

Abstract
The way cells respond to DNA damage is important since inefficient repair or misrepair of lesions can have deleterious consequences, including mutation, genomic instability, neurodegenerative disorders, premature aging, cancer or death. Whether damage occurs spontaneously as a byproduct of normal metabolic processes, or after exposure to exogenous agents, cells muster a coordinated, complex DNA damage response (DDR) to mitigate potential harmful effects. A variety of activities are involved to promote cell survival, and include DNA repair, DNA damage tolerance, as well as transient cell cycle arrest to provide time for repair before entry into critical cell cycle phases, an event that could be lethal if traversal occurs while damage is present. When such damage is prolonged or not repairable, senescence, apoptosis or autophagy is induced. One major level of DDR regulation occurs via the orchestrated transcriptional control of select sets of genes encoding proteins that mediate the response. p53 is a transcription factor that transactivates specific DDR downstream genes through binding DNA consensus sequences usually in or near target gene promoter regions. The profile of p53-regulated genes activated at any given time varies, and is dependent upon type of DNA damage or stress experienced, exact composition of the consensus DNA binding sequence, presence of other DNA binding proteins, as well as cell context. RAD9 is another protein critical for the response of cells to DNA damage, and can also selectively regulate gene transcription. The limited studies addressing the role of RAD9 in transcription regulation indicate that the protein transactivates at least one of its target genes, p21/waf1/cip1, by binding to DNA sequences demonstrated to be a p53 response element. NEIL1 is also regulated by RAD9 through a similar DNA sequence, though not yet directly verified as a bonafide p53 response element. These findings suggest a novel pathway whereby p53 and RAD9 control the DDR through a shared mechanism involving an overlapping network of downstream target genes. Details and unresolved questions about how these proteins coordinate or compete to execute the DDR through transcriptional reprogramming, as well as biological implications, are discussed.
INTRODUCTION
DNA damage can occur after exposure to exogenous agents, or as a result of normal metabolic processes. If that damage is not properly repaired, harmful effects can ensue. Cells have a variety of pathways capable of repairing the damage, or causing senescence or death if damage lingers. p53 regulates the global cellular response to DNA damage, primarily by controlling transcription of a select set of downstream target genes through binding consensus sequences in or near gene promoter regions. RAD9 also plays a prominent role in the DNA damage response, through a variety of mechanisms, but notably also by transactivating genes. Moreover, studies thus far indicate that at least one and likely many target genes are regulated by both p53 and RAD9 interacting with the same DNA consensus sequences.
In this review, we summarize the myriad of mechanisms by which cells acquire DNA damage, the pathways used by cells to respond, the role of p53 in regulating the transcription of select target genes that participate in these pathways, and the similarities by which RAD9 can transactivate an apparently overlapping subset of p53 target genes to control genomic integrity. Future work to define the relationships between p53 and RAD9, in the context of regulating critical DNA damage responses, is also posed.
Acquisition of DNA Damage
DNA aberrations occur as a byproduct of normal physiological processes or after exposure to exogenous agents. Due to normal metabolic events alone, each cell incurs on average 104–105 DNA lesions per day (1). During DNA replication, errors such as DNA base pair mismatches can arise (2). Reactive oxygen species produced from oxidative respiration or redox and Fenton reactions (3), and reactive oxygen and nitrogen species generated by macrophages and neutrophils as part of the inflammatory response (4) cause DNA base damage or loss, as well as DNA strand breaks. Furthermore, DNA strand breaks occur during different processes such as immunoglobulin class switch recombination to produce immune receptor diversity (5), and gametogenesis in meiotic prophase, specifically in pachytene, to generate genetic diversity (6).
Besides damage generated as part of these inherent activities, environmental exposures to radiations or chemicals also cause DNA aberrations, with potential to inhibit transcription or DNA replication and lead to deleterious effects. Ultraviolet light emitted from the sun is highly prevalent on earth. The ozone layer completely filters out the very harmful UV-C (100–280 nm) component, and most of UV-B (280–315 nm), but a large fraction of UV-A (315– 400 nm) reaches the earth’s surface (7). UV primarily causes two types of DNA aberrations, cyclobutane pyrimidine dimers (CPDs) and 6–4 photoproducts (6–4 PPs), bulky lesions that block critical DNA transactions (8). Ionizing radiation from radon in rocks and soil, diagnostic and therapeutic medical procedures, airline travel, and nuclear power plant accidents, such as in Chernobyl in 1986 (9) and more recently in Fukushima Daiichi (10) causes a wide spectrum of DNA aberrations including single and double DNA strand breaks. Industrial waste products containing heavy metals such as arsenic, cadmium, chromium and nickel cause oxidative stress and DNA damage (11). Even food can contain DNA damaging agents. Heterocyclic aromatic amines are produced when meats are heated above 1808C and can react with purine bases, resulting in bulky DNA adducts that have potential to cause mutations and cancer (12). Aflatoxins are produced by the molds Aspergillus flavus and Aspergillus parasiticus, which are sometimes found in peanuts, cottonseed, corn, rice, tree nuts or other foods when improperly stored (13).
Metabolites of aflatoxins intercalate into DNA, aberrantly alkylate bases, and cause mutations as well as cancer (14, 15). Therefore, since genomic DNA frequently incurs damage induced by agents emanating from a large array of sources, both intracellular and extracellular, repair must occur to insure cell survival and the well being of each organism.
The DNA Damage Response (DDR) and Genomic Stability
Multiple mechanisms are available for cells to repair DNA damage. A large array of DNA repair processes mend damage directly through homologous recombination repair, non-homologous end joining (16), alternative non-homologous end joining (17), such as through microhomology-mediated DNA strand annealing (18), nucleotide excision repair (19), base excision repair (20, 21), transcription-coupled repair (22) and mismatch repair (23). Damage tolerance mechanisms, such as translesion synthesis (TLS) and recombination-dependent daughter-strand gap repair (DSGR) (24, 25), as well as direct reversal of UV-induced pyrimidine dimers by photoreactivation (26) also occur.
Furthermore, cell cycle checkpoint mechanisms transiently delay cell cycle progression at specific junctures to provide extra time for restoration of DNA integrity before entry into critical cell cycle phases (27). If damage remains unrepaired or lingers for a prolonged period of time, senescence (28), or death by apoptosis or autophagy (29) will take place. All of these processes have been studied independently and in isolation, yet there is a higher level of organization and coordination, which is referred to as the DNA damage response [(DDR (30–33)].
When cells incur DNA damage, a complex, global cellular process becomes activated, which regulates activities that promote repair and address difficulties with DNA replication. Generally, it consists of sensors that detect damage, transducers capable of rapidly sending signals, and downstream effectors that perform functions to enhance genomic integrity (30). DNA strand breaks are highly potent inducers of the DDR (34, 35). The kinases ATM and ATR are activated early in the signaling response cascade to damage (36). It was originally thought these two proteins mediate signaling independently. For example, ionizing radiation-induced DNA double-strand breaks lead to activation of ATM and then cell cycle checkpoint control in a CHK2 dependent manner, after this latter protein is phosphorylated by ATM at Thr-68 (37, 38). Single- or double-strand breaks and other types of damage cause DNA replication forks to stall. This activates ATR, which then activates CHK1 via phosphorylation on Ser-317 and Ser-345 (39–41). Subsequently, both of these signaling events lead to cell cycle delays. The cell cycle can be blocked at the G1/S transition point, within S phase, or at the G2/M transition, depending upon the nature of the DNA damage and the cell cycle phase at the time damage is encountered. CHK1 and CHK2 modify cell cycle progression through reduction of cyclin dependent kinase activity by several mechanisms (42). However, it is clear that ATM and ATR have overlapping but nonredundant functions. ATM along with the nuclease activity of MRE11 act to initiate resection at radiation-induced DNA double-strand breaks, generating single-stranded DNA coated with Replication Protein A (RPA), which is required for ATR recruitment and CHK1 phosphorylation. This latter activity is restricted to the S and G2 phases of the cell cycle (43).
Aside from damage-induced signaling to produce cell cycle delays, ATM and ATR have numerous targets that also promote increased repair of damaged DNA, or induce senescence or cell death. Downstream events occur by diverse mechanisms that include transcriptional regulation of genes (44), protein–protein interactions (45), or post-translational modulation of proteins by acetylation, methylation, neddylation, phosphorylation, poly-ADP-ribosylation, sumoylation, and mono- as well as poly-ubiquitylation that alter their activities or modulate recruitment to sites of DNA damage (30, 45, 46).
P53 AND THE TRANSCRIPTIONAL RESPONSE TO DNA DAMAGE
The p53 protein is an important and predominant tumor suppressor, as it is inactivated most often by a gene mutation in more than 50% of human cancers (47). A significant component of the response of cells to DNA damage involves transcriptional reprogramming, and p53 plays a major role as a transactivator of gene expression under these circumstances (34, 48). It directly controls expression of several hundred RNA polymerase II transcribed genes, and thousands of others indirectly (49). p53 functions primarily as a transcription factor with a defined yet extensive set of downstream target genes whose encoded proteins mediate critical DNA damage response activities. For example, p53 regulates autophagy by transactivating expression of DRAM (50). Apoptosis is controlled by direct transcriptional regulation of BAD (51), BAX (52), BID (53), ECK (54), PUMA (55, 56, 57) and several other genes. Cell cycle arrest in response to DNA damage occurs through the ability of p53 to activate transcription of p21 (58) and 14-3-3sigma (59). DNA repair activity is altered by p53 through regulation of genes such as DDB2 (60) and XPC (61, 62). The direct transactivation by p53 of PAI-1 controls senescence (63). p53 regulates the response of cells to a wide array of stress through transactivation of overlapping subsets of its target genes, dependent upon the kind of stress or damage incurred, as well as the type or origin of host cell or tissue. Nevertheless, it is not fully clear how p53 selects the specific, limited set of genes transactivated when certain stress conditions prevail. Certainly, target gene transcription regulatory sequences bound by p53, post-translational modifications of the p53 protein, and an array of binding partners all contribute to the profile of genes expressed under different conditions (49).
Of note, p53 can also repress expression of certain genes when DNA damage is encountered, with great specificity as well (64, 65).
Gene Transactivation by p53 through DNA Consensus Sequence Binding
p53 recognizes and binds specific DNA sequences usually upstream of the transcription start site in target genes, resulting in transcriptional activation. However, these p53 response elements are not restricted to promoter regions, and can also be found within early intronic sequences, as well as within exons (49). Analyses of several independent p53 binding sites in the human genome revealed consensus sequences with internal symmetry, made up of 2 copies of 5′-PuPuPuC(A/T)(T/A)GPyPyPy-3′ separated by 0–13 base pairs (66). However, the effects of p53 binding on transcriptional activity are not always predictable, even when consensus sequences are present. Wang et al. (67) analyzed the role of specific nucleotides within the sequence for impact on p53-mediated transcription. They found that a specific dinucleotide core combination within the CWWG motif (i.e., W is a purine base) is critical for determining whether p53 will activate or, in contrast, repress transcription. They also demonstrated that the triplet RRR and YYY sequences (i.e., R is an adenine or guanine base and Y is a pyrimidine) bordering the core motif could modulate the transcriptional activity of p53. Another group reported the analysis of canonical and noncanonical p53 response elements in the entire human genome, and ranked them according to predicted transactivation potential (68). They assessed not only full standard canonical sites but also partial motifs, and demonstrated that all of these sequences could function as p53 response elements to activate transcription of associated coding sequences.
Cooperative and Competitive Binding of p53 to DNA
p53 protein binds its target gene DNA consensus sequences in a 4:1 ratio, and in a cooperative manner (69, 70). Interestingly, Schlereth and co-workers (71) found that the ability of p53 to bind cooperatively to a specific consensus site could be used to predict which genes will be transactivated. For example, they reported that low-cooperativity consensus sequences are enriched in genes that encode cell cycle functions, whereas high-cooperativity consensus sequences are enriched in apoptosis-related genes. Cis-overlapping motifs specific to different transcription factors can influence p53 binding. For example, there are sites in the genome where p53 and c-MYC binding motifs share sequences, so competition for binding and gene transactivation is clearly an issue (72). This is important biologically, as p53 is a tumor suppressor and c-MYC is an oncogene.
Regulation of p53 Stability and Activity
Under normal metabolic nonstress conditions, p53 has a short half-life in cells. However, under stress, p53 must be stabilized to ensure proper transactivation of downstream target genes. There is much known about the regulation of p53 stability (47, 73). MDM2 is an E3 ubiquitin ligase and a major directly acting, key negative regulator of p53 (74, 75). MDM2 ubiquitinates p53, and this post-translational event triggers rapid p53 nuclear export, and proteosomal degradation, efficiently neutralizing p53-mediated transactivity function. Levels and function of MDM2 are tightly controlled in response to specific stress, helping ensure p53 stability when appropriate, and thus proper cellular responses after DNA damaging agent exposures. Furthermore, the p53 and MDM2 relationship is even more complex since they form a critical autoregulatory loop, as the MDM2 gene is a specific transcriptional target of p53 (76–78). Phosphorylation plays an important role in the p53-MDM2 interaction, and ultimately p53 stability and function.
JNK-mediated phosphorylation of p53 inhibits the p53-MDM2 interaction, stabilizes p53, and permits transactivity (79). c-ABL is a tyrosine kinase that binds p53 in response to stress and antagonizes the MDM2 inhibitory effect, resulting in enhanced p53 stability and function (73). The phosphorylation of p53 by ATM at serine-15 after ionizing radiation exposure reduces the affinity of p53 for MDM2, also resulting in reduced p53 degradation facilitated by MDM2 and thus enhanced p53 protein stability and function (74). Phosphorylation of p53 on serine-20 by Chk2 and Chk1 stabilizes the protein as well (80, 81). In addition, phosphorylation of p53 serine-15 and serine-37 by DNA-dependent protein kinase (DNA-PK), in response to DNA damage, impairs the inhibition of the p53 transactivation function by MDM2 (82).
MDMX (also called MDM4) is another major negative regulator of p53, and like MDM2 it is a direct transcriptional target of p53 (78). MDMX has no intrinsic E3-ligase activity, but it regulates p53 abundance indirectly by modulating the levels and activity of MDM2 (83). In addition, through direct binding to p53, MDMX strongly inhibits p53 transactivation function without appreciably affecting stability (84). Although p53 is a significant, established regulator of MDM2 and MDMX transcription, other signaling pathways and elements that control their expression are emerging (85–88).
An analysis of single cells after gamma-ray exposure revealed that p53 is expressed as a series of pulses, either zero, one, two or more (89). Although the average height and duration of each pulse is independent of amount of DNA damage, pulse number is directly related to damage level. Pulses of the upstream signaling kinases, ATM and CHK2, control these p53 pulses and uniform pulse shape is controlled by negative feedback between p53 and ATM, through Wip1 activity (90). Furthermore, ATM could repeatedly initiate p53 pulses in response to lingering DNA damage. In contrast, 254 nm UV light induces a single p53 pulse and the amplitude as well as duration are proportional to the UV dose (91). Transient damage is spontaneously produced during normal metabolic processes, including passage through the cell cycle, and pulses of p53 can be detected in those cells even without exposure to exogenous DNA damaging agents (92). However, such intrinsic damage is not sustained, and post-translational modifications prevent p53 from being activated and in turn inducing the DDR.
DIVERSE ROLES OF RAD9 IN THE DNA DAMAGE RESPONSE AND MAINTAINING GENOMIC STABILITY
RAD9 has multiple functions that play critical roles in the response of cells to DNA damage, whether caused by normal metabolic processes or exposure to exogenous DNA damaging agents (93, 94). The human protein can function as part of a complex with HUS1 and RAD1 [i.e., 9-1-1 complex (95)], but also independently (96, 97). Knock out or knock down of RAD9 in mammalian cells causes extreme cellular sensitivity to a large variety of radiations and chemicals that damage DNA, and influences genomic stability through very complex and diverse activities. RAD9 is essential for maintaining genomic integrity even in the absence of exogenous DNA damaging agents, as loss of function causes increased frequencies of spontaneous chromosome and chromatid breaks, gene mutation and micronuclei formation (93, 98).
RAD9 plays a number of key roles that promote resistance to radiation-induced DNA damage. For example, the protein is critical for cell cycle arrest (99). After DNA damage is incurred, DNA-bound RPA binds ATRIP, which stimulates the RAD17-RFC2-5 complex to recruit 9-1-1 to the damaged site, perhaps containing inappropriate single-stranded DNA, primed single-stranded DNA or DNA with a gap (100). Binding of 9-1-1 at the damaged site brings TOPBP1 to that location via an interaction with the phosphorylated C-terminal tail of RAD9. TOPBP1 then activates ATR, an event that depends upon ATRIP and is facilitated also by RHINO (101, 102). This in turn leads to the phosphorylation and activation of the CHK1 kinase, and a variety of other downstream target proteins. The C-terminus of RAD9 is also important for bringing CLASPIN to the damaged site, which aids in CHK1 activation as well (103, 104). This complex signaling cascade culminates in cell cycle arrest and the stabilization as well as restart of replication forks as needed (105).
In addition to having a major role in cell cycle checkpoint control, RAD9 participates in five DNA repair pathways, mainly through protein–protein interactions that stimulate the activity of binding partners. RAD9 functions in homologous recombination repair (106), base excision repair (107–111), nucleotide excision repair (112), mismatch repair (113), and alternative non-homologous end joining [such as microhomology-mediated DNA strand annealing (114, 115)]. Post-translational processing of RAD9 can influence repair. For example, phosphorylation of RAD9 Ser-272 by ATM can impact recombinational repair efficiency and genomic stability (116).
RAD9 functions as a pro-apoptotic (117, 118) or anti-apoptotic protein (98), depending upon cell context and level of abundance. RAD9 has a BH3-like domain in its N-terminal region that binds the anti-apoptotic proteins BCL-XL and BCL-2 to promote programmed cell death. c-ABL can phosphorylate tyrosine-28 in the RAD9 BH3-like domain, which promotes binding of the anti-apoptotic protein BCL-XL to that domain and consequently apoptosis (119). Phosphorylation of RAD9 by protein kinase Cδ (PKCd) regulates the interaction between RAD9 and BCL-2, and consequently the apoptotic response to DNA damage (120).
RAD9 REGULATES TRANSCRIPTION OF GENES THAT CONTRIBUTE TO THE DNA DAMAGE RESPONSE
RAD9 and p53 are important for executing the proper cellular response to DNA damage, including cell cycle checkpoint control and DNA repair. p53 functions primarily in this regard as a transcriptional reprogrammer, to activate or repress expression of genes central to the DDR (34, 49). In contrast, most DDR functions reported thus far for RAD9 involve protein–protein interactions that facilitate the phosphorylation and, consequently, activation of other proteins, or stimulate their activity directly through physical interactions. These activities often involve RAD9 bound to HUS1 and RAD1. However, like p53, RAD9 can transactivate the expression of genes whose encoded proteins mediate the DDR, as well as other cellular processes, and there is no evidence that HUS1 and RAD1 are involved (97, 111). p53 regulates the radiation-induced G1 checkpoint in part by transactivation of p21waf1/cip1 (121–123), which encodes a protein that halts progression of the cell cycle by inhibiting cyclins and cyclin-dependent kinases, such as CDK4 and CDK6 (124). RAD9 also regulates expression of p21 by binding to the same p53 consensus DNA sequences in the promoter region of the gene, even in the absence of p53 (97, 125). Ishikawa et al. (126) confirmed that RAD9 can control p21 expression. These investigators further showed that p53 and RAD9 physically interact, and that UV exposure enhances binding of RAD9 to the p53 response element in the p21 promoter. RAD9 also functions as a tumor suppressor by inducing p21-dependent senescence, and suppressing epithelial to mesenchymal transition by binding to the SLUG promoter and inhibiting transcription (127). The ability of p53 to act as a tumor suppressor is also rooted in its ability to control p21 expression (128). In addition to p21, RAD9 regulates transcription of the base excision repair gene NEIL1 through binding p53 response-like elements in the gene promoter, and consequentially regulates BER via this transactivation function (111); However, the ability of p53 to recognize those same sequences and transactivate NEIL1 as well has not yet been reported. A limited microarray gene expression screen revealed that RAD9 can regulate expression of 92 genes, and it will be important to determine whether that control is direct and through p53 response elements (97). Regardless, it is becoming clear that RAD9 and p53 impact carcinogenesis and the DDR via transcriptional control of an overlapping set of genes encoding proteins critical for maintaining genomic stability.
This model of RAD9 and p53 regulating DDR by transactivation of an overlapping set of genes is illustrated in Fig. 1.
FIG. 1.
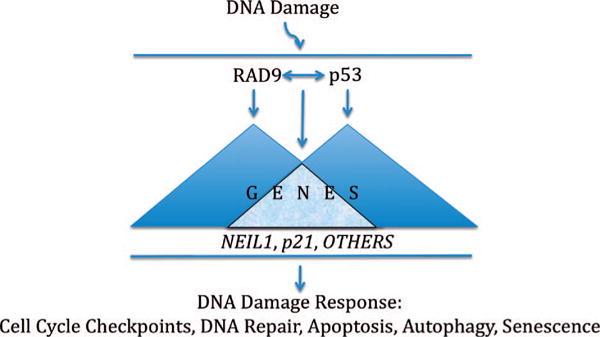
RAD9 and p53 regulation of the transcriptional response to DNA damage. In brief, the model proposes that DNA damage induces a signaling cascade, within which p53 and RAD9 transactivate an overlapping set of genes that control the response of cells to DNA damage. Exactly how RAD9 and p53 interact, whether independently, coordinately or competitively to activate transcription has yet to be determined.
CONCLUSIONS ANF FUTURE GOALS
When cells incur DNA damage, a complex global cellular system known as the DNA damage response is activated to promote survival and minimize deleterious biological effects, or activate senescence or a cell death program if damage is extensive or lingers. The system involves sensors, transducers and effectors that orchestrate DNA repair and cell cycle arrest, to ensure genomic stability and normal replication fork function. p53 and RAD9 are key players in the DDR, and each regulates multiple response phenotypes. p53 functions primarily as a transactivator, although sometimes also as a repressor of select subsets of downstream genes in response to specific types of stress, and binding to response elements usually near transcription start sites allows precise control of target gene expression. Most studies describing the mechanism by which RAD9 regulates the DDR involve protein–protein interactions that stimulate binding partner activity as a result of physical proximity, or that facilitate acquisition of post-translational modifications to enhance or focus activity. However, RAD9 also contributes to the DDR by regulating transcription of genes encoding proteins critical for DNA repair or cell cycle progression.
The fact that RAD9 and p53 can control expression of genes by binding common DNA sequences raises a number of questions, and can form the foundation for future work. Do these proteins bind shared response elements in a coordinated or competitive manner? Are their activities independent, antagonistic or synergistic with respect to controlling gene expression? What is the role of co-factors, binding partners, post-translational modifications, epigenetic changes, or chromatin structure in determining the relationships between RAD9 and p53? Do the relationships change in response to different types of stress? These are important questions since an improper DDR can lead to genomic instability, mutation and cancer. In this regard, p53 is a key tumor suppressor (47) and RAD9 can function as either a tumor suppressor or oncogene (127, 129, 130–132).
Understanding how they mediate the response of cells to DNA damage, and significance with respect to carcinogenesis, could reveal novel strategies and targets for anti-cancer therapy.
Acknowledgments
This work is supported by National Institutes of Health (NIH) grant R01CA130536.
References
- 1.Bernstein C, Prasad AR, Nfonsam V, Bernstein H. DNA Damage, DNA Repair and Cancer. In: Chen C, editor. New Research Directions in DNA Repair. Rijeka, Croatia: InTech; 2013. pp. 413–465. [Google Scholar]
- 2.Chen J, Furano AV. Breaking bad: The mutagenic effect of DNA repair. DNA Repair (Amst) 2015;32:43–51. doi: 10.1016/j.dnarep.2015.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Valko M, Rhodes CJ, Moncol J, Izakovic M, Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact. 2006;160:1–40. doi: 10.1016/j.cbi.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 4.Kawanishi S, Hiraku Y, Pinlaor S, Ma N. Oxidative and nitrative DNA damage in animals and patients with inflammatory diseases in relation to inflammation-related carcinogenesis. Biol Chem. 2006;387:365–372. doi: 10.1515/BC.2006.049. [DOI] [PubMed] [Google Scholar]
- 5.Canela A, Sridharan S, Sciascia N, Tubbs A, Meltzer P, Sleckman BP, et al. DNA Breaks and End Resection Measured Genome-wide by End Sequencing. Mol Cell. 2016;63:898–911. doi: 10.1016/j.molcel.2016.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cooper TJ, Garcia V, Neale MJ. Meiotic DSB patterning: A multifaceted process. Cell Cycle. 2016;15:13–21. doi: 10.1080/15384101.2015.1093709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Matsumi Y, Kawasaki M. Photolysis of atmospheric ozone in the ultraviolet region. Chem Rev. 2003;103:4767–82. doi: 10.1021/cr0205255. [DOI] [PubMed] [Google Scholar]
- 8.Chan GL, Doetsch PW, Haseltine WA. Cyclobutane pyrimidine dimers and (6-4) photoproducts block polymerization by DNA polymerase I. Biochemistry. 1985;24:5723–8. doi: 10.1021/bi00342a006. [DOI] [PubMed] [Google Scholar]
- 9.Saenko V, Ivanov V, Tsyb A, Bogdanova T, Tronko M, Demidchik Y, et al. The Chernobyl accident and its consequences. Clin Oncol (R Coll Radiol) 2011;23:234–43. doi: 10.1016/j.clon.2011.01.502. [DOI] [PubMed] [Google Scholar]
- 10.Akiba S. Epidemiological studies of Fukushima residents exposed to ionising radiation from the Fukushima Daiichi nuclear power plant prefecture–a preliminary review of current plans. J Radiol Prot. 2012;32:1–10. doi: 10.1088/0952-4746/32/1/1. [DOI] [PubMed] [Google Scholar]
- 11.Kim HS, Kim YJ, Seo YR. An Overview of Carcinogenic Heavy Metal: Molecular Toxicity Mechanism and Prevention. J Cancer Prev. 2015;2:232–40. doi: 10.15430/JCP.2015.20.4.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wogan GN, Hecht SS, Felton JS, Conney AH, Loeb LA. Environmental and chemical carcinogenesis. Semin Cancer Biol. 2004;14:473–86. doi: 10.1016/j.semcancer.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 13.Wilson DM, Mubatanhema W, Jurjevic Z. Biology and ecology of mycotoxigenic Aspergillus species as related to economic and health concerns. Adv Exp Med Biol. 2002;504:3–17. doi: 10.1007/978-1-4615-0629-4_2. [DOI] [PubMed] [Google Scholar]
- 14.Hamilton PL, Arya DP. Natural product DNA major groove binders. Nat Prod Rep. 2012;29:134–43. doi: 10.1039/c1np00054c. [DOI] [PubMed] [Google Scholar]
- 15.Ames BN, Durston WE, Yamasaki E, Lee FD. Carcinogens are mutagens: a simple test system combining liver homogenates for activation and bacteria for detection. Proc Natl Acad Sci U S A. 1973;70:2281–5. doi: 10.1073/pnas.70.8.2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Symington LS, Gautier J. Double-strand break end resection and repair pathway choice. Annu Rev Genet. 2011;45:247–71. doi: 10.1146/annurev-genet-110410-132435. [DOI] [PubMed] [Google Scholar]
- 17.Ceccaldi R, Rondinelli B, D’Andrea AD. Repair pathway choices and consequences at the double-strand break. Trends Cell Biol. 2016;26(1):52–64. doi: 10.1016/j.tcb.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sinha S, Villarreal D, Shim EY, Lee SE. Risky business: Microhomology-mediated end joining. Mutat Res. 2016;788:17–24. doi: 10.1016/j.mrfmmm.2015.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schärer OD. Nucleotide excision repair in eukaryotes. Cold Spring Harb Perspect Biol. 2013;5:a012609. doi: 10.1101/cshperspect.a012609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wallace SS. Base excision repair: a critical player in many games. DNA Repair (Amst) 2014;19:14–26. doi: 10.1016/j.dnarep.2014.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bauer NC, Corbett AH, Doetsch PW. The current state of eukaryotic DNA base damage and repair. Nucleic Acids Res. 2015;43:10083–101. doi: 10.1093/nar/gkv1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Spivak G. Transcription-coupled repair: an update. Arch Toxicol. 2016;90:2583–94. doi: 10.1007/s00204-016-1820-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kunkel TA, Erie DA. Eukaryotic mismatch repair in relation to DNA replication. Annu Rev Genet. 2015;49:291–313. doi: 10.1146/annurev-genet-112414-054722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scully R, Puget N, Vlasakova K. DNA polymerase stalling, sister chromatid recombination and the BRCA genes. Oncogene. 2000;19:6176–83. doi: 10.1038/sj.onc.1203971. [DOI] [PubMed] [Google Scholar]
- 25.Li Z, Xiao W, McCormick JJ, Maher VM. Identification of a protein essential for a major pathway used by human cells to avoid UV-induced DNA damage. Proc Natl Acad Sci. 2002;99:4459–64. doi: 10.1073/pnas.062047799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rastogi RP, Richa, Kumar A, Tyagi MB, Sinha RP. Molecular mechanisms of ultraviolet radiation-induced DNA damage and repair. J Nucleic Acids. 2010;2010:592980. doi: 10.4061/2010/592980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shiloh Y. ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer. 2003;3:155–68. doi: 10.1038/nrc1011. [DOI] [PubMed] [Google Scholar]
- 28.Lujambio A. To clear, or not to clear (senescent cells)? That is the question. Bioessays. 2016;38(Suppl 1):S56–64. doi: 10.1002/bies.201670910. [DOI] [PubMed] [Google Scholar]
- 29.Roos WP, Thomas AD, Kaina B. DNA damage and the balance between survival and death in cancer biology. Nat Rev Cancer. 2016;16:20–33. doi: 10.1038/nrc.2015.2. [DOI] [PubMed] [Google Scholar]
- 30.Harper JW, Elledge SJ. The DNA damage response: ten years after. Mol Cell. 2007;28:739–45. doi: 10.1016/j.molcel.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 31.Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40(2):179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yan S, Sorrell M, Berman Z. Functional interplay between ATM/ATR-mediated DNA damage response and DNA repair pathways in oxidative stress. Cell Mol Life Sci. 2014;71:3951–67. doi: 10.1007/s00018-014-1666-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hanawalt PC. Historical perspective on the DNA damage response. DNA Repair (Amst) 2015;36:2–7. doi: 10.1016/j.dnarep.2015.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rashi-Elkeles S, Elkon R, Shavit S, Lerenthal Y, Linhart C, Kupershtein A, et al. Transcriptional modulation induced by ionizing radiation: p53 remains a central player. Mol Oncol. 2011;5:336–48. doi: 10.1016/j.molonc.2011.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mladenov E, Magin S, Soni A, Iliakis G. DNA double-strand-break repair in higher eukaryotes and its role in genomic instability and cancer: Cell cycle and proliferation-dependent regulation. Semin Cancer Biol. 2016;37–38:51–64. doi: 10.1016/j.semcancer.2016.03.003. [DOI] [PubMed] [Google Scholar]
- 36.Maréchal A, Zou L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb Perspect Biol. 2013;5:1–17. doi: 10.1101/cshperspect.a012716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matsuoka S, Huang M, Elledge SJ. Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science. 1998;282:1893–7. doi: 10.1126/science.282.5395.1893. [DOI] [PubMed] [Google Scholar]
- 38.Melchionna R, Chen XB, Blasina A, McGowan CH. Threonine 68 is required for radiation-induced phosphorylation and activation of Cds1. Nat Cell Biol. 2000;2:762–5. doi: 10.1038/35036406. [DOI] [PubMed] [Google Scholar]
- 39.Walworth NC, Bernards R. rad-dependent response of the chk1-encoded protein kinase at the DNA damage checkpoint. Science. 1996;271:353–6. doi: 10.1126/science.271.5247.353. [DOI] [PubMed] [Google Scholar]
- 40.Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, et al. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 2000;14:1448–59. [PMC free article] [PubMed] [Google Scholar]
- 41.Lopez-Girona A, Tanaka K, Chen XB, Baber BA, McGowan CH, Russell P. Serine-345 is required for Rad3-dependent phosphorylation and function of checkpoint kinase Chk1 in fission yeast. Proc Natl Acad Sci U S A. 2001;98:11289–94. doi: 10.1073/pnas.191557598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smith J, Tho LM, Xu N, Gillespie DA. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv Cancer Res. 2010;108:73–112. doi: 10.1016/B978-0-12-380888-2.00003-0. [DOI] [PubMed] [Google Scholar]
- 43.Jazayeri A, Falck J, Lukas C, Bartek J, Smith GC, Lukas J, et al. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat Cell Biol. 2006;8:37–45. doi: 10.1038/ncb1337. [DOI] [PubMed] [Google Scholar]
- 44.Giono LE, Nieto Moreno N, Cambindo Botto AE, Dujardin G, Muñoz MJ, Kornblihtt AR. The RNA Response to DNA Damage. J Mol Biol. 2016;428:2636–51. doi: 10.1016/j.jmb.2016.03.004. [DOI] [PubMed] [Google Scholar]
- 45.Dantuma NP, van Attikum H. Spatiotemporal regulation of posttranslational modifications in the DNA damage response. EMBO J. 2016;35:6–23. doi: 10.15252/embj.201592595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huen MS, Chen J. The DNA damage response pathways: at the crossroad of protein modifications. Cell Res. 2008;18:8–16. doi: 10.1038/cr.2007.109. [DOI] [PubMed] [Google Scholar]
- 47.Joerger AC, Fersht AR. The p53 Pathway: Origins, Inactivation in Cancer, and Emerging Therapeutic Approaches. Annu Rev Biochem. 2016;85:375–404. doi: 10.1146/annurev-biochem-060815-014710. [DOI] [PubMed] [Google Scholar]
- 48.Derks KW, Hoeijmakers JH, Pothof J. The DNA damage response: the omics era and its impact. DNA Repair (Amst) 2014;19:214–20. doi: 10.1016/j.dnarep.2014.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Beckerman R, Prives C. Transcriptional regulation by p53. Cold Spring Harb Perspect Biol. 2010;2:a000935. doi: 10.1101/cshperspect.a000935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Crighton D, Wilkinson S, O’Prey J, Syed N, Smith P, Harrison PR, et al. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell. 2006;126:121–34. doi: 10.1016/j.cell.2006.05.034. [DOI] [PubMed] [Google Scholar]
- 51.Jiang P, Du W, Heese K, Wu M. The Bad guy cooperates with good cop p53: Bad is transcriptionally up-regulated by p53 and forms a Bad/p53 complex at the mitochondria to induce apoptosis. Mol Cell Biol. 2006;26:9071–82. doi: 10.1128/MCB.01025-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Miyashita T, Reed JC. The tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell. 1995;80:293–9. doi: 10.1016/0092-8674(95)90412-3. [DOI] [PubMed] [Google Scholar]
- 53.Sax JK, Fei P, Murphy ME, Bernhard E, Korsmeyer SJ, el-Deiry WS. BID regulation by p53 contributes to chemosensitivity. Nat Cell Biol. 2002;4:842–9. doi: 10.1038/ncb866. [DOI] [PubMed] [Google Scholar]
- 54.Jin YJ, Wang J, Qiao C, Hei TK, Brandt-Rauf PW, Yin Y. A novel mechanism for p53 to regulate its target gene ECK in signaling apoptosis. Mol Cancer Res. 2006;4:769–78. doi: 10.1158/1541-7786.MCR-06-0178. [DOI] [PubMed] [Google Scholar]
- 55.Nakano K, Vousden KH. PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell. 2001;7:683–694. doi: 10.1016/s1097-2765(01)00214-3. [DOI] [PubMed] [Google Scholar]
- 56.Jeffers JR, Parganas E, Lee Y, Yang C, Wang J, Brennan J, et al. Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell. 2003;4:321–8. doi: 10.1016/s1535-6108(03)00244-7. [DOI] [PubMed] [Google Scholar]
- 57.Yu J, Zhang L. No PUMA, no death: implications for p53-dependent apoptosis. Cancer Cell. 2003;4:248–249. doi: 10.1016/s1535-6108(03)00249-6. [DOI] [PubMed] [Google Scholar]
- 58.Dulic V, Kaufmann WK, Wilson SJ, Tlsty TD, Lees E, Harper JW, et al. p53-dependent inhibition of cyclin-dependent kinase activities in human fibroblasts during radiation-induced G1 arrest. Cell. 1994;76:1013–23. doi: 10.1016/0092-8674(94)90379-4. [DOI] [PubMed] [Google Scholar]
- 59.Hermeking H, Lengauer C, Polyak K, He TC, Zhang L, Thiagalingam S, et al. 14-3-3sigma is a p53-regulated inhibitor of G2/M progression. Mol Cell. 1997;1:3–11. doi: 10.1016/s1097-2765(00)80002-7. [DOI] [PubMed] [Google Scholar]
- 60.Tan T, Chu G. p53 Binds and activates the xeroderma pigmentosum DDB2 gene in humans but not mice. Mol Cell Biol. 2002;22:3247–54. doi: 10.1128/MCB.22.10.3247-3254.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Amundson SA, Patterson A, Do KT, Fornace AJ., Jr A nucleotide excision repair master-switch: p53 regulated coordinate induction of global genomic repair genes. Cancer Biol Ther. 2002;1:145–9. doi: 10.4161/cbt.59. [DOI] [PubMed] [Google Scholar]
- 62.Adimoolam S, Ford JM. p53 and DNA damage-inducible expression of the xeroderma pigmentosum group C gene. Proc Natl Acad Sci U S A. 2002;99:12985–90. doi: 10.1073/pnas.202485699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kortlever RM, Higgins PJ, Bernards R. Plasminogen activator inhibitor-1 is a critical downstream target of p53 in the induction of replicative senescence. Nat Cell Biol. 2006;8:877–84. doi: 10.1038/ncb1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Murray-Zmijewski F, Slee EA, Lu X. A complex barcode underlies the heterogeneous response of p53 to stress. Nat Rev. 2008;9:702–12. doi: 10.1038/nrm2451. [DOI] [PubMed] [Google Scholar]
- 65.Vousden KH, Prives C. Blinded by the light: the growing complexity of p53. Cell. 2009;137:413–31. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- 66.el-Deiry WS, Kern SE, Pietenpol JA, Kinzler KW, Vogelstein B. Definition of a consensus binding site for p53. Nat Genet. 1992;1:45–9. doi: 10.1038/ng0492-45. [DOI] [PubMed] [Google Scholar]
- 67.Wang B, Xiao Z, Ren EC. Redefining the p53 response element. Proc Natl Acad Sci U S A. 2009;106:14373–8. doi: 10.1073/pnas.0903284106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tebaldi T, Zaccara S, Alessandrini F, Bisio A, Ciribilli Y, Inga A. Whole-genome cartography of p53 response elements ranked on transactivation potential. BMC Genomics. 2015;16:464. doi: 10.1186/s12864-015-1643-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Balagurumoorthy P, Sakamoto H, Lewis MS, Zambrano N, Clore GM, Gronenborn AM, et al. Four p53 DNA-binding domain peptides bind natural p53-response elements and bend the DNA. Proc Natl Acad Sci U S A. 1995;92:8591–5. doi: 10.1073/pnas.92.19.8591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McLure KG, Lee PW. How p53 binds DNA as a tetramer. EMBO J. 1998;17:3342–50. doi: 10.1093/emboj/17.12.3342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schlereth K, Heyl C, Krampitz AM, Mernberger M, Finkernagel F, Scharfe M, et al. Characterization of the p53 cistrome–DNA binding cooperativity dissects p53’s tumor suppressor functions. PLoS Genet. 2013;9:e1003726. doi: 10.1371/journal.pgen.1003726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kin K, Chen X, Gonzalez-Garay M, Fakhouri WD. The effect of non-coding DNA variations on P53 and cMYC competitive inhibition at cis-overlapping motifs. Hum Mol Genet. 2016;25:1517–27. doi: 10.1093/hmg/ddw030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Levav-Cohen Y, Goldberg Z, Zuckerman V, Grossman T, Haupt S, Haupt Y. C-Abl as a modulator of p53. Biochem Biophys Res Commun. 2005;331:737–49. doi: 10.1016/j.bbrc.2005.03.152. [DOI] [PubMed] [Google Scholar]
- 74.Hu W, Feng Z, Levine AJ. The regulation of multiple p53 stress responses is mediated through MDM2. Genes Cancer. 2012;3:199–208. doi: 10.1177/1947601912454734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Inoue K, Fry EA, Frazier DP. Transcription factors that interact with p53 and Mdm2. Int J Cancer. 2016;138:1577–85. doi: 10.1002/ijc.29663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Barak Y, Juven T, Haffner R, Oren M. mdm2 expression is induced by wild type p53 activity. EMBO J. 1993;12:461–8. doi: 10.1002/j.1460-2075.1993.tb05678.x. (1993) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wu X, Bayle JH, Olson D, Levine AJ. The p53-mdm-2 autoregulatory feedback loop. Genes Dev. 1993;7:1126–32. doi: 10.1101/gad.7.7a.1126. [DOI] [PubMed] [Google Scholar]
- 78.Zhang Q, Zeng SX, Lu H. Targeting p53-MDM2-MDMX loop for cancer therapy. Subcell Biochem. 2014;85:281–319. doi: 10.1007/978-94-017-9211-0_16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fuchs SY, Adler V, Pincus MR, Ronai Z. MEKK1/JNK signaling stabilizes and activates p53. Proc Natl Acad Sci U S A. 1998;95:10541–6. doi: 10.1073/pnas.95.18.10541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hirao A, Kong YY, Matsuoka S, Wakeham A, Ruland J, Yoshida H, et al. DNA damage-induced activation of p53 by the checkpoint kinase Chk2. Science. 2000;287:1824–7. doi: 10.1126/science.287.5459.1824. [DOI] [PubMed] [Google Scholar]
- 81.Shieh SY, Ahn J, Tamai K, Taya Y, Prives C. The human homologs of checkpoint kinases Chk1 and Cds1 (Chk2) phosphorylate p53 at multiple DNA damage-inducible sites. Genes Dev. 2000;14:289–300. [PMC free article] [PubMed] [Google Scholar]
- 82.Shieh SY, Ikeda M, Taya Y, Prives C. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell. 1997;91:325–34. doi: 10.1016/s0092-8674(00)80416-x. [DOI] [PubMed] [Google Scholar]
- 83.Linke K, Mace PD, Smith CA, Vaux DL, Silke J, Day CL. Structure of the MDM2/MDMX RING domain heterodimer reveals dimerization is required for their ubiquitylation in trans. Cell Death Differ. 2008;15:841–8. doi: 10.1038/sj.cdd.4402309. [DOI] [PubMed] [Google Scholar]
- 84.Popowicz GM, Czarna A, Holak TA. Structure of the human Mdmx protein bound to the p53 tumor suppressor transactivation domain. Cell Cycle. 2008;7:2441–3. doi: 10.4161/cc.6365. [DOI] [PubMed] [Google Scholar]
- 85.Gilkes DM, Pan Y, Coppola D, Yeatman T, Reuther GW, Chen J. Regulation of MDMX expression by mitogenic signaling. Mol Cell Biol. 2008;28:1999–2010. doi: 10.1128/MCB.01633-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ries S, Biederer C, Woods D, Shifman O, Shirasawa S, Sasazuki T, et al. Opposing effects of Ras on p53: transcriptional activation of mdm2 and induction of p19ARF. Cell. 2000;103:321–30. doi: 10.1016/s0092-8674(00)00123-9. [DOI] [PubMed] [Google Scholar]
- 87.Tarocchi M, Hannivoort R, Hoshida Y, Lee UE, Vetter D, Narla G, et al. Carcinogen-induced hepatic tumors in KLF6+/− mice recapitulate aggressive human hepatocellular carcinoma associated with p53 pathway deregulation. Hepatology. 2011;54:522–31. doi: 10.1002/hep.24413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhang X, Zhang Z, Cheng J, Li M, Wang W, Xu W, et al. Transcription factor NFAT1 activates the mdm2 oncogene independent of p53. J Biol Chem. 2012;287:30468–76. doi: 10.1074/jbc.M112.373738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lahav G, Rosenfeld N, Sigal A, Geva-Zatorsky N, Levine AJ, Elowitz MB, et al. Dynamics of the p53–Mdm2 feedback loop in individual cells. Nat Genet. 2004;36:147–50. doi: 10.1038/ng1293. [DOI] [PubMed] [Google Scholar]
- 90.Batchelor E, Mock CS, Bhan I, Loewer A, Lahav G. Recurrent initiation: a mechanism for triggering p53 pulses in response to DNA damage. Mol Cell. 2008;30:277–89. doi: 10.1016/j.molcel.2008.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Batchelor E, Loewer A, Mock C, Lahav G. Stimulus dependent dynamics of p53 in single cells. Mol Syst Biol. 2011;7:488. doi: 10.1038/msb.2011.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Loewer A, Batchelor E, Gaglia G, Lahav G. Basal dynamics of p53 reveal transcriptionally attenuated pulses in cycling cells. Cell. 2010;142:89–100. doi: 10.1016/j.cell.2010.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hopkins KM, Auerbach W, Wang XY, Hande MP, Hang H, Wolgemuth DJ, et al. Deletion of mouse rad9 causes abnormal cellular responses to DNA damage, genomic instability, and embryonic lethality. Mol Cell Biol. 2004;24:7235–48. doi: 10.1128/MCB.24.16.7235-7248.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lieberman HB. Rad9, an evolutionarily conserved gene with multiple functions for preserving genomic integrity. J Cell Biochem. 2006;97:690–7. doi: 10.1002/jcb.20759. [DOI] [PubMed] [Google Scholar]
- 95.Hang H, Lieberman HB. Physical interactions among human checkpoint control proteins HUS1p, RAD1p, and RAD9p, and implications for the regulation of cell cycle progression. Genomics. 2000;65:24–33. doi: 10.1006/geno.2000.6142. [DOI] [PubMed] [Google Scholar]
- 96.Broustas CG, Lieberman HB. RAD9 enhances radioresistance of human prostate cancer cells through regulation of ITGB1 protein levels. Prostate. 2014;74:1359–70. doi: 10.1002/pros.22842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yin Y, Zhu A, Jin YJ, Liu YX, Zhang X, Hopkins KM, et al. Human RAD9 checkpoint control/proapoptotic protein can activate transcription of p21. Proc Natl Acad Sci U S A. 2002;101:8864–9. doi: 10.1073/pnas.0403130101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zhu A, Zhou H, Leloup C, Marino SA, Geard CR, Hei TK, et al. Differential impact of mouse Rad9 deletion on ionizing radiation-induced bystander effects. Radiat Res. 2005;164:655–61. doi: 10.1667/rr3458.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lieberman HB, Hopkins KM, Nass M, Demetrick D, Davey S. A human homolog of the Schizosaccharomyces pombe rad9+ checkpoint control gene. Proc Natl Acad Sci U S A. 1996;93:13890–5. doi: 10.1073/pnas.93.24.13890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zou L, Liu D, Elledge SJ. Replication protein A-mediated recruitment and activation of Rad17 complexes. Proc Natl Acad Sci U S A. 2003;100:13827–32. doi: 10.1073/pnas.2336100100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Cotta-Ramusino C, McDonald ER, 3rd, Hurov K, Sowa ME, Harper JW, Elledge SJ. A DNA damage response screen identifies RHINO, a 9-1-1 and TopBP1 interacting protein required for ATR signaling. Science. 2011;332:1313–7. doi: 10.1126/science.1203430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lindsey-Boltz LA, Kemp MG, Capp C, Sancar A. RHINO forms a stoichiometric complex with the 9-1-1 checkpoint clamp and mediates ATR-Chk1 signaling. Cell Cycle. 2015;14:99–108. doi: 10.4161/15384101.2014.967076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sierant ML, Archer NE, Davey SK. The Rad9A checkpoint protein is required for nuclear localization of the claspin adaptor protein. Cell Cycle. 2010;9:548–56. doi: 10.4161/cc.9.3.10553. [DOI] [PubMed] [Google Scholar]
- 104.Liu S, Song N, Zou L. The conserved C terminus of Claspin interacts with Rad9 and promotes rapid activation of Chk1. Cell Cycle. 2012;11:2711–6. doi: 10.4161/cc.21041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cimprich KA, Cortez D. ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol. 2008;9:616–27. doi: 10.1038/nrm2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Pandita RK, Sharma GG, Laszlo A, Hopkins KM, Davey S, Chakhparonian M, et al. Mammalian Rad9 plays a role in telomere stability, S- and G2-phase-specific cell survival, and homologous recombinational repair. Mol Cell Biol. 2006;26:1850–64. doi: 10.1128/MCB.26.5.1850-1864.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Balakrishnan L, Brandt PD, Lindsey-Boltz LA, Sancar A, Bambara RA. Long patch base excision repair proceeds via coordinated stimulation of the multienzyme DNA repair complex. J Biol Chem. 2009;284:15158–72. doi: 10.1074/jbc.M109.000505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gembka A, Toueille M, Smirnova E, Poltz R, Ferrari E, Villani G, et al. The checkpoint clamp, Rad9-Rad1-Hus1 complex, preferentially stimulates the activity of apurinic/apyrimidinic endonuclease 1 and DNA polymerase beta in long patch base excision repair. Nucleic Acids Res. 2007;35:2596–608. doi: 10.1093/nar/gkl1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Helt CE, Wang W, Keng PC, Bambara RA. Evidence that DNA damage detection machinery participates in DNA repair. Cell Cycle. 2005;4:529–32. doi: 10.4161/cc.4.4.1598. [DOI] [PubMed] [Google Scholar]
- 110.Hwang BJ, Jin J, Gunther R, Madabushi A, Shi G, Wilson GM, Lu AL. Association of the Rad9-Rad1-Hus1 checkpoint clamp with MYH DNA glycosylase and DNA. DNA Repair (Amst) 2015;31:80–90. doi: 10.1016/j.dnarep.2015.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Panigrahi SK, Hopkins KM, Lieberman HB. Regulation of NEIL1 protein abundance by RAD9 is important for efficient base excision repair. Nucleic Acids Res. 2015;43:4531–46. doi: 10.1093/nar/gkv327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Li T, Wang Z, Zhao Y, He W, An L, Liu S, Liu Y, Wang H, Hang H. Checkpoint protein Rad9 plays an important role in nucleotide excision repair. DNA Repair (Amst) 2013;12:284–92. doi: 10.1016/j.dnarep.2013.01.006. [DOI] [PubMed] [Google Scholar]
- 113.He W, Zhao Y, Zhang C, An L, Hu Z, Liu Y, et al. Rad9 plays an important role in DNA mismatch repair through physical interaction with MLH1. Nucleic Acids Res. 2008;36:6406–17. doi: 10.1093/nar/gkn686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Crespan E, Czabany T, Maga G, Hübscher U. Microhomology-mediated DNA strand annealing and elongation by human DNA polymerases λ and β on normal and repetitive DNA sequences. Nucleic Acids Res. 2012;40:5577–90. doi: 10.1093/nar/gks186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Tsai FL, Kai M. The checkpoint clamp protein Rad9 facilitates DNA-end resection and prevents alternative non-homologous end joining. Cell Cycle. 2014;13:3460–4. doi: 10.4161/15384101.2014.958386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Shin MH, Yuan M, Zhang H, Margolick JB, Kai M. ATM-dependent phosphorylation of the checkpoint clamp regulates repair pathways and maintains genomic stability. Cell Cycle. 2012;11:1796–803. doi: 10.4161/cc.20161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Komatsu K, Hopkins KM, Lieberman HB, Wang H. Schizosaccharomyces pombe Rad9 contains a BH3-like region and interacts with the anti-apoptotic protein Bcl-2. FEBS Lett. 2000;481:122–6. doi: 10.1016/s0014-5793(00)01975-x. [DOI] [PubMed] [Google Scholar]
- 118.Komatsu K, Miyashita T, Hang H, Hopkins KM, Zheng W, Cuddeback S, Yamada M, Lieberman HB, Wang HG. Human homologue of S. pombe Rad9 interacts with BCL-2/BCL-xL and promotes apoptosis. Nat Cell Biol. 2000;2:1–6. doi: 10.1038/71316. [DOI] [PubMed] [Google Scholar]
- 119.Yoshida K, Komatsu K, Wang HG, Kufe D. c-Abl tyrosine kinase regulates the human Rad9 checkpoint protein in response to DNA damage. Mol Cell Biol. 2002;22:3292–300. doi: 10.1128/MCB.22.10.3292-3300.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Yoshida K, Wang HG, Miki Y, Kufe D. Protein kinase Cdelta is responsible for constitutive and DNA damage-induced phosphorylation of Rad9. EMBO J. 2003;22:1431–41. doi: 10.1093/emboj/cdg134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Brugarolas J, Chandrasekaran C, Gordon JI, Beach D, Jacks T, Hannon GJ. Radiation-induced cell cycle arrest compromised by p21 deficiency. Nature. 1995;377:552–7. doi: 10.1038/377552a0. [DOI] [PubMed] [Google Scholar]
- 122.Deng C, Zhang P, Harper JW, Elledge SJ, Leder P. Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell. 1995;82:675–84. doi: 10.1016/0092-8674(95)90039-x. [DOI] [PubMed] [Google Scholar]
- 123.el-Deiry WS, Tokino T, Waldman T, Oliner JD, Velculescu VE, Burrell M, et al. Topological control of p21WAF1/CIP1 expression in normal and neoplastic tissues. Cancer Res. 1995;55:2910–9. [PubMed] [Google Scholar]
- 124.Xiong Y, Hannon GJ, Zhang H, Casso D, Kobayashi R, Beach D. p21 is a universal inhibitor of cyclin kinases. Nature. 1993;366:701–4. doi: 10.1038/366701a0. [DOI] [PubMed] [Google Scholar]
- 125.Lieberman HB, Yin Y. A novel function for human Rad9 protein as a transcriptional activator of gene expression. Cell Cycle. 2004;3:1008–10. [PubMed] [Google Scholar]
- 126.Ishikawa K, Ishii H, Murakumo Y, Mimori K, Kobayashi M, Yamamoto K, Mori M, et al. Rad9 modulates the P21WAF1 pathway by direct association with p53. BMC Mol Biol. 2007;8:37. doi: 10.1186/1471-2199-8-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wen FC, Chang TW, Tseng YL, Lee JC, Chang MC. hRAD9 functions as a tumor suppressor by inducing p21-dependent senescence and suppressing epithelial-mesenchymal transition through inhibition of Slug transcription. Carcinogenesis. 2014;35:1481–90. doi: 10.1093/carcin/bgu009. [DOI] [PubMed] [Google Scholar]
- 128.el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, et al. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–25. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- 129.Zhu A, Zhang CX, Lieberman HB. Rad9 has a functional role in human prostate carcinogenesis. Cancer Res. 2008;68:1267–74. doi: 10.1158/0008-5472.CAN-07-2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Hu Z, Liu Y, Zhang C, Zhao Y, He W, Han L, et al. Targeted deletion of Rad9 in mouse skin keratinocytes enhances genotoxin-induced tumor development. Cancer Res. 2008;68:5552–61. doi: 10.1158/0008-5472.CAN-07-5670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Broustas CG, Lieberman HB. Contributions of Rad9 to tumorigenesis. J Cell Biochem. 2012;113:742–51. doi: 10.1002/jcb.23424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Broustas CG, Lieberman HB. DNA damage response genes and the development of cancer metastasis. Radiat Res. 2014;181:111–30. doi: 10.1667/RR13515.1. [DOI] [PMC free article] [PubMed] [Google Scholar]