

Abstract
CRISPR/Cas9 has emerged as a revolutionary tool for fast and efficient targeted gene knockouts and genome editing in almost any organism. The laboratory model tunicate Ciona is no exception. Here we describe our latest protocol for the design, implementation, and evaluation of successful CRISPR/Cas9-mediated gene knockouts in somatic cells of electroporated Ciona embryos. Using commercially available reagents, publically accessible plasmids, and free web-based software applications, any Ciona researcher can easily knock out any gene of interest in their favorite embryonic cell lineage.
Developmental biologists have always been interested in targeted loss-of-function mutations to probe the role of specific genes in embryogenesis and regeneration. One approach towards this goal has been to engineer the sequence-specificity of DNA-binding domains found in natural transcription factors. When these customized DNA-binding proteins are fused to DNA nuclease domains, they are capable of inducing site-specific double-stranded breaks (DSBs), resulting in mutations through improper repair of these breaks by non-homologous end joining (NHEJ). Among these engineered reagents are the Zinc Finger Nucleases (ZFNs)(Beerli and Barbas 2002; Bibikova et al. 2003; Maeder et al. 2008) and Transcription Activator-Like Effector Nucleases (TALENs)(Christian et al. 2010; Miller et al. 2011). Both ZFNs and TALENs have been used for targeted mutagenesis in Ciona embryos (Kawai et al. 2012; Treen et al. 2014; Yoshida et al. 2014).
While these programmable nucleases made it possible to cause site-directed DSBs at any part of the genome, even in a tissue- or cell lineage-specific manner, expensive and tedious cloning procedures posed as a barrier to their widespread adoption and hampered their scaling for higher-throughput applications such as genome-wide reverse genetic screens. More recently, a targeted platform known as Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/Cas9 was developed, based on the immune response mechanism of Streptococcus bacteria (Barrangou et al. 2007; Jinek et al. 2012; Cong et al. 2013; Jinek et al. 2013; Mali et al. 2013). In these bacteria, processed short CRISPR RNA sequences guide the Cas9 protein to specific target sites on foreign DNA. Cas9 is characterized by two signature nuclease domains, and interacts with a DNA sequence (‘NGG’ for S. pyogenes Cas9) known as the Protospacer Adjacent Motif (PAM). Sequence-specific base-pairing between the Cas9-associated short RNAs and protospacer DNA sequence of 20 bp adjacent to the PAM then triggers the protein’s nuclease activity, resulting in cleavage of both strands of the target sequence (Garneau et al. 2010; Deltcheva et al. 2011; Gasiunas et al. 2012; Anders et al. 2014; Jinek et al. 2014).
In its native context, two distinct short RNAs guide Cas9: CRISPR RNA (crRNA) and trans-activating crRNA (tracrRNA). However, a chimeric “single-guide RNA” (sgRNA) is sufficient to mimic the roles of these two components (Jinek et al. 2012). This small but profound improvement has helped launch CRISPR/Cas9 as a cheap, simple, and efficient system for targeted mutagenesis in a remarkably wide variety of organisms (Perry and Henry 2015; Iaffaldano et al. 2016; Long et al. 2016; Nomura et al. 2016; Nymark et al. 2016; Tian et al. 2016), as well as in tunicates (Sasaki et al. 2014; Stolfi et al. 2014; Abdul-Wajid et al. 2015; Cota and Davidson 2015; Gandhi et al. 2016; Segade et al. 2016; Tolkin and Christiaen 2016).
Modifications to the CRISPR/Cas9 system have allowed for further applications, such as targeted knock-ins (Wang et al. 2013), transcriptional activation or repression (Maeder et al. 2013; Perez-Pinera et al. 2013; Qi et al. 2013), chromatin modifications (Hilton et al. 2015), and the visualization of genome organization and dynamics (Chen et al. 2013), although these approaches have yet to be adapted to tunicates. Similarly, other CRISPR variants such as CRISPR/Cpf1 have been developed for targeted mutagenesis in mammalians (Kleinstiver et al. 2015; Zetsche et al. 2015), but their effects have not yet been tested in Ciona.
In Ciona, the most widely used application of CRISPR to date is for targeted mutagenesis in somatic cells of transiently-transfected (electroporated) embryos. In this method, in vitro-fertilized embryos are electroporated at the one-cell stage with plasmids that drive the zygotic expression of Cas9 protein and sgRNAs. While sgRNAs are transcribed ubiquitously from a U6 small RNA promoter (Nishiyama and Fujiwara 2008), by RNA polymerase III (RNAPolIII), Cas9 can be expressed in a cell-specific manner by using a lineage-specific promoter. We use a humanized Cas9 flanked by nuclear localization signals (NLS::Cas9::NLS)(Chen et al. 2013; Stolfi et al. 2014), though other Cas9 variants have not been thoroughly evaluated in Ciona. Targeted mutations will occur only when both Cas9 and the sgRNA are present, and can happen on different sister chromatids in different cells at different times. This means that each embryo is actually a mosaic composed of cells bearing a combination of wildtype and/or distinct mutant alleles. In spite of this mosaicism, somatic knockouts are a powerful means to dissect the tissue-specific functions of a gene in development.
Here we present our latest protocols for generating successful CRISPR/Cas9-mediated mutagenesis (hereinafter referred to as “CRISPR knockouts”) in somatic cells of Ciona embryos, based on our published and unpublished reports (Stolfi et al. 2014; Gandhi et al. 2016). The aim of this chapter is to empower laboratories working on Ciona (and other tunicates) to harness the power of this simple but very effective tool. The protocols presented here only use widely available commercial reagents, and all plasmids can be ordered from Addgene (https://www.addgene.org/Lionel_Christiaen/).
sgRNA design
Perhaps nothing is more important for successful CRISPR knockouts in Ciona than selecting the right sgRNAs, which vary widely in their ability to actually induce Cas9-mediated DSBs. We refer to this as sgRNA mutagenesis “activity” or, more precisely, efficacy. Some sgRNAs will be highly active, while others may not yield detectable mutations. Predicting which sgRNAs will cause either frequent or rare mutations is a arduous and potentially frustrating task. Many high-throughput studies have sought to create predictive algorithms to distinguish, a priori, “good” vs. “bad” sgRNAs. A recent meta-study of these methods (Haeussler et al. 2016) concluded that most available algorithms do not accurately predict the activity of sgRNAs outside a narrow range of organisms, cell types, or experimental conditions. The authors recommended two such algorithms, depending on the method of sgRNA transcription (in vivo by RNA polymerase III, or in vitro by viral T7 RNA polymerase). This is because the efficacy of an sgRNA is probably contingent upon its expression level and stability, which will vary depending on the methods used to transcribe it. According to their comparisons, Fusi/Doench is the more accurate predictive algorithm for in vivo-transcribed sgRNAs in metazoans including Ciona (Fusi et al. 2015; Doench et al. 2016), while CRISPRScan (Moreno-Mateos et al. 2015) is recommended for predicting the activity of T7-transcribed sgRNAs.
The CRISPOR portal incorporates these findings and features into a useful web-based CRISPR sgRNA design tool (http://crispor.tefor.net/)(Haeussler et al. 2016). The input is any sequence from the Ciona genome (three different assembly versions are supported), and the output is every valid sgRNA target, their scores by the various algorithms used to predict efficacy and specificity, and primer sequences for constructing an expression vector.
Important considerations for sgRNA design and selection include not only predicted cutting efficiency, but also off-target effects and possible escape by polymorphisms in the target sequence. Ideally, an sgRNA should match extensively only one site in the genome (the target site) and no other site, which could be potentially cleaved as a result. On the other hand, single nucleotide polymorphisms (SNPs) and other naturally occurring mutations can prevent sgRNA pairing to the intended target, precluding efficient cleavage by Cas9. While the compact genome of Ciona depresses off-target effects, SNPs are extremely frequent in genetically diverse wild Ciona populations (Satou et al. 2012). CRISPOR v4.0 takes both off-targets and SNPs into account. Individual SNPs and sites of potential off-target effect are shown for each candidate sgRNA, which allows the user to choose whether the sgRNA is worth using or not.
Considerable attention must also be paid to selecting the location of the sgRNA target within a locus of interest. Our analysis of CRISPR/Cas9 knockouts in Ciona indicates that, as in other organisms, NHEJ repair of targets cleaved by Cas9 overwhelmingly favors short indels (Gandhi et al. 2016). If targeting coding sequence, there is a 2-in-3 chance that the indel will result in a frameshift, and likely premature stop codon. Conversely, there is a 1-in-3 chance that an in-frame indel will be generated, which may or may not affect the function of the resulting protein. Bear in mind that, once an indel is generated, the sgRNA will no longer match to the target site. This means that CRISPR/Cas9-generated mutations are all-or-nothing and irreversible. If deleting a few amino acid residues from the target region does not affect the function of your protein of interest, then 1/3 of the alleles in your embryo will be virtually wild-type, even assuming a 100% mutagenesis rate.
While a short out-of-frame indel can result in a loss-of-function allele, in certain cases the truncated protein may act as a neomorphic variant, like a “dominant-negative”. The further the target is from the translation start site, the higher the chance that a CRISPR/Cas9-generated indel will result in a truncated protein. However, if the indel is too close to the translation start, translation initiation may simply shift to a downstream start codon, with little impact on resulting protein function. Thus, selecting a good sgRNA also depends on finding this “sweet spot”, which will vary from protein to protein.
An effective strategy to circumvent all these potential pitfalls is to use two or more highly active sgRNAs in combination. This increases the odds of generating at least one out-of-frame indel, and the large deletions spanning multiple targets have been consistently observed in Ciona embryos (Gandhi et al. 2016), the largest deletion reported being ~13 kb (Abdul-Wajid et al. 2015).
sgRNA expression cassette construction by One-step Overlap PCR (OSO-PCR)
CRISPOR will return a list of sgRNA targets and their relevant efficacy and specificity scores and information. A link is provided for each target to a page that lists the oligonucleotide sequences one needs to order to construct the sgRNA expression vector according to a variety of strategies. For Ciona, the relevant primers are for One-Step Overlap PCR (OSO-PCR)(Urban et al. 1997), which allows for the rapid synthesis of a U6>sgRNA cassette in a single PCR reaction (Gandhi et al. 2016). The target-specific sequence (the “protospacer”) of any sgRNA cassette is only 19 bp. Thus, in OSO-PCR, limiting amounts of unique overlap primers generate a protospacer “bridge” between universal U6 promoter and sgRNA scaffold sequences, which are amplified from separate template molecules. In Ciona, a modified sgRNAF+E scaffold is used to increase stability and decrease premature termination of transcription (Orioli et al. 2011; Chen et al. 2013; Stolfi et al. 2014).
sgRNA expression cassettes can then be electroporated directly into Ciona embryos as unpurified PCR products for in vivo transcription, or further processed/purified for cloning into plasmid for long-term storage/propagation. We can reliably detect mutagenesis activity of sgRNAs transcribed in embryos electroporated with as little as 20 μl of unpurified OSO-PCR reaction per 700 μl electroporation volume (see Peakshift assay, below). This makes it possible to test a large number of candidate sgRNAs quickly.
Step-by-step protocol (adapted from Gandhi et al. 2016):
1- If selecting target using CRISPOR, select those with high Fusi/Doench scores (>60) and no known SNPs or off-targets. Click on “PCR primers” link underneath the target sequence and you will find the pre-designed primers for OSO-PCR ready to be ordered from your preferred oligonucleotide vendor. With oligos in hand, skip ahead to step 5.
If you have to identify targets and design primers manually, look for candidate targets of N(19) + PAM (“NGG”) sequence.
![]()
2- Add a “G” to 5’ end of target sequence, to obtain a G+(N)19 sequence. Initial “G” is important for transcription start by PolIII.
![]()
3- Append “GTTTAAGAGCTATGCTGGAAACAG” to the 3’ end of the G+N(19) sequence. This is now the forward primer used to amplify the sgRNA scaffold part of the cassette
![]()
4- Copy reverse complement of G+N(19), append  to the 3’ end of this now. This is the reverse primer to amplify the U6 promoter part of the cassette
to the 3’ end of this now. This is the reverse primer to amplify the U6 promoter part of the cassette
![]()
5- Set up the following PCR reaction.Template plasmids are available from Addgene (https://www.addgene.org/Lionel_Christiaen/):

6- Check 2 μl of the PCR reaction on a gel. There should be a strong band at ~1.2 kbp. If the band is only 1 kbp, the fusion did not occur. In our hands, the success rate is 94%.
Cloning OSO-PCR cassette using In-Fusion
Although OSO-PCR cassettes can be directly tested in Ciona by co-electroporation with Cas9 expression plasmid, they can also be processed for cloning into an empty plasmid vector. This allows for their replication and long-term propagation in E. coli cells, and preparation of pure, highly concentrated sgRNA expression vector plasmid DNA for electroporations. We recommend using the In-Fusion restriction enzyme-free cloning system from Clontech/Takara (https://www.clontech.com/), though restriction enzyme cloning and other systems can be used as well.
Step-by-step procol:
1- Set up a “Boost” PCR reaction to add 15-nt overhangs to the ends of the cassette required for cloning into the empty vector:

2- Add 2 μl DpnI enzyme to the reaction and incubate for 2 hours at 37°C. This will digest any remaining template plasmid.
3- Gel-purify boost PCR band, elute in 50 μl water.
4- Set up In-Fusion reaction and incubate at 50°C for 20 minutes:
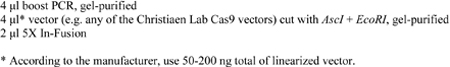
5- Transform 1 μl in 25 μl of Stellar competent E. coli cells, which come with In-Fusion kit, and plate on LB ampicillin agar plate.
6- Pick and grow at least 4 colonies, and screen for positive clones by colony PCR directly on cultured E. coli cells using the U6 forward primer (5’- TGGCGGGTGTATTAAACCAC -3’) and the In-Fusion reverse primer. The correct band should be ~1 kb in length.
Conventional sgRNA expression vector assembly
sgRNAs expression vectors can also be directly assembled in plasmid form by traditional ligation of annealed oligonucleotides into linearized vector. Our initial sgRNA vectors were constructed this way and this T4-ligase based method is indeed a faster and more reliable approach for obtaining sgRNA expression plasmids. The obvious downside is that colony selection and plasmid preparation must be performed before testing sgRNA efficacy, which is notoriously difficult to predict a priori. As a result, we do not recommend the following method to assemble untested sgRNAs. However, this is a suitable approach to recreate expression vectors for sgRNAs that have already been tested and validated.
Step-by-step protocol (adapted from Stolfi et al. 2014):
1- Given the same N(19) + PAM (“NGG”) target sequence that was provided as an example for OSO-PCR design:
![]()
2- Add a “G” to 5’ end of target sequence, to obtain a G+(N)19 sequence.
![]()
3- Append  to the 5’ end of the G+N(19) sequence. This is now the sense oligonucleotide to be ordered:
to the 5’ end of the G+N(19) sequence. This is now the sense oligonucleotide to be ordered:
![]()
4- Copy reverse complement of G+N(19), append  to the 5’ end of this now. This is the antisense oligonucleotide:
to the 5’ end of this now. This is the antisense oligonucleotide:
![]()
5- Anneal the oligonucleotides at 10 μM by boiling for 5 minutes in 10 mM Tris pH 7.5, 50 mM NaCl and then cooling naturally to room temperature.
![]()
6- Dilute the annealed oligos 1:1000 and ligate this into U6>sgRNA(F+E) linearized with BsaI:
![]()
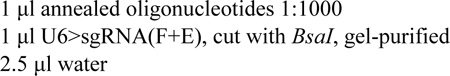

7- Transform this ligation into E. coli cells, and screen colonies by PCR using U6 forward primer and the antisense oligonucleotide detailed above as a reverse primer.
Assaying CRISPR knockouts
Either in plasmid or unpurified, PCR product format, sgRNA expression constructs should be assayed for their ability to cause on-target CRISPR knockouts. We have encountered a wide range of mutagenesis efficacies, from 0% to >60%, estimated by next-generation sequencing (Gandhi et al. 2016). Thus, it is advised that one test 4 to 8 candidate sgRNAs per target in order to identify the most effective ones to use in further experiments.
It is not absolutely necessary to use an sgRNA expression plasmid to assay its efficacy. We have verified highly active sgRNAs expressed from unpurified OSO-PCR products electroporated into Ciona embryos. This has allowed us to quickly test the efficacies of large numbers of sgRNAs, either by target sequence analysis or by phenotypic assay (Gandhi et al. 2016). Typically, 15 to 45 µl of unpurified products can be added to a single 700 µl electroporation solution, together with the Cas9 vector. However, the linear nature of the PCR product, and the reagents present in the reaction may interfere with normal development. Therefore, our current strategy is to assay sgRNA efficacy using OSO-PCR products, but then clone those products that prove most effective into a plasmid for use for publication-quality experiments.
There are different methods to estimate sgRNA efficacies in a quantitative manner. A very basic approach consists of amplifying target regions by PCR and cloning these products into a plasmid vector, then sequencing a handful of clones and counting the number of mutant clones (Sasaki et al. 2014; Stolfi et al. 2014). However, this approach is very time consuming, labor-intensive, and not accurate since a very large number of clones would need to be sequenced to approach a reliable sample size.
sgRNA efficacies have also been measured in Ciona by Cel-I nuclease assay (Sasaki et al. 2014) or Thermo Fisher Scientific GeneArt Genomic Cleavage Detection kit (Stolfi et al. 2014). These methods depend on nucleases that recognize and cleave DNA bulges resulting from hybridization of DNA strands bearing distinct indels. The result is smaller “cleavage bands” that can be measured by fluorescence intensity on an agarose gel. However, the nuclease will also cleave bulges resulting from single-nucleotide mismatches, which is extremely problematic when using this assay on animals from a highly polymorphic population, as we do for Ciona.
More recently, we have employed next-generation sequencing to calculate the ratio of mutant and wild-type sequences amplified by PCR (Gandhi et al. 2016). This approach allowed us to assay the efficacies of over 80 sgRNAs in parallel, by pooling PCR products amplified from embryos electroporated with different sgRNA vectors. However, the cost and depth of this method of sequencing would not be justified if you were only measuring a handful sgRNAs at a time. Therefore, we only recommend the next-generation sequencing route for large-scale assays (>100 sgRNAs).
Sanger sequencing-based “peakshift” assay for sgRNA activity
Currently, our recommended approach for estimating the efficacies of a few sgRNAs at a time is to use Sanger sequencing of target sequence PCR products. This is a relatively simple and cost-effective method that returns highly consistent, fairly quantitative estimates of sgRNA efficacy. Unlike next-generation sequencing, Sanger sequencing cannot resolve the sequences of individual molecules, but rather returns a composite of all the molecules sequenced in the reaction. Normally, the sequence is readable because all the molecules are identical. However, when you have many products bearing short indels due to CRISPR, the peaks in a typical Sanger sequencing trace will appear mixed, with signal for more than one nucleotide base at the same position in the sequence (Figure 1). This “peakshift” can be quantified by algorithms such as the ab1 Peak Reporter by Thermo Fisher Scientific (https://apps.thermofisher.com/ab1peakreporter/) (Roy and Schreiber 2014). We have shown a nearly linear correlation between CRISPR knockout peakshifts measured by ab1 Peak Reporter and frequency of a loss-of-function phenotype in F0 (Gandhi et al. 2016). This suggests that the sgRNAs that produce the highest peakshifts are the most effective at generating loss-of-function alleles, which is ultimately the goal of CRISPR knockout experiments.
Fig.1. CRISPR indels.
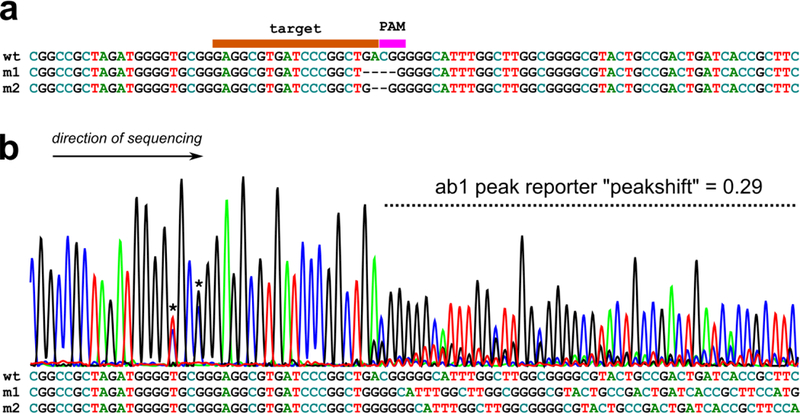
a) Wild-type (“wt”) target sequence aligned with two CRISPR knockout mutant sequences (“m1” and “m2”) generated by imprecise repair of CRISPR/Cas9-mediated double-stranded breaks. Alignment shows gaps (−) in place of missing nucleotides in target or PAM sequence. b) When sequenced by Sanger sequencing, pools of wild-type and mutant sequences will produce a “peakshift”, which can be quantified by ab1 Peak Reporter web app (see text for details). Below, the same sequences in (a) aligned without gaps, showing the cause of the overlapping peaks seen in the peakshift area. Asterisks denote naturally-occurring single-nucleotide polymorphisms.
Up to three sgRNA cassettes targeting different genes have been electroporated in the same embryos and assayed in this manner, and their efficacies do not seem to be hampered by this multiplexing (A.S., unpublished observation). However, one must pay attention not to test targets that are on the same chromosome, since large deletions or chromosomal breaks may occur as a result. What follows is a protocol for electroporating a given sgRNA construct (plasmid or OSO-PCR) and assaying its mutagenesis efficacy by peakshift.
Step-by-step protocol (adapted from Gandhi et al. 2016):
1- Following the standard electroporation protocol (Christiaen
et al. 2009), prepare an electroporation mix:
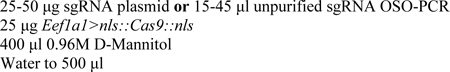
This solution is then mixed with 200 μl sea water containing fertilized Ciona eggs for electroporation.
2- Grow embryos at 18–24°C until hatching. Collect hatched larvae and extract genomic DNA using the QIAamp DNA Micro Kit (Qiagen) following a modified protocol.
-
Modifications to manufacturer’s protocol:
Lyse embryos in 180 μl Buffer ATL + 5 μl proteinase K for 30 minutes
Use carrier RNA (as supplied by kit)
Elute DNA in 20 μl water
3- Measure the extracted DNA using a spectrophotometer. Prepare the following PCR reaction to amplify the target sequence. For best results, you should aim to design primers to amplify a fragment 300–1500 bp long, with the target site(s) at least 150 bp away from either end of the fragment. We prefer Pfx platinum from Thermo Fisher Scientific, but any proof-reading polymerase should suffice.

4- Column- or gel-purify the resulting PCR product, and send off for Sanger sequencing. The primers used for sequencing can be the same used for PCR, provided the target is at least 150 bp and at most 500 bp away from the primer. This ensures large enough stretches of “normal” and “shifted” peaks for a proper quantification by ab1 Peak Reporter. The orientation of sequencing does not matter, but it is critically important to avoid sequencing reads that may encounter naturally occurring indels before the target site, which can cause a natural peakshift and mask the effect of CRISPR. You may have to design and test several internal primers specifically for sequencing, if the PCR primers are not suitable.
5- The resulting .ab1 sequencing file are then uploaded to Thermo Fisher Scientific’s ab1 Peak Reporter (https://apps.thermofisher.com/ab1peakreporter/), which may require registering/logging in to the Thermo Fisher website. The program will return a .csv file, which can be opened in Microsoft Excel and saved as an .xlsx file.
6- The data should first be filtered as to only display the values at each peak called. This is because the data contain signal reads at every position measured by the instrument, including in between peaks (in between individual basepairs in the sequence). To do this, create a filter for the “BaseCall” column (column B) and exclude “-”. You will be left with only the peaks, represented by “calls” indicating G, A, T, C, or N.
7- After filtering this way, you can now search for your target sequence and PAM in column B, displayed as 5’ to 3’ from top to bottom (Figure 2). Once you have found your target sequence, color-coding it may help you keep track of your position in the file.
Fig.2. ab1 Peak Reporter spreadsheet.

Annotated example of an excel spreadsheet generated by the ab1 Peak Reporter web app. Each row represents a called peak, or nucleotide, of the sequence, from 5’ to 3’ (top to bottom, respectively). Cells of interest color coded or outlined manually. In yellow, the sgRNA target and in red, the PAM. In light blue, the MaxSig7Scan ratios for 30 nucleotides upstream of the Cas9 cut site, and in pink, the MaxSig7Scan ratios of 30 nucleotides downstream of the Cas9 cut site. Cas9 tend to cut in the target, ~3 basepairs from the PAM. Outlined in red box: the sum of secondary MaxSig7Scan ratios for each nucleotide, using the formula indicated. The average of these values after the Cas9 cut site represents the “peakshift”, the amount of secondary peak calling due to presence of sequences with short indels in the target. The average of the value before the Cas9 cut site is the background signal. See text for details.
8- In column U, calculate the sum of the secondary peaks by adding the values in columns H-K (“MaxSig7Scan Filtered Ratios”) and subtracting 1. Subtracting 1 is to remove the contribution of the primary peak, which is always 1 regardless of its actual identity.
9- To get a quantitative estimate of the peakshift resulting from mutant reads, calculate the average value in column U, over 30 positions donwstream of (3’ to) the Cas9 cleavage site, usually around the 3rd basepair in the target from the PAM. To get a sense of the secondary signal background of your read, calculate the average in column U over 30 positions upstream of the cleavage site. Subtracting this background average from the peakshift average, you can obtain a corrected peakshift value.
Bear in mind that the peakshift can be suppressed by sequence homogeneity near the target site. Because CRISPR knockouts are usually short indels, shifting peaks of the same identity will not be detected. For instance, a 1-bp deletion in the sequence GGGGAAAA will only produce secondary peaks at one position, while a 1-bp deletion in the sequence GAGAGAGA will result in secondary peaks at all positions.
Conclusion
As more Ciona research groups adopt CRISPR, more data will emerge on the best practices to ensure optimal CRISPR activity, including sgRNA efficacy prediction. We hope the above protocols will speed up this adoption and bring about exciting improvements to CRISPR knockout strategies in Ciona.
Acknowledgments
Research in the laboratory of L.C. is supported by R01 awards HL108643 and GM096032 from the NIH/NHLBI and NIH/NIGMS, respectively; and by grant 15CVD01 from the Leducq Foundation. A.S. is supported by K99 award HD084814 from the NIH/NICHD.
References
- Abdul-Wajid S, Morales-Diaz H, Stephanie M. Khairallah and William C. Smith, 2015. T-type Calcium Channel Regulation of Neural Tube Closure and EphrinA/EPHA Expression. Cell Reports 13: 829–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders C, Niewoehner O, Duerst A and Jinek M, 2014. Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature 513: 569–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P et al. , 2007. CRISPR provides acquired resistance against viruses in prokaryotes. Science 315: 1709–1712. [DOI] [PubMed] [Google Scholar]
- Beerli RR, and Barbas CF, 2002. Engineering polydactyl zinc-finger transcription factors. Nature biotechnology 20: 135–141. [DOI] [PubMed] [Google Scholar]
- Bibikova M, Beumer K, Trautman JK and Carroll D, 2003. Enhancing gene targeting with designed zinc finger nucleases. Science 300: 764–764. [DOI] [PubMed] [Google Scholar]
- Chen B, Gilbert LA, Cimini BA, Schnitzbauer J, Zhang W et al. , 2013. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell 155: 1479–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christiaen L, Wagner E, Shi W and Levine M, 2009 Electroporation of transgenic DNAs in the sea squirt Ciona. Cold Spring Harbor protocols 2009: pdb. prot5345.. [DOI] [PubMed] [Google Scholar]
- Christian M, Cermak T, Doyle EL, Schmidt C, Zhang F et al. , 2010. Targeting DNA double-strand breaks with TAL effector nucleases. Genetics 186: 757–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R et al. , 2013. Multiplex genome engineering using CRISPR/Cas systems. Science 339: 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cota CD, and Davidson B, 2015. Mitotic Membrane Turnover Coordinates Differential Induction of the Heart Progenitor Lineage. Developmental cell 34: 505–519. [DOI] [PubMed] [Google Scholar]
- Deltcheva E, Chylinski K, Sharma CM, Gonzales K, Chao Y et al. , 2011. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 471: 602–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW et al. , 2016. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nature biotechnology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusi N, Smith I, Doench J and Listgarten J, 2015. In Silico Predictive Modeling of CRISPR/Cas9 guide efficiency. bioRxiv : 021568. [Google Scholar]
- Gandhi S, Haeussler M, Razy-Krajka F, Christiaen L and Stolfi A, 2016. Evaluation and rational design of guide RNAs for efficient CRISPR/Cas9-mediated mutagenesis in Ciona. bioRxiv : 041632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garneau JE, Dupuis M-È, Villion M, Romero DA, Barrangou R et al. , 2010. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 468: 67–71. [DOI] [PubMed] [Google Scholar]
- Gasiunas G, Barrangou R, Horvath P and Siksnys V, 2012. Cas9–crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proceedings of the National Academy of Sciences 109: E2579–E2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeussler M, Schönig K, Eckert H, Eschstruth A, Mianné J et al. , 2016. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biology 17: 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilton IB, D’Ippolito AM, Vockley CM, Thakore PI, Crawford GE et al. , 2015. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nature biotechnology 33: 510–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iaffaldano B, Zhang Y and Cornish K, 2016. CRISPR/Cas9 genome editing of rubber producing dandelion Taraxacum kok-saghyz using Agrobacterium rhizogenes without selection. Industrial Crops and Products 89: 356–362. [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA et al. , 2012. A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science 337: 816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, East A, Cheng A, Lin S, Ma E et al. , 2013. RNA-programmed genome editing in human cells. eLife 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Jiang F, Taylor DW, Sternberg SH, Kaya E et al. , 2014. Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science 343: 1247997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai N, Ochiai H, Sakuma T, Yamada L, Sawada H et al. , 2012. Efficient targeted mutagenesis of the chordate Ciona intestinalis genome with zinc‐finger nucleases. Development, growth & differentiation 54: 535–545. [DOI] [PubMed] [Google Scholar]
- Kleinstiver BP, Prew MS, Tsai SQ, Topkar VV, Nguyen NT et al. , 2015. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature 523: 481–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long S, Wang Q and Sibley LD, 2016. Analysis of noncanonical calcium-dependent protein kinases in Toxoplasma gondii by targeted gene deletion using CRISPR/Cas9. Infection and immunity 84: 1262–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeder ML, Linder SJ, Cascio VM, Fu Y, Ho QH et al. , 2013. CRISPR RNA-guided activation of endogenous human genes. Nature methods 10: 977–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeder ML, Thibodeau-Beganny S, Osiak A, Wright DA, Anthony RM et al. , 2008. Rapid “open-source” engineering of customized zinc-finger nucleases for highly efficient gene modification. Molecular cell 31: 294–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M et al. , 2013. RNA-guided human genome engineering via Cas9. Science 339: 823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JC, Tan S, Qiao G, Barlow KA, Wang J et al. , 2011. A TALE nuclease architecture for efficient genome editing. Nature biotechnology 29: 143–148. [DOI] [PubMed] [Google Scholar]
- Moreno-Mateos MA, Vejnar CE, Beaudoin J-D, Fernandez JP, Mis EK et al. , 2015. CRISPRscan: designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nat Meth 12: 982–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama A, and Fujiwara S, 2008. RNA interference by expressing short hairpin RNA in the Ciona intestinalis embryo. Development, growth & differentiation 50: 521–529. [DOI] [PubMed] [Google Scholar]
- Nomura T, Sakurai T, Osakabe Y, Osakabe K and Sakakibara H, 2016. Efficient and Heritable Targeted Mutagenesis in Mosses Using the CRISPR/Cas9 System. Plant and Cell Physiology 57: 2600–2610. [DOI] [PubMed] [Google Scholar]
- Nymark M, Sharma AK, Sparstad T, Bones AM and Winge P, 2016. A CRISPR/Cas9 system adapted for gene editing in marine algae. Scientific reports 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orioli A, Pascali C, Quartararo J, Diebel KW, Praz V et al. , 2011. Widespread occurrence of non-canonical transcription termination by human RNA polymerase III. Nucleic acids research 39: 5499–5512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Pinera P, Kocak DD, Vockley CM, Adler AF, Kabadi AM et al. , 2013. RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nature methods 10: 973–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry KJ, and Henry JQ, 2015. CRISPR/Cas9‐mediated genome modification in the mollusc, Crepidula fornicata. genesis 53: 237–244. [DOI] [PubMed] [Google Scholar]
- Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS et al. , 2013. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152: 1173–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy S, and Schreiber E, 2014. Detecting and quantifying low level gene variants in Sanger sequencing traces using the ab1 Peak Reporter tool. Journal of biomolecular techniques: JBT 25: S13. [Google Scholar]
- Sasaki H, Yoshida K, Hozumi A and Sasakura Y, 2014. CRISPR/Cas9‐mediated gene knockout in the ascidian Ciona intestinalis. Development, growth & differentiation 56: 499–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satou Y, Shin-i T, Kohara Y, Satoh N and Chiba S, 2012. A genomic overview of short genetic variations in a basal chordate, Ciona intestinalis. BMC genomics 13: 208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segade F, Cota C, Famiglietti A, Cha A and Davidson B, 2016. Fibronectin contributes to notochord intercalation in the invertebrate chordate, Ciona intestinalis. EvoDevo 7: 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolfi A, Gandhi S, Salek F and Christiaen L, 2014. Tissue-specific genome editing in Ciona embryos by CRISPR/Cas9. Development 141: 4115–4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian S, Jiang L, Gao Q, Zhang J, Zong M et al. , 2016. Efficient CRISPR/Cas9-based gene knockout in watermelon. Plant Cell Reports : 1–8. [DOI] [PubMed] [Google Scholar]
- Tolkin T, and Christiaen L, 2016. Rewiring of an ancestral Tbx1/10-Ebf-Mrf network for pharyngeal muscle specification in distinct embryonic lineages. Development 143: 3852–3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treen N, Yoshida K, Sakuma T, Sasaki H, Kawai N et al. , 2014. Tissue-specific and ubiquitous gene knockouts by TALEN electroporation provide new approaches to investigating gene function in Ciona. Development 141: 481–487. [DOI] [PubMed] [Google Scholar]
- Urban A, Neukirchen S and Jaeger K-E, 1997. A rapid and efficient method for site-directed mutagenesis using one-step overlap extension PCR. Nucleic acids research 25: 2227–2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW et al. , 2013. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 153: 910–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida K, Treen N, Hozumi A, Sakuma T, Yamamoto T et al. , 2014. Germ cell mutations of the ascidian Ciona intestinalis with TALE nucleases. genesis 52: 431–439. [DOI] [PubMed] [Google Scholar]
- Zetsche B, Gootenberg JS, Abudayyeh OO, Slaymaker IM, Makarova KS et al. , 2015. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 163: 759–771. [DOI] [PMC free article] [PubMed] [Google Scholar]