


Abstract
(6–4) Photolyases ((6–4)PLs) are flavoenzymes that repair the carcinogenic UV-induced DNA damage, pyrimidine(6–4)pyrimidone photoproducts ((6–4)PPs), in a light-dependent manner. Although the reaction mechanism of DNA photorepair by (6–4)PLs has been intensively investigated, the molecular mechanism of the lesion recognition remains obscure. We show that a well-conserved arginine residue in Xenopus laevis (6–4)PL (Xl64) participates in DNA binding, through Coulomb and CH–π interactions. Fragment molecular orbital calculations estimated attractive interaction energies of –80–100 kcal mol–1 for the Coulomb interaction and –6 kcal mol–1 for the CH–π interaction, and the loss of either of them significantly reduced the affinity for (6–4)PP-containing oligonucleotides, as well as the quantum yield of DNA photorepair. From experimental and theoretical observations, we formulated a DNA binding model of (6–4)PLs. Based on the binding model, we mutated this Arg in Xl64 to His, which is well conserved among the animal cryptochromes (CRYs), and found that the CRY-type mutant exhibited reduced affinity for the (6–4)PP-containing oligonucleotides, implying the possible molecular origin of the functional diversity of the photolyase/cryptochrome superfamily.
INTRODUCTION
Photolyases (PLs) are flavoproteins that harness the blue light in sunlight to restore UV-induced DNA damage, namely cyclobutane pyrimidine dimers (CPDs) and pyrimidine(6–4)pyrimidone photoproducts ((6–4) photoproducts, (6–4)PPs), to the two intact, adjacent pyrimidine nucleobases (1). PLs share amino acid sequences and tertiary structures with cryptochromes (CRYs), which participate in signal transduction in plants (2) and circadian clock regulation in vertebrates (3), and therefore, PLs and CRYs are considered to be evolutionarily linked, despite their diverse biological functions. Understanding the molecular origin of the functional differentiation of these two closely-related proteins with similar architectures is an interesting and challenging issue.
The primary reaction in the photorepair of CPDs and (6–4)PPs by their specific PLs (referred to as CPD-PLs and (6–4)PLs, respectively) is the light-driven electron transfer from the excited state of the fully-reduced form of the non-covalently bound flavin adenine dinucleotide (FAD) to the lesions (Figure 1A) (4,5). In CPD photorepair, the two covalent bonds linking the two pyrimidine bases in the CPD lesion are cleaved within 100 ps after the electron injection, and the electron returns within ∼2 ns to the transiently formed FADH•, resulting in the recovery of the catalytic FADH– and the restoration of the intact bases (6–8). In (6–4)PP photorepair, a more complicated mechanism using two photons in the entire repair process has been suggested (9,10).
Figure 1.

UV-damaged DNA and its photorepair by photolyases. (A) Photorepair reaction scheme of CPDs and (6–4)PPs by CPD-PL and (6–4)PL, respectively. (B and C) Close-up views of the complex structure of Methanosarcina mazei class II CPD-PL (B) and Drosophila melanogaster (6–4)PL (C). The coordinates were taken from Protein Data Bank with the accession numbers of 2XRZ (17) and 3CVU (18). (D) Sequence alignment of a selected region among CPD-PL and (6–4)PL. Abbreviations: Ec, Escherichia coli; An, Anacystis nidulans; At, Arabidopsis thaliana; Os, Oryza sativa; Mm, Methanosarcina mazei; Dm, Drosophila melanogaster; Xl, Xenopus laevis.
As electron transfer is the primary photorepair reaction by the PLs, the enzymes need to accommodate the UV lesions as close as possible to the light-absorbing catalytic center. However, the CPD lesion is stably located in the DNA duplex by forming hydrogen bonds with the complementary adenine bases (11,12), with a theoretical total interaction energy around the CPD of –60 kcal mol–1 (13). Upon binding of PLs, the lesions in the duplex are flipped out of the helix (14), and the flipped bipyrimidine lesions are captured within the FAD-buried cavity in the C-terminal domain (15). This accommodation of the lesion in the active site has clearly been observed in the crystal structures of Anacystis nidulans CPD-PL (16), Methanosarcina mazei class II CPD-PL (MmCPDII) (17), and Drosophila melanogaster (6–4)PL (Dm64) (18), in the complexes with their double-stranded substrates containing the CPD or the (6–4)PP. In these complex structures, the flipped lesions are recognized by the surrounding amino acid residues, as well as a water cluster, in the active site through hydrogen bonds, which keep the lesion flipped. Indeed, a recent theoretical study demonstrated that the amino acid residues around the active site play a determinant role in the DNA binding ability of CPD-PL (19).
Considering the very beginning of the enzyme binding process, however, the enzymes first need to recognize the lesion from the outside of the DNA helix in the proper direction, since the lesions, especially (6–4)PPs, have an asymmetric structure. How do the PLs recognize the lesion in a direction-dependent manner, and what kind of interaction mediates the recognition? In a survey of the reported structures around the active sites of MmCPDII and Dm64, an arginine side chain (Arg441 of MmCPDII and Arg421 of Dm64) protruded into the space in the DNA helix, which was occupied by the photoproduct before the enzyme binding (Figure 1B and C). This Arg is highly conserved among PLs (Figure 1D), and thus is considered to play a key role in the lesion recognition at the primary stage of the binding. Maul et al. indicated that Arg421 in Dm64 forms a salt bridge with the phosphate groups in the DNA backbone, and even more interestingly, stacks on the guanine base 3′ flanking to the (6–4)PP (18), whereas these interactions were not detected in the CPD-PL complex structure (Figure 1B), probably due to the electrically-neutral internucleoside formacetal linkage in the CPD analog (16,17). In addition to the conserved Arg residue, some other amino acid side chains also occlude the space. In the MmCPDII structure, Trp431, which is conserved among the class II CPD-PLs (Figure 1D), is located adjacent to Arg441. However, Trp431 in MmCPDII is replaced with Gln418 in Dm64 (Figure 1D), but Gln418 occupies a different position from that of Trp431 in MmCPDII (Figure 1B and C). This Gln is only conserved among (6–4)PLs (Figure 1D), implying a specific functional role in the (6–4)PP recognition. Although structural (18) and theoretical (20) studies indicated the electrostatic interaction between Arg and the phosphate groups, the other interactions between DNA and the enzyme and the importance of such interactions in the biological function of the (6–4)PL have not yet been described.
Here, we have comprehensively investigated the binding of Xenopus laevis (6–4)PL (Xl64) to (6–4)PP-containing substrates, by using mutants of Xl64 and chemically-modified oligonucleotides. In combination with theoretical investigations, we identified two important interactions between DNA and Xl64, which strikingly impacted the DNA binding and even the DNA repair abilities. Based on our experimental and theoretical observations, we propose a binding model of Xl64. This formulated binding model explains the reason why the CRYs cannot bind to the (6–4)PP-containing DNA.
MATERIALS AND METHODS
Reagents for DNA synthesis were purchased from Thermo Fisher Scientific (Tokyo, Japan) and Glen Research (Sterling, VA). All other reagents were purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan), except for 6-carboxytetramethylrhodamine N-hydroxysuccinimide ester, which was purchased from Berry & Associates, Inc. (Dexter, MI, USA). HPLC analyses were performed on a Gilson gradient-type analytical system, equipped with a Waters 2998 photodiode-array detector. A Waters μBondasphere C18 5 μm 300 Å column (3.9 mm × 150 mm) was used on this system, at a flow rate of 1.0 ml min–1 with a linear gradient of acetonitrile in 0.1 M triethylammonium acetate (TEAA) (pH 7.0) generated over 20 min. Absorption and fluorescent spectra were measured on a PerkinElmer Lambda 35 UV/Vis spectrometer and a JASCO FP-6500 spectrofluorometer, respectively.
Solid-phase DNA synthesis
Oligonucleotides used in this study were synthesized on an Applied Biosystems 3400 DNA synthesizer, using phosphoramidite building blocks of the (6–4)PP (21), 5,6-dihydrothymidine (Glen Research), 5′-amino linker (5′-amino modifier C3-TFA, Glen Research), dSpacer (Glen Research), and deoxyribonucleosides for ultra-mild DNA synthesis (Glen Research), which were assembled on a Universal Support III PS (Glen Research). Phenoxyacetic anhydride (Glen Research), benzimidazolium triflate (22,23), and iodine (Glen Research) were used as capping, activation, and oxidation reagents, respectively. The synthesized oligonucleotides were cleaved from the support by a treatment with 2 M ammonia in methanol for 1 h, and then deprotected by a treatment with 28% ammonia water at room temperature for 2 h. The products were purified by HPLC.
The oligonucleotides containing the 5′-amino linker were dissolved in 500 μl of 0.1 M carbonate buffer (pH 9.0), and 6 μl of 0.17 M 6-carboxytetramethylrhodamine (TAMRA) N-hydroxysuccinimide ester, dissolved in anhydrous DMSO, was added to the solution. The mixture was incubated at room temperature overnight. After ethanol precipitation, the fluorescently-labeled oligonucleotides were analyzed and purified by HPLC, using a gradient of 7.3–25.3% acetonitrile in 0.1 M TEAA. The amounts of the oligonucleotides were determined by the TAMRA absorption (molar extinction coefficient at 556 nm of 89 000 M–1 cm–1).
Site-directed mutagenesis
The site-directed mutagenesis of WT-Xl64 for the Escherichia coli UV survival assay and the overexpression of the mutant genes were performed by polymerase chain reaction (PCR), using either a PrimeSTAR Mutagenesis Basal kit (Takara-Bio, Inc., Shiga, Japan) or a QuickChange II Site-Directed Mutagenesis kit (Agilent Technologies, Inc., La Jolla, CA, USA), with either the pGEX4T-2 or pET28a plasmid encoding Xl64 as the template. The primer sets for the mutagenesis are shown in Supplementary Table S1. After the PCR amplification, E. coli DH5α competent cells were transformed with the PCR products. The purified plasmids were sequenced and used for the following experiments.
UV survival assays and purification of the Xl64 mutants
UV survival assays of the E. coli SY32/pRT2 strain producing WT or mutants of Xl64 were performed with the pGEX4T-2 plasmids, as described previously (24,25). All survival rates were normalized to that measured without the UV irradiation (set to 1). The experiments were independently performed in triplicate, and the data were analyzed by the two-sided Student's t-test. Statistical significance was set to P < 0.01.
Overexpression and purification of the mutants were performed with the pET28a plasmids, as described previously (9,26). The concentrations of the obtained enzymes were determined by the absorption of oxidized FAD at 450 nm, using its absorption coefficient of 11 200 M –1 cm–1.
Binding of the wildtype and mutants of Xl64 analyzed by electrophoretic mobility shift assays
Electrophoretic mobility shift assays on polyacrylamide gels were performed, as described previously (27). The 49-mer oligonucleotide containing the (6–4)PP, d(AGCTACCATGCCTGCTGCACGAAT(6–4)TAAGCAATTCGTAATCATGGTCATAGCT), where T(6–4)T represents the (6–4)PP formed between two thymidines, was treated with T4 polynucleotide kinase (Takara-Bio, Japan) in the presence of 32P-ATP (∼400 kBq, Perkin Elmer), at 37°C for 30 min. The 32P-labeled oligonucleotide was passed through a G-25 Microspin column (GE Healthcare), and was hybridized to a complementary strand, d(AGCTATGACCATGATTACGAATTGCTTAATTCGTGCAGCAGGCATGGTAGCT). The duplex was incubated at 4°C with the enzymes, in a buffer containing 50 mM Tris–HCl (pH 8.0), 50 mM NaCl and 5% glycerol, for 30 min under the yellow light, and the samples were subjected to electrophoresis on a 5% polyacrylamide gel. The dried gels were visualized with an FLA-7000 image analyzer (Fuji-Film, Japan), and the bound and unbound fractions were quantified by the MultiGauge software. The experiments were performed in triplicate, and the averaged bound concentrations were plotted over the enzyme concentrations. The dissociation constants were obtained by fitting the data with a single binding site model.
Quantum yield measurement
The steady-state quantum yields of the (6–4)PP repair by the wildtype or mutants of Xl64 were determined in vitro, as described previously (9). A single-stranded 8-mer containing the (6–4)PP, d(CGAT(6–4)TGAT), was generally used in this study. A typical reaction solution (250 μl) consisted of 40–50 μM substrate, ∼2 μM enzyme, 10 mM phosphate (pH 7.0), 100 mM NaCl, 6 mM cysteine, and 5% glycerol, and the photorepair was performed with an LQX1800 xenon lamp under the same setup conditions as described previously (9). Quantum yields were calculated from the absorbance of the enzyme at 384 nm, the slope of the diminishment at 325 nm, Δϵ at 325 nm (–6000 M–1 cm–1), and the slope of the bleaching of the p-(dimethylamino)-benzenediazonium tetrafluoroborate actinometer, as described previously (9,28). The quantum yields of the wildtype Xl64 for the DHT-containing substrates, d(THHHT(6–4)THHHH), d(HHHTT(6–4)THHHH), d(HHHHT(6–4)TTHHH), and d(HHHHT(6–4)THHHT) were obtained in the same manner.
Fluorescence anisotropy measurements
Fluorescence anisotropy was measured on an FP-6500 fluorometer (JASCO), equipped with polarizers for both the excitation and emission windows. Excitation and emission wavelengths were set to 556 and 580 nm, respectively. All measurements were performed at 10°C, under the control of a Peltier device (EHC-573T, JASCO). To 200 μl of the DNA binding solution, containing 40 nM DNA, 10 mM phosphate, 100 mM NaCl, and 5% glycerol (pH 7.0), was added a diluted protein solution, containing up to 20 μM Xl64 in total. After 5 min incubation of each solution, the fluorescence anisotropy was measured. The experiments were performed in triplicate, and the mean values and standard deviations were plotted over the enzyme concentrations ([E] in Equation (1)). The data were fitted with the following equation:
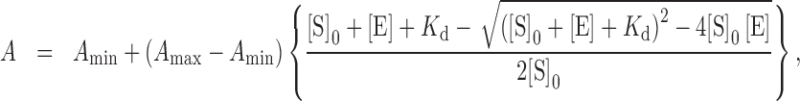 |
(1) |
where Amax and Amin represent the ideal anisotropies for the bound and free DNA, respectively, and Kd represents the dissociation constant. These values were considered as variables, while the total concentration of DNA ([S]0) was extrapolated.
Setups of the theoretical calculations
Theoretical calculations were performed using the reported complex structure of Drosophila melanogaster (6–4)PL (Dm64) and duplex DNA (PDB ID: 3CVU), referred to as PLWT, and its mutants (PLR421A, PLR421K and PLR421L), which were prepared with the Winmostar software (29) by converting the corresponding arginine residue (421 for Dm64, instead of 410 for Xl64) into alanine, lysine and leucine, respectively. The geometric optimization and the electronic structure calculations for the (6–4)PP and FADH– (Supplementary Figure S1) were performed using Gaussian09 (30), at the B3LYP/6-31G(d) level. The obtained results were converted into the Amber force field, using the Antechamber module of the AMBER 14 program package (31) (shown in Supplementary Tables S2 and S3). Throughout the following calculations, the Amber force field 14 SB (32) and the created force fields for the (6–4)PP and FADH– were used. The calculation systems were neutralized with Na+ counter ions, and solvated using the TIP3P water model (33) provided in the tLeap module. Note that the total atom numbers of each system (for PLWT, PLR421A, PLR421K and PLR421L) were 81 565, 81 549, 81 563 and 81 558, respectively. Energy minimization was performed using the Sander module at 5000 iterations with restraints on all heavy atoms (the force constant of which was set to 10 kcal mol–1), followed by 10 000 iterations without restraint.
The obtained structures were further optimized by QM/MM calculations with ONIOM(B3LYP/6-31G(d)/AMBER)-EE (see Supporting Information) (34–39), in which the QM region covers Gln418, the amino acid residue at 421st position, (6–4)PP, and the nucleotides flanking the (6–4)PP, whereas the other part belongs to the MM region. The optimized structures are shown in Supplementary Figure S3.
Fragment molecular orbital (FMO) method
To estimate the interaction energy between amino acid residues and nucleic acids, the fragment molecular orbital (FMO) calculation (40–42) (see Supplementary data) was performed in the GAMESS program package (43), and the electronic structure was calculated at the MP2/6-31G(d,p) level with the two-body FMO (FMO2) expansion. The optimized structures of the QM region were extracted and divided into eight fragments, by replacing covalent bond(s) with hydrogen atom(s), in order to obtain the interfragment interaction energies (IFIEs) (40,44,45) between the two fragments of interest. Using the fragmented systems, FMO calculations were performed. To evaluate the type of attractive/repulsive energies, the pair interaction energy decomposition analysis (PIEDA (44), see Supplementary data) was performed. The decomposed IFIEs for PLWT, PLR421A, PLR421K and PLR421L are summarized in Supplementary Tables S4–S7.
RESULTS AND DISCUSSIONS
Mutations of Gln407 and Arg410 in Xenopus (6–4) photolyase
To investigate the involvement of the Gln and Arg residues (Figure 2A) in the recognition of the (6–4)PP, we first performed UV survival assays of E. coli producing the wild-type (WT) or mutants (Q407A and R410A) of Xl64. If these residues function in the recognition and/or the DNA repair, then the survival of the strain producing the mutant is expected to be reduced, as compared to that of the WT-producing strain. The results (Figure 2B) indicated that the survival of the strain producing the R410A mutant decreased significantly, while the Q407A mutation did not cause such a decrease in the survival, indicating that Arg410 in Xl64 plays a key role in its enzymatic function.
Figure 2.

Binding of the WT and mutants of Xl64 to the (6–4)PP containing substrate. (A) Selected view of the structure of Dm64 after the QM/MM optimization (see Materials and Methods). (B) UV survival assays of bacteria producing the WT or mutants of Xl64. Null means no production of Xl64 from the empty vector. The experiments were independently repeated three times (n = 3), and statistical analysis was carried out by the Student's t-test, where the significance was set to P < 0.01. Asterisks indicated the P-values of 0.0048 (*) and 0.0033 (**). (C) Electromobility shift assays of WT-Xl64. The upper bands represent the complex between 32P-DNA and the enzyme. Aliquots (0.5 nmol) of the 32P-labeled double-stranded 49-mer oligonucleotides were incubated with WT-Xl64 for 30 min at increasing concentrations of 0, 0.5, 1, 2, 5, 10, 20, 50, 100, 150 and 200 nM (lanes 1–11), and free- and bound-DNA were separated on a 5% polyacrylamide gel. The experiments were performed in triplicate, and the band intensities were quantified. (D) Binding of the WT (black square), Q407A (blue circle), R410A (red triangle), and R410K (open red triangle) of Xl64 to the (6–4)PP-containing double-stranded DNA. The complex concentrations were calculated from the quantified band intensities, and the mean values were plotted against enzyme concentrations. Their fitting curves are also shown.
Next, the binding and DNA repair abilities of the recombinant Xl64 mutants of Gln407 and Arg410 were investigated. As the decrease in the survival was observed for the R410A mutant-producing E. coli strain, we exclusively focused on the arginine side chain, and replaced Arg410 with either lysine or alanine. The guanidine moiety in Arg410 is in principle positively charged at physiological pH, forming salt bridges with the phosphate groups proximal to the (6–4)PP (18). Lysine consists of an alkyl chain and a terminal positively-charged amino group, similar to the arginine side chain, and hence the R410K mutant would help to clarify the importance of the salt bridge. The Q407A mutant was also prepared, with the expectation of no significant mutation effect.
The binding abilities of WT-Xl64 and the mutants were evaluated by electrophoretic mobility shift assays, using 32P-labeled 49-mer double-stranded oligonucleotides containing a single (6–4)PP (Figure 2C). The bound fractions were quantified and plotted against the enzyme concentrations (Figure 2D), and the dissociation constants were obtained by the non-linear fitting of the data points. The Kd values are summarized in Table 1. The Kd value of the WT enzyme was 30 ± 3 nM under our experimental conditions, while the Kd of the Q407A mutant (45 ± 6 nM) was 1.5-fold larger than that of the WT, suggesting that the loss of the glutamine residue slightly lowers the affinity for the substrate. The Kd value for the R410A mutant (71 ± 6 nM) was 2.4-fold larger than that of the WT. Interestingly, the mutation of Arg410 to lysine restored its binding ability, and the Kd for R410K (14 ± 1 nM) was actually 2-times smaller than that of the WT. These results indicated that the arginine residue that is highly conserved among photolyases (i.e. Arg410 in Xl64) plays an important role in the enzyme binding to the substrate, which is dominated by an electrostatic interaction between Arg410 and the DNA, as suggested in the previous report on the complex structure (18).
Table 1.
Binding and photorepair properties of WT and mutants of Xl64
| Enzyme | K d (nM) | Obtained ηss | Ideal ηssa |
|---|---|---|---|
| WT | 30 ± 3 | 5.0% | 4.9% |
| Q407A | 45 ± 6 | 5.6% | 5.5% |
| R410A | 71 ± 6 | 1.3% | 5.5% |
| R410K | 14 ±1 | 5.1% | 5.0% |
aEstimated by using η1 = 6.7%, η2 = 83%, kx = 0.01 s–1 and respective kexc, as described previously9.
Next, we compared the DNA repair activities of the mutants. The enzymes containing fully-reduced FADH– were prepared anaerobically, and in the presence of the substrate, the enzyme solution was illuminated with monochromic light at 384 nm, while the photon flux was counted by chemical actinometry. In this assay, we used a single-stranded 8-mer substrate, because the steady-state quantum yield (η ss) of photorepair of the 8-mer substrate was almost identical to that of the double-stranded 14-mer substrate (Supplementary Figure S3). By monitoring the disappearance of the 325 nm absorption band of the (6–4)PP (Figure 3A, Δϵ = –6 mM–1 cm–1), the repaired substrate per enzyme (S/p0) was plotted against the incident photon numbers for the excitation of FADH– (Figure 3B), and the obtained η ss values are summarized in Table 1. WT and Q407A repaired the substrate at a similar rates, and their η ss values for the DNA photorepair were 5.0% for WT and 5.6% for Q407A. In a previous study, η ss reportedly depends on the excitation rate of FADH– (kexc) (9). By taking into account the kexc in each experiment (0.044 s–1 for WT and 0.082 s–1 for Q407A), the quantum yields of the first and second photoreactions by WT (η 1 and η 2 as 6.7% and 83%, respectively), and the rate constant of the reversion of the (6–4)PP from the intermediate at 10°C (kx as 0.01 s–1) (9), the ideal η ss values (the extrapolated η ss for the photorepair by the WT under these conditions) are estimated to be 4.9% and 5.5%, respectively, indicating that the repair activity of Q407A is equivalent to that of WT. This result, in addition to the DNA binding ability of Q407A, demonstrated that the Gln407 residue does not play any role in the entire repair process of the (6–4)PP.
Figure 3.

Photorepair of (6–4)PP by the WT and mutants of Xl64. (A) Spectral bleaching of the (6–4)PP band upon photorepair of the single-stranded 8-mer substrate by WT-Xl64. The inset represents the time course of the absorption change at 325 nm upon 384-nm illumination. (B) Photorepair of the 8-mer substrate by the WT (black square) and the Xl64 mutants (Q407A: blue circle, R410A: red triangle, and R410K: red open triangle). The numbers of the repaired substrate per enzyme were plotted against the numbers of the incident photons absorbed by FADH–.
In contrast, R410A repaired the substrate quite inefficiently. The calculated η ss for R410A was 1.3%, while the ideal η ss at kexc of 0.082 s–1 is 5.5%. The reduced activity for R410A was expected from the reduced survival of the R410A-producing E. coli strain (Figure 2B), and again confirmed that Arg410 plays a key role in the photorepair process of the (6–4)PL. Interestingly, the repair activity of R410K was recovered to the same level as that of WT, and the η ss for R410K was 5.1%, which is in nice agreement with the ideal η ss at kexc of 0.047 s–1 (5.0%). One may consider that the presence of the positive charge interacting with the phosphate group would be required for the proper electron transfer from the excited state of FADH– to the (6–4)PP. However, a recent large-QM/MM study demonstrated that the electron transfer to the 5′ moiety of the (6–4)PP is a downhill reaction, without taking into account the presence of the Arg residue in the calculation (46), and thus the reduction in η ss for R410A would mainly be caused by the substrate binding. These results indicated that a positive charge in the amino acid residue at the 410th position in Xl64 is required for the formation of a catalytically active DNA-protein complex.
Theoretical estimation of interaction energy between Arg and DNA
To characterize the types of interactions that the arginine residue contributed to the binding and the repair, the interaction energies between Arg and its proximate residues in the DNA were evaluated by the fragment molecular orbital (FMO) method. We used the coordinates of the structure of Dm64 in complex with the (6–4)PP-containing DNA for the calculation, since Dm64 and Xl64 share high amino acid sequence similarity (58% identity and 70% similarity). In the search for the proximate residues to Arg421 in Dm64 (corresponding to Arg410 in Xl64), Gln418, (6–4)PP, and the nucleosides flanking the (6–4)PP were found. After the replacement of the arginine by alanine or lysine, followed by the energy minimization, the geometric optimization was performed by the QM/MM calculations (see the results in Supplementary Figure S2), in which the QM region covered the above-mentioned molecules, while the others were in the MM region without water molecules. The molecules in the QM region were then extracted and divided into eight parts (eight fragment monomers), as shown in Figure 4A, and the inter-fragment interaction energies (IFIEs) (45,47,48) between the two fragment monomers among them were calculated at the FMO2-MP2/6-31G(d,p) level. In order to analyze which type of interaction is prominent, the pair interaction energy decomposition analysis (PIEDA) was performed, and the electrostatic energy (Ees) and the dispersion energy (Edisp) between the two fragment monomers obtained by PIEDA were mapped (Figure 4B).
Figure 4.

Pair interaction energy decomposition analysis (PIEDA) in the FMO calculation. (A) Chemical structures of the fragments in the FMO calculation for PLWT. In the calculation, the molecules were divided into eight fragments; the Gln residue (fragment 1), the Arg residue (fragment 2), dGMP 5′ next to the (6–4)PP (fragment 3), the 5′ side deoxyribose moiety of the (6–4)PP with the internucleoside phosphate within the (6–4)PP (fragment 4), the 3′ side deoxyribose moiety of the (6–4)PP with the 3′ flanking phosphate (fragment 5), the base moiety of the (6–4)PP (fragment 6), the deoxyribose moiety of the dG 3′ flanking the (6–4)PP (fragment 7), and the base moiety of the 3′ dG. In the case of the mutants (PLR421A and PLR421K), the fragment 2 was replaced with the corresponding amino acid residue. The IFIEs for these fragments were calculated at the FMO2-MP2/6-31G(d,p) level with the PIEDA option. Red and blue dotted lines represent remarkable electrostatic and dispersive interactions, respectively. Note that we simulated the ideal contribution of the electrostatic and dispersive interaction energies without taking into account the solvation, in order to exclude the solvation-induced stabilizing effect. (B) Heat maps of the electrostatic and dispersion energies (Ees and Edisp, shown in red and blue, respectively) between the two individual fragments in PLWT, PLR421A, or PLR421K. The detailed values of the simulated interaction energies are summarized in Supplementary Tables S4–S6. In PIEDA, Ees and Edisp for the fragment pair originally linked through a covalent bond (e.g. fragments 4 and 5) were found to be largely negative (see Supplementary Tables S4–S6). These interactions are shown in gray, not to mix them up with the spatial interactions in the fragments of interest. (C) Selected interaction energies. Full lists of the interaction energies are summarized in Supplementary Tables S4–S6.
Regarding the Ees for WT, the strongest interaction was observed for the 2–5 pair; namely, between Arg and the 3′ side of the deoxyribose moiety of the (6–4)PP with the phosphate group 3′ to the (6–4)PP, and its interaction energy was estimated to be –97.5 kcal mol–1. In addition, a pair between Arg and the 5′ side deoxyribose moiety of the (6–4)PP with the internucleoside phosphate in the (6–4)PP exhibited the great stabilization energy of –83.1 kcal mol–1. These are likely to be attributable to the Coulomb interactions between the positively-charged Arg residue and the negatively-charged phosphate groups. By contrast, these interactions were absent in PLR421A (Figure 4B), explaining the lowered affinity of the R410A mutant of Xl64 for the (6–4)PP-containing oligonucleotide, as shown in Figure 2D. In the same manner as observed in the previous section, these interactions were recovered in PLR421K (Figure 4B), and the interaction energies of Lys with the internucleoside phosphate in the (6–4)PP and the phosphate group 3′ to the (6–4)PP were even more negative than those in WT (–98.9 and –124.5 kcal mol–1, respectively), reasonably explaining the smaller Kd (better binding) obtained for R410K of Xl64 than that of WT-Xl64.
We then exclusively focused on the dispersion energies (Edisp) between the two individual fragment monomers, and found the two distinct dispersive interactions of Arg, with the 3′ side deoxyribose moiety of the (6–4)PP (–6.5 kcal mol–1) and the nucleobase of dG 3′ flanking the (6–4)PP (–6.3 kcal mol–1). These interactions were absent in PLR421A, but recovered in PLR421K (Figure 4B and C), in the same manner as the Coulomb interactions with the phosphate groups. Arg and Lys commonly possess a long alkyl chain and a positive charge at the terminus of the alkyl chain, which would contribute to form attractive dispersive interactions between the amino acid side chain and the sugar moiety of the (6–4)PP. As the alkyl chains of Arg and Lys are located over the 3′ nucleobase in the optimized structures of PLWT and PLR421K, the dispersive interaction between Arg (Lys) and the 3′ nucleobase is most likely attributable to a CH–π interaction. In their optimized structures, the Hα atom in the amino acid residues at the 421st position is directed to the planar guanine base, in a distance range of 2.5–3.0 Å. As the stabilization energy of the CH–π interaction between benzene and carbohydrate is reportedly 0.5–2.7 kcal mol–1 per single interaction (49–51), our calculation thus implicated that several CH–π interactions are involved in the DNA–Xl64 complex.
Role of the CH–π interactions in the substrate binding and the DNA repair by Xl64
Our FMO calculations for the Dm64 complex structure suggested the presence of the CH–π interaction between Arg and a nucleobase 3′ flanking the (6–4)PP, and this interaction contributed to the stabilization of the protein-DNA complex by about –6 kcal mol–1, which is apparently not quite as important, as compared to the strong Coulomb interaction between Arg and the phosphate groups (–80–100 kcal mol–1). To verify how and to what extent the suggested CH–π interaction impacts the enzymatic activity of Xl64, we synthesized a series of 10-mer substrates containing dihydrothymidines (DHT) on both sides of the (6–4)PP, in which a normal thymidine is incorporated at different positions in the sequence; namely at the 5′ terminus (DHT-1), 5′ flanking the (6–4)PP (DHT-2), 3′ flanking the (6–4)PP (DHT-3), and the 3′ terminus (DHT-4) (Figure 5A). The base moiety of the DHT is puckered due to saturation and thus is no longer π–planar, and therefore is not expected to form the CH–π interaction with Arg. As DHT-3 possesses a planar aromatic nucleobase at the 3′ position flanking the (6–4)PP, it is considered to be the only substrate able to form the CH–π interaction with Arg, and thus it can be used to test the DHT-3-specific behavior.
Figure 5.

Binding and photorepair properties of WT-Xl64 using chemically modified substrates. (A) Structures of the DHT and AP substrates. The 5′ terminus of each oligonucleotide for the anisotropy measurement was modified with the C3 amino linker on a DNA synthesizer, followed by labeling with TAMRA, while the DHT substrates used for the DNA repair assays were unlabeled, and had the intact 5′-hydroxy group. (B) Fluorescence anisotropy measurements of the fluorescently labeled DHT-1 (black), DHT-2 (blue), DHT-3 (red), and DHT-4 (green) upon titration with increasing amounts of WT-Xl64. The measurements were performed independently in triplicate, and the mean ± SD values are shown. (C) Fluorescence anisotropy measurements of fluorescently labeled (6–4)T8 (black square), 3AP (red triangle), and 5AP (blue triangle) upon titration with increasing amounts of WT-Xl64. (D) Photorepair of the DHT substrates by WT-Xl64.
First, the binding of Xl64 to the DHT substrates was investigated by fluorescent anisotropy measurements, using the tetramethylrhodamine-labeled DHT substrates (Figure 5B), and the Kd values of the enzyme for the substrates were obtained by non-linear fitting. The dissociation constants of WT-Xl64 for the DHT substrates were on a μM order (Table 2), and much larger than those obtained in the EMSA experiments. This is simply caused by the weaker binding of Xl64 to single-stranded DNA than to double-stranded DNA, as described previously (27,52). Interestingly, the Kd value for DHT-3 was found to be the lowest (0.73 μM) among the tested DHT substrates, suggesting that the presence of the aromatic nucleobase at the position 3′ flanking the (6–4)PP is required for the proper DNA binding of Xl64, likely through the CH–π interaction between the alkyl chain of the arginine side chain and the aromatic nucleobase 3′ flanking to the (6–4)PP.
Table 2.
Binding and photorepair properties of WT-Xl64 for the DHT substrates
| Entry | K d (μM) | ηss |
|---|---|---|
| DHT-1 | 3.39 ± 0.39 | 1.6% |
| DHT-2 | 2.73 ± 0.13 | 1.7% |
| DHT-3 | 0.73 ± 0.09 | 5.3% |
| DHT-4 | 1.87 ± 0.17 | 1.6% |
To corroborate the presence of the CH–π interaction, another set of substrates was prepared; d(TTTTT(6–4)TTTTT) ((6–4)T8), d(TTT_T(6–4)TTTTT) (5AP), and d(TTTTT(6–4)T_TTT) (3AP), where the underscore in the sequences represents a stable analog of the apyrimidinic/apurinic (AP) site (Figure 5A). WT-Xl64 was expected to exhibit the weakest binding affinity for 3AP, which lacks a nucleobase 3′ flanking the (6–4)PP, due to the loss of the CH–π interaction. These substrates were also fluorescently-labeled, and the Kd values were obtained in the same manner as described above (Figure 5C and Table 3). The Kd for (6–4)T8 was 154 ± 18 nM, which was much smaller than that for DHT-3. Since the base moiety of DHT is puckered, the steric hindrance of the enzyme-DNA interaction might be more prominent in DHT-3 than in (6–4)T8, leading to the large Kd value for DHT-3. The introduction of the AP site analog into the sequence drastically changed the binding affinity, and the Kd value for 5AP (53 ± 6.2 nM) was 3-fold smaller than that for (6–4)T8, while the value for 3AP (468 ± 45 nM) was 3-fold larger than that for (6–4)T8. This result clearly demonstrated that the loss of an aromatic base at the position 3′ adjacent to the (6–4)PP significantly reduces the affinity, indicating the key role of the CH–π interaction in the substrate binding of Xl64. The larger affinity for 5AP than for (6–4)T8 was unexpected, but this may simply be caused by the absence of the steric hindrance on the 5′ side of the (6–4)PP, and the enzyme would be able to access the lesion more easily in 5AP than in (6–4)T8. Comprehensively, we have demonstrated that the aromatic nucleobase 3′ adjacent to the (6–4)PP participates in the CH–π interaction that contributes to the tight binding of Xl64 to the (6–4)PP-containing substrate.
Table 3.
Binding of WT-Xl64 to the substrates containing an AP site
| Entry | K d (nM) |
|---|---|
| (6–4)T8 | 154 ±18 |
| 5AP | 53 ± 6.2 |
| 3AP | 468 ± 45 |
Next, the photorepair of the DHT substrates by WT-Xl64 was investigated, and the S/p0 values for the DHT substrates were plotted against the incident photon numbers (Figure 5D). The results clearly demonstrated that the photorepair of DHT-3 exhibited the steepest slope, while those of DHT-1, DHT-2, and DHT-4 proceeded much more slowly, with almost the same inefficiency as the photorepair of the normal substrate by R410A (Figure 3B). Their η ss values were calculated as 1.6% for DHT-1, 1.7% for DHT-2, 5.3% for DHT-3, and 1.6% for DHT-4, indicating that DHT-3 was the sole substrate to be repaired as efficiently as the standard substrate (see Figure 3). In fact, DHT-3 was previously utilized for the detection of the photorepair induced by single-turnover laser flashes (9), in order to reduce the background of the UV absorption. This substrate design idea was originally proposed in the study on CPD photorepair by Thiagarajan et al., in which they showed that a substrate containing the cis-syn CPD, in place of the (6–4)PP, in the DHT-4 sequence was repaired in a comparable manner to the standard substrate (28). In contrast, our results showed that Xl64 was not able to repair the DHT-4 substrate as efficiently as the standard substrate. The difference in the substrate preferences between CPD-PL and (6–4)PL indicated their diverse substrate binding mechanisms. The DNA repair experiments using Xl64 also revealed that the CH–π interaction plays a very significant role in the stabilization of a robust repair-active complex. The repair of the (6–4)PP by (6–4)PL reportedly requires two photons, associated with the formation of the intermediate (9). Thus, the (6–4)PL should keep the lesion and the intermediate accommodated in the active site, until the enzyme releases the repaired product. As the reduced binding affinity for the substrates lacking the aromatic nucleobase at the position 3′ flanking the (6–4)PP was observed, the complex lacking the CH–π interaction would not be stable enough to hold the lesion (and probably the intermediate, as well), leading to the reduced η ss values in the CH–π interaction-lacking substrates.
Binding of the mutants of Xl64 to the DHT substrates
We next investigated the binding of the Arg mutants of Xl64 to DHT-1 and DHT-3, in addition to (6–4)T8, and the results are summarized in Supplementary Figure S4 and Table 4. A comparison of the WT and R410A results for (6–4)T8 indicated that the loss of Arg410 lowered the affinity for (6–4)T8, with a Kd value of 3 μM. A similar increase of the Kd was observed for DHT-3, but not for DHT-1. These results indicated that the loss of Arg410 significantly reduces the affinity for the substrates containing an aromatic nucleobase 3′ flanking the (6–4)PP, and also that the low affinities with the Kd values of 3–5 μM reflect the non-specific binding to the substrate. The binding of R410K to (6–4)T8 and DHT-3 exhibited similar affinities to that of the WT, while the Kd value for DHT-1 was 3.08 μM. To evaluate the importance of the CH–π interaction, we prepared the R410L mutant, which possesses an alkyl side chain potentially capable of forming a CH–π interaction with the 3′ flanking aromatic nucleobase, but lacks a positive charge. The binding of R410L was found to be in the same range as R410A, and the Kd values for these substrates were 2–3.5 μM. Due to a concern that the Kd value of R410L for DHT-3 (2.28 μM) was smaller than that of R410A (3.72 μM), presumably due to the presence of the CH–π interaction, the simulated interaction energies for PLR421L were evaluated in the same manner as described in the previous section. The results (shown in Supplementary Figure S5 and Supplementary Table S7) indicated that the Leu mutant showed no significant dispersive interaction between the Leu residue and the 3′ nucleobase. In addition, the photorepair of the 8-mer substrate by R410L was as inefficient as that by R410A (Supplementary Figure S6), indicating that the Leu residue does not participate in the formation of the CH–π interaction and therefore R410L cannot stabilize the repair-active complex.
Table 4.
Binding of the Xl64 mutants to the DHT substrates
| K d (μM) | ||||
|---|---|---|---|---|
| Entry | R410A | R410K | R410L | WTa |
| (6–4)T8 | 3.05 ± 0.40 | 0.14 ± 0.02 | 3.18 ± 0.14 | 0.15 ± 0.02 |
| DHT-1 | 4.68 ± 1.20 | 3.08 ± 0.15 | 3.53 ± 1.24 | 3.39 ± 0.39 |
| DHT-3 | 3.72 ± 0.25 | 0.56 ± 0.05 | 2.28 ± 0.55 | 0.73 ± 0.09 |
aRecalled from the results shown in Figure 5.
‘Ship docking’ model for the binding of (6–4)PL
We propose the general requirements for the formation of the stable and repair-active DNA-Xl64 complex (Figure 6). First, the presence of the positive charge at the 410th position in Xl64 is mandatory, and the loss of the Coulomb interaction between the guanidine moiety and the phosphate groups in DNA makes the DNA-Xl64 complex fragile. Secondly, the CH–π interaction between the alkyl side chain of the 410th amino acid and the aromatic nucleobase 3′ flanking the (6–4)PP is also required for the formation of the robust DNA-Xl64 complex. In contrast, the presence of an alkyl chain capable of forming the CH–π interaction is not sufficient to form a stable complex. As observed in the FMO calculations, the stabilization energy of the CH–π interaction is only –6 kcal mol–1, and thus the Coulomb interaction, which is much stronger than the CH–π interaction, would exclusively anchor the protein to the DNA, spatially placing the alkyl moiety of the side chain of the 410th amino acid at the location proximal to the aromatic nucleobase 3′ flanking the (6–4)PP. The CH–π interaction then ties up the Arg side chain and DNA. If the CH–π interaction cannot be formed there, then the DNA-protein complex would no longer be stable, as shown by the weak binding and the reduced η ss values observed for DHT-1, DHT-2 and DHT-4. Our results demonstrated that both the Coulomb and CH–π interactions ensure the robust repair-active DNA–protein complex, even though the stabilization energy of the CH–π interaction is small.
Figure 6.
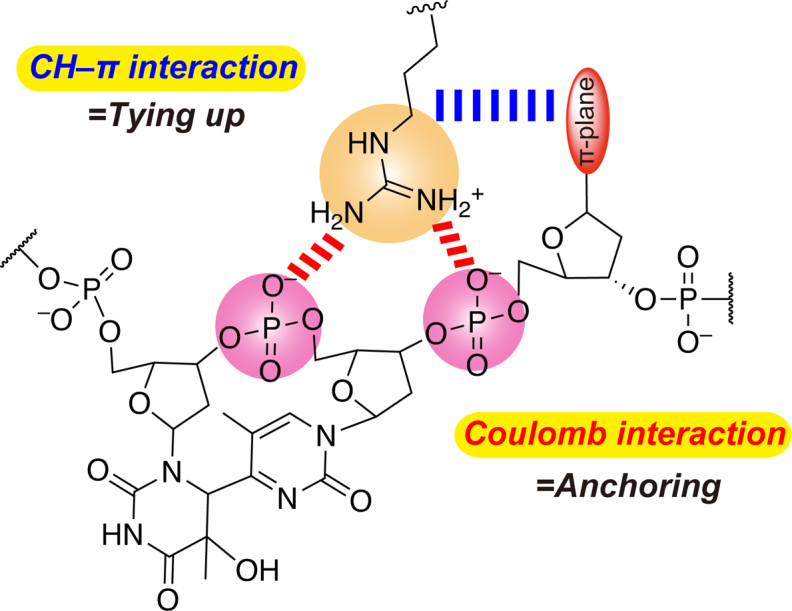
Important interactions for and their roles in the stabilization of the repair-active DNA-(6–4)PL complex.
The binding of (6–4)PL can be likened to ship docking. An anchor is cast prior to ship docking, but the passengers in the ship cannot land by only fixing the ship with the anchor, because the ship will become unstable when the wind blows. The ship crew then throws ropes to fix the ship to bollards on the dock, so the ship does not move away from the pier. Tethering the ship to the bollards with the ropes, without the anchor, cannot guarantee the safe landing because the ropes can easily break. Both the casting of the anchor and the tethering with ropes are required. In the DNA repair by the (6–4)PL, the enzyme first needs to be anchored to the DNA by the interaction between the positively-charged Arg residue and the phosphate groups at the lesion site via the Coulomb interactions, and then the Arg-DNA interaction is reinforced by the CH–π interaction between the Arg alkyl chain and the 3′ flanking nucleobase, ensuring a tightly-bound DNA-protein complex capable of robust DNA repair. The Arg residue thus plays a crucial role in the function of DNA repair by the (6–4)PLs.
Animal cryptochromes possess His in place of Arg
Animal CRYs share high sequence similarity to the (6–4)PLs, but their functions are diverse. Although the amino acid residues responsible for the (6–4)PL activity, such as the His-His-Tyr motif in the active site (18), are conserved even in the animal CRYs, they neither bind nor repair the (6–4)PP-containing oligonucleotides (3,53). Interestingly, the Arg residue highlighted in this study is replaced with His in most animal CRYs (Figure 7A). Although the imidazole moiety of histidine can partially be protonated in a neutral solution, in a similar manner to the Arg and Lys residues, the length of the alkyl chain connecting the imidazole ring to the Cα atom seems to be too short to form the Coulomb interaction with the phosphate groups in the DNA backbone, and this may be the reason why the animal CRYs have lost the specific binding ability to the (6–4)PP-containing DNA.
Figure 7.

Binding of the animal CRY-type mutant of Xl64 to the (6–4)PP-containing substrates. (A) Sequence alignment of the (6–4)PLs and animal CRYs. The arginine and histidine residues in question are highlighted in red and blue, respectively. Abbreviations: Dr, Danio rerio; Er, Erithacus rubecula; Mm, Mus musculus; Hs, Homo sapiens. (B) Fluorescence anisotropy measurements of the fluorescently labeled (6–4)T8 (black square), DHT-1 (black circle), and DHT-3 (red circle) upon titration with increasing amounts of R410H-Xl64. The measurements were performed independently in triplicate, and the mean ± SD values are shown. (C) Dissociation constants (Kd) for the binding of R410H-Xl64 to the tested substrates.
To address this possibility, we prepared another mutant of Xl64, in which Arg410 was replaced with His (R410H), and the binding abilities of R410H to (6–4)T8, DHT-1, and DHT-3 were evaluated in the same manner (Figure 7B). The anisotropy changes of DHT-1 and DHT-3 upon titration with the R410H mutant exhibited almost the same curves as those obtained for R410A and R410L (Supplementary Figure S4), and the Kd values for the DHT substrates were found to be on the order of 3 μM (Figure 7C), suggesting a reduced binding ability. Unlike R410A and R410L, however, R410H bound (6–4)T8 with a smaller Kd value (1.22 μM) than those obtained for DHT-1 and DHT-3. A comparison of the Kd values of WT-Xl64 and R410H for (6–4)T8 revealed that the mutation of Arg to His clearly reduced the affinity, by a factor of 8. In fact, the FAD-binding cavity of mouse CRY (54,55) is obviously wider and shallower than that of Dm64, and thus the animal CRY seems to be no longer capable of accommodating the flipped lesion, due to the structural difference. However, our result demonstrated that the single mutation of Arg in Xl64 to His drastically changes its specific DNA binding ability to the (6–4)PP-containing substrate, even though the DNA binding site is preserved, implying that this Arg residue could be the turning point for the biological roles of (6–4)PLs and CRYs.
In conclusion, we have comprehensively demonstrated that the Coulomb and CH–π interactions between DNA and the Arg residue in (6–4)PL are critical for its biological functions. The loss of either of them reduces the affinity for the substrate, even though the stabilization energy of the CH–π interaction is much smaller than that of the Coulomb interaction. The CH–π interaction networks facilitate the construction and the stabilization of protein structures (50), but there are very few examples in which the CH–π interaction governs the enzymatic function (56), especially in between DNA and proteins. The replacement of the Arg residue with His in Xl64, which is found in animal CRYs, also reduces the affinity for the substrates, implying the functional differentiation of the (6–4)PLs and CRYs, despite the similarities in their structures and amino acid sequences.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Drs Klaus Brettel (CEA Saclay, France), Pascal Plaza (ENS Paris/CNRS, France), Motohiro Nishio (CHPI Institute, Japan), and Keiji Hirose (Osaka University, Japan) for fruitful discussions. The computations were performed at the Research Center for Computational Science (RCCS) at the Institute of Molecular Science (IMS), the Institute of Solid State Physics (ISSP) at the University of Tokyo, and the Center for Computational Sciences (CCS) at University of Tsukuba.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Japan Society for the Promotion of Science [JP25870400, JP16K07321 to J.Y., JP16H06164, JP1503797 to R.H.]; Grants-in-Aid for Scientific Research of the Innovative Areas ‘Photosynergetics’ [JP26107004] from Ministry of Education, Culture, Sports, Science and Technology, Japan; and a support for cooperative work by IMS. Funding for open access charge: Japan Society for the Promotion of Science.
Conflict of interest statement. None declared.
REFERENCES
- 1. Sancar A. Structure and function of DNA photolyase and cryptochrome blue-light photoreceptors. Chem. Rev. 2003; 103:2203–2237. [DOI] [PubMed] [Google Scholar]
- 2. Lin C., Shalitin D.. Cryptochrome structure and signal transduction. Annu. Rev. Plant Biol. 2003; 54:469–496. [DOI] [PubMed] [Google Scholar]
- 3. Chaves I., Pokorny R., Byrdin M., Hoang N., Ritz T., Brettel K., Essen L.-O., van der Horst G.T.J., Batschauer A., Ahmad M.. The cryptochromes: Blue light photoreceptors in plants and animals. Annu. Rev. Plant Biol. 2011; 62:335–364. [DOI] [PubMed] [Google Scholar]
- 4. Kao Y.-T., Saxena C., Wang L., Sancar A., Zhong D.. Direct observation of thymine dimer repair in DNA by photolyase. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:16128–16132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Li J., Liu Z., Tan C., Guo X., Wang L., Sancar A., Zhong D.. Dynamics and mechanism of repair of ultraviolet-induced (6-4) photoproduct by photolyase. Nature. 2010; 466:887–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. MacFarlane A.W., Stanley R.J.. Cis–syn thymidine dimer repair by DNA photolyase in real time. Biochemistry. 2003; 42:8558–8568. [DOI] [PubMed] [Google Scholar]
- 7. Thiagarajan V., Byrdin M., Eker A.P.M., Müller P., Brettel K.. Kinetics of cyclobutane thymine dimer splitting by DNA photolyase directly monitored in the UV. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:9402–9407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Liu Z., Tan C., Guo X., Kao Y.-T., Li J., Wang L., Sancar A., Zhong D.. Dynamics and mechanism of cyclobutane pyrimidine dimer repair by DNA photolyase. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:14831–14836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yamamoto J., Martin R., Iwai S., Plaza P., Brettel K.. Repair of the (6–4) photoproduct by DNA photolyase requires two photons. Angew. Chem. Int. Ed. 2013; 52:7432–7436. [DOI] [PubMed] [Google Scholar]
- 10. Yamamoto J., Plaza P., Brettel K.. Repair of (6-4) lesions in DNA by (6-4) photolyase: 20 years of quest for the photoreaction mechanism. Photochem. Photobiol. 2017; 93:51–66. [DOI] [PubMed] [Google Scholar]
- 11. Park H., Zhang K., Ren Y., Nadji S., Sinha N., Taylor J.-S., Kang C.. Crystal structure of a DNA decamer containing a cis-syn thymine dimer. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:15965–15970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bdour H.M., Kao J.L.-F., Taylor J.-S.. Synthesis and characterization of a [3-15N]-labeled cis-cyn thymine dimer-containing DNA duplex. J. Org. Chem. 2006; 71:1640–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sato R., Harada R., Shigeta Y.. Theoretical analyses on a flipping mechanism of UV-induced DNA damage. Biophys. Physicobiol. 2016; 13:311–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Christine K.S., MacFarlane A.W., Yang K., Stanley R.J.. Cyclobutylpyrimidine dimer base flipping by DNA photolyase. J. Biol. Chem. 2002; 277:38339–38344. [DOI] [PubMed] [Google Scholar]
- 15. Torizawa T., Ueda T., Kuramitsu S., Hitomi K., Todo T., Iwai S., Morikawa K., Shimada I.. Investigation of the cyclobutane pyrimidine dimer (CPD) photolyase DNA recognition mechanism by NMR analyses. J. Biol. Chem. 2004; 279:32950–32956. [DOI] [PubMed] [Google Scholar]
- 16. Mees A., Klar T., Gnau P., Hennecke U., Eker A.P.M., Carell T., Essen L.-O.. Crystal structure of a photolyase bound to a CPD-like DNA lesion after in situ repair. Science. 2004; 306:1789–1793. [DOI] [PubMed] [Google Scholar]
- 17. Kiontke S., Geisselbrecht Y., Pokorny R., Carell T., Batschauer A., Essen L.-O.. Crystal structure of an archaeal class II DNA photolyase and its complex with UV-damaged duplex DNA. EMBO J. 2011; 30:4437–4449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Maul M.J., Barends T.R.M., Glas A.F., Cryle M.J., Domratcheva T., Schneider S., Schlichting I., Carell T.. Crystal structure and mechanism of a DNA (6-4) photolyase. Angew. Chem. Int. Ed. 2008; 47:10076–10080. [DOI] [PubMed] [Google Scholar]
- 19. Sato R., Harada R., Shigeta Y.. The binding structure and affinity of photodamaged duplex DNA with members of the photolyase/cryptochrome family: A computational study. Biophys. Physicobiol. 2018; 15:18–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jepsen K.A., Solov’yov I.A.. On binding specificity of (6-4) photolyase to a T(6-4)T DNA photoproduct. Eur. Phys. J. D. 2017; 71:155. [Google Scholar]
- 21. Iwai S., Shimizu M., Kamiya H., Ohtsuka E.. Synthesis of a phosphoramidite coupling unit of the pyrimidine (6–4) pyrimidone photoproduct and its incorporation into oligodeoxynucleotides. J. Am. Chem. Soc. 1996; 118:7642–7643. [Google Scholar]
- 22. Iwai S., Mizukoshi T., Fujiwara Y., Masutani C., Hanaoka F., Hayakawa Y.. Benzimidazolium triflate-activated synthesis of (6–4) photoproduct-containing oligonucleotides and its application. Nucleic Acids Res. 1999; 27:2299–2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yamamoto J., Hitomi K., Todo T., Iwai S.. Chemical synthesis of oligodeoxyribonucleotides containing the Dewar valence isomer of the (6–4) photoproduct and their use in (6–4) photolyase studies. Nucleic Acids Res. 2006; 34:4406–4415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Todo T., Ryo H., Yamamoto K., Toh H., Inui T., Ayaki H., Nomura T., Ikenaga M.. Similarity among the Drosophila (6-4)photolyase, a human photolyase homolog, and the DNA photolyase-blue-light photoreceptor family. Science. 1996; 272:109–112. [DOI] [PubMed] [Google Scholar]
- 25. Yamamoto J., Shimizu K., Kanda T., Hosokawa Y., Iwai S., Plaza P., Müller P.. Loss of fourth electron-transferring tryptophan in animal (6–4) photolyase impairs DNA repair activity in bacterial cells. Biochemistry. 2017; 56:5356–5364. [DOI] [PubMed] [Google Scholar]
- 26. Müller P., Yamamoto J., Martin R., Iwai S., Brettel K.. Discovery and functional analysis of a 4th electron-transferring tryptophan conserved exclusively in animal cryptochromes and (6-4) photolyases. Chem. Commun. 2015; 51:15502–15505. [DOI] [PubMed] [Google Scholar]
- 27. Hitomi K., Kim S.-T., Iwai S., Harima N., Otoshi E., Ikenaga M., Todo T.. Binding and catalytic properties of Xenopus (6-4) photolyase. J. Biol. Chem. 1997; 272:32591–32598. [DOI] [PubMed] [Google Scholar]
- 28. Thiagarajan V., Villette S., Espagne A., Eker A.P.M., Brettel K., Byrdin M.. DNA repair by photolyase: a novel substrate with low background absorption around 265 nm for transient absorption studies in the UV. Biochemistry. 2010; 49:297–303. [DOI] [PubMed] [Google Scholar]
- 29. Senda N. Winmostar Software, Version 7.020, by Delphi. [Google Scholar]
- 30. Frisch M.J., Trucks G.W., Schlegel H.B., Scuseria G.E., Robb M.A., Cheeseman J.R., Scalmani G., Barone V., Mennucci B., Petersson G.A. et al. . Gaussian09, revision D01. 2016; Wallingford: Gaussian, Inc. [Google Scholar]
- 31. Case D.A., Babin V., Berryman J.T., Betz R.M., Cai Q., Cerutti D.S., Cheatham T.E., Darden T.A., Duke R.E., Gohlke H. et al. . AMBER14. 2014; San Francisco: University of California. [Google Scholar]
- 32. Maier J.A., Martinez C., Kasavajhala K., Wickstrom L., Hauser K.E., Simmerling C.. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015; 11:3696–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jorgensen R.W., Chandrasekhar J., Madura J.D., Impey R.W., Klein M.L.. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983; 79:926–935. [Google Scholar]
- 34. Maseras F., Morokuma K.. IMOMM: A new integrated ab initio + molecular mechanics geometry optimization scheme of equilibrium structures and transition states. J. Comput. Chem. 1995; 16:1170–1179. [Google Scholar]
- 35. Humbel S., Sieber S., Morokuma K.. The IMOMO method: Integration of different levels of molecular orbital approximations for geometry optimization of large systems: Test for n-butane conformation and SN2 reaction: RCl+Cl–. J. Chem. Phys. 1996; 105:1959–1967. [Google Scholar]
- 36. Matsubara T., Maseras F., Koga N., Morokuma K.. Application of the new “integrated MO + MM” (IMOMM) method to the organometallic reaction Pt(PR3)2 + H2 (R = H, Me, t-Bu, and Ph). J. Phys. Chem. 1996; 100:2573–2580. [Google Scholar]
- 37. Vreven T., Morokuma K., Farkas Ö., Schlegel H.B., Frisch M.J.. Geometry optimization with QM/MM, ONIOM, and other combined methods. I. Microiterations and constraints. J. Comput. Chem. 2003; 24:760–769. [DOI] [PubMed] [Google Scholar]
- 38. Svensson M., Humbel S., Froese R.D.J., Matsubara T., Sieber S., Morokuma K.. ONIOM: A multilayered integrated MO + MM method for geometry optimizations and single point energy predictions. A test for Diels-Alder reactions and Pt(P(t-Bu)3)2 + H2 oxidative addition. J. Phys. Chem. 1996; 100:19357–19363. [Google Scholar]
- 39. Chung L.W., Sameera W.M.C., Ramozzi R., Page A.J., Hatanaka M., Petrova G.P., Harris T.V., Li X., Ke Z., Liu F. et al. . The ONIOM method and its applications. Chem. Rev. 2015; 115:5678–5796. [DOI] [PubMed] [Google Scholar]
- 40. Kitaura K., Ikeo E., Asada T., Nakano T., Uebayashi M.. Fragment molecular orbital method: an approximate computational method for large molecules. Chem. Phys. Lett. 1999; 313:701–706. [Google Scholar]
- 41. Kitaura K., Sugiki S.-I., Nakano T., Komeiji Y., Uebayashi M.. Fragment molecular orbital method: analytical energy gradients. Chem. Phys. Lett. 2001; 336:163–170. [Google Scholar]
- 42. Nakata H., Fedorov D.G., Nagata T., Yokojima S., Ogata K., Kitaura K., Nakamura S.. Unrestricted Hartree-Fock based on the fragment molecular orbital method: Energy and its analytic gradient. J. Chem. Phys. 2012; 137:044110. [DOI] [PubMed] [Google Scholar]
- 43. Schmidt M.W., Baldridge K.K., Boatz J.A., Elbert S.T., Gordon M.S., Jensen J.H., Koseki S., Matsunaga N., Nguyen K.A., Su S. et al. . General atomic and molecular electronic structure system. J. Comput. Chem. 1993; 14:1347–1363. [Google Scholar]
- 44. Fedorov D.G., Kitaura K.. Extending the power of quantum chemistry to large systems with the fragment molecular orbital method. J. Phys. Chem. A. 2007; 111:6904–6914. [DOI] [PubMed] [Google Scholar]
- 45. Nakano T., Kaminuma T., Sato T., Fukuzawa K., Akiyama Y., Uebayashi M., Kitaura K.. Fragment molecular orbital method: use of approximate electrostatic potential. Chem. Phys. Lett. 2002; 351:475–480. [Google Scholar]
- 46. Dokainish H.M., Yamada D., Iwata T., Kandori H., Kitao A.. Electron fate and mutational robustness in the mechanism of (6-4)photolyase-mediated DNA repair. ACS Catal. 2017; 7:4835–4845. [Google Scholar]
- 47. Amari S., Aizawa M., Zhang J., Fukuzawa K., Mochizuki Y., Iwasawa Y., Nakata K., Chuman H., Nakano T.. VISCANA: Visualized cluster analysis of protein–ligand interaction based on the ab initio fragment molecular orbital method for virtual ligand screening. J. Chem. Inf. Model. 2006; 46:221–230. [DOI] [PubMed] [Google Scholar]
- 48. Kurisaki I., Fukuzawa K., Komeiji Y., Mochizuki Y., Nakano T., Imada J., Chmielewski A., Rothstein S.M., Watanabe H., Tanaka S.. Visualization analysis of inter-fragment interaction energies of CRP-cAMP-DNA complex based on the fragment molecular orbital method. Biophys. Chem. 2007; 130:1–9. [DOI] [PubMed] [Google Scholar]
- 49. Tsuzuki S., Fujii A.. Nature and physical origin of CH/π interaction: significant difference from conventional hydrogen bonds. Phys. Chem. Chem. Phys. 2008; 10:2584–2594. [DOI] [PubMed] [Google Scholar]
- 50. Nishio M., Umezawa Y., Fantini J., Weiss M.S., Chakrabarti P.. CH–π hydrogen bonds in biological macromolecules. Phys. Chem. Chem. Phys. 2014; 16:12648–12683. [DOI] [PubMed] [Google Scholar]
- 51. Ishikawa T., Mochizuki Y., Amari S., Nakano T., Tokiwa H., Tanaka T., Tanaka K.. Fragment interaction analysis based on local MP2. Theor. Chem. Acc. 2007; 118:937–945. [Google Scholar]
- 52. Zhao X., Liu J., Hsu D.S., Zhao S., Taylor J.-S., Sancar A.. Reaction mechanis of (6-4) photolyase. J. Biol. Chem. 1997; 272:32580–32590. [DOI] [PubMed] [Google Scholar]
- 53. Heijde M., Zabulon G., Corellou F., Ishikawa T., Brazard J., Usman A., Sanchez F., Plaza P., Martin M., Falciatore A. et al. . Characterization of two members of the cryptochrome/photoyase family from Ostreococcus tauri provides insights into the origin and evolution of cryptochromes. Plant Cell Environ. 2010; 33:1614–1626. [DOI] [PubMed] [Google Scholar]
- 54. Xing W., Busino L., Hinds T.R., Marionni S.T., Saifee N.H., Bush M.F., Pagano M., Zheng N.. SCFFBXL3 ubiquitin ligase targets cryptochromes at their cofactor pocket. Nature. 2013; 496:64–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Czarna A., Berndt A., Singh H.R., Grudziecki A., Ladurner A.G., Timinszky G., Kramer A., Wolf E.. Structures of Drosophila cryptochrome and mouse cryptochrome1 provide insight into circadian function. Cell. 2013; 153:1394–1405. [DOI] [PubMed] [Google Scholar]
- 56. Harigai M., Kataoka M., Imamoto Y.. A single CH/π weak hydrogen bond governs stability and the photocycle of the photoactive yellow protein. J. Am. Chem. Soc. 2006; 128:10646–10647. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.