

SUMMARY
Breast cancer cells relocate to bone and activate osteoclast-induced bone resorption. Soluble factors secreted by breast cancer cells trigger a cascade of events that stimulate osteoclast differentiation in the bone microenvironment. MacroH2A is a unique histone variant with a C-terminal non-histone domain and plays a crucial role in modulating chromatin organization and gene transcription. Here, we show that macroH2A1.2, one of the macroH2A isoforms, has an intrinsic ability to inhibit breast cancer- derived osteoclastogenesis. This repressive effect requires macroH2A1.2-dependent attenuation of expression and secretion of lysyl oxidase (LOX) in breast cancer cells. Furthermore, our mechanistic studies reveal that macroH2A1.2 physically and functionally interacts with the histone methyltrans- ferase EZH2 and elevates H3K27me3 levels to keep LOX gene in a repressed state. Collectively, this study unravels a role for macroH2A1.2 in regulating osteoclastogenic potential of breast cancer cells, suggesting possibilities for developing therapeutic tools to treat osteolytic bone destruction.
Graphical Abstract
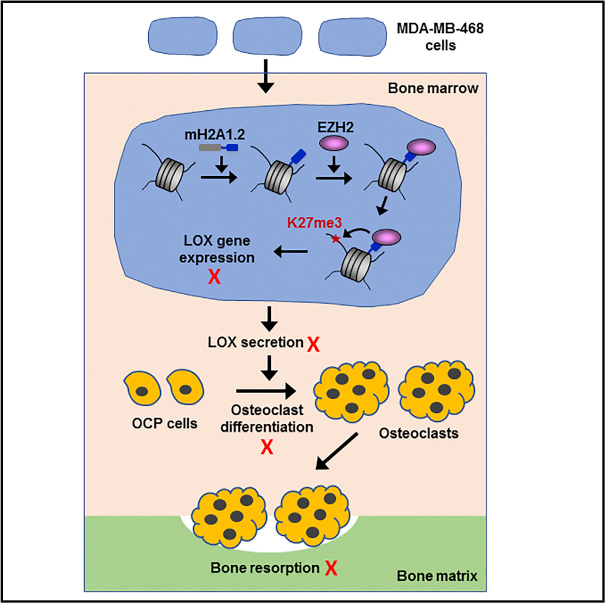
In Brief
Kim et al. demonstrate that mH2A1.2 attenuates breast cancer-induced osteoclastogenesis by maintaining the LOX gene in an inactive state. Mechanistically, mH2A1.2 recruits EZH2 to induce H3K27me3 and create a repressive barrier to LOX transcription.
INTRODUCTION
Histone variants are non-allelic isoforms of canonical histones and play important roles in mediating dynamic changes in chromatin structure and gene transcription (Henikoff and Smith, 2015; Maze et al., 2014). One such histone variant is macroH2A (mH2A). mH2A has a tripartite structure consisting of an N-termi- nal histone-fold, an unstructured linker domain, and a unique C-terminal macrodomain (Pehrson and Fried, 1992). In vertebrates, there are two mH2A isoforms, mH2A1 and mH2A2, which are encoded by separate genes. Two closely related subtypes of mH2A1, mH2A1.1 and mH2A1.2, are produced by alternative splicing in the macrodomain. mH2A is widely enriched in the inactive X chromosome and is nonrandomly distributed in specific chromosomal regions such as pseudoautosomal region and scaffold attachment region (Henikoff and Smith, 2015; Turner et al., 2001). This accumulation of mH2A has been proposed to contribute to long-term maintenance of gene silencing in these genomic regions. Beside its role in X chromosome inactivation, mH2A can also modulate specific gene transcription both negatively and positively (Dell’Orso et al., 2016; Gamble et al., 2010; Kapoor et al., 2010; Kim et al., 2013). These properties of mH2A may be generated through physical and functional interactions with gene specific regulators, as supported by previous studies showing that HDAC1/2, PARP1, and Pbx1 are recruited by mH2A1 and necessary for establishing distinct transcription states (Buschbeck et al., 2009; Chakravarthy et al., 2005; Chen et al., 2014; Dell’Orso et al., 2016; Kim et al., 2013).
Bone remodeling is a tightly regulated process responsible for bone resorption and formation through a series of steps that depend on the coordinated action of two cell lineages, osteoclasts and osteoblasts (Karsenty et al., 2009). Osteoclasts are large multinucleated hematopoietic cells responsible for bone resorption, whereas osteoblasts are bone-forming cells with a single nucleus (Raggatt and Partridge, 2010). Osteoclast precursor (OCP) cells are progressively differentiated into mature osteoclasts by fusion over a period of several days. The interaction of receptor activator of nuclear factor κB (NF-κB) ligand (RANKL), which is expressed as a membrane-bound protein in osteoblasts, with RANK on OCP cell membrane induces the initial expression of master transcription factors such as NF-kB, c-Fos, and NFATc1 (Karsenty et al., 2009; Raggatt and Partridge, 2010). These osteoclastogenic factors, then, trigger major signaling pathways to turn on multiple downstream genes encoding key determinants of osteoclast differentiation (Boyle et al., 2003; Teitelbaum and Ross, 2003). It is becoming evident that the deregulation of osteoclastogenic activity under certain pathological conditions leads to abnormal bone remodeling and contributes to the pathogenesis of bone disorders such as osteoporosis, rheumatoid arthritis, and bone metastases (Zaidi, 2007).
Breast cancer is the most common cancer in women and frequently metastasizes to bone and disrupts the normal bone remodeling process (Weilbaecher et al., 2011). While breast cancer bone metastases can be classified as osteolytic or osteoblastic, osteolytic bony changes are most frequently observed during the pathogenic processes (Weilbaecher et al., 2011). Breast cancer cells residing in bone express and secrete a plethora of osteolytic factors that stimulate osteoclast differentiation and maturation. (Kang et al., 2003; Weilbaecher et al., 2011). This unbalanced generation of osteoclasts by breast cancer-secreted factors leads to a massive bone resorption and causes osteoclast-mediated bone destruction. In turn, this destruction causes the release of bone matrix-stored growth factors, which act on cancer cells to produce osteoclastogenic factors and fuel a feed-forward vicious cycle in the bone. Thus, the identification of secreted factors capable of promoting osteoclast formation, activation, and survival is crucial in preventing and reducing the osteolytic bone metastases of breast cancer (Clézardin, 2011; Yin et al., 2005).
Because genes encoding secreted factors are stored in the nucleus by chromatinization, a fundamental mechanism underlying breast cancer-induced osteoclastogenesis might be regulated through chromatin-dependent pathways. In fact, previous studies including ours identified some of histone modifications as possible mechanisms underlying epigenetic regulation of osteoclastogenic transcription programs (Kim et al., 2016b; Zhang et al., 2015). Despite these advances, however, nothing is currently known about the effects of histone variants on the process of osteoclast differentiation. Here, we show that mH2A1.2 inhibits the expression and secretion of soluble factors influencing osteoclast differentiation and function in the process of breast cancer bone metas- tases. We then demonstrate that EZH2 histone methyltransfer- ase is required for mH2A1.2-dependent generation of inactive chromatin domain and suppression of osteoclastogenic genes in breast cancer cells.
RESULTS
mH2A1.2 Suppresses Osteoclastogenic Potential of Breast Cancer Cell-Secreted Factors
As an initial step in our investigation, we set up an assay system to determine the effects of breast cancer cell conditioned media (CMs) on osteoclast differentiation (Figure 1A). The suitability of this assay system was confirmed by the finding that osteoclast precursor (OCP) cells are synchronously differentiated into TRAP-positive multinuclear osteoclasts in α-minimal essential medium (a-MEM) following treatment with M-CSF, RANKL and CMs (Figure 1B). Under these conditions, higher numbers of mature osteoclast were generated, when OCP cells were cultured for 6 days in a 1:1 mixture of α-MEM and CMs prepared from moderately metastatic MDA-MB-468 breast cancer cells (Figure 1B). To analyze possible roles played by histone variants in regulating CM-induced osteoclastogenesis, analogous assays were carried out using CMs from MDA-MB-468 cells infected with lentiviruses encoding a short hairpin RNA (shRNA) against each of histone variants. The fact that shRNA-mediated downregulation of one histone variant did not affect the expression of other histone variants made it possible to functionally evaluate each of the histone variants (Figure S1A). When the effects of depletion of endogenous H2AX, H2A.Z, mH2A2, H2ABbd, H3.3, or CENP-A in MDA-MB-468 cells were examined in differentiation assays with MDA-MB-468 CMs, undetectable or only minor changes were observed in the formation of mature osteoclasts (Figure 1E). On the other hand, the treatment of OCP cells with CMs from mH2A1-depleted MDA-MB-468 cells significantly increased the number of differentiated osteoclasts (Figure 1E).
Figure 1. Inhibition of Osteoclastogenic Activity of Breast Cancer Cell CMs by mH2A1.2.
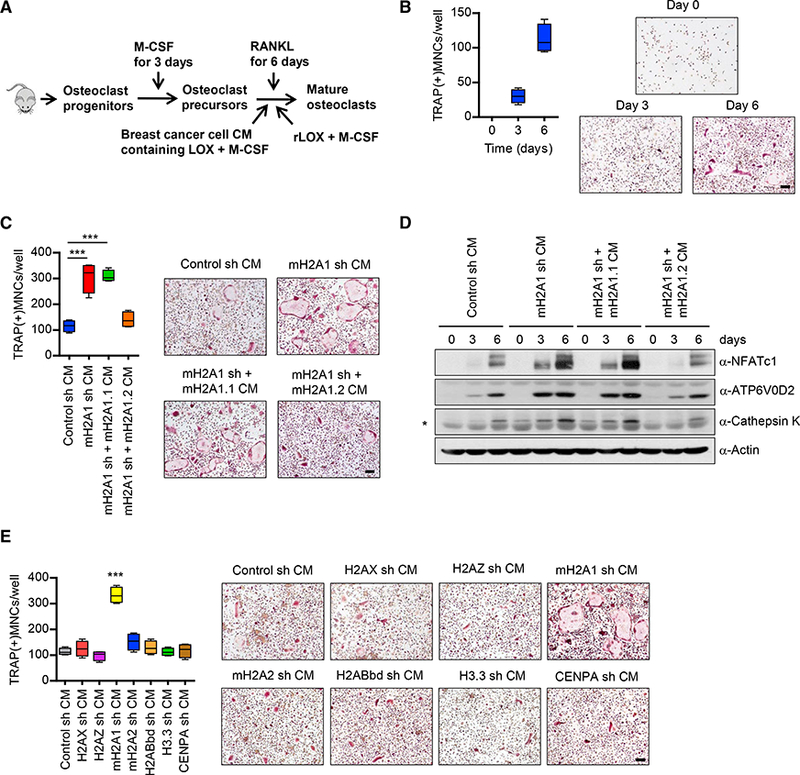
(A) Shown are the steps for analysis of osteoclastogenic properties of breast cancer cell-derived conditioned media (CMs) and recombinant LOX (rLOX).
(B) OCP cells were incubated with RANKL and MDA-MB-468 CMs for 0, 3, or6 days, fixed, stained for TRAP, and photographed under a light microscope (10×) (right). Representative images of osteoclasts are shown (scale bar, 100 μm). TRAP positive multinucleated cells (TRAP(+)MNCs) containing three or more nuclei and a full actin ring were counted as osteoclasts (left). Scale bar, 100 μm.
(C) CMs were prepared from mH2A1 -depleted MDA-MB-468 cells expressing exogenous shRNA-resistant mH2A1.1 or mH2A1.2 and analyzed for osteoclastogenic activity as in (B). Scale bar, 100 μm. Error bars are the means ± SD (n = 4) of three independent experiments; ***p < 0.001 (ANOVA analysis).
(D) Cell lysates were collected from OCP cells after treating with MDA-MB-468 CMs for the indicated time periods and analyzed for the expression of three osteoclast markers (NFATc1, ATP6V0D2, and cathepsin K) by western blot. β-Actin was used as a loading control. Non-specific band was marked by asterisk.
(E) OCP cells were cultured with CMs collected from MDA-MB-468 breast cancer cells depleted of the indicated histone variants. At the end of day 6, OCP- induced cells were stained for TRAP (right) to measure osteoclast differentiation (left). Error bars represent the means ± SD (n = 4); ***p < 0.001 versus Control sh CM (ANOVA analysis).
Because mH2A1.1 and mH2A1.2 isoforms were co-depleted by transfecting OCP cells with mH2A1 shRNA in our assays, we tried to individually knockdown them. However, the high degree of sequence homology between mH2A1.1 and mH2A1.2 mRNAs made it difficult. Hence, we took the alternative approach of transfecting RNAi-resistant, FLAG-tagged mH2A1.1, or mH2A1.2 in mH2A1-depleted MDA-MB-468 cells and determining if there is a change in osteoclastogenic effects of CMs (Figure S1B). As indicated by TRAP staining, the ectopic expression of mH2A1.2 in mH2A1-depleted cells significantly reduced the osteoclastogenic property of CMs (Figure 1C). However, ectopic mH2A1.1 was unable to substitute for the anti-os- teoclastogenic function of mH2A1.2 within the scope of this analysis (Figure 1C). Consistent with these data, treatment of OCP cells with CMs from mH2A1-depleted cells expressing FLAG-tagged mH2A1.2, but not mH2A1.1, blocked the induction of NFATc1 and its downstream target genes ATP6V0D2 and cathepsin K (Figure 1D). We also found that mH2A1.2 overexpression in MDA-MB-468 cells inhibits CM-induced osteoclast formation by ~70% (Figures S1C and S1D). Expectedly, however, ectopic expression of mH2A1.1 in MDA-MB-468 cells resulted in no detectable effects on CM-induced osteoclasto- genesis (Figures S1C and S1D). These observations are in consonance with our knockdown experiment data and underscore the notion that mH2A1.2 has repressive effects on CM os- teoclastogenic activity.
Beside CMs from moderately metastatic MDA-MB-468 cells, we also examined the osteoclastogenic effects of CMs from nonmetastatic MCF-7 and highly metastatic MDA-MB-231 breast cancer cells as well as MCF-10–2A normal breast cells. As confirmed by western blot, highly metastatic MDA-MB-231 cancer cells expressed less mH2A1 than moderately metastatic MDA-MB-468 and nonmetastatic MCF-7 cancer cells (Figure S2A). In contrast, mH2A1 expression level was much higher in MCF-10–2A normal breast cells compared with three other breast cancer cell lines (Figure S2A). When CMs from these cell lines were analyzed in osteoclast differentiation assays, MDA-MB-231 CMs were far more osteoclastogenic, generating 3 times higher numbers of TRAP+ mature osteoclasts compared with MDA-MB-468 CMs (Figure S2B). Conversely, addition of MCF-7 and MCF-10–2A CMs to OCP cell culture failed to generate multinucleated cells with phenotypic features of osteoclasts (Figure S2B).
mH2A1.2 Inactivates the Genes Encoding Secreted Osteoclastogenic Factors in Breast Cancer Cells
The data presented above strongly suggest a potential role of mH2A1.2 in the regulation of osteoclastogenic activity of CMs from breast cancer cells, particularly moderately metastatic cells. To assess whether this reflects transcriptional regulation of genes encoding secreted factors, we examined the transcrip- tome of secretory protein genes by comparing the mRNA profiles of mock-depleted and mH2A1-depleted MDA-MB-468 breast cancer cells. Recent technological advances provide unique opportunities to globally identify potential candidate genes whose products could regulate the progression of bone metastasis. Although such genome-wide studies produced several databases summarizing known or predicted secreted factors, those databases are incomplete or in inconsistent formats that use different types of protein and gene identifiers. To overcome these limitations, we collected public databases as well as published papers providing lists of secreted proteins, transmembrane proteins, and signal peptides, and constructed a breast cancer secretome through in silico approaches. As detailed in the Experimental Procedures, we used a bioinformatics approach and curated secretory proteins in GRCh37 genome in each database (Table S2). In total, 3,948 genes were selected for quantitative comparison of mRNA levels in mock-depleted control and mH2A1-depleted MDA-MB-468 cells.
In conventional microarray data analysis, genes exhibiting significantly different expression levels between two phenotypes are selected and analyzed for biological functions. However, it is becoming increasingly clear that smaller but coordinated expression changes in a set of genes involved in a particular biological process may dictate the biological pathway associated with a specific phenotype (Bild et al., 2006). Therefore, we decided to analyze microarray data at the level of pathways without including any pre-determined fold change filtering in our analysis. We found that pathways involved in metastasis as well as the crosstalk between bone cells and cancer cells are enriched in mH2A1-depleted MDA-MB-468 cells (Figure 2A; Table S3; false discovery rate [FDR] p < 0.05). In supervised two-way hierarchical clustering of gene expression data, two representative pathways, osteoclastogenesis and metastasis, showed a distinct signature profile of mH2A1-depleted cells over control cells, suggesting that mH2A1 plays a critical role in governing the expression of osteoclastogenic and metastatic genes in MDA-MB-468 cells (Figure 2B). When we further focus our analysis on osteoclastogenesis and metastasis pathways, lysyl oxidase (LOX) gene was ranked at the top in these categories (Figure 2C). The datasets resulting from microarray-based se- cretome analysis were further confirmed by qRT-PCR (Figure 2D). As we made use of mH2A1-depleted MDA-MB-468 cells to generate microarray data, mH2A1.1 or mH2A1.2 was again transfected into these cells to distinguish mH2A1.1-specific versus mH2A1.2-specific target genes. Expression of ectopic mH2A1.2 inhibited three out of five genes encoding secreted bone remodeling factors that were most highly expressed in mH2A1-depleted MDA-MB-468 cells (Figure 2D). Among the two remaining genes, one gene was inactivated by ectopic mH2A1.1, while the other gene was slightly affected by both ectopic mH2A1.1 and mH2A1.2 (Figure 2D).
Figure 2. Identification of mH2A1.2 Target Genes in Breast Cancer Cells.
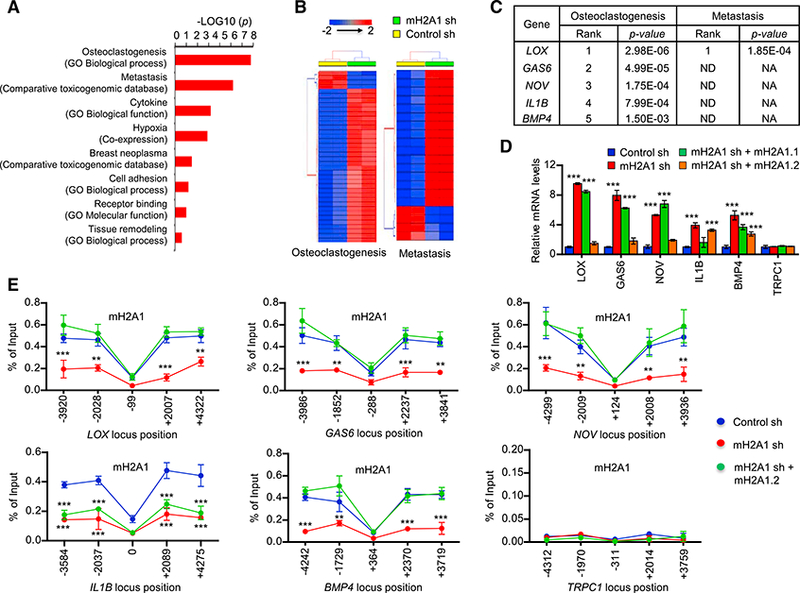
(A) Pathway analysis of breast cancer secretome identified the enrichment of pathways related to breast cancer associated bone metastasis and osteoclas- togenesis in 3,948 secretome genes. A ranked p value was computed for each pathway based on hypergeometric distribution along with Benjamini Hochberg correction (p < 0.05). Datasets used to extract the pathway terms are indicated in parentheses. The genes identified in each pathway are listed in Table S3.
(B) A two-way hierarchical clustering of two representative pathways—osteoclast differentiation and metastasis-showing distinct signature expression profile in mH2A1 sh knockdown and wild-type MDA-MB-486 cells. Euclidean distance and Average linkage were used for clustering.
(C) Prioritization of candidate genes in two important pathways differentially regulated in control and mH2A1-depleted MDA-MB-486 cells. Genes are ranked in the order of their functional statistical significance in a particular pathway. Top five ranked genes in osteoclast differentiation pathway and their corresponding rank in metastatic pathway are shown.
(D) qRT-PCR was performed to quantify relative mRNA levels of the top five genes using primers listed in Table S4. Error bars denote the SD from triplicate reactions by real-time PCR; ***p < 0.001 versus Control sh (ANOVA analysis).
(E) ChIP assays were performed at five different regions of the five mH2A1.2-repressed and one control genes using mH2A1 antibody and primers listed in Table S4. Error bars denote the means ± SD obtained from triplicate real-time PCR reactions; **p < 0.01, ***p < 0.001 versus Control sh (ANOVA analysis).
Whereas the above results suggested a role for mH2A1.2 in suppressing osteoclastogenic and metastasis-promoting genes, we could not discriminate between direct and indirect effects of mH2A1.2 knockdown on target genes in this analysis. We therefore used chromatin immunoprecipitation (ChIP) assays to investigate whether mH2A1.2 localizes along the potential target genes. Because ChIP-grade mH2A1.2-specific antibody is not available, a mH2A1 antibody detecting both mH2A1.1 and mH2A1.2 was used in these experiments. DNA obtained from immunoprecipitation with this antibody was subjected to real-time PCR using primers targeting five different regions of the target genes. Primer pairs amplifying regions ~2 and 4 kb upstream or downstream of the transcription start site (TSS) revealed a substantial enrichment of mH2A1 (Figure 2E). As a control, mH2A1 was not detected at TRPC1 gene and failed to show any changes in response to mH2A1 knockdown. ChIP analysis using H3 antibody showed little fluctuation in signals along the five different regions of the target genes, indicating similar levels of nucleosome occupancy at those regions (Figure S2C). When ChIP experiments were performed in mH2A1-depleted MDA- MB-468 cells, almost complete loss of mH2A1 ChIP signals was detected at the five target genes (Figure 2E). Similar analysis after expressing ectopic mH2A1.2 in mH2A1-depleted cells failed to show the recovery of ChIP signals at IL1B gene, results indicative of the incapability of mH2A1.2 to localize at this gene (Figure 2E). Expectedly, however, rescue expression of mH2A1.2 largely overrides the ChIP signal defects caused by mH2A1 knockdown at LOX, GAS6, NOV, and BMP4 genes (Figure 2E), strongly suggesting that mH2A1.2 is responsible for mH2A1 ChIP signals observed in these four target genes.
LOX Is a mH2A1.2 Target Gene and Is Necessary for mH2A1.2 Function
A noteworthy observation emerged from our secretome-based microarray data analysis was that mH2A1 depletion in MDA- MB-468 breast cancer cells most significantly activated the expression of LOX. Because LOX is known to facilitate the dissemination of cancer cells in the bone marrow, formation of mature osteoclasts, and generation of osteolytic lesions (Cox et al., 2015; Reynaud et al., 2017; Tsukasaki et al., 2017), we decided to examine whether LOX is responsible for the major osteoclastogenic activity of CMs and behaves as a primary downstream target for mH2A1.2. Accordingly, we transfected MDA-MB-468 cells with specific shRNAs against LOX and mH2A1 alone or together (Figure 3A). Upon stable knockdown of LOX, LOX activity in CMs was drastically reduced as shown by in vitro oxidase assays (Figure 3B). LOX gene expression was fully activated under mH2A1 knockdown condition (Figure 3A). When OCP cells were treated with CMs from MDA-MB-468 cells depleted of LOX, the effects of CMs on RANKL-induced osteoclast differentiation and osteoclast-specific gene expression were significantly attenuated (Figure 3C). Furthermore, ectopic expression of LOX wild-type, but not LOX oxidase-dead K320A mutant (Erler et al., 2009), in MDA- MB-468 cells substantially rescued the osteoclastogenic potential and LOX activity of CMs (Figures 3D and S3). In accordance with these findings, our western blot analysis showed that osteoclast differentiation marker genes (NFATc1, ATP6V0D2, and cathepsin K) were more highly expressed in control CM-treated OCP cells compared to LOX-depleted OCP cells (Figure 3E). These results argue against the possibility that the observed effects of LOX shRNA are generated by off-target effects and indicate that osteoclastogenic function of LOX is indeed accurately established in our assays. Importantly, knockdown of mH2A1 in LOX-deficient MDA-MB-468 cells was unable to augment osteoclastogenic potential of CMs (Figure 3C), reinforcing the conclusion that anti-osteoclastogenic function of mH2A1.2 is dependent on its inhibitory activity against LOX expression. Further support for our conclusion came from the observed enhancement of osteoclastogenesis after treatment with CMs from LOX-transfected MCF-7 cells that have low levels of endogenous LOX (Figures S2 and S3).
Figure 3. Dependence of mH2A1.2 Anti-osteoclastogenic Function on LOX.
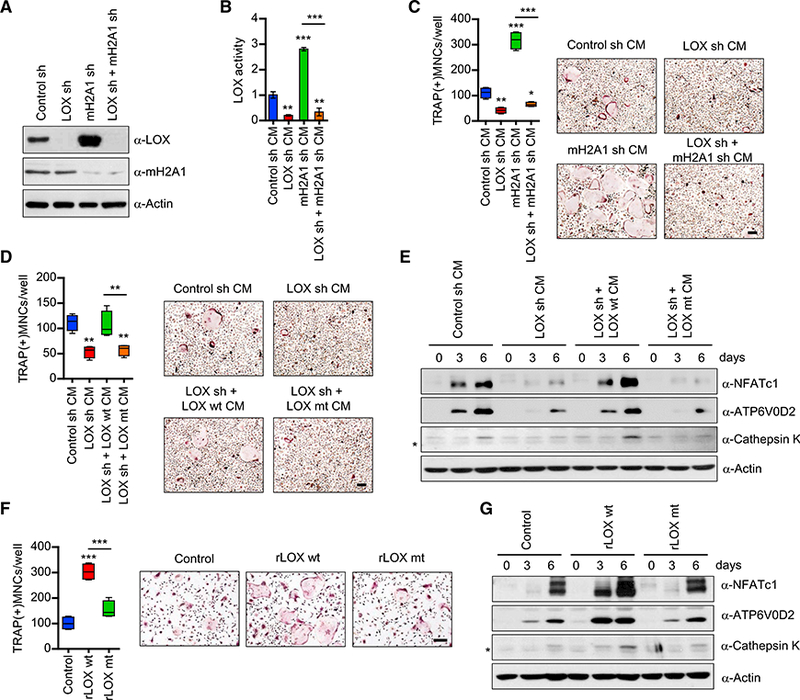
(A) Western blot analysis of lysates from MDA-MB-468 breast cancer cells expressing a control, LOX, or mH2A1 shRNA using the indicated antibodies.
(B) CMs were collected from MDA-MB-468 breast cancer cells depleted of LOX and/or mH2A1 and were used to measure LOX enzymatic activity as described in the Experimental Procedures.
(C) OCP cells were treated with CMs from MDA-MB-468 cells depleted of LOX and mH2A1 individually or together for 6 days. TRAP-positive multinucleated osteoclasts were stained (right) and counted (left). Scale bar, 100 μm.
(D) Osteoclast differentiation assays were carried out as in (C), but using CMs from LOX-depleted MDA-MB-468 cells expressing shRNA-resistant LOX wild-type (LOX wt) or oxidase-dead mutant (LOX mt). Scale bar, 100 μm.
(E) After treating OCP cells with MDA-MB-468 CMs as in (D), altered expression of NFATc1, ATP6V0D2, and cathepsin K was analyzed by western blot. β-Actin was used as a loading control. Non-specific band was marked by asterisk.
(F) OCP cells were treated with recombinant LOX (rLOX) wt or rLOX mt for 0, 3, and 6 days. Changes in osteoclast differentiation were assessed by TRAP staining (right) and counting (left). Scale bar, 100 μm.
(G) Western blot analyses of expression levels of NFATc1, ATP6V0D2, and cathepsin K after treating OCP cells with rLOX wt or rLOX mt. Error bars in (B)-(D) and (F) represent the means ± SD (n = 4) of three independent experiments; *p < 0.05, **p < 0.01, ***p < 0.001 (ANOVA analysis).
Given that breast cancer-mediated osteoclastogenesis involves several distinct steps, we also investigated the impact of altering LOX levels on OCP cell migration and MDA-MB-468 cell invasion. It was apparent in these experiments that knockdown and ectopic expression of LOX reduced and restored, respectively, the capacity of CMs to stimulate OCP cell migration (Figures S3E and S3F, left). Similar results were obtained from invasion assays using MDA-MB-468 cells (Figures S3E and S3F, right). Because knocking down mH2A1 in LOX-depleted MDA- MB468 cells failed to generate stimulatory effects in our assays (Figure S3E), these findings indicate that mH2A1.2 constitutes a repressive barrier to OCP and MDA-MB-468 cells in a LOX- dependent fashion.
For the purpose of further validating the stimulatory role of LOX in the osteoclastogenesis process, OCP cells were treated with recombinant LOX (rLOX) in the presence of M-CSF and RANKL. When incubated with rLOX wild-type (rLOX wt) for 6 days, OCP cells were differentiated into osteoclasts and expressed osteoclast marker genes (NFATc1, ATP6V0D2, and cathepsin K) at much higher levels compared with mock-treated control cells (Figures 3F and 3G). In contrast, however, no changes in osteoclast differentiation and marker gene expression were detected in OCP cells treated with rLOX oxidase-dead mutant (rLOX mt) (Figures 3F and 3G). In agreement with the knockdown data (Figures S3E and S3F), treatment with rLOX also potentiates transwell movements of OCP cells, again linking LOX to OCP cell migration and MDA-MB-468 invasion (Figure S3G).
LOX Is a Positive Regulator of Osteoclast Differentiation and Bone Resorption
As an extension of the above-described study, we plated osteoclasts on dentin slices and evaluated the effects of rLoX treatment on their capacity to resorb bone. Consistent with previous observations (Cox et al., 2015; Tsukasaki et al., 2017), rLOX-enhanced osteoclast formation increased the rate of bone resorption, stimulating the formation of resorption pits (Figure 4A). To further characterize the involvement of LOX in osteo- clastogenesis and investigate the effect of rLOX treatment in vivo, we injected rLOX into the periosteal regions of female mouse calvaria once daily for 10 days. Our microscopic analysis of bone sections from rLOX-treated mice showed that treating mice with rLOX wt generated an apparent increase in the size of bone cavities (Figure 4B). Moreover, the fact that mice treated with rLOX mt display minimal changes in bone cavities strongly argues that LOX enzymatic function is required for enhancing bone destruction (Figure 4B). The observed changes in bone cavity were due to enhanced osteoclast differentiation, because higher numbers of TRAP-positive osteoclasts were detected in rLOX wt-injected mice compared to control and rLOX mt-treated mice in our histomorphometric analysis (Figure 4C). Consistent with these observations and to further confirm the repressive function of mH2A1.2 in LOX expression, the mice treated with mH2A1-depleted MDA-MB-468 CMs showed higher levels of mature osteoclast formation and bone cavity as compared to the control mice group treated with mock-depleted MDA-MB- 468 CMs (Figure S4).
Figure 4. LOX-Mediated Stimulation of Osteoclastogenesis and Bone Resorption.
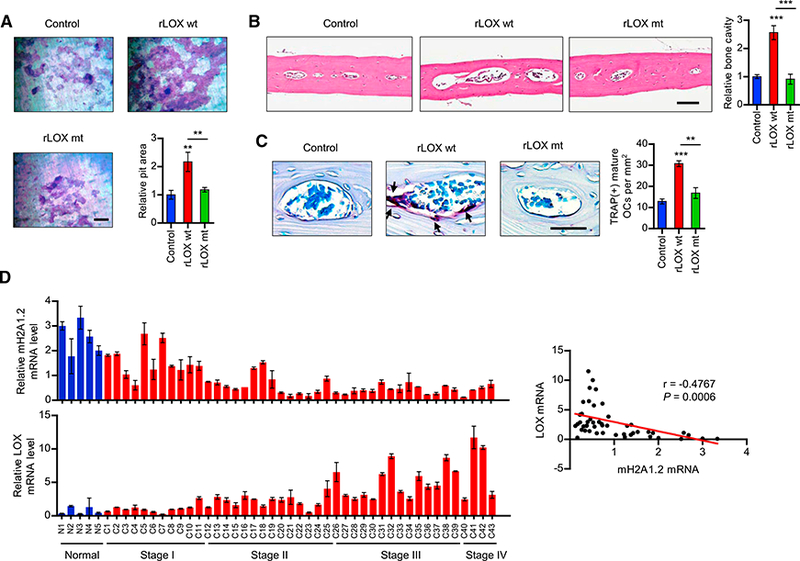
(A) Osteoclasts were placed on dentin slices and cultured with MDA-MB-468 CMs in the presence of vehicle (control), rLOX wt, or rLOX mt for 48 hr. The dentin slices were stained for toluidine blue and analyzed for resorption pit area (lower right). Scale bar, 100 μm.
(B and C) Calvarial bones from C57BL/6 mice treated with vehicle (control), rLOX wt or rLOX mt were fixed, embedded, and subjected to sectioning, TRAP staining, and histological analysis. H&E staining analyses of bone lesions in C57BL/6 mice treated with vehicle (control), rLOX wt or rLOX mt are shown in (B). Representative images of TRAP-staining (scale bar, 100 mm) and quantification of TRAP-positive cells are shown in (C). Arrows point to TRAP-positive mature osteoclasts.
(D) Breast cancer TissueScan cDNA arrays (OriGene) were analyzed for mH2A1.2 (upper) and LOX (lower) expression by qRT-PCR. The scatterplot diagram on the right shows the Pearson correlation analysis of the relationship between mH2A1.2 and LOX expression. Error bars in (A)-(D) represent the means ± SD (A and D; n = 3, B and C; n = 5); **p < 0.01, ***p < 0.001 versus Control (ANOVA analysis).
Having established the importance of LOX gene silencing in mH2A1.2 function, we then measured the expression levels of mH2A1.2 and LOX in a cDNA array of 39 breast tumor and 9 normal samples by qPCR. Our analysis revealed that expression levels of mH2A1.2 were lower in ~90% of tumor samples compared with normal samples (Figure 4D). By contrast, ~80% of the tumor samples exhibited higher levels of LOX expression than their normal counterparts. When the two expression data were compared, we found that mH2A1.2 and LOX expression levels are inversely correlated in the large majority of the examined samples. It is also noteworthy that mH2A1.2 and LOX show more significant expression changes in advanced stages of breast cancer (Figure 4D). Collectively, these data indicate that dysregulated expression of mH2A1.2 and LOX is a common feature in breast cancer and could be used to predict a risk for breast cancer-induced bone loss.
c-Src Signaling Pathway Is a Critical Target of LOX
LOX activity is associated with the phosphorylation status of c-Src, which is a key regulator of cytoskeletal reorganization, migration, and bone resorption in osteoclasts (Payne et al., 2005; Soriano et al., 1991; Tanaka et al., 1996). Given that rLOX treatment facilitates osteoclast differentiation, we wondered whether c-Src phosphorylation is also affected by rLOX treatment. In order to check this possibility, we analyzed the levels of phosphorylated c-Src in OCP cells after rLOX treatment by western blot. OCP cells treated with rLOX wt exhibited higher levels of c-Src phosphorylation over a 30-min time period, as compared with those observed in mock-treated cells (Figure 5A). In the same experiment, c-Src phosphorylation levels were unchanged following the treatment with rLOX mt. We next wanted to know how LOX-mediated extracellular matrix remodeling can stimulate phosphorylation of c-Src. As integrins are known to mediate interactions between the extracellular and intracellular environments (Guo and Giancotti, 2004), they are good candidates for transducing LOX-mediated signals. Activation of integrin αvβ3 is known to promote c-Src phosphorylation in osteoclasts via an “outside-in” signaling mechanism (Faccio et al., 2003; Geiger et al., 2001). Therefore, we investigated whether integrin αvβ3 was involved in transducing the effects of extracellular LOX activity to intracellular c-Src phosphorylation using a function-blocking antibody against integrin β3 subunit. Treatment with this blocking antibody reduced the levels of phosphorylated c-Src in OCP cells seeded onto the culture wells coated with rLOX wt or rLOX mt in MDA-MB-468 CMs (Figure 5B). These results suggest that LOX-mediated ECM remodeling may induce activation of integrin β3 and phosphorylation of c-Src kinase.
Figure 5. An Important Role for LOX in Facilitating c-Src Phosphorylation and Function.
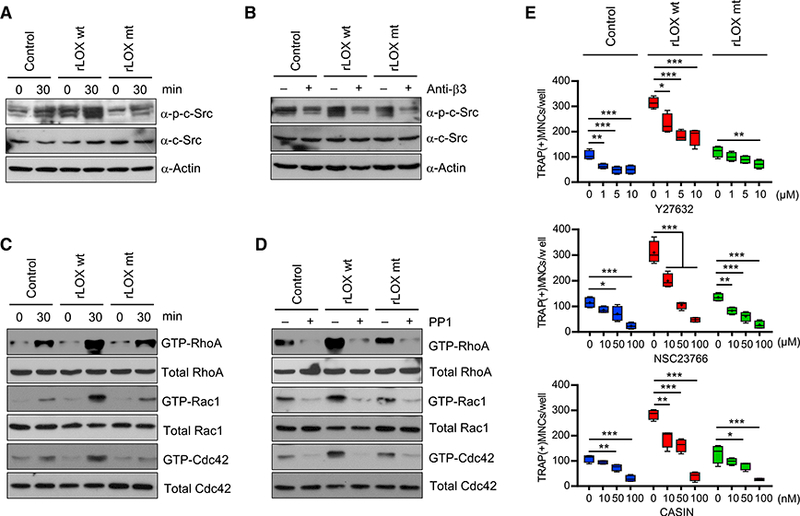
(A) OCP cells were seeded onto the culture dishes coated with rLOX wt or mt in MDA-MB-468 CMs for 30 min. Whole cell lysates were prepared and analyzed by western blot using antibodies recognizing c-Src and c-Src phosphorylation.
(B) After pretreating with function-blocking antibody against integrin β3 subunit, OCP cells were plated on the culture dishes coated with rLOX wt or rLOX mt in MDA-MB-468 CMs and incubated for 30 min. Whole cell lysates were prepared and analyzed by western blot as in (A).
(C) Small GTPase (RhoA/Rac1/Cdc42) assays were performed as in (A), and subjected to western blot analysis with antibodies against RhoA, Rac1, and Cdc42.
(D) Small GTPases assays were done as in (B, but using the Src inhibitor PP1.
(E) OCP cells were treated with rLOX wt or rLOX mt for 6 days in the presence of the indicated concentrations of RhoA inhibitor Y27632, Rac1 inhibitor NSC23766, or Cdc42 inhibitor CASIN. TRAP-positive multinucleated osteoclasts were stained and counted. Error bars represent the means ± SD (n = 4); *p < 0.05, **p <0.01, ***p < 0.001 (ANOVA analysis).
Considering that the Rho family small GTPases (RhoA, Rac1, and Cdc42) are downstream effectors of integrin αvβ3 activation and c-Src signaling (Faccio et al., 2003), we also investigated the levels of active GTP-bound forms of RhoA, Rac1, and Cdc42 in osteoclasts seeded onto on the culture plates coated with rLOX wt or rLOX mt in MDA-MB-468 CMs. The results of this effort indicated that levels of GTP-bound RhoA, Rac1, and Cdc42 are significantly increased after treatments with rLOX wt, but not rLOX mt (Figure 5C). Our western blot analysis also revealed lower levels of active GTP-bound RhoA, Rac1, and Cdc42 after treatments with c-Src inhibitor PP1 (Figure 5D), suggesting that small GTPase signaling pathways are downstream effectors of LOX-mediated c-Src activation. Consistent with these results, when the effects of small molecule inhibitors targeting RhoA (Y27632), Rac1 (NSC23766), and Cdc42 (CASIN) on rLOX-enhanced osteoclastogenesis were evaluated, OCP cell differentiation was reduced significantly after NSC23766 or CASIN treatment and moderately after Y27632 treatment (Figure 5E). These results support the view that LOX-stimulated osteoclastogenesis is dependent on signaling pathways involving RhoA, Rac1, and Cdc42. As mH2A1.2 has a role in inhibiting LOX gene expression and osteoclastogenic function, mH2A1-depleted MDA-MB-468 CMs might therefore be expected to exhibit upregulation in c-Src phosphorylation and function. Indeed, when we repeated the above experiments using control and mH2A1-depleted MDA-MB-468 CMs and compared their effects, we clearly observed stronger responses to mH2A1-depleted MDA-MB-468 CMs as compared with control MDA-MB-468 CMs (Figure S5).
OCP cell migration is a prerequisite for cell-cell contact and fusion prior to the formation of multinuclear osteoclasts. Because small GTPases are important players in controlling this differentiation process (Kim et al., 2016a), a potential role for LOX in OCP cell migration was investigated by using RhoA, Rac1, and Cdc42 inhibitors. The results showed that treating OCP cells with rLOX wt stimulated migration, but antagonizing RhoA, Rac1, and Cdc42 GTPase activities with their inhibitors impeded OCP cell migration (Figure S3H). Because small GTPases play important roles in controlling invasion properties of cancer cells, which is linked to LOX function (Erler et al., 2006; Gómez del Pulgar et al., 2005), rLOX-treated MDA-MB- 468 cells were subjected to cell invasion assays after exposure to RhoA, Rac1, and Cdc42 inhibitors. Transfection of rLOX wt promoted MDA-MB-468 cell invasion, but rLOX mt showed no invasion-promoting activity (Figure S3I). Importantly, exposure of rLOX-treated cells to RhoA, Rac1, and Cdc42 inhibitors attenuated their invasion capacity, albeit to different extents (Figure S3I). These repressive patterns were also observable after exposing control or rLOX mt-treated cells to the inhibitors, but to a much less severe degree. These data overall support the notion that the small GTPases signaling plays a critical role in LOX-mediated OCP cell migration and breast cancer cell invasion.
EZH2 Binds to mH2A1.2 Nucleosomes and Establishes LOX G Silencing in Breast Cancer Cells
Several recent studies, including ours, suggested that mH2A incorporation into nucleosomes influences histone modification patterns associated with transcriptional silencing or activation (Chen et al., 2014; Douet et al., 2017; Gamble et al., 2010; Gaspar-Maia et al., 2013; Kim et al., 2013). Thus, we asked whether any histone modifications are altered in mH2A1.2 nucleosomes and play a role, if any, mH2A1.2-induced gene silencing in MDA-MB-468 breast cancer cells. To this end, MDA-MB-468 cells were stably transfected with expression vectors encoding FLAG-tagged H2A or mH2A1.2 (Figure 6A). After confirming the comparable expression of ectopic H2A and mH2A1.2 in transfected cells (data not shown), soluble chromatin was prepared and digested with micrococcal nuclease to yield mainly mononucleosomes. Mononucleo- somes containing ectopic histones were then purified by immu- noprecipitations with anti-FLAG antibody under stringent conditions (300 mM KCl and 0.2% NP40). Coomassie blue staining confirmed stoichiometry of H2A, H2B, H3, and ectopic H2A/mH2A1.2 in the purified nucleosomes (Figure 6A). In checking the states of histone acetylation and methylation of purified nucleosomes by western blot, we detected much higher levels of H3K27me3 in mH2A1.2 nucleosomes than in H2A nucleosomes (Figure 6A). Consistent with its role in transcriptional repression, mH2A1.2 nucleosomes retained lower levels of three active histone marks, H3ac, H4ac, and H3K4me3. Contrarily, other histone modifications did not show any significant differences between H2A and mH2A1.2 nucleosomes. These results are consistent with the previous observation that H3K27me3 is a dominant histone modification in mH2A nucleosomes and mH2A-enriched genomic regions (Buschbeck et al., 2009; Gamble et al., 2010). Along with a recent demonstration of H3K27me3 being most highly increased in breast cancer cells (Leroy et al., 2013), this finding also suggests a possible role for H3K27me3 in mH2A1.2- induced gene silencing in MDA-MB-468 cells. To investigate this possibility, we suppressed the expression of EZH2 that is mainly responsible for H3K27me3 in breast cancer cells (Leroy et al., 2013) and evaluated its impact on LOX gene transcription. Relative to non-targeting control shRNA, shRNA directed against EZH2 efficiently depleted EZH2 and almost completely abrogated H3K27me3 in MDA-MB-468 cells (Figure 6B). This decrease in EZH2-mediated H3K27me3 generated a significant enhancement of LOX expression in the cells and thus LOX enzymatic activity in CMs (Figures 6B and 6C). The observed effects of EZH2 knockdown could be rescued by EZH2 wildtype, but not a methyltransferase-dead mutant (Figures 6B and 6C), strongly supporting that EZH2 enzymatic activity is critical for mH2A1.2 function in repressing LOX expression.
Figure 6. Selective Recognition and Modification of mH2A1.2 Nucleosomes by EZH2.
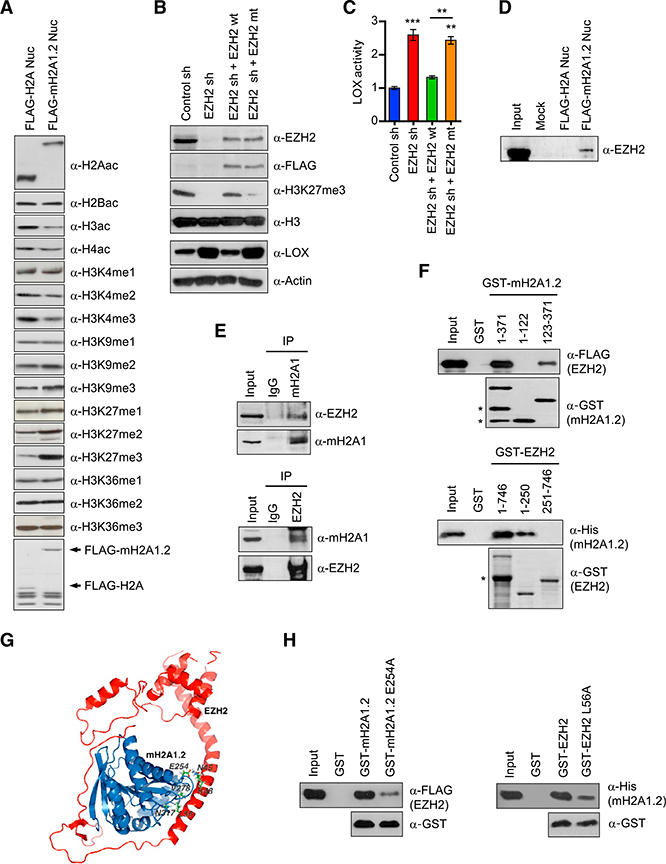
(A) Mononucleosomes were prepared from MDA- MB-468 cells transfected with FLAG-H2A or FLAG- mH2A1.2 expression vector as described (Kim et al., 2013). FLAG-H2A and FLAG-mH2A1.2 nu- cleosomes were immunoprecipitated and analyzed by western blotting with antibodies specific for the indicated histone modifications. The bottom panel shows a Coomassie-stained gel of the purified nucleosomes.
(B) EZH2-depleted MDA-MB-468 cells were complemented with EZH2 wild-type (EZH2 wt) or methyltransferase-dead mutant (EZH2 mt), and relative levels of H3K27me3 and LOX expression were determined by western blot.
(C) LOX activity was measured in CMs collected from mock-depleted control or EZH2-depleted MDA-MB-468 cells. The rescue effects of EZH2 wt and mt on LOX activity were also analyzed. Error bars represent the means ± SD (n = 3); **p < 0.01, ***p < 0.001 versus Control sh (ANOVA analysis).
(D) Mononucleosomes containing ectopic H2A or mH2A1.2 were purified as in (A) and subjected to western blotting with EZH2 antibody.
(E) MDA-MB-468 cell extracts were immunopre- cipitated with mH2A1 and EZH2 antibodies and analyzed by western blot. Ten percent of the input proteins were also examined by western blotting.
(F) FLAG-EZH2 and His-mH2A1.2 were incubated with the indicated GST-mH2A1.2 and GST-EZH2 fusions. After extensive washing, bound EZH2 and mH2A1.2 proteins were analyzed by western blot. Non-specific bands were marked by asterisks.
(G) The model of mH2A1.2-EZH2 interaction was built on the basis of PDB entries 1zr5 (mH2A1.2) and 5hyn (EZH2) in docking simulations using the program Cluspro 2.0. The chosen docking prediction is shown with the EZH2 (L56) and mH2A1.2 (V278, N317) side chains in ball-and-stick representation.
(H) GST pull-down assays were conducted as in(F), except that GST-mH2A1.2 and GST-EZH2 carrying the indicated point mutations were used.
The fact that EZH2 catalyzes H3K27me3 in mH2A1.2-depen- dent manner led us to propose that EZH2 can recognize and bind mH2A1.2 nucleosomes in MDA-MB-468 cells. An initial western blot analysis showed, as predicted, that EZH2 is co-purified with mH2A1.2 nucleosomes from MDA-MB-468 breast cancer cells (Figure 6D). Because there was no detectable binding of EZH2 to H2A nucleosomes, the observed binding was specific and occurred through mH2A1.2. These data were supported by our finding that mH2A1 antibody is able to coimmuno- precipitate endogenous EZH2 from lysates of MDA-MB-468 breast cancer cells (Figure 6E). The observed interaction was further verified by reciprocal immunoprecipitation using EZH2 antibody (Figure 6E). To more precisely determine the nature of the observed interactions, we also carried out a series of glutathione S-transferase (GST) pull down assays. In clear confirmation of a direct interaction, EZH2 bound strongly to GST-mH2A1.2 but not to GST (Figure 6F). In similar binding experiments with truncated versions of mH2A1.2, the C-terminal macrodomain of mH2A1.2 (residues 123–371) still interacted with EZH2 (Figure 6F). However, N-terminal histone domain (residues 1–122) failed to show any affinity for EZH2, indicating that the binding of mH2A1.2 to EZH2 is dependent upon its C-terminal macrodomain. In mapping mH2A1.2-interacting region of EZH2, the binding of the N-terminal region (residues 1–250) of EZH2 was readily detectable, but its central and C-terminal regions (residues 251–500 and 501–746) showed no interactions with mH2A1.2 under the same assay conditions (Figure 6F). To identify amino acid residues critical for the mH2A1.2-EZH2 interaction, we next generated a structural model of the mH2A1.2- EZH2 complex based on their crystal structures (Justin et al., 2016; Kustatscher et al., 2005) using the program Cluspro 2.0 (Kozakov et al., 2017). Our model suggested that E254 of mH2A1.2 is positioned to contact N45 and K48 of EZH2. Moreover, L56 of EZH2 was predicted to be in hydrophobic contacts with V278 and N317 of mH2A1.2 (Figure 6G). Consistent with this model, in vitro binding assays employing mH2A1.2 E254A and EZH2 L56A mutant proteins demonstrated that the mH2A1.2- EZH2 interaction was significantly inhibited by either mutation (Figure 6H).
EZH2 Localizes at the LOX Locus and Impairs Osteoclastogenesis in a mH2A1.2-Dependent Fashion
We next initiated efforts to examine the significance of EZH2- mH2A1.2 interaction in establishing and maintaining a repressive chromatin environment over the LOX locus. We hypothesized that this LOX gene-targeted activity in MDA-MB-468 breast cancer cells confers anti-osteoclastogenic properties to EZH2. In an attempt to test this hypothesis, we prepared CMs from MDA-MB-468 cells depleted of EZH2 and explored their effects on OCP cell differentiation into osteoclasts. Our analysis clearly showed that knockdown of EZH2 in MDA-MB-468 cells endowed CMs with the ability to stimulate osteoclast differentiation (Figure 7A). Ectopic expression of EZH2 wild-type, but not the enzymatically dead mutant, in EZH2-depleted MDA-MB-468 cells restored OCP cell differentiation to levels quantitatively similar to those observed with mock-depleted control cell CMs—indicative of the importance of EZH2 methyltransferase activity (Figure 7B). Importantly, knockdown of EZH2 in mH2A1-depleted cells failed to increase the osteoclastogenic activity of CMs, indicative of mH2A1-dependent function of EZH2 in breast cancer cells (Figure 7A). It was also apparent that depleting EZH2 accelerated OCP cell migration and MDA- MB-468 cell invasion dependently of mH2A1 (Figure S6). The inhibitory effects of EZH2 on osteoclastogenesis were also verified by monitoring changes in the expression of NFATc1, ATP6V0D2, and cathepsin K genes in response to knockdown and rescue of EZH2 in MDA-MB-468 cells (Figure 7C). Moreover, and as shown in Figures 7D and 7E, we confirmed by ChIP assay that LOX gene harbors high levels of EZH2 and H3K27me3 and loses H3K27me3 following EZH2 knockdown. The observed enrichment of EZH2 largely disappeared upon concomitant depletion of mH2A1 (Figure 7D), again confirming the functional link between EZH2 and mH2A1.2 in LOX gene silencing.
Figure 7. mH2A1.2-Dependent Repression of LOX Expression and Osteoclast Differentiation by EZH2.
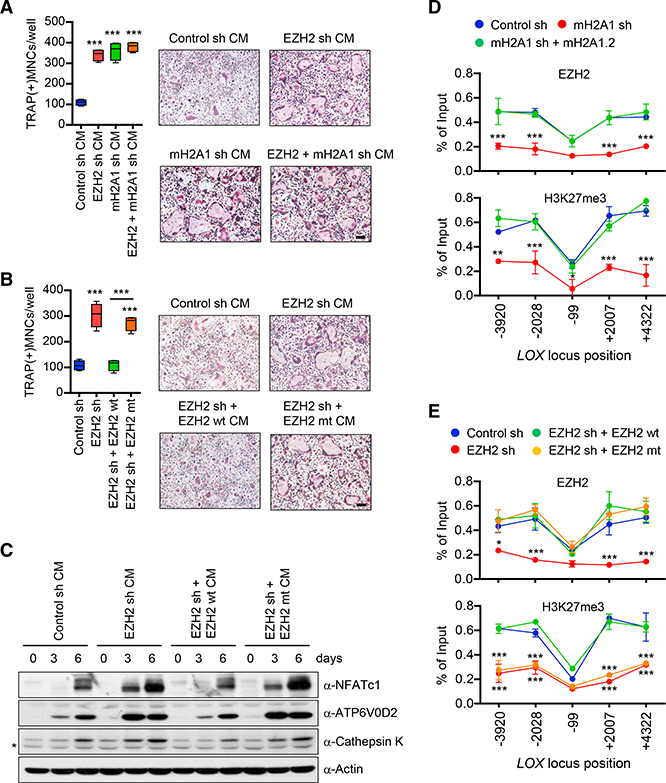
(A) CMs were harvested from MDA-MB-468 breast cancer cells depleted of EZH2 or/and mH2A1 and analyzed for osteoclastogenic activity. Scale bar, 100 μm.
(B) OCP cells were treated with CMs from EZH2- depleted MDA-MB-468 cells expressing shRNA- resistant EZH2 wt or mt. Scale bar, 100 μm.
(C) After treating OCP cells with indicated CMs, expression levels of three osteoclast marker genes were analyzed by western blot. β-Actin was used as a loading control. Non-specific band was marked by asterisk.
(D) ChIP assays on mock-depleted or mH2A1- depleted MDA-MB-468 cells complemented with shRNA-resistant mH2A1.2 were performed to assess the levels of EZH2 and H3K27me3 at the LOX gene. Primer sets locating at five different regions of the LOX gene were used.
(E) ChIP assays were carried out as in (D), but using mock-depleted or EZH2-depleted MDA-MB-468 cells. For rescue experiments, shRNA-resistant EZH2 wt or mt was expressed. Error bars in (A), (B), (D), and (E) represent the means ± SD (A and B; n = 4, D and E; n = 3); *p < 0.05, **p < 0.01, ***p < 0.001 versus Control sh (ANOVA analysis).
DISCUSSION
While a considerable amount of data have accumulated over the last several years regarding osteoclastogenic potentials of cancer cell-secreted factors, a detailed mechanistic description for how the expression of these bone destructive factors is regulated in cancer cells is lacking. We undertook a systematic investigation of the mechanisms that are used to regulate these processes in breast cancer cells. As a model system for our study, we selected the ER+ MDA-MB-468 cell, which is a moderately metastatic breast cancer cell. We also took advantage of an in vitro CM-based assay that circumvents some of the complications linked to in vivo studies. One important initial result obtained using this assay system was that mH2A1.2 significantly decreases osteoclastogenic potential of soluble factors secreted by MDA-MB-468 breast cancer cells. The observation that two other mH2A isoforms, mH2A1.1 and mH2A2, do not participate in regulating osteoclastogenesis is important in light of several recent reports suggesting that mH2A isoforms are dedicated to regulatory roles in specific gene expression (Dell’Orso et al., 2016; Gamble et al., 2010; Kapoor et al., 2010; Kim et al., 2013). In agreement with these observations, our secretome analysis of microarray data demonstrates that mH2A1.2 is essential for keeping inactive the genes encoding the factors that influence osteo- clastogenesis and metastasis in MDA- MB-468 breast cancer cells. This study represents the first genome-wide expression profiling and secretome analysis of mH2A target genes, and establishes mH2A1.2 as a key player in stable epigenetic silencing of genes encoding secreted factors. Our ChIP and qRT-PCR assays using mH2A1 -depleted cells rescued with ectopic mH2A1.2 also suggest that accurate enrichment of mH2A1.2 in target genes is vital to the control of secretory factor gene expression in MDA-MB-468 cells.
Because our secretome analysis of microarray data ranked LOX gene highest, and because LOX has been implicated in bone metastasis, we investigated the mechanism underlying the observed mH2A1.2-dependent phenomena by studying a role for mH2A1.2 in regulating LOX gene. It was shown that LOX is overexpressed and secreted from MDA-MB-231 breast cancer cells with high metastatic potential, and this secreted LOX is a critical player in this cancer cell bone metastasis (Cox et al., 2015). In highly metastatic MDA-MB-231 cells, mH2A1.2 is minimally expressed—contrary to what was observed in moderately metastatic MDA-MB-468 cells—and thus CMs from these cells may well facilitate osteoclast differentiation. In MDA-MB-468 cells, however, mH2A1.2 is highly expressed and constitute a repressive barrier to LOX expression, and it needs to be removed from the LOX gene locus in order to activate LOX transcription. The exchange of canonical histones with histone variants is highly dynamic and can be modulated through a number of different mechanisms. Although the precise mechanism is unknown, the exchange of specific histone variants is shown to be promoted by specific patterns of posttrans- lational modifications. Thus, we are tempted to speculate that the ability of exchange factors to selectively incorporate mH2A1.2 into the LOX locus might be dependent on specific modification status of mH2A1.2. The osteoclastogenic function of LOX seems to be dependent on LOX-mediated collagen cross-linking, because the expression of LOX wild-type, but not LOX oxidase-dead mutant, in LOX-depleted cells restored CM-induced osteoclast differentiation rates. Notably, and consistent with recent studies (Cox et al., 2015; Tsukasaki et al., 2017), our investigation demonstrated that rLOX can recapitulate the osteoclastogenic properties in vitro and stimulate bone resorption in vivo. These findings underscore the importance of mH2A1.2 in the regulation of LOX expression and also suggest that accurate targeting of mH2A1.2 within the LOX gene locus may be vital for the control of breast cancer-induced osteoclastogenesis and its related physiological responses. In this regard, LOX is a promising target for novel therapeutic strategies, as rLOX treatments significantly stimulated bone resorption and osteoporosis. Our data also demonstrate that LOX likely facilitates breast cancer-induced osteoclast differentiation through the activation of extracellular matrix remodeling processes by upregulating Src phosphorylation and integrin signaling. Further, by inhibitor and blocking antibody treatments, we were able to confirm that Src phosphorylation and integrin signaling processes are mutually dependent in LOX-dependent osteoclastogenic reactions.
Another intriguing finding of our study is that mH2A1.2 nucle- osomes are enriched with H3K27me3 in MDA-MB-468 breast cancer cells, underscoring the connection between mH2A1.2 and H3K27me3 in regulating breast cancer-induced generation of mature osteoclasts. Such a connection between mH2A1 and H3K27me3 has been also observed in previous studies (Buschbeck et al., 2009; Chen et al., 2014; Gamble et al., 2010); however, the mechanistic details for this observation have not been explored. Functionally, we demonstrate that EZH2 is responsible for generating H3K27me3 and acts as a negative regulator of LOX gene in MDA-MB-468 cells. EZH2 directly interacts and colocalizes with mH2A1.2 in the LOX gene locus and does so in a mH2A1.2 macrodomaindependent fashion. Depletion of EZH2 substantially increased the osteoclastogenic capacity of CMs from MDA-MB-468 cells. mH2A1.2 fails to control LOX-triggered activation of osteoclast differentiation after EZH2 knockdown in MDA-MB-468 cells, indicating that the suppressive behavior of mH2A1.2 on CM- induced osteoclast differentiation is EZH2-dependent. Thus, mH2A1.2 constitutes a recruitment platform for EZH2 to mediate H3K27me3 and generate LOX inactivation, and it needs to be dissociated from LOX locus in order to remove H3K27me3 and activate LOX transcription during osteoclastogenic event. Mechanistically, such cooperative actions of mH2A1.2 and EZH2 should provide additional levels of efficiency and specificity to transcriptional manipulation of LOX gene in MDA-MB-468 cells.
Because the expression of mH2A1.2 is mis-regulated in cell lines for other cancer types (Kim et al., 2013), it would be interesting to investigate whether a similar EZH2-dependent mechanism of mH2A1.2 action may also apply to other bone metastatic cancers.
Based on our observations, together with findings from previous studies, we present the following working model for how mH2A1.2 negatively regulates breast cancer-induced osteoclastogenesis (Figure S7). In an initial state, mH2A1.2 is highly expressed and localizes at LOX gene. Once incorporated into LOX gene, mH2A1.2 utilizes its macrodomain to recruit EZH2 through direct protein-protein interactions. An epigenetic gene silencing process involving H3K27me3 then takes place to keep LOX gene in an inactive state. In breast cancer cells, EZH2 functionally cooperates with mH2A1.2, such that facilitating its binding to mH2A could be a novel strategy for controlling LOX expression and thus breast cancer-induced osteoclas- togenesis. Moreover, stimulating mH2A1.2 exchange reactions can enhance mH2A1.2-based localization of EZH2 over LOX gene locus and thus disrupt the active state of LOX expression and pathological oseteoclastogenesis.
EXPERIMENTAL PROCEDURES
Cell Culture and Osteoclast Differentiation
MDA-MB-468, MCF-7, MCF-10–2A, and MDA-MB-231 cells were purchased from the American Type Culture Collection. MDA-MB-468, MCF-7, and MDA-MB-231 cells were cultured in DMEM supplemented with 10% fetal bovine serum (FBS). MCF-10–2A cells were maintained in DMEM/F12 media with 20 ng/mL epidermal growth factor, 100 ng/mL cholera toxin, 0.01 mg/mL insulin, 500 ng/mL hydrocortisone, and 5% FBS. CMs were prepared by culturing cells to ~80% confluence, changing media to α-minimum essential medium (α-MEM) containing 0.5% FBS, and collecting media after 24 hr incubation. For osteoclast differentiation assays, bone marrow cells were isolated from 6-week-old C57BL/6 mice (The Jackson Laboratory) as recently described (Kim et al., 2016b) and in accordance with the University of Southern California guidelines. After priming the harvested cells with a-MEM containing 10% FBS and macrophage colony stimulating factor (M-CSF, 5 ng/mL) for 16 hr, non-adherent mononuclear cells were collected and cultured for an additional 3 days with α-MEM containing M-CSF (30 ng/mL) to generate primary osteoclast precursor (OCP) cells. OCP cells were then seed into a 48-well plate (2 × 104 cells/well) and grown in a 1:1 mixture of α-MEM culture medium and CMs supplemented with M-CSF (30 ng/mL) and RANKL (30 ng/mL) for 0, 3, or 6 days. The cells were fixed and stained with tartrate-resistant acid phosphatase (TRAP) using a leukocyte acid phosphatase staining kit (Sigma-Aldrich) according to manufacturer’s instructions. TRAP-positive multinuclear cells with more than 3 nuclei were counted as osteoclasts under a light microscope.
Treatment and Analysis of Mice
Animal studies were conducted according to the guidelines set by the Institutional Animal Care and Use Committee. Female 6- to 8-week-old C57BL/6 mice were injected with 0.2 mg/kg rLOX or PBS alone onto their calvarias subcutaneously every other day. Five mice were injected for each group. 10 days after peptide injection, mice were euthanized, and calvaria were collected and immediately fixed with 3.7% formaldehyde. Paraffin-embedded samples were sectioned (6 μm thickness) and subjected to H&E and TRAP staining. Images were acquired with an Aperio ScanScope Model T3 and were analyzed with ImageScope software (Aperio Technologies). OCP cells (2 × 104 cells/well) were also treated with rLOX (100 ng/mL) along with RANKL (15 ng/mL) and M-CSF (30 ng/mL) and used to evaluate in vitro response to rLOX proteins.
For additional experimental materials and methods, see Supplemental Experimental Procedures.
Supplementary Material
Highlights.
mH2A1.2 inhibits the expression of genes encoding osteoclastogenic soluble factors
LOX is necessary for mH2A1.2 to attenuate breast cancer- induced osteoclastogenesis
LOX targets the c-Src signal transduction pathway
mH2A1.2 cooperates with EZH2 to inhibit LOX expression and osteoclastogenesis
ACKNOWLEDGMENTS
This work was supported by the NIH (grant CA201561 awarded to W.A.). The study was also funded in part by pilot project grants from the Keck School of Medicine of USC.
Footnotes
DATA AND SOFTWARE AVAILABILITY
The accession number for the gene expression array data reported in this study is GEO: GSE107570.
AUTHOR CONTRIBUTIONS
J. Kim designed the experiments. W.A. provided guidance throughout. J. Kim, Y.S., S.L., M.K., V.P., J.F.L., H.S., K.K., T.S.U., J. Koh, D.J., and W.A. performed experiments and analyzed data. J. Kim and W.A. wrote the manuscript.
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures, seven figures, and five tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.06.020.
REFERENCES
- Bild AH, Potti A, and Nevins JR (2006). Linking oncogenic pathways with therapeutic opportunities. Nat. Rev. Cancer 6, 735–741. [DOI] [PubMed] [Google Scholar]
- Boyle WJ, Simonet WS, and Lacey DL (2003). Osteoclast differentiation and activation. Nature 423, 337–342. [DOI] [PubMed] [Google Scholar]
- Buschbeck M, Uribesalgo I, Wibowo I, Rué P, Martin D, Gutierrez A, Morey L, Guigo R, Lopez-Schier H, and Di Croce L (2009). The histone variant macroH2A is an epigenetic regulator of key developmental genes. Nat. Struct. Mol. Biol 16, 1074–1079. [DOI] [PubMed] [Google Scholar]
- Chakravarthy S, Gundimella SK, Caron C, Perche PY, Pehrson JR, Khochbin S, and Luger K (2005). Structural characterization of the histone variant macroH2A. Mol. Cell. Biol 25, 7616–7624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Ruiz PD, Novikov L, Casill AD, Park JW, and Gamble MJ (2014). MacroH2A1.1 and PARP-1 cooperate to regulatetranscription by promoting CBP-mediated H2B acetylation. Nat. Struct. Mol. Biol 21, 981–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clézardin P (2011).Therapeutic targets for bone metastases in breast cancer. Breast Cancer Res. 13, 207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox TR, Rumney RMH, Schoof EM, Perryman L, H∅ye AM, Agrawal A.,Bird D, Latif NA, Forrest H, Evans HR,et al. (2015). The hypoxic cancer secretome induces pre-metastatic bone lesions through lysyl oxidase. Nature 522, 106–110. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Dell’Orso S, Wang AH, Shih HY, Saso K, Berghella L, Gutierrez-Cruz G, Ladurner AG, O’Shea JJ, Sartorelli V, and Zare H (2016). The histone variant macroH2A1.2 is necessary for the activation of muscle enhancers and recruitment of the transcription factor Pbx1. Cell Rep. 14, 1156–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douet J, Corujo D, Malinverni R, Renauld J, Sansoni V, Posavec Marjanovic M, Cantariño N, Valero V, Mongelard F, Bouvet P, et al. (2017). MacroH2A histone variants maintain nuclear organization and heterochromatin architecture. J. Cell Sci 130, 1570–1582. [DOI] [PubMed] [Google Scholar]
- Erler JT, Bennewith KL, Nicolau M, Dornhöfer N, Kong C, Le QT, Chi JT, Jeffrey SS, and Giaccia AJ (2006). Lysyl oxidase is essential for hypoxia-induced metastasis. Nature 440, 1222–1226. [DOI] [PubMed] [Google Scholar]
- Erler JT, Bennewith KL, Cox TR, Lang G, Bird D, Koong A, Le QT, and Giaccia AJ (2009). Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell 15, 35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faccio R, Novack DV, Zallone A, Ross FP, and Teitelbaum SL (2003). Dynamic changes in the osteoclast cytoskeleton in response to growth factors and cell attachment are controlled by beta3 integrin. J. Cell Biol 162, 499–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamble MJ, Frizzell KM, Yang C, Krishnakumar R, and Kraus WL (2010). The histone variant macroH2A1 marks repressed autosomal chromatin, but protects a subset of its target genes from silencing. Genes Dev. 24, 21–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaspar-Maia A, Qadeer ZA, Hasson D, Ratnakumar K, Leu NA, Leroy G, Liu S, Costanzi C, Valle-Garcia D, Schaniel C, et al. (2013). MacroH2A histone variants act as a barrier upon reprogramming towards pluripotency. Nat. Commun 4, 1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger B, Bershadsky A, Pankov R, and Yamada KM (2001). Transmembrane crosstalk between the extracellular matrix-cytoskeleton crosstalk. Nat. Rev. Mol. Cell Biol 2, 793–805. [DOI] [PubMed] [Google Scholar]
- Gómez del Pulgar T, Benitah SA, Valerón PF, Espina C, and Lacal JC (2005). Rho GTPase expression in tumourigenesis: evidence for a significant link. BioEssays 27, 602–613. [DOI] [PubMed] [Google Scholar]
- Guo W, and Giancotti FG (2004). Integrin signalling during tumour progression. Nat. Rev. Mol. Cell Biol 5, 816–826. [DOI] [PubMed] [Google Scholar]
- Henikoff S, and Smith MM (2015). Histone variants and epigenetics. Cold Spring Harb. Perspect. Biol 7, a019364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justin N, Zhang Y, Tarricone C, Martin SR, Chen S, Underwood E, De Marco V, Haire LF, Walker PA, Reinberg D, et al. (2016). Structural basis of oncogenic histone H3K27M inhibition of human polycomb repressive complex 2. Nat. Commun 7, 11316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y, Siegel PM, Shu W, Drobnjak M, Kakonen SM, Cordón-Cardo C, Guise TA, and Massagué J (2003). A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 3, 537–549. [DOI] [PubMed] [Google Scholar]
- Kapoor A, Goldberg MS, Cumberland LK, Ratnakumar K, Segura MF, Emanuel PO, Menendez S, Vardabasso C, Leroy G, Vidal CI, et al. (2010). The histone variant macroH2A suppresses melanoma progression through regulation of CDK8. Nature 468, 1105–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karsenty G, Kronenberg HM, and Settembre C (2009). Genetic control of bone formation. Annu. Rev. Cell Dev. Biol 25, 629–648. [DOI] [PubMed] [Google Scholar]
- Kim JM, Heo K, Choi J, Kim K, and An W (2013). The histone variant MacroH2A regulates Ca(2+) influx through TRPC3 and TRPC6 channels. Oncogenesis 2, e77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JM, Kim MY, Lee K, and Jeong D (2016a). Distinctive and selective routeofPI3K/PKCα-PKCδ/RhoA-Rac1 signaling in osteoclastic cell migration. Mol. Cell. Endocrinol 437, 261–267. [DOI] [PubMed] [Google Scholar]
- Kim K, Punj V, Kim JM, Lee S, Ulmer TS, Lu W, Rice JC, and An W (2016b). MMP-9 facilitates selective proteolysis of the histone H3 tail at genes necessary for proficient osteoclastogenesis. Genes Dev. 30, 208–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozakov D, Hall DR, Xia B, Porter KA, Padhorny D, Yueh C, Beglov D, and Vajda S (2017). The ClusPro web server for protein-protein docking. Nat. Protoc 12, 255–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kustatscher G, Hothorn M, Pugieux C, Scheffzek K, and Ladurner AG (2005). Splicing regulates NAD metabolite binding to histone macroH2A. Nat. Struct. Mol. Biol 12, 624–625. [DOI] [PubMed] [Google Scholar]
- Leroy G, Dimaggio PA, Chan EY, Zee BM, Blanco MA, Bryant B, Fla- niken IZ, Liu S, Kang Y, Trojer P, and Garcia BA (2013). A quantitative atlas of histone modification signatures from human cancer cells. Epigenetics Chromatin 6, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maze I, Noh KM, Soshnev AA, and Allis CD (2014). Every amino acid matters: essential contributions of histone variants to mammalian development and disease. Nat. Rev. Genet 15, 259–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne SL, Fogelgren B, Hess AR, Seftor EA, Wiley EL, Fong SF, Csiszar K, Hendrix MJ, and Kirschmann DA (2005). Lysyl oxidase regulates breast cancer cell migration and adhesion through a hydrogen peroxide-mediated mechanism. Cancer Res. 65, 11429–11436. [DOI] [PubMed] [Google Scholar]; Pehrson JR, and Fried VA (1992). MacroH2A, a core histone containing a large nonhistone region. Science 257, 1398–1400. [DOI] [PubMed] [Google Scholar]
- Raggatt LJ, and Partridge NC (2010). Cellular and molecular mechanisms of bone remodeling. J. Biol. Chem 285, 25103–25108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynaud C, Ferreras L, Di Mauro P, Kan C, Croset M, Bonnelye E, Pez F, Thomas C, Aimond G, Karnoub AE, et al. (2017). Lysyl oxidase is a strong determinant of tumor cell colonization in bone. Cancer Res. 77, 268–278. [DOI] [PubMed] [Google Scholar]
- Soriano P, Montgomery C, Geske R, and Bradley A (1991). Targeted disruption of the c-src proto-oncogene leads to osteopetrosis in mice. Cell 64, 693–702. [DOI] [PubMed] [Google Scholar]; Tanaka S, Amling M, Neff L, Peyman A, Uhlmann E, Levy JB, and Baron R (1996). c-Cbl is downstream of c-Src in a signalling pathway necessary for bone resorption. Nature 383, 528–531. [DOI] [PubMed] [Google Scholar]
- Teitelbaum SL, and Ross FP (2003). Genetic regulation of osteoclast development and function. Nat. Rev. Genet 4, 638–649. [DOI] [PubMed] [Google Scholar]
- Tsukasaki M, Hamada K, Okamoto K, Nagashima K, Terashima A, Komatsu N, Win SJ, Okamura T, Nitta T, Yasuda H, et al. (2017). LOX fails to substitute for RANKL in osteoclastogenesis. J. Bone Miner. Res 32, 434–439. [DOI] [PubMed] [Google Scholar]
- Turner JM, Burgoyne PS, and Singh PB (2001). M31 and macroH2A1.2 colocalise at the pseudoautosomal region during mouse meiosis. J. Cell Sci 114, 3367–3375. [DOI] [PubMed] [Google Scholar]
- Weilbaecher KN, Guise TA, and McCauley LK (2011). Cancer to bone: a fatal attraction. Nat. Rev. Cancer 11, 411–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin JJ, Pollock CB, and Kelly K (2005). Mechanisms of cancer metastasis to the bone. Cell Res. 15, 57–62. [DOI] [PubMed] [Google Scholar]
- Zaidi M (2007). Skeletal remodeling in health and disease. Nat. Med 13, 791–801. [DOI] [PubMed] [Google Scholar]
- Zhang YX, Sun HL, Liang H, Li K, Fan QM, and Zhao QH (2015). Dynamic and distinct histone modifications of osteogenic genes during osteogenic differentiation. J. Biochem 158, 445–457. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.