

Abstract
Membrane contact sites between organelles serve as molecular hubs for the exchange of metabolites and signals. In yeast, the Endoplasmic Reticulum − Mitochondrion Encounter Structure (ERMES) tethers these two organelles likely to facilitate the non-vesicular exchange of essential phospholipids. Present in Fungi and Amoebas but not in Metazoans, ERMES is composed of five distinct subunits; among those, Mdm12, Mmm1 and Mdm34 each contain an SMP domain functioning as a lipid transfer module. We previously showed that the SMP domains of Mdm12 and Mmm1 form a hetero-tetramer. Here we describe our strategy to diversify the number of Mdm12/Mmm1 complexes suited for structural studies. We use sequence analysis of orthologues combined to protein engineering of disordered regions to guide the design of protein constructs and expand the repertoire of Mdm12/Mmm1 complexes more likely to crystallize. Using this combinatorial approach we report crystals of Mdm12/Mmm1 ERMES complexes currently diffracting to 4.5 Å resolution and a new structure of Mdm12 solved at 4.1 Å resolution. Our structure reveals a monomeric form of Mdm12 with a conformationally dynamic N-terminal β-strand; it differs from a previously reported homodimeric structure where the N-terminal β strands where swapped to promote dimerization. Based on our electron microscopy data, we propose a refined pseudo-atomic model of the Mdm12/Mmm1 complex that agrees with our crystallographic and small-angle X-ray scattering (SAXS) solution data.
Keywords: ERMES, Membrane contact sites, Mdm12/Mmm1 complex reconstitution, SMP domain, Crystal structure, SAXS
1. Introduction
Eukaryotic cells are characterized by their exquisite compartmentalization with a multitude of organelles each fulfilling specific functions essential to cellular life. Membrane contact sites (MCSs), regions where two organelles come in close proximity to one another, act as molecular hubs for the exchange of small molecules (e.g. lipids) and signals (e.g. calcium ions) [1,2]. Lipid exchange between organelles is important for the establishment of organelle identity and proper function. While the endoplasmic reticulum (ER) is the main site for phospholipid synthesis, other organelles such as the mitochondrion rely on inter-organelle lipid exchange processes for their biogenesis. Mitochondria attached membranes (MAMs) in particular have been involved in the exchange and transfer of phospholipids between organelles [3,4]. In yeast, the endoplasmic reticulum −mitochondrion encounter structure (ERMES) is one of the well-characterized inter-organelle tethering complexes [5]. Still in yeast, other tethers have been since discovered such as the mitochondrion-vacuole tether vCLAMP [6,7] and the conserved ER membrane protein complex EMC [8], another ER-mitochondrion tether.
The ERMES is composed of five subunits: The cytosolic protein Mdm12, the ER-anchored Mmm1 subunit and the three outer-mitochondrial membrane proteins Mdm34, Mdm10 and Gem1 [9,10]. Mdm12, Mmm1 and Mdm34 all contain a synaptotagmin-like mitochondrial lipid-binding domain (SMP) (Fig. 1A and B); SMP domains are exclusively found at MCSs between different organelles such as ER-Mitochondrion, ER-Plasma Membrane and Nucleus-Vacuole junctions [11]. The crystal structure of the extended synaptotagmin-2 (E-SYT2) [12], involved in ER to plasma membrane contact sites [13], revealed that the SMP domain belongs to the TULIP (for TUbular LIPid-binding) protein superfamily of lipid transfer proteins [14−16]. Biophysical studies using pro-teoliposomes also demonstrated that the SMP domain present in E-SYTs is required for the exchange of glycerophospholipids [17]. Last, a study using a novel in vitro assay system with isolated yeast membrane fractions suggested a phospholipid transfer function for ERMES [18]. We have shown that the SMP domains of Mdm12 and Mmm1 bind glycerophospholipids and assemble into a heterotetrameric complex. Our 17 A resolution negative staining electron microscopy (NS-EM) structure revealed a distinctive architecture where two monomers of Mdm12 bind separately to a central ER-anchored Mmm1 homodimer [19]. These studies suggest that at MAMs, the SMP domains of ERMES directly mediate lipid transfer between the two organelles.
Fig. 1. ERMES and the SMP domain.
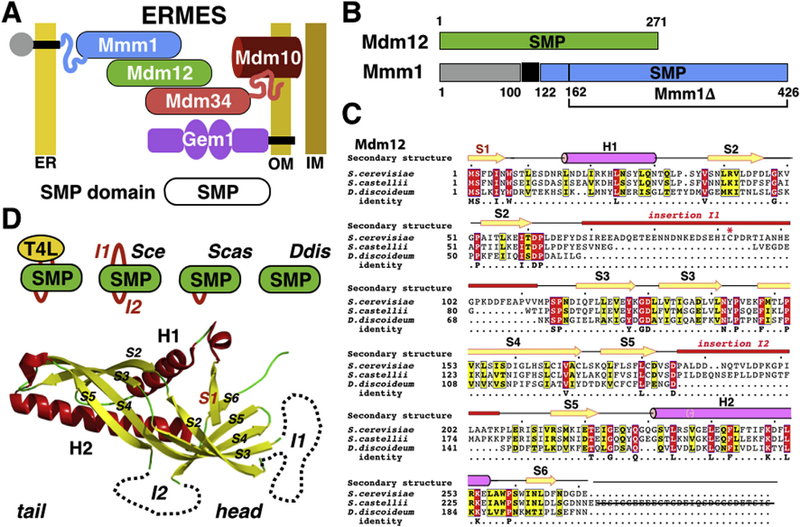
(A) Schematic of the yeast ERMES bridging the endoplasmic reticulum (ER) and mitochondria (Outer and Inner) membranes. (B) Domain organization of yeast Mdm12 and Mmm1. Mdm12 consists of an SMP domain while Mmm1 contains a luminal domain (grey), one transmembrane anchor and a single cytoplasmic SMP domain (blue). (C) Protein sequence alignments of Mdm12 in Saccharomyces cerevisiae, Saccharomyces castellii and Dictyostelium discoideum. Non-conserved insertions (I1 and I2) are highlighted. Secondary structure elements are labeled. (D) Two variable insertions in the SMP fold of Mdm12: I1 (absent in Scas and Ddis) and I2 (absent in Ddis). The T4L insert replaces the longest insertion I1. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Our structural understanding of ERMES remains limited; we thus crystallized Mdm12 and the Mdm12/Mmm1 complex previously characterized in Saccharomyces cerevisiae (Sce). To grow suitable crystals we describe here the purification, characterization and reconstitution of several Mdm12 proteins and Mdm12/Mmm1 complexes by expanding the repertoire of Mdm12 proteins available through the combined use of orthologues and protein engineering to reduce disorder. We obtained diffracting crystals of Mdm12 and Mdm12/Mmm1 complex and solved a 4.1 Å resolution crystal structure of Sce-Mdm12 revealing the monomeric nature of the SMP domain and the structural plasticity of its N-terminus.
2. Materials and methods
2.1. Protein expression and complex reconstitution
Saccharomyces castellii (Scas) Mdm12 (residues S2-E244) and Dictyostelium discoideum (Ddis) Mdm12 (residues S2-N202) proteins were expressed as His-MBP fusions using a pCDF vector. The sequence coding the 162 residues of T4 lysozyme (T4L) was inserted in the Sce-Mdm12 gene between positions S88 and S115 in the non-conserved insertion I1 (Fig. 1C and D). The chimeric protein was expressed using a pJexpress411 plasmid (DNA2.0 Inc.) (Supplementary Fig. S1). All proteins were expressed and purified following protocols described in AhYoung et al. [19]. Complexes and proteins were purified by one final size exclusion chromatography (SEC) step on a Superdex S200 HR10/30 analytical SEC column (GE Healthcare) equilibrated in 200 mM Nacl, 20 mM Tris-HCl pH = 8.0, 2% glycerol, 0.1 mM TCEP and 0.1 mM PMSF (Fig. 2).
Fig. 2. Characterization of proteins and complexes by SDS-PAGE and SEC.
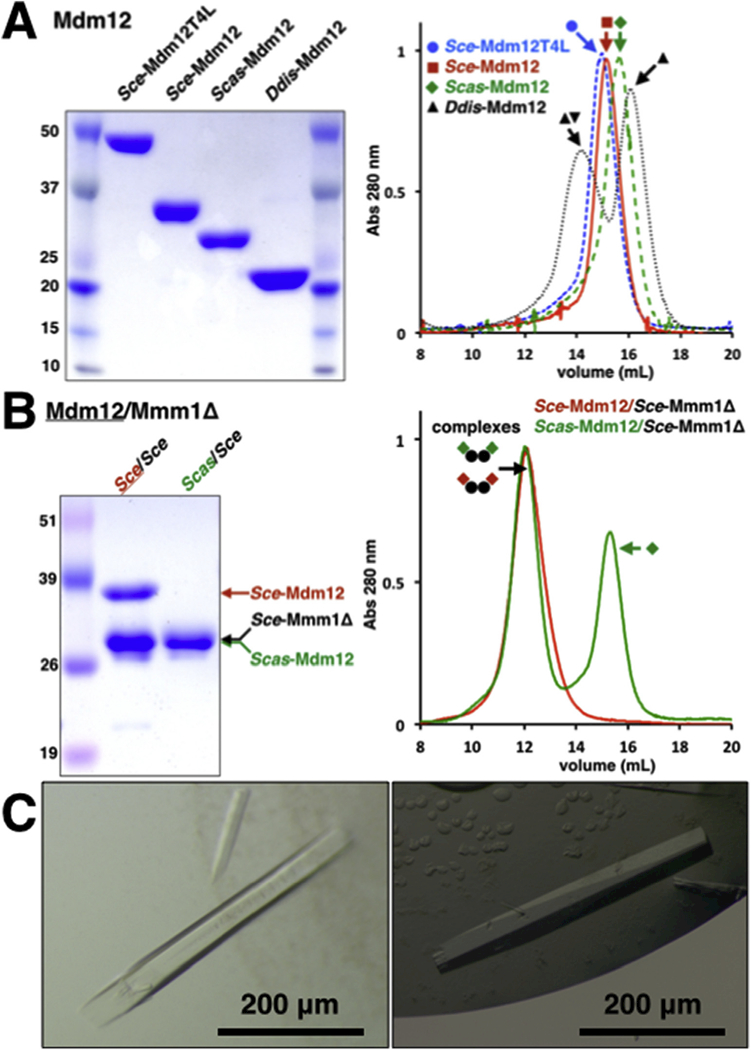
(A) Mdm12 proteins. Sce, Scas and Ddis together with the Sce-Mdm12T4L internal fusion protein. The two Ddis-Mdm12 peaks correspond to a dimer/monomer mixture, all other proteins are monomeric. (B) Mdm12/Mmm1Δ heterotetrameric complexes. Scas-Mdm12 and Sce- Mmm 1Δ have identical molecular weight and cannot be resolved on this gel. For Scas-Mdm12/Sce-Mmm1Δ complex, excess of free monomeric Scas-Mdm12 is separated from the complex. (C) Crystals of Mdm12/Mmm1Δ complexes.
2.2. Mdm12 and Mmd12/Mmm1Δ complex crystallization
Crystallization screenings were performed by vapor diffusion at 4 °C in hanging drops using protein solutions concentrated at 15 mg/ml. Protein-to-reservoir ratios of 2-to-1, 1-to-1 and 1-to-2 were tested. Crystals of Sce-Mdm12 grew in 15−25% PEG 3,350, 400 mM ammonium phosphate and 3.5 mM Mega-10 (Anatrace); they diffracted to 4.1 Å resolution and belong to rhombohedral space group P3221 with unit cell parameters a = b = 116.0 A and c = 161.7 Å with two molecules in the asymmetric unit and 78% solvent. Crystals of complex diffracted to 4.5 Å resolution and belong to one of the tetragonal space groups in the P4/mmm Laue group with unit cell parameters a = b = 167 Å and c = 89.2 Å with 1 molecule of complex in the asymmetric unit and 54% solvent.
2.3. Diffraction data collection, structure determination, and refinement
Diffraction data were collected at the Advanced Photon Source in the Argonne National Laboratory and at the Advanced Light Source in the Lawrence Berkeley National Laboratory. Crystals were cryoprotected in mother liquor supplemented with 20−25% glycerol. Data were processed in XDS [20]. The structure of Mdm12 was solved by molecular replacement with Phaser [21] using the Sce-Mmd12 crystal structures described by Jeong et al. [22] (PDB accession codes 5GYD and 5GYK) as search probes. To minimize model bias, the search model consisted in the monomer where the 14 first residues, corresponding to the swapped N-terminal β-strand S1 and the connecting loop preceding helix H1 were removed; two copies of Sce-Mdm12 were located in the asymmetric unit. Inspection of the initial unbiased Fo-Fc map revealed that only one N-terminal β-strand S1, assigned to monomer Å, could be located (Supplementary Fig. S2); the corresponding β-strand in monomer B cannot be located, likely disordered and flipped out towards the solvent. Given the low resolution of our diffraction data, we applied a negative thermal factor of −129 A2 estimated using the UCLA-DOE diffraction anisotropy server at services.mbi.ucla.edu/anisoscale [23], to sharpen the experimental electron density maps [24,25]. B-sharpened electron density maps and data were exclusively used to guide model building but not used to refine the structure. We previously used a similar approach to refine a membrane protein structure [26]. In this case, original phases are based on a molecular replacement solution using an identical structure solved at a higher resolution (3.1 Å) and in a different space group. To avoid over-fitting, three refinement cycles were done, one in Phenix [27], and two in Buster [28]. Model building was done in COOT [29]. The final model is refined to Rfree and Rcryst values of 26.3% and 24.8%, respectively, with acceptable stereochemistry and electron density maps (Supplementary Figs. S4 and S5). Crystallographic statistics are detailed in Supplementary Table 1.
2.4. Small-angle X-ray solution scattering
SAXS data were collected at the Advanced Light Source at the Lawrence Berkeley National Laboratory. Experimental conditions were as previously described [30,31]. Scattering curves for the complex model were calculated using CRYSOL [32] and pair distance distributions -P(r) - derived by Fourier inversion using GNOM [33] to estimate Dmax, the longest distance occurring in the particle, and RG, its radius of gyration.
3. Results and discussion
3.1. Identification of Mdm12 orthologues with fewer and/or shorter insertions
Bioinformatic analyses have identified ERMES in lineages outside Fungi [34]. The TULIP/SMP fold consists into a highly twisted β-sheet sandwiched between two α-helices; the resulting elongated barrel-shaped cylindrical structure harbors a lateral opening and a central hydrophobic cavity where phospholipids can bind. Sequence analysis of diverse Mdm12 protein sequences (Fig. 1C) and homology modeling reveal the presence of two non-conserved insertions 11 and 12 (Fig. 1D) located at the so-called ‘head’ region of the domain. The presence of long and/or disordered regions is a poor predictor of crystallization. Following this rationale, we sought to identify orthologues of Sce-Mdm12 with shorter insertions or no insertions. We identified two other Mdm12 proteins in Saccharomyces castellii and Dictyostelium discoideum. While Sce-Mdm12 harbors the two insertions, Mdm12 from the closely related yeast Scas only contains insertion 12 while its orthologue in the evolutionarily distant amoeba Ddis does not contain any of those insertions; Ddis-Mdm12 thus appears to represent a minimalistic version of the TULIP/SMP domain in the ERMES component Mdm12. To further expand our repertoire of constructs and improve the odds to grow diffracting crystals, we also applied an internal fusion protein engineering strategy [35] by replacing part of insertion I1 of Sce-Mdm12 with T4L.
3.2. Purification of Mdm12 orthologues and combinatorial reconstitution of Mdm12/Mmm1Δ complexes
While Sce-Mdm12 robustly expressed by itself in E. coli [19], it was necessary to express its orthologues from Scas and Ddis as N-terminal MBP fusions. Based on previous analyses [19], the orthologue from Scas and the T4L internal fusion protein behave as exclusive monomers in solution while the orthologue from Ddis is a mixture of dimers and monomers. While Sce-Mdm12 expressed in E. coli yields a mixture of dimers and monomers (although the monomer is more prominent), the same protein purified from its native organism (yeast) is only observed under its monomeric form [19]. Furthermore, the Sce-Mdm12T4L internal fusion protein is exclusively monomeric in solution (Fig. 2A).
We were able to purify the heterologous complex formed between Sce-Mmm1Δ and the Mdm12 protein from Scas but not from Ddis (Fig. 2B). This is not that surprising since the two proteins from the two different species of Saccharomyces are 58% identical while the Mdm12 from the amoeba Dictyostelium only shares ~20% sequence identity with its orthologues in Saccharomyces (Fig. 1D). We were also unable to form a complex between the SMP domains of Sce-Mmm1Δ and Sce-Mdm12T4L; this indicates that the presence of a bulky protein domain replacing most of the first non-conserved insertion I1 (Fig. 1D) does prevent complex formation. The two new complexes characterized in this study are heterotetramers of equimolecular stoichiometry.
3.3. Crystallization trials
Despite extensive effort, crystallization trials on Mdm12 orthologues met limited success, yielding numerous crystallization conditions with large but overall poorly diffracting crystals in the case of Sce-Mdm12 and, surprisingly, no crystals for the shorter Mdm12 versions from Scas and Ddis although we predicted them to be more amenable to crystallization. Crystals of Sce-Mdm12T4L proved difficult to reproduce. We eventually grew crystals of Sce-Mdm12 diffracting to 4.1 Å resolution. Sce-Mdm12/Sce-Mmm1Δ and Scas-Mdm12/Sce-Mmm1Δ complexes yielded also numerous crystals forms (Fig. 2C) that we are currently optimizing for diffraction data collection; the current diffraction limit is about 4.5 Å (Supplementary Table 1).
3.4. The crystal structure of a monomeric form of yeast Mdm12 reveals a dynamic N-terminus
We describe a new crystal structure of Sce-Mdm12 solved by molecular replacement at 4.1 A resolution using the crystal structures of Sce-Mdm12 recently published by Jeong et al. [22] as search models. Our structure corresponds to a different crystal form (i.e. rhombohedral vs orthorhombic) and crystallization condition. The high solvent (~78%) content and weak crystal packing explain the poor diffraction and high estimated Wilson B thermal factor (~143 Å2). We do not observe bound phospholipids. Our structure thus corresponds to an apo state of Mdm12 in contrast with Jeong et al. [22]; the use of detergent MEGA-10 for crystallization might explain the apo-state.
The presence of a N-terminal β-strand (S1) is a distinctive feature of the SMP fold of Mdm12 (and by homology also Mdm34) in opposition with the SMP domain of Mmm1 that is predicted to be structurally more similar to the SMP of E-SYT2 [19,22]. Differences between the Mdm12 and the E-SYT2 SMP structures were significant enough to prevent solving the structure by molecular replacement using the E-SYT2 crystal structure or homology models based on all available structures from other TULIP proteins (Supplemental Fig. S3).
In our case, Mdm12 crystallized in a rhombohedral space group in contrast with the orthorhombic crystal forms previously reported. These different crystalline form and packing reveal the dynamic behavior of the TULIP/SMP domain in Mdm12. Although two molecules of Mdm12 are present in the asymmetric unit, we do not observe a swapped dimer where the first N-terminal β strands S1 are exchanged to complete the ‘head-to-head’ dimerization interface (Fig. 3A) reported by Jeong et al. [22]. Furthermore, within the asymmetric unit, the two monomers differ in the conformation adopted by their N-terminal β-strands S1. In monomer A, β-strand S1 is well resolved in the electron density map and hydrogen bonds with β-strand S2 of the same monomer; thus it adopts a non-swapped conformation. On the other hand, the N-terminal β-strand S1 of monomer B cannot be traced and is likely to be flipped towards the solvent; it is clearly not engaged in the same inter-molecular interactions observed in monomer A (Fig. 3B). Despite the modest resolution of our data, this structural difference is unambiguous as demonstrated by the maximum likelihood weighted mFo-DFc Fourier difference map obtained after molecular replacement using a search model consisting of the monomer of Mdm12 where the first 14 residues were omitted (Supplemental Fig. S2). The non-crystallographic dimer observed in our crystal form corresponds to an ‘anti-parallel’ arrangement along the long α-helix H2 that is part of the TULIP/SMP fold; this large crystal contact interface is also observed in the Mdm12 structures recently published [22] although it does not involve residues conserved among all Mdm12 sequences.
Fig. 3. A new crystal structure of Sce-Mdm12.
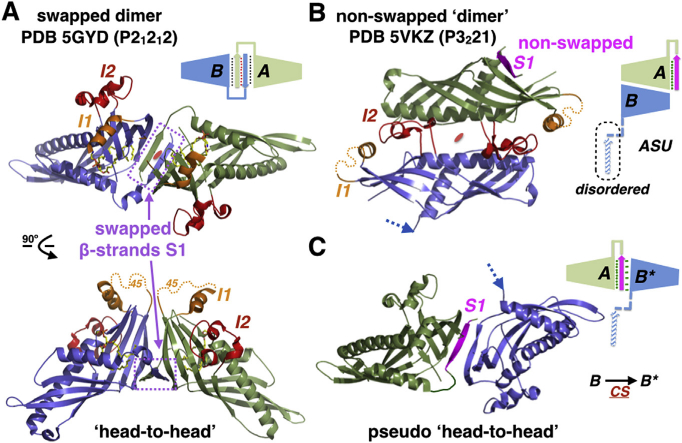
(A) Swapped ‘head-to-head’ dimer of Sce-Mdm12 observed in the 3.1 Å resolution orthorhombic structure from Jeong et al. [22]. (B) Non-swapped dimer of Sce-Mdm12 observed in our 4.1 A resolution rhombohedral structure. Two monomers (A and B) are observed in the asymmetric unit. In monomer A, the N-terminal β-strand S1 (magenta) is resolved in the electron density but does not swap. The N-terminal β-strand S1 of monomer B has flipped into a solvent-exposed conformation and cannot be resolved in the electron density maps. Insertions I1 and I2 are colored in gold and red, respectively. (C) A crystallographic symmetry-related copy of monomer B (note B*), forms a pseudo ‘head-to-head’ dimer where only one β-strand S1 (from monomer A) sits at the interface between the two SMP/TULIP domains. CS indicates that B and B* are related by a P3221crystallographic symmetry operator. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
The most peculiar aspect of the crystal packing in our rhombohedral crystal form resides in the fact that two Mdm12 monomers (i.e. monomer A and a crystallographic-symmetry related copy of monomer B, labeled B*) form a pseudo-dimer where the two molecules associate in a ‘head-to-head’ arrangement (Fig. 3C); although the two resulting ‘head-to-head’ Mdm12 dimers might look identical, they are not (Supplementary Fig. S6). Thus, although there is no swapping within the asymmetric unit there is partial swapping within the unit cell as one β-strand S1 from one SMP displaces and replaces the β-strand S1 of another SMP while still interacting with its own SMP. Within the non-swapped monomer A of our structure, β-strands S1 and S2 adopt an anti-parallel arrangement, this is the opposite of what is observed in the swapped dimers from Jeong et al. [22] where the swapped β-strands S1 and S2 run parallel to each other; however, in our asymmetric pseudo-dimer interface (A/B*) the two β-strands, S1 from monomer A and S2 from monomer B*, associate in a parallel arrangement.
This unusual case of ‘broken’ symmetry underlines two important functional aspects of the SMP domain of Mdm12 and also potentially of Mdm34 that shares a similar N-terminal sequence: First, the N-terminal β-strand of the SMP domain of Mdm12 is dynamic; second, the putative ‘head-to-head’ dimerization interface of Mdm12 appears to be somehow promiscuous. The basis for that promiscuity is rooted in the type of interactions that mediates association between the two SMP domains: Mdm12 SMP ‘dimerization’ is essentially driven by strand S1-to-strand S1 interactions through backbone-to-backbone hydrogen bonds (Supplementary Fig. S7). Within our non-swapped monomer A, N-terminal β-strand S1 completes the canonical TULIP/SMP anti-parallel β-barrel through its association with β-strand S2. The anti-parallel arrangement of β-strands into β-sheets is thermodynamically favored because it allows the inter-strand hydrogen bonds between carbonyls and amines to be planar, which is their preferred orientation; this arrangement results in a strongest inter-strand stability.
3.5. Solution conformation and improved pseudo-model of the Mdm12/Mmm1 heterotetramer of SMP domains
We published a 17 A resolution NS-EM structure of the Sce- Mdm12/Mmm1Å hetero-tetramer [19] where we established the number of Mmm1 and Mdm12 subunits present, together with their relative positions, within the elongated crescent-shaped complex (Fig. 4A). Our predictions supported a model where two Mmm1 SMP domains associate to form a homodimer similar to the ‘head-to-head’ E-SYT2 homodimer of SMP domains [12] (Fig. 4B). EM electron density maps display a distinct mass of density near the putative Mmm1-to-Mdm12 interface (Fig. 4B). The best fit between EM density and crystal structures can only be achieved through a ‘tail-to-head’ association between the ‘tail’ of Mmm1 and the ‘head’ of Mdm12 (Fig. 4). A last argument in favor of this model resides in the observation that the Sce-Mdm12T4L protein cannot form a complex; given the position of the fused T4L near the ‘head’ region (Fig. 1D), the resulting steric hindrance could prevent association with Mmm1.
Fig. 4. Pseudo-atomic model of the Sce-Mdm12/Mmm1Δ hetero-tetramer and solution scattering analysis of its average conformation.
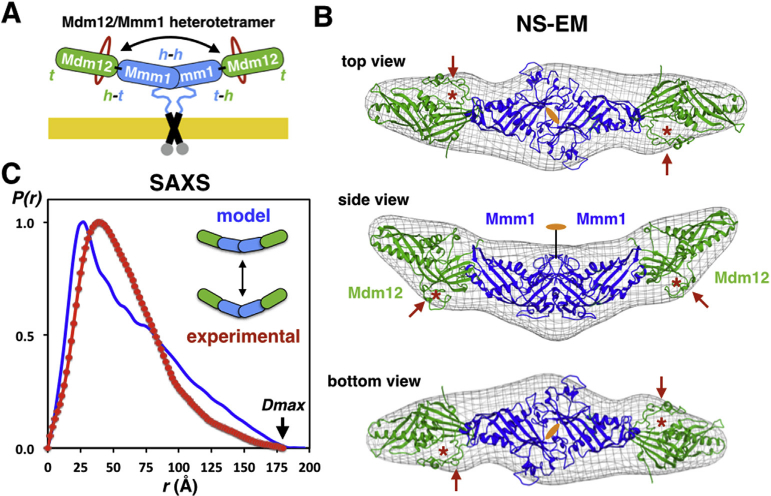
(A). Schematic model of the Mdm12/ Mmm1 complex. Insertions in Mdm12 are depicted in red; h and t correspond to the ‘head’ and ‘tail’ regions of each of the four SMP domains, respectively. The ‘head-to-head’ dimer of Mmm1 SMP domains is anchored to the ER membrane. The double arrow highlights the curvature/bent of the complex. (B) Fitting of our Mdm12/Mmm1Δ model using the crystal structure of Sce-Mdm12 in the EM density maps [19]. Red arrows and asterisks indicate the two insertions located next to the ‘head’ in yeast Mdm12. Three views are shown. (C) SAXS analysis of the Sce-Mdm12/Mmm1Δ complex. Comparison of the pair distance distributions determined from experimental scattering data (red) or calculated using our NS-EM/crystallographic mode (blue). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
We characterized the solution conformation of the Sce-Mdm12/ Mmm1Δ complex using SAXS [36] by comparing its experimental pair distance distribution with the one calculated using our pseudo-model based on NS-EM and crystallographic data. The experimental curve is characteristic of a rod-like elongated structure [37]; the longest distance Dmax of ~185 Å is close to the one inferred from the model and can only result from four SMP domains aligning along their longest axis. Variation in the curvature/bend of the complex could explain the discrepancy between experimental and calculated and Dmax values (Fig. 4C and Supplementary Table S2).
3.6. Interactions between SMP domains and assembly of ERMES
Interactions between the SMP domains of Mdm12 and Mmm1 are strong. We previously showed that the three SMP domains of Mmm1, Mdm12, and Mdm34 form a weak ternary complex and that Mdm12 and Mdm34 are interacting directly [19] suggesting that Mdm12 acts as a bridging subunit between the ER-bound Mmm1 and the mitochondria-bound subunits Mdm34 and Mdm10. Jeong et al. [22] showed evidence for a weak interaction between Mdm12 and a fragment of Mdm34 corresponding to the first residues of its SMP domain and observed that the N-termini of Mdm12 and Mdm34 share common sequence and secondary structure features. Thus, the conformational dynamics and plasticity of the N-terminus of the SMP domain of Mdm12 revealed by two distinct crystal structures might also apply to Mdm34. Although both monomeric and dimeric forms of Mdm12 have been observed in solution and their crystal structures determined, their biological significance needs to be further investigated. Our model predicts that the N-terminus of Mdm12 is engaged at the Mdm12/ Mmm1 interface, thus formation of “our” complex implies dissociation of a swapped Mdm12 homodimer. The structure of the Mdm12/Mmm1 complex will provide valuable insights into the molecular mechanisms for the transfer of phospholipids by the SMP domains of ERMES at MAMs.
Supplementary Material
Acknowledgements
This work was supported by the UCLA Geffen School of Medicine, the Stein-Oppenheimer Seed Grant Award and the Alexander and Renée Kolin Endowed Chair in Molecular Biology and Biophysics to P.F.E. A.P.A.Y. was supported by a Gates Millennium Fellowship and the UCLA Dissertation of the Year Fellowship. We thank the staffs from beamlines 24-ID (APS), 8.3.1 and 12.3.1 (ALS) for their assistance.
Footnotes
Appendix A. Supplementary data
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.bbrc.2017.05.021.
Transparency document
Transparency document related to this article can be found online at http://dx.doi.org/10.1016/j.bbrc.2017.05.018.
Accession number
Coordinates and structure factors have been deposited under the Protein Data Bank accession code: 5VKZ.
References
- [1].Lahiri S, Toulmay A, Prinz WA, Membrane contact sites, gateways for lipid homeostasis, Curr. Opin. Cell Biol 33 (2015) 82–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Gonzalez Montoro A, Ungermann C, StARTing to understand membrane contact sites, Trends Cell Biol. 25-9 (2015) 497–498. [DOI] [PubMed] [Google Scholar]
- [3].Vance JE, MAM (mitochondria-associated membranes) in mammalian cells: lipids and beyond, Biochim. Biophys. Acta 1841 (2014) 595–609. [DOI] [PubMed] [Google Scholar]
- [4].Vance JE, Phospholipid synthesis and transport in mammalian cells, Traffic 16 (2015) 1–18. [DOI] [PubMed] [Google Scholar]
- [5].Kornmann B, Currie E, Collins SR, Schuldiner M, Nunnari J, Weissman JS, Walter P, An ER-mitochondria tethering complex revealed by a synthetic biology screen, Science 325 (2009) 477–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Elbaz-Alon Y, Rosenfeld-Gur E, Shinder V, Futerman AH, Geiger T, Schuldiner M, A dynamic interface between vacuoles and mitochondria in yeast, Dev. Cell 30 (2014) 95–102. [DOI] [PubMed] [Google Scholar]
- [7].Honscher C, Mari M, Auffarth K, Bohnert M, Griffith J, Geerts W, van der Laan M, Cabrera M, Reggiori F, Ungermann C, Cellular metabolism regulates contact sites between vacuoles and mitochondria, Dev. Cell 30 (2014) 86–94. [DOI] [PubMed] [Google Scholar]
- [8].Lahiri S, Chao JT, Tavassoli S, Wong AK, Choudhary V, Young BP, Loewen CJ, Prinz WA, A conserved endoplasmic reticulum membrane protein complex (EMC) facilitates phospholipid transfer from the ER to mitochondria, PLoS Biol. 12 (2014) e1001969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kornmann B, Osman C, Walter P, The conserved GTPase Gem1 regulates endoplasmic reticulum-mitochondria connections, Proc. Natl. Acad. Sci. U. S. A 108 (2011) 14151–14156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Stroud DA, Oeljeklaus S, Wiese S, Bohnert M, Lewandrowski U, Sickmann A, Guiard B, van der Laan M, Warscheid B, Wiedemann N, Composition and topology of the endoplasmic reticulum-mitochondria encounter structure, J. Mol. Biol 413 (2011) 743–750. [DOI] [PubMed] [Google Scholar]
- [11].Toulmay A, Prinz WA, A conserved membrane-binding domain targets proteins to organelle contact sites, J. Cell Sci 125 (2012) 49–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Schauder CM, Wu X, Saheki Y, Narayanaswamy P, Torta F, Wenk MR, De Camilli P, Reinisch KM, Structure of a lipid-bound extended synaptotagmin indicates a role in lipid transfer, Nature 510 (2014) 552–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Fernandez-Busnadiego R, Saheki Y, De Camilli P, Three-dimensional architecture of extended synaptotagmin-mediated endoplasmic reticulum-plasma membrane contact sites, Proc. Natl. Acad. Sci. U. S. A 112 (2015) E2004–E2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kopec KO, Alva V, Lupas AN, Homology of SMP domains to the TULIP superfamily of lipid-binding proteins provides a structural basis for lipid exchange between ER and mitochondria, Bioinformatics 26 (2010) 1927–1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Reinisch KM, De Camilli P, SMP-domain proteins at membrane contact sites: structure and function, Biochim. Biophys. Acta 18-5 (2015) 504–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Alva V, Lupas AN, The TULIP superfamily of eukaryotic lipid-binding proteins as a mediator of lipid sensing and transport, Biochim. Biophys. Acta 1861 (2016) 913–923. [DOI] [PubMed] [Google Scholar]
- [17].Saheki Y, Bian X, Schauder CM, Sawaki Y, Surma MA, Klose C, Pincet F, Reinisch KM, De Camilli P, Control of plasma membrane lipid homeostasis by the extended synaptotagmins, Nat. Cell Biol 18 (2016) 504–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kojima R, Endo T, Tamura Y, A phospholipid transfer function of ER-mitochondria encounter structure revealed in vitro, Sci. Rep 6 (2016) 30777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].AhYoung AP, Jiang J, Zhang J, Khoi Dang X, Loo JA, Zhou ZH, Egea PF, Conserved SMP domains of the ERMES complex bind phospholipids and mediate tether assembly, Proc. Natl. Acad. Sci. U. S. A 112 (2015) E3179–E3188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kabsch W, Xds, Acta Crystallogr. D. Biol. Crystallogr 66 (2010) 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ, Phaser crystallographic software, J. Appl. Crystallogr 40 (2007) 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Jeong H, Park J, Lee C, Crystal structure of Mdm12 reveals the architecture and dynamic organization of the ERMES complex, EMBO Rep 17 (2016) 1857–1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Strong M, Sawaya MR, Wang S, Phillips M, Cascio D, Eisenberg D, Toward the structural genomics of complexes: crystal structure of a PE/PPE protein complex from Mycobacterium tuberculosis, Proc. Natl. Acad. Sci. U. S. A 103 (2006) 8060–8065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].DeLaBarre B, Brunger AT, Considerations for the refinement of low-resolution crystal structures, Acta Crystallogr. Sect. D-Biol. Crystallogr 62 (2006) 923–932. [DOI] [PubMed] [Google Scholar]
- [25].Brunger AT, DeLaBarre B, Davies JM, Weis WI, X-ray structure determination at low resolution, Acta Crystallogr. Sect. D-Biol. Crystallogr 65 (2009) 128–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Egea PF, Stroud RM, Lateral opening of a translocon upon entry of protein suggests the mechanism of insertion into membranes, Proc. Natl. Acad. Sci. U. S. A 107 (2010) 17182–17187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Adams PD, Afonine PV, Bunkoczi G, Chen VB, Echols N, Headd JJ, Hung LW, Jain S, Kapral GJ, Grosse Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner RD, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH, The Phenix software for automated determination of macromolecular structures, Methods 55 (2011) 94–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Blanc E, Roversi P, Vonrhein C, Flensburg C, Lea SM, Bricogne G, Refinement of severely incomplete structures with maximum likelihood in BUSTER-TNT, Acta Crystallogr. Sect. D-Biol. Crystallogr 60 (2004) 2210–2221. [DOI] [PubMed] [Google Scholar]
- [29].Emsley P, Cowtan K, Coot: model-building tools for molecular graphics, Acta Crystallogr. D. Biol. Crystallogr 60 (2004) 2126–2132. [DOI] [PubMed] [Google Scholar]
- [30].Peng M, Cascio D, Egea PF, Crystal structure and solution characterization of the thioredoxin-2 from Plasmodium falciparum, a constituent of an essential parasitic protein export complex, Biochem. Biophys. Res. Commun 456 (2015) 403–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].AhYoung AP, Koehl A, Cascio D, Egea PF, Structural mapping of the ClpB ATPases of Plasmodium falciparum: targeting protein folding and secretion for antimalarial drug design, Protein Sci. 24 (2015) 1508–1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Svergun DI, Barberato C, Koch MH, CRYSOL: a program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates, J. Appl. Crystallogr 28 (1995) 768–773. [Google Scholar]
- [33].Svergun DI, Determination of the regularization parameter in indirect transform methods using perceptual criteria, J. Appl. Crystallogr 25 (1992) 495–503. [Google Scholar]
- [34].Wideman JG, Gawryluk RMR, Gray MW, Dacks JB, The ancient and widespread nature of the ER-mitochondria encounter structure, Mol. Biol. Evol 30 (2013) 2044–2049. [DOI] [PubMed] [Google Scholar]
- [35].Prive GG, Verner GE, Weitzman C, Zen KH, Eisenberg D, Kaback HR, Fusion proteins as tools for crystallization: the lactose permease from Escherichia coli, Acta Crystallogr. D. Biol. Crystallogr 50 (1994) 375–379. [DOI] [PubMed] [Google Scholar]
- [36].Blanchet CE, Svergun DI, Small-angle X-ray scattering on biological macromolecules and nanocomposites in solution, Annu. Rev. Phys. Chem 64 (2013) 37–54. [DOI] [PubMed] [Google Scholar]
- [37].Ortega E, Manso JA, Buey RM, Carballido AM, Carabias A, Sonnenberg A, de Pereda JM, The structure of the plakin domain of plectin reveals an extended rod-like shape, J. Biol. Chem 291 (2016) 18643–18662. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.