cigarette smoke has always been considered as a risk factor for chronic obstructive pulmonary diseases (COPD).
Cigarette smoke has always been considered as a risk factor for chronic obstructive pulmonary diseases (COPD).
Abstract
Cigarette smoke has always been considered as a risk factor for chronic obstructive pulmonary diseases (COPD). In this study, we have examined the effect of ten individual cigarette smoke compounds (nicotine, benzo[a]pyrene, naphthalene, formaldehyde, ammonia, acrylic acid, toluene, benzene, m-xylene, and hexamine) on glutathione S transferase (GST) activity, an important Phase II metabolic enzyme and their possible role in inflammatory pathophysiology leading to COPD. Lower Glutathione (GSH) levels and GST activity and higher CRP, TNF-α, and IL-6 levels were observed in COPD patients compared to age and gender-matched controls. Using human recombinant GST and plasma as well as erythrocytes collected from normal subjects this study demonstrates that out of the ten compounds, nicotine (5 mg mL–1), benzo[a]pyrene (10 ng mL–1), naphthalene (250 μg mL–1), and formaldehyde (5 pg mL–1) caused a significant decrease in recombinant, plasma, and erythrocyte GST activity. Further cell culture studies show that exposure to nicotine, benzo[a]pyrene, naphthalene, and formaldehyde caused a significant decrease in GSH levels and GST activity and its protein expression and an increase in intracellular ROS production in THP-1 monocytes. Interestingly, treatment with benzo[a]pyrene and naphthalene significantly up regulated the phosphorylation of the p65 subunit of NF-κB and increased the secretion of TNF-α and CRP compared to control. This study suggests the potential role of benzo[a]pyrene and naphthalene in the activation of the inflammatory signaling pathway leading to cigarette smoke-induced COPD.
Introduction
Chronic Obstructive Pulmonary Disease (COPD) is one of the foremost cigarette smoke (CS) mediated diseases. Several studies have found a link between cigarette smoke exposure and COPD disease progression.1 CS has been reported to induce chronic inflammation2 as well as a protease-anti protease imbalance.3 Exposure to CS leads to the release of several inflammatory cells including macrophages in the lungs, which release pro-inflammatory cytokines as well as chemokines.4 COPD has been found to be associated with a higher level of TNF-α in sputum and serum samples.5,6 An increased level of IL-6, IL-17A, and IL-22 and decreased levels of IL-10 have been observed among cigarette smoke-induced COPD patients.7,8 CS exposure causes the activation of monocytes to macrophages by a variety of mechanisms, such as inducible bronchus-associated lymphoid tissue (iBALT) and activator protein 1 (AP-1) mediated pathways.9–11 Monocytes are in the intermediate development stage between the bone marrow precursors and tissue macrophages.12 Macrophages play central roles in the initiation of inflammation, release of pro-inflammatory cytokines, and the production of reactive oxygen species (ROS).13
Oxidative stress is generated as a result of various metabolic reactions involving xenobiotics. Among the various factors, particulate matter (PM2.5) and CS exposure contribute significantly in the pathogenesis of oxidative stress.14,15 Under normal conditions the lung has the efficient regulatory system to up regulate the gene expression of antioxidant enzymes to neutralize oxidative stress.16 However, in smokers, oxidative stress causes the inactivation of antioxidant enzymes, such as superoxide dismutase leading to the over production of ROS.16 Excessive ROS may also in turn lead to a pro-oxidant imbalance.17
CS is a mixture of more than 4000 chemicals containing carcinogenic compounds, free radicals and oxidants.18 Cigarette smoke extracts (CSE) are quite regularly being used to study its immunomodulatory effect relating to COPD.19–21 Recently a few investigators studied the inflammatory effects of individual cigarette smoke compounds.22,23 However, basic molecular mechanism studies are needed to decipher the signal transduction pathways relating to inflammation caused by individual cigarette smoke compounds. Among the various defense mechanisms against CS-induced oxidative stress, the glutathione S-transferase (GST) family of enzymes is quite well known. The prime responsibility of these enzymes is to neutralize the xenobiotic compounds as well as the ROS generated during various metabolic reactions.24–26 The alterations in the cellular GSH level and the GST activity influence many signaling pathways leading to inflammation.27 Thus it is important to study the effect of individual cigarette smoke compounds (aldehydes, poly aromatic hydrocarbons, aromatic, alkaloid, heterocyclic organic, unsaturated carboxylic acid) on cellular GSH levels and GST activity, if any. Therefore, the aim of the present study is to determine the effect of individual cigarette smoke compounds on cellular GSH levels as well as on GST activity and their possible signaling mechanisms underlying the inflammation leading to COPD.
Methodology
Materials
Human specific antibodies were purchased from Abcam, Inc. (Cambridge, MA). All other chemicals were purchased from Sigma (Saint Louis, USA) unless otherwise mentioned.
Patient enrollment and blood sample collection
Informed written consent was obtained from all patients according to the protocol approved by the Indian Council of Medical Research (ICMR). All patients included in this study were adults with COPD who were admitted to Assam Medical College and Hospital, Dibrugarh, and Jorhat Medical College, Jorhat, Assam. The age and gender-matched control samples were collected from the Clinical Centre, CSIR-NEIST, Jorhat, Assam. Women with a positive pregnancy test or those nursing infants were excluded from the study. All patients who gave written consent were included in the study. The blood samples were collected in EDTA tubes. Clear plasma was separated via centrifugation of blood at 3000 rpm (1500g) for 15 min.
Human monocytic cell line
The human THP-1 monocytic cell line was obtained from the National Centre for Cell Sciences (Pune, India). These cells were maintained at 37 °C in RPMI 1640 medium containing 7 mM glucose, 10% (v/v) heat inactivated FBS, 100 units per ml penicillin, 100 μg per ml streptomycin, 12 mM sodium carbonate, 25 mM HEPES, and 2 mM l-glutamine in a humidified atmosphere containing 5% (v/v) CO2. For treatments, cells were washed once in plain RPMI before being suspended in fresh medium (complete) containing serum and other supplements.
Cigarette smoke compounds taken for study
The cigarette smoke compounds selected for the study were nicotine, benzo[a]pyrene, formaldehyde, naphthalene, and ammonia. The concentrations taken under consideration for the compounds were – nicotine (0.5–10 mg mL–1), benzo[a]pyrene (10–25 ng mL–1), naphthalene (100–500 μg mL–1), formaldehyde (5–20 pg mL–1), ammonia (10–50 μg mL–1), acrylic acid (0.5–1 pg mL–1), toluene (10–50 μg mL–1), benzene (10–100 μg mL–1), m-xylene (5–20 μg mL–1), and hexamine (30–50 μg mL–1). The solvent system 0.5% dimethyl sulphoxide (DMSO) was used for benzo[a]pyrene, naphthalene, acrylic acid, toluene, benzene, m-xylene and hexamine. However, water was used as the solvent for nicotine, ammonia and formaldehyde.
Recombinant human glutathione S-transferase assay
Glutathione S-transferase recombinant expressed in E. coli (Human gene GSTM1) was purchased from Sigma Aldrich. The selected cigarette smoke compounds [nicotine (5 mg mL–1), benzo[a]pyrene (10 ng mL–1), naphthalene (250 μg mL–1) and formaldehyde (5 pg mL–1)] were incubated with recombinant GST (72 μg mL–1) for 2 h. Kinetic analysis was performed for 3 minutes at 37 °C. One unit of GST activity is defined as the 1 μmol of adduct formation per minute per mL of protein sample.
Treatment of cells with the cigarette smoke compounds
Cells were treated with the selected cigarette smoke compounds [(nicotine (5 mg mL–1), benzo[a]pyrene (10 ng mL–1), naphthalene (250 μg mL–1), and formaldehyde (5 pg mL–1)] for 4 h. After treatment, cells were lysed in radio immuno precipitation assay (RIPA) buffer (50 mM Tris in pH 8, 150 mM NaCl, 1% NP-40, 0.5% deoxycholic acid, and 0.1% SDS) supplemented with protease and phosphatase inhibitors (1 mM PMSF, 5 mg per mL leupeptin, 2 mg mL–1, aprotinin, 1 mM EDTA, 10 mM NaF, and 1 mM Na3VO4). Lysates were cleared using centrifugation and total protein concentrations were determined using the BCA assay as per the manufacturer's protocol (Pierce/Thermo Scientific, Rockford, IL).
Treatment of human plasma and erythrocytes with the cigarette smoke compounds
The cigarette smoke compounds such as nicotine (5 mg mL–1), benzo[a]pyrene(10 ng mL–1), naphthalene (250 μg mL–1) and formaldehyde (5 pg mL–1) were incubated with 100 μl of plasma (control subject of age 45 years) for 2 h at 37 °C. The erythrocyte lysates were prepared according to the method of Orhan et al.28 The cigarette smoke compounds were then incubated with 100 μl of erythrocyte lysates. After the incubation, an assessment of GST activity was undertaken using the method of Habig and Jakoby.29 The reaction mixture contained a suitable amount of the enzyme, KH2PO4 buffer (pH 7.4), EDTA (1 mM), CDNB (1 mM) and GSH (6 mM). The reaction was carried out at 37 °C and monitored spectrophotometrically by the increase in absorbance of the conjugate of GSH and CDNB at 340 nm for 3 minutes. One unit of GST activity is defined as 1 μmol of adduct formation per minute per mL of plasma or erythrocyte lysate.
Reduced glutathione (GSH) assay
Reduced GSH levels were measured using the method described by Ellman.30 To precipitate the protein content, 150 μl of precipitating solution (5% TCA and 1 mM EDTA) was added to 100 μl of plasma and allowed to precipitate for 5 minutes at 4 °C. After centrifugation (10 000g for 15 minutes) the supernatant was removed, DTNB solution (Ellman's reagent) was added to it and the absorbance was measured at 412 nm.
Detection of intracellular ROS levels
Intracellular reactive oxygen species (ROS) levels were measured using the fluorescent dye DCFDA (2′,7′-dichlorofluoresceindiacetate). After treatment, cells were washed once with PBS and then loaded with 5μM DCFDA in PBS with 4% FBS. The cells were incubated at 37 °C for 30 min in the dark and subsequently washed with PBS, and centrifuged at 12 000g for 10 min at 37 °C. After washing, the cells were mounted onto microscope slides using mounting medium and images were collected using a fluorescence microscope (Leica DM3000LED).
Cell viability and cytokine studies
Cell viability was determined using the Alamar Blue reduction bioassay (Himedia, India). This method is based upon Alamar Blue dye reduction by live cells. IL-6, TNF-α, CRP, and MCP-4 levels in the cell culture sample as well as in the human plasma were determined by ELISA using commercially available kits from Sigma Aldrich, USA as per manufacturer's protocol. In the cytokine assay, control samples were analyzed each time to check the variation from plate to plate on different days of analysis.
Immunoblotting
All samples, which contained approximately the same amount of protein (20–40 μg), were run on a 8–10% SDS PAGE gel and transferred to a nitrocellulose membrane. Membranes were blocked at room temperature for 2 h in blocking buffer containing 1% BSA to prevent nonspecific binding and then incubated with anti-NF-κB (p65), anti-phosphorylated NF-κB (serine 276) (1 : 500), anti-GSTT1 antibody (1 : 500), and anti β-actin (1 : 500) primary antibodies at 4 °C overnight. The membranes were washed in TBS-T (50 mmol L–1 Tris HCl, pH 7.6, 150 mmol L–1 NaCl, and 0.1% Tween 20) for 30 min and incubated with the appropriate HRP conjugated secondary antibody (1 : 5000) for 2 h at room temperature, then images were developed using the ultrasensitive ECL substrate (Bio-Rad). The intensity of each immunoblotting band was measured using the histogram tool of Adobe Photoshop CS5.
Statistical analysis
Data were analyzed statistically using one way analysis of variance (ANOVA) with Sigma Stat statistical software (Jandel Scientific, San Rafael, CA). When data passed a normality test, all groups were compared using the Student–Newman–Keuls post hoc method. A difference was considered significant at the P < 0.05 level. The data were represented as mean ± SE.
Results
GST activity, GSH levels, and the secretion of pro-inflammatory cytokines in the plasma of COPD patients, and healthy subjects
In the present study we have measured the GST activity, GSH levels, and the pro-inflammatory cytokines, such as CRP, IL-6, TNF-α, and MCP-4 in plasma samples of COPD patients (n = 23) and healthy subjects (n = 23). The results demonstrate a significant decrease in GST activity and GSH levels among COPD patients compared to those recorded in age and gender-matched healthy subjects. In addition, significantly higher levels of CRP, IL-6, and TNF-α have also been observed among COPD patients compared to controls (Fig. 1). However, we did not observe any significant change in MCP-4 levels between COPD patients and healthy subjects.
Fig. 1. Plasma levels of GST activity (μmol mL–1 min–1) (A), GSH (B), TNF-α (C), IL-6 (D), CRP (E), and MCP-4 (F) in age and gender-matched control subjects (n = 23) and chronic obstructive pulmonary disease (COPD) patients (n = 23). Values are expressed as mean ± SE.

Effect of cigarette smoke compounds on GST activity using recombinant GST enzyme, human plasma, erythrocyte lysate and THP-1 monocyte cells
Cigarette smoking is a primary risk factor in the development of COPD. Orhan et al. reported the role of cigarette smoke extract in the down regulation of the erythrocyte GST activity.28 However, there is no report so far examining the effect of individual cigarette smoke compounds on GST activity. In the present study we have examined the effect of different cigarette smoke compounds on GST activity using human recombinant GST enzyme, plasma samples from healthy donors, and a monocyte cell culture model.
Fig. 2 and 3 demonstrate the effect of different cigarette smoke compounds on the activity of human recombinant GST enzyme. The results show that treatment with nicotine, benzo[a]pyrene, naphthalene, and formaldehyde causes decreases in recombinant GST activity. Treatment with nicotine at a dose of 5 mg mL–1 caused a significant (p < 0.05) reduction in GST activity compared to control (8.5 ± 0.75 vs. 12.04 ± 0.56 μmol–1 mL–1 min–1). Benzo[a]pyrene at a dose of 10 ng mL–1 significantly (p < 0.05) decreased the GST activity compared to control (11.58 ± 0.73 vs. 14.67 ± 0.63 μmol–1 mL–1 min–1). Naphthalene exposure at a dose of 250 μg mL–1, significantly down regulated the GST activity compared to control (9.83 ± 1.39 vs. 13.41 ± 0.21 μmol–1 mL–1 min–1). Treatment with formaldehyde at a dose of 5 pg mL–1 also caused a significant decrease in GST activity compared to control (8.91 ± 1.02 vs. 11.17 ± 0.62 μmol–1 mL–1 min–1). However, treatment with ammonia, acrylic acid, toluene, benzene, m-xylene, and hexamine did not cause any decrease in GST activity compared to the respective controls as shown in Fig. 3. The doses of all cigarette smoke compounds used in the study are within the physiological range as reported in the literature.31–39 This study suggests that among all the different cigarette smoke compounds nicotine, benzo[a]pyrene, naphthalene, and formaldehyde play an important role in reducing the activity of human recombinant GST enzyme. The enzyme assay was performed at different time points (15, 30, 60, 90, 120, 150, and 180 min) (data not included) with the 10 cigarette smoke compounds and only 4 compounds produced significant changes in the GST activity compared to control at the 2 h time point. Beyond this 2 h time point we didn't find any significant changes in the enzyme activities. Thus we have chosen these 4 compounds and a 2 h time point for the experiment with human plasma and erythrocytes.
Fig. 2. Effect of nicotine (A), benzo[a]pyrene (B), naphthalene (C), and formaldehyde (D) on GST activity (μmol mL–1 min–1). Human recombinant GST (72 μg mL–1) was incubated with various compounds (Conc. as mentioned in the respective panels) for 2 h at 37 °C. Values are expressed as mean ± SE (n = 4).

Fig. 3. Effect of acrylic acid (A), toluene (B), benzene (C), m-xylene (D), hexamine (E) and ammonia (F) on GST activity (μmol mL–1 min–1). Human recombinant GST (72 μg mL–1) was incubated with various cigarette smoke compounds (conc. as mentioned in the respective panels) for 2 h at 37 °C. Values are expressed as mean ± SE (n = 4).

In further studies with human plasma and erythrocytes, collected from healthy volunteers, we have also examined the direct effect of nicotine, benzo[a]pyrene, naphthalene, and formaldehyde on the activity of the GST enzyme. Different cigarette smoke compounds were incubated with human plasma and erythrocytes for 2 h at 37 °C. Results show a significant decrease in GST activity caused by the treatment with nicotine (5 mg mL–1), benzo[a]pyrene (10 ng mL–1), naphthalene (250 μg mL–1), and formaldehyde (5 pg mL–1) compared to those recorded in controls (Fig. 4A and B).
Fig. 4. Effect of nicotine (5 mg mL–1), benzo[a]pyrene (10 ng mL–1), naphthalene (250 μg mL–1), formaldehyde (5 pg mL–1) on human plasma (A) and erythrocyte lysate (B) GST activity (μmol mL–1 min–1). Human plasma and erythrocyte lysate (100 μL) were incubated with various cigarette smoke compounds (conc. as mentioned in the respective panels) for 2 h at 37 °C. Values are expressed as mean ± SE (n = 4).

Using a THP-1 human monocyte cell culture model this study further examined the effect of different cigarette smoke compounds, such as nicotine, benzo[a]pyrene, naphthalene, and formaldehyde on the activity of the intracellular GST enzyme. Results demonstrate that incubation of monocytes with nicotine (5 mg mL–1), benzo[a]pyrene (10 ng mL–1), naphthalene (125 μg mL–1), and formaldehyde (5 pg mL–1) for 4 h at 37 °C significantly decreased the intracellular GST activity and its protein expression compared to control (Fig. 5A). This cell culture study demonstrates a direct effect of individual cigarette smoke compounds on the reduction of GST activity, which is in agreement with the above findings (Fig. 5C). None of the compounds were found to reduce the cell viability as compared to control (Fig. 5D).
Fig. 5. Effect of nicotine (5 mg mL–1), benzo[a]pyrene (10 ng mL–1), naphthalene (125 μg mL–1), and formaldehyde (20 pg mL–1) on GST protein expression (A), GSH level (nmol mL–1) (B), GST activity (μmol mL–1 min–1) (C), and cell viability (D) in THP-1 monocytic cells. Cells (2 million mL–1) were incubated with various compounds (conc. as mentioned in the respective panels) for 4 h at 37 °C. Values are expressed as mean ± SE (n = 8).

Effect of cigarette smoke compounds (nicotine, benzo[a]pyrene, naphthalene, and formaldehyde) on GSH levels and ROS production in THP-1 monocytes
Oxidative stress plays an important role in the progression of COPD.40 A decrease in intracellular antioxidant defense and an increase in ROS production lead to the oxidative stress pathophysiology. The present study examined the effect of cigarette smoke compounds (nicotine, benzo[a]pyrene, naphthalene, and formaldehyde) on intracellular GSH levels (Fig. 5B) and ROS production in THP-1 monocyte cells. Results suggest that treatment with nicotine (5 mg mL–1), benzo[a]pyrene (10 ng mL–1), naphthalene (125 μg mL–1), and formaldehyde (5 pg mL–1) for 4 h at 37 °C significantly decreased the intracellular GSH levels and increased the ROS production compared to control (Fig. 6). The effect of benzo[a]pyrene and naphthalene on ROS production is significantly higher than those seen in nicotine and formaldehyde-treated cells. These results suggest a role of cigarette smoke compounds in the development of oxidative stress, which may mediate the pathogenesis of COPD.
Fig. 6. Effect of benzo[a]pyrene (10 ng mL–1), naphthalene (125 μg mL–1), nicotine (5 mg mL–1) and formaldehyde (20 pg mL–1) on intercellular ROS production in THP-1 monocytic cells. H2O2 (10 mM) was used as a positive control. Cells were incubated with the respective compounds for 4 h at 37 °C. Intracellular ROS production was measured using DCFDA (5 mM).

Effect of cigarette smoke compounds (nicotine, benzo[a]pyrene, naphthalene, and formaldehyde) on NF-κB activation and the secretion of pro-inflammatory cytokines in THP-1 monocytes
The activation of transcription factor NF-κB has been well recognized in the development of inflammatory lung disorders.41 Upon stimulation, NF-κB induces the secretion of various pro-inflammatory molecules, such as TNFα, CRP, IL-6, etc.42,43 The activated form of NF-κB consists of two subunits, p65 and p50, and the transcriptional activity of NF-κB is regulated by the phosphorylation of its p65 subunits.44 The present study examined whether the exposure to individual cigarette smoke compounds (nicotine, benzo[a]pyrene, naphthalene, and formaldehyde) can activate NF-κB and stimulate the secretion of pro-inflammatory cytokines in THP-1 monocyte cells. Results demonstrate that treatment with benzo[a]pyrene and naphthalene significantly up regulated the phosphorylation of the p65 subunit of NF-κB and increased the secretion of TNFα and CRP compared to control (Fig. 7A–C). However, treatment with nicotine and formaldehyde did not cause any activation of NF-κB and the secretion of TNFα and CRP. This study suggests that among the various cigarette smoke compounds, benzo[a]pyrene and naphthalene play an important role in the activation of inflammatory signaling pathways, which may mediate the inflammatory pathogenesis in COPD.
Fig. 7. Effect of benzo[a]pyrene (10 ng mL–1), naphthalene (125 μg mL–1), nicotine (5 mg mL–1) and formaldehyde (20 pg mL–1) on the protein expression of phospho NF-κB (S276) and total NF-κB (A) and the levels of TNF-α (B) and CRP (C) in THP-1 monocytic cells.

Discussion
Our earlier study has shown that smokers with the GSTM1 (null genotype) gene polymorphism are more susceptible to developing COPD than the non-smokers.45 Moreover, one earlier study also reported the effect of cigarette smoke extract in down-regulating the erythrocyte GST activity.30 These studies suggest a possible role of cigarette smoke exposure in decreasing GST activity, which may play an important role in the pathogenesis of COPD. However, so far, no study in the literature has investigated a direct effect of individual cigarette smoke compounds on GST activity and also the underlying mechanism leading to the development of inflammatory lung diseases. The present study demonstrated that COPD patients have a significantly lower GST activity compared to age and gender-matched controls. In addition, the present study, for the first time, reported that treatment with different cigarette smoke compounds (nicotine, benzo[a]pyrene, naphthalene, and formaldehyde) significantly decreased the activity of both the recombinant and plasma GST enzyme. Cell culture studies further demonstrated that treatment with nicotine, benzo[a]pyrene, naphthalene, and formaldehyde also cause a decrease in intracellular GST activity as well as its protein expression. Combining these data, this study suggests a role of individual cigarette smoke compounds in the impairment of GST activity, which may be linked to the development of COPD.
Impairment in intracellular redox status and development of oxidative stress play an important role in inflammatory lung diseases like COPD.40 Elevated levels of superoxide anions and lipid peroxidation have been observed in the plasma sample of COPD patients.46,47 Data from our clinical study demonstrated that COPD patients have lower plasma GSH levels as compared to controls. GSH is a physiological antioxidant, a co-factor of many enzymes and plays an important role in the reduction of cellular oxidative stress.48 However, it was not clear whether cigarette smoke-induced oxidative stress may have a role in the progression of COPD pathophysiology. Among the various cell types associated with the immune system, monocytes play an important role in the pathogenesis of the inflammatory cascade.49 Using THP-1 monocyte cells this study reported that treatment with different cigarette smoke compounds, such as nicotine, benzo[a]pyrene, naphthalene, and formaldehyde caused a decrease in GSH levels and an increase in intracellular ROS production. This study suggests a direct effect of individual cigarette smoke compounds including nicotine, benzo[a]pyrene, naphthalene, and formaldehyde in the development of oxidative stress which may have a role in the progression of COPD.
TNF-α is an important pro-inflammatory cytokine which plays a major role in innate and adaptive immunity, cell proliferation, and apoptotic processes.50 Higher IL-6 levels have been found to be associated with poor lung function among patients with COPD.51 Elevated CRP level is another important bio-marker of the inflammatory process that occurs in patients with COPD.52 In line with the earlier studies, we have also observed an increased plasma level of TNF-α, IL-6, and CRP among COPD patients compared to those seen in age and gender-matched controls, which suggests the development of inflammatory pathophysiology in COPD.
Activation of NF-κB stimulates secretion of various pro-inflammatory molecules, such as, TNF-α, IL-6 etc.42,43 Using TNF-α receptor knock-out mice exposed to cigarette smoke, Churg et al. (2002) showed that TNF-α plays a central role in cigarette smoke-induced inflammation and breakdown of connective tissues, a precursor of emphysema.53 A positive association has also been observed between smoking status and CRP levels among adolescent smokers.54 Cytokines, such as TNF-α, IL-6, and IL-1β have been reported as the primary regulators of CRP.55 Using THP-1 monocyte cells, the present study shows that treatment with individual cigarette smoke compounds, such as benzo[a]pyrene and naphthalene significantly activated the transcription factor NF-κB and increased the secretion of TNF-α and CRP. However, treatment with nicotine and formaldehyde did not cause any activation of NF-κB and the secretion of TNF-α and CRP. This study suggests that among the various cigarette smoke compounds, benzo[a]pyrene and naphthalene play a major role in the activation of the inflammatory signaling pathway, which may mediate the progression of inflammatory lung disease like COPD.
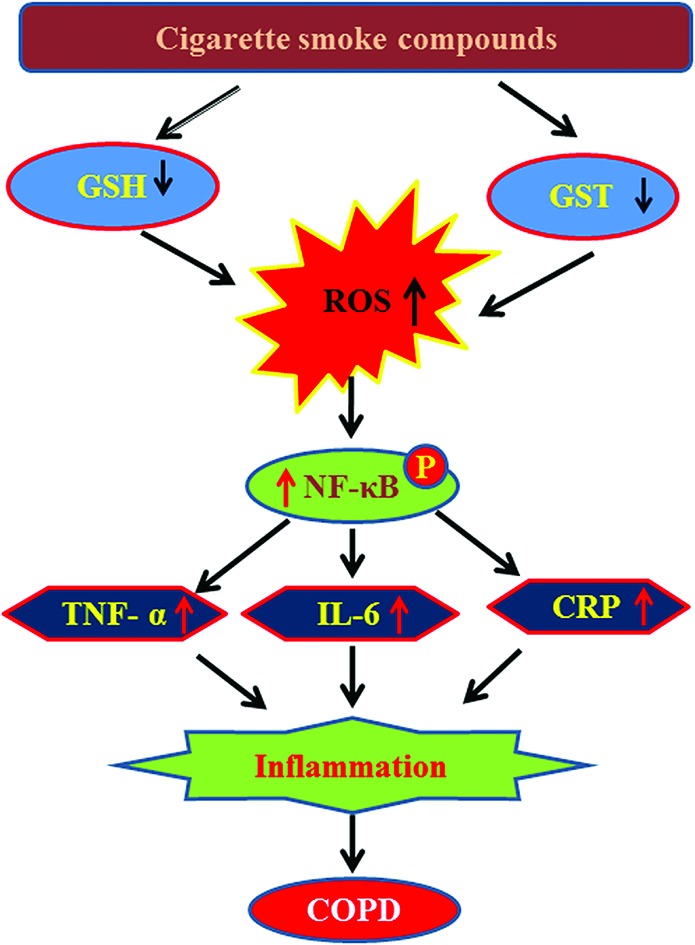
The present study demonstrates a decrease in plasma GSH levels and GST activity and an increase in pro-inflammatory cytokine levels, such as TNF-α, IL-6, and CRP among COPD patients compared to controls. This study for the first time examined the effect of individual cigarette smoke compounds on GST activity using human recombinant GST enzyme. Results revealed that out of ten cigarette smoke compounds, nicotine, benzo[a]pyrene, naphthalene, and formaldehyde cause a significant decrease in GST activity. Cell culture studies with THP-1 monocytes showed that treatment with nicotine, benzo[a]pyrene, naphthalene, and formaldehyde also caused a decrease in GST activity and its protein expression, and GSH levels and an increase in intracellular ROS production. Interestingly, treatment with benzo[a]pyrene and naphthalene activated the transcription factor NF-κB, increased the secretion of TNF-α and CRP, which suggests a role for these two cigarette smoke compounds in the development of inflammatory pathophysiology in COPD (Fig. 8).
Fig. 8. Probable mechanistic pathway underlying the role of cigarette smoke compounds-mediated inflammatory pathophysiology leading to COPD.

Conclusion
This study demonstrates the effect of individual cigarette smoke compounds in the development of intracellular redox imbalance and the activation of inflammatory signaling pathways which may possibly lead to the progression of COPD. However, in vivo studies are further required to understand the detailed molecular mechanism underlying the role of individual cigarette smoke compounds in the development of COPD.
Abbreviations
- COPD
Chronic obstructive pulmonary disease
- CRP
C-reactive protein
- GSH
Glutathione
- GST
Glutathione S-transferase
- IL-6
Interleukin 6
- ROS
Reactive oxygen species
- MCP-4
Monocyte chemo-attractant protein-4
- TNF-α
Tumor necrosis factor α
Acknowledgments
This work was supported by the Council of Scientific and Industrial Research, New Delhi, under the network project grant TREAT-COPD (BSC-0116). Authors would like to thank the Director, CSIR-North East Institute of Science & Technology, Jorhat for all round support to carry out this work. Authors also acknowledge the contribution of Dr Avisek Chakroborty and Dr Arnav Sarmah from Assam Medical College and Hospital, Dibrugarh for their kind help during sample collection.
References
- Zuo L., He F., Sergakis G. G., Koozehchian M. S., Stimpfl J. N., Rong Y., Diaz P. T., Best T. M. Am. J. Physiol.: Lung Cell. Mol. Physiol. 2014;307:L205–L218. doi: 10.1152/ajplung.00330.2013. [DOI] [PubMed] [Google Scholar]
- Barnes P. J. Nat. Rev. Drug Discovery. 2013;12:543–559. doi: 10.1038/nrd4025. [DOI] [PubMed] [Google Scholar]
- Malhotra D., Thimmulappa R., Vij N., Navas-Acien A., Sussan T., Merali S., Zhang L., Kelsen S. G., Myers A., Wise R., Tuder R., Biswal S. Am. J. Respir. Crit. Care Med. 2009;180:1196–1207. doi: 10.1164/rccm.200903-0324OC. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Sopori M. Nat. Rev. Immunol. 2002;2:372–377. doi: 10.1038/nri803. [DOI] [PubMed] [Google Scholar]
- Tangedal S., Aanerud M., Persson L. J., Brokstad K. A., Bakke P. S., Eagan T. M. Respir. Res. 2014;15:138. doi: 10.1186/s12931-014-0138-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelegrino N. R., Tanni S. E., Amaral R. A., Angeleli A. Y., Correa C., Godoy I. Am. J. Med. Sci. 2013;345:440–445. doi: 10.1097/MAJ.0b013e31825f32a7. [DOI] [PubMed] [Google Scholar]
- de Moraes M. R., da Costa A. C., Correa Kde S., Junqueira-Kipnis A. P., Rabahi M. F. Int. J. Chron. Obstruct. Pulmon. Dis. 2014;9:735–743. doi: 10.2147/COPD.S64135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L., Cheng Z., Liu W., Wu K. COPD. 2013;10:459–465. doi: 10.3109/15412555.2013.770456. [DOI] [PubMed] [Google Scholar]
- John-Schuster G., Hager K., Conlon T. M., Irmler M., Beckers J., Eickelberg O., Yildirim A. O. Am. J. Physiol.: Lung Cell. Mol. Physiol. 2014;307:L692–L706. doi: 10.1152/ajplung.00092.2014. [DOI] [PubMed] [Google Scholar]
- Walters M. J., Paul-Clark M. J., McMaster S. K., Ito K., Adcock I. M., Mitchell J. A. Mol. Pharmacol. 2005;68:1343–1353. doi: 10.1124/mol.105.012591. [DOI] [PubMed] [Google Scholar]
- Kirkham P. A., Spooner G., Ffoulkes-Jones C., Calvez R. Free Radicals Biol. Med. 2003;35:697–710. doi: 10.1016/s0891-5849(03)00390-3. [DOI] [PubMed] [Google Scholar]
- Lewis J. S., Lee J. A., Underwood J. C., Harris A. L., Lewis C. E. J. Leukocyte Biol. 1999;66:889–900. doi: 10.1002/jlb.66.6.889. [DOI] [PubMed] [Google Scholar]
- Auffray C., Sieweke M. H., Geissmann F. Annu. Rev. Immunol. 2009;27:669–692. doi: 10.1146/annurev.immunol.021908.132557. [DOI] [PubMed] [Google Scholar]
- Ni L., Chuang C. C., Zuo L. Front. Physiol. 2015;6:294. doi: 10.3389/fphys.2015.00294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L., Diaz P. T., Best T. M., Stimpfl J. N., He F., Zuo L. Ann. Allergy Asthma Immunol. 2014;113:137–142. doi: 10.1016/j.anai.2014.05.030. [DOI] [PubMed] [Google Scholar]
- Lee H., Park J. R., Kim E. J., Kim W. J., Hong S. H., Park S. M., Yang S. R. Toxicol. Lett. 2016;240:140–148. doi: 10.1016/j.toxlet.2015.10.030. [DOI] [PubMed] [Google Scholar]
- Zuo L., Hallman A. H., Yousif M. K., Chien M. T. Front. Biol. 2012;6:506–513. [Google Scholar]
- Pryor W. A., Stone K. Ann. N. Y. Acad. Sci. 1993;686:12–27. doi: 10.1111/j.1749-6632.1993.tb39148.x. [DOI] [PubMed] [Google Scholar]
- Baglole C. J., Bushinsky S. M., Garcia T. M., Kode A., Rahman I., Sime P. J., Phipps R. P. Am. J. Physiol.: Lung Cell. Mol. Physiol. 2006;291:L19–L29. doi: 10.1152/ajplung.00306.2005. [DOI] [PubMed] [Google Scholar]
- Kent L., Smyth L., Clayton C., Scott L., Cook T., Stephens R., Fox S., Hext P., Farrow S., Singh D. Cytokine. 2008;42:205–216. doi: 10.1016/j.cyto.2008.02.001. [DOI] [PubMed] [Google Scholar]
- Chen L., Ge Q., Tjin G., Alkhouri H., Deng L., Brandsma C. A., Adcock I., Timens W., Postma D., Burgess J. K., Black J. L., Oliver B. G. Eur. Respir. J. 2014;44:634–646. doi: 10.1183/09031936.00171313. [DOI] [PubMed] [Google Scholar]
- McMaster S. K., Paul-Clark M. J., Walters M., Fleet M., Anandarajah J., Sriskandan S., Mitchell J. A. Br. J. Pharmacol. 2008;153:536–543. doi: 10.1038/sj.bjp.0707595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comer D. M., Elborn J. S., Ennis M. BMC Pulm. Med. 2014;14:32. doi: 10.1186/1471-2466-14-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend D. M., Findlay V. L., Tew K. D. Methods Enzymol. 2005;401:287–307. doi: 10.1016/S0076-6879(05)01019-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G., Fang Y. Z., Yang S., Lupton J. R., Turner N. D. J. Nutr. 2004;134:489–492. doi: 10.1093/jn/134.3.489. [DOI] [PubMed] [Google Scholar]
- Tirona R. G., Pang K. S. J. Pharmacol. Exp. Ther. 1999;290:1230–1241. [PubMed] [Google Scholar]
- Sen C. K. Curr. Top. Cell. Regul. 2000;36:1–30. doi: 10.1016/s0070-2137(01)80001-7. [DOI] [PubMed] [Google Scholar]
- Orhan H., Evelo C. T., Sahin G. J. Biochem. Mol. Toxicol. 2005;19:226–233. doi: 10.1002/jbt.20088. [DOI] [PubMed] [Google Scholar]
- Habig W. H., Pabst M. J., Jakoby W. B. J. Biol. Chem. 1974;249:7130–7139. [PubMed] [Google Scholar]
- Ellman G. L. Arch. Biochem. Biophys. 1959;82:70–77. doi: 10.1016/0003-9861(59)90090-6. [DOI] [PubMed] [Google Scholar]
- Brunnemann K. D., Masaryk J., Hoffmann D. J. Agric. Food Chem. 1983;31(6):1221–1224. [Google Scholar]
- Ding Y. S., Ashley D. L., Watson C. H. J. Agric. Food Chem. 2007;55:5966–5973. doi: 10.1021/jf070649o. [DOI] [PubMed] [Google Scholar]
- Ding Y. S., Yan X. J., Jain R. B., Lopp E., Tavakoli A., Polzin G. M., Stanfill S. B., Ashley D. L., Watson C. H. Environ. Sci. Technol. 2006;40:1133–1138. doi: 10.1021/es0517320. [DOI] [PubMed] [Google Scholar]
- Talhout R., Schulz T., Florek E., van Benthem J., Wester P., Opperhuizen A. Int. J. Environ. Res. Public Health. 2011;8:613–628. doi: 10.3390/ijerph8020613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verschueren K., Handbook of Environmental Data of Organic Chemicals, John Wiley and Sons Inc, New York, NY, 4th edn, 2001, 1170–1174. [Google Scholar]
- Rom W. N., Environmental and Occupational Medicine, Brown and Company, Boston, MA, Little, 2nd edn, 1992, p. 1212. [Google Scholar]
- Clayton G. D. and Clayton F. E., Patty's Industrial Hygiene and Toxicology, John Wiley & Sons, New York, NY, 4th edn, 1994, vol. IIB, p. 1302. [Google Scholar]
- NAS, The Alkyl Benzenes, 1980, pp. I-1–I-99. [Google Scholar]
- The Health Consequences of Involuntary Exposure to Tobacco Smoke: A Report of the Surgeon General, U.S. Department of Health and Human Services, Atlanta, GA, USA, 2006, pp. 1–709
- Mak J. C. Int. J. Tuberc. Lung Dis. 2008;12:368–374. [PubMed] [Google Scholar]
- Edwards M. R., Bartlett N. W., Clarke D., Birrell M., Belvisi M., Johnston S. L. Pharmacol. Ther. 2009;121:1–13. doi: 10.1016/j.pharmthera.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silswal N., Singh A. K., Aruna B., Mukhopadhyay S., Ghosh S., Ehtesham N. Z. Biochem. Biophys. Res. Commun. 2005;334:1092–1101. doi: 10.1016/j.bbrc.2005.06.202. [DOI] [PubMed] [Google Scholar]
- Wellen K. E., Hotamisligil G. S. J. Clin. Invest. 2005;115:1111–1119. doi: 10.1172/JCI25102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes P. J., Karin M. N. Engl. J. Med. 1997;336:1066–1071. doi: 10.1056/NEJM199704103361506. [DOI] [PubMed] [Google Scholar]
- Dey T., Gogoi K., Unni B. G., Kalita M., Bharadwaz M., Bhattacharjee M., Boruah P. K., Bora T., Ozah D. PLoS One. 2014;9:e96739. doi: 10.1371/journal.pone.0096739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman I., Morrison D., Donaldson K., MacNee W. Am. J. Respir. Crit. Care Med. 1996;154:1055–1060. doi: 10.1164/ajrccm.154.4.8887607. [DOI] [PubMed] [Google Scholar]
- van Antwerpen V. L., Theron A. J., Richards G. A., Steenkamp K. J., van der Merwe C. A., van der Walt R., Anderson R. Free Radicals Biol. Med. 1995;18:935–941. doi: 10.1016/0891-5849(94)00225-9. [DOI] [PubMed] [Google Scholar]
- Rahman T., Hosen I., Towhidul Islam M. M., Shekhar H. U. Adv. Biosci. Biotechnol. 2012;3:997–1019. [Google Scholar]
- Pappas K., Papaioannou A. I., Kostikas K., Tzanakis N. Cytokine. 2013;64:613–625. doi: 10.1016/j.cyto.2013.09.010. [DOI] [PubMed] [Google Scholar]
- Popa C., Netea M. G., van Riel P. L., van der Meer J. W., Stalenhoef A. F. J. Lipid Res. 2007;48:751–762. doi: 10.1194/jlr.R600021-JLR200. [DOI] [PubMed] [Google Scholar]
- Hubeau C., Kubera J. E., Masek-Hammerman K., Williams C. M. Clin. Sci. 2013;125:483–493. doi: 10.1042/CS20130110. [DOI] [PubMed] [Google Scholar]
- Pinto-Plata V. M., Mullerova H., Toso J. F., Feudjo-Tepie M., Soriano J. B., Vessey R. S., Celli B. R. Thorax. 2006;61:23–28. doi: 10.1136/thx.2005.042200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churg A., Dai J., Tai H., Xie C., Wright J. L. Am. J. Respir. Crit. Care Med. 2002;15(166):849–54. doi: 10.1164/rccm.200202-097OC. [DOI] [PubMed] [Google Scholar]
- O'Loughlin J., Lambert M., Karp I., McGrath J., Gray-Donald K., Barnett T. A., Delvin E. E., Levy E., Paradis G. Nicotine Tob. Res. 2008;10:525–532. doi: 10.1080/14622200801901997. [DOI] [PubMed] [Google Scholar]
- Tonstad S., Cowan J. L. Int. J. Clin. Pract. 2009;63:1634–1641. doi: 10.1111/j.1742-1241.2009.02179.x. [DOI] [PMC free article] [PubMed] [Google Scholar]