

 Non-alcoholic fatty liver disease is a major cause of chronic liver pathology in humans.
Non-alcoholic fatty liver disease is a major cause of chronic liver pathology in humans.
Abstract
Non-alcoholic fatty liver disease is a major cause of chronic liver pathology in humans. Fatty liver disease involves the accumulation of hepatocellular fat in hepatocytes that can progress to hepatitis. Steatohepatitis is categorized into alcoholic (ASH) or non-alcoholic (NASH) steatohepatitis based on the etiology of the insult. Both pathologies involve an initial steatosis followed by a progressive inflammation of the liver and eventual hepatic fibrosis (steatohepatitis) and cirrhosis. The involvement of pharmaceuticals and other chemicals in the initiation and progression of fatty liver disease has received increased study. This review will examine not only how xenobiotics initiate hepatic steatosis and steatohepatitis but also how the presence of fatty liver may modify the metabolism and pathologic effects of xenobiotics. The feeding of a high fat diet results in changes in the expression of nuclear receptors that are involved in adaptive and adverse liver effects following xenobiotic exposure. High fat diets also modulate cellular and molecular pathways involved in inflammation, metabolism, oxidative phosphorylation and cell growth. Understanding the role of hepatic steatosis and steatohepatitis on the sequelae of toxic and pathologic changes seen following xenobiotic exposure has importance in defining proper and meaningful human risk characterization of the drugs and other chemical agents.
Pathogenesis and etiology of non-alcoholic fatty liver disease
Etiology
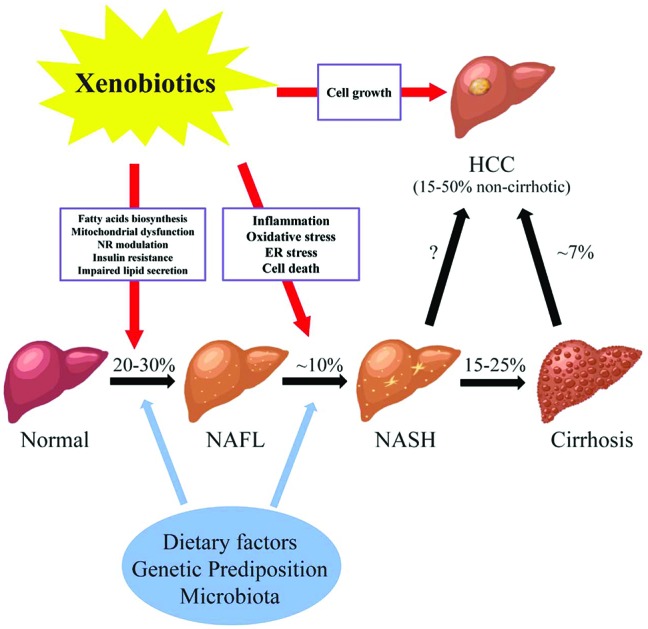
Fatty liver disease is a major public health burden affecting up to one-third of the general population in Western Countries. Fatty liver disease is the result of accumulation of fat (steatosis) in hepatocytes. The diagnosis of steatosis is made when fat in the liver exceeds 5–10% by weight. Fatty liver disease has been classified into two general categories; alcoholic (ASH) or non-alcoholic (NASH) steatohepatitis.1 In the case of the former, chronic alcohol consumption results in fatty liver due to production of toxic metabolites of alcohol including aldehydes. In NASH, multiple (non-alcohol) factors including nutritional, drugs and hepatic-toxicants and metabolic disorders have been identified in the initiation and progression of the disease. While different in etiology, the pathologic sequelae from the accumulation of hepatocellular fat in alcoholic or non-alcoholic steatosis are commonly accompanied by a progressive inflammation of the liver and resulting hepatic fibrosis (steatohepatitis). Non-alcoholic fatty liver disease consists of a spectrum of liver damage from simple steatosis (non-alcoholic fatty liver disease), to steatohepatitis (NASH), fibrosis and cirrhosis, and hepatocellular carcinoma (Fig. 1). The early stage of fat accumulation is reversible and is characterized by simple steatosis. Continual fat accumulation results in hepatocyte damage, inflammation and fibrosis producing the nonalcoholic steatohepatitis (NASH) phenotype. Non-alcoholic fatty liver disease progresses to NASH in approximately 10% of patients, and cirrhosis in 2–3% (15–25% of those that have NASH) patients usually over a period of 10–20 years.2 Some patients with advanced NASH may eventually develop hepatocellular carcinoma (HCC). Multiple factors are involved in the progression from simple steatosis to nonalcoholic fatty liver to NASH to cirrhosis including diet, genetic predisposition and the gut microbiota. These factors form the background by which xenobiotics, including drugs and environmental agents, may influence the progression to steatohepatitis through modulation of multiple pathways including oxidative stress, mitochondrial function, fatty acid biosynthesis and inflammation.
Fig. 1. The potential roles of xenobiotics in non-alcoholic fatty liver diseases. Abbreviations: NAFL, non-alcoholic fatty liver; NASH, non-alcoholic steatohepatitis; HCC, hepatocellular carcinoma; NR, nuclear receptor; ER, endoplasmic reticulum; % prevalence/incidence; missing value (“?”), the magnitude of non-cirrhotic liver cancer developed from NASH is not well-defined. NAFLD consists of a wide spectrum of liver damages from simple steatosis (NAFL), to steatohepatitis (NASH), fibrosis and cirrhosis, and hepatocellular carcinoma (HCC). The pathogenesis of NAFLD is multi-factorial. Genetic background, dietary factors, gut microbiota and other factors act simultaneously in the initiation and progression of NAFLD. In addition, recently studies have shown the link between xenobiotics and the courses of NAFLD. Liver is the major organ for the metabolism and transportation of drugs and environmental agents. Exposure to these compounds may lead to the lipid accumulation in the liver possibly through increased fatty acid biosynthesis, mitochondrial dysfunction, modulation of nuclear receptors, insulin resistance, and impaired lipid excretion. Moreover, the progression of steatohepatitis from simple steatosis may be induced by xenobiotics via oxidative stress, inflammation, ER stress, and cell death. Finally, chemical exposure may stimulate the cell growth in the liver, which further increases the chance of developing HCC in NAFLD.

Pathogenesis
The pathogenesis of non-alcoholic fatty liver disease is multi-factorial. Genetic background, dietary factors, gut microbiota and other factors act simultaneously in the initiation and progression of non-alcoholic fatty liver disease. In addition, recent studies have suggested a link between xenobiotic exposure (including pharmaceuticals and non-pharmaceutical chemicals) and the initiation and progression of the clinical course of non-alcoholic fatty liver disease.3,4 The liver is the major organ for the metabolism of drugs and environmental agents and it has been demonstrated that exposure to some of these agents may lead to the lipid accumulation in hepatocytes through increased fatty acid biosynthesis, mitochondrial dysfunction, modulation of nuclear receptor activation, insulin resistance, and impaired lipid excretion (Fig. 1). Moreover, the progression from simple steatosis to steatohepatitis may be induced by xenobiotics via oxidative stress, inflammation, endoplasmic reticulum (ER) stress, and cell death. Finally, exposure to chemicals may stimulate the cell growth in the liver, which further increases the chance of developing HCC in patients with NASH. The initiation and progression of non-alcoholic fatty liver disease to NASH involves multiple pathways. Initially a 2-hit hypothesis for NASH was proposed.5 The first hit is hepatic steatosis which sensitizes the hepatocytes to further injury (second hits) from inflammatory cytokines, oxidative stress and mitochondrial dysfunction. The second hit eventually leading to steatohepatitis and fibrosis. The two-hit hypothesis has been expanded to a multi-hit model (the modified two hit hypothesis) which takes into account genetic predisposition, diet, and environmental factors functioning together during the progression of non-alcohol fatty liver disease. As with the two hit hypothesis, a number of parallel mechanisms including insulin resistance, lipotoxicity, inflammation, mitochondria malfunction, oxidative stress, and endoplasmic reticulum stress may all be involved at various stages of the disease.6,7
Inflammation
The hallmarks of NASH include inflammation, hepatocyte injury, fibrosis, and cell death. Accumulation of fatty acids in hepatocytes can induce hepatotoxicity exclusive of other pathways. Hepatocytes injured by lipid accumulation can recruit innate immune cells involving Toll-like receptors (TLRs), Kupffer cells, dendritic cells, natural killer cells, lymphocytes, and neutrophils, triggering the inflammatory response, which further promotes simple steatosis to NASH.8 Changes in the gut microbiota from dietary fat may lead to the dysfunction of the tight junction of epithelial cells in the intestine. This increased gut permeability facilitates the delivery of endotoxin, bacteria, and virus to the liver, and induces inflammation.9,10 Reactive oxygen species (ROS) generation are also involved in the pathogenesis of NASH. One hypothesis put forth is that free oxygen radicals produced by lipid peroxidation may damage the mitochondria DNA and result in the apoptosis and necrosis of hepatocytes.11 In addition, accumulated fatty acids in hepatocytes may induce lipotoxicity by generating excessive lipid free radicals that induce oxidative stress. Although rare, hepatocellular neoplasms may be developed from advanced NASH or cirrhosis. In general, the sustained cell death and compensatory proliferation in response to stress and injury in NASH appears to make the liver more susceptible to neoplasm development.12 In non-alcohol fatty liver disease several molecular mechanisms linked to liver tumor promotion have been described. Cytokines such as TNFα and interleukin 6 (IL-6) released from expanded adipose tissues are key activators of pro-oncogenic signaling pathways (NFκB, JNK, etc.). These cytokines, induced by high fat diet (HFD) feeding, have been demonstrated to promote liver tumor formation in diethyl nitrosamine initiated mice.13 Secondly, Kupffer cells are a key regulator in liver injury and hepatocarcinogenesis.14 Leptin, a hormone elevated in non-alcohol fatty liver disease, activates Kupffer cells resulting in further inflammatory and fibrogenic factors being released.15,16 While the endotoxin-mediated inflammatory response in Kupffer cells is normally suppressed by adiponectin,17 reduced adiponectin mRNA and adiponectin protein levels were found in NASH patients that may amplify the ongoing inflammation.18
Fibrosis
Hepatic fibrosis involves the extracellular accumulation of collagen in the sinusoidal and extra hepatocyte areas of the liver lobule. Fibrosis occurs in 10–15% of NASH patients and the presence of fibrosis constitutes a poorer prognosis for liver function and increased mortality.19–21 Multiple causes for the formation of hepatic fibrosis including excessive alcohol consumption, hepatitis viral infection, and cholestasis have been demonstrated. In contrast, studies that have examined the role of xenobiotic exposure in the induction of hepatic fibrosis is limited.22 Vinyl chloride is an organochloride that is used as an intermediate in chemical processing and has been linked to hepatotoxicity (hepatomegaly, hepatic fibrosis) as well as hepatic hemangiosarcomas in workers.23 Vinyl chloride has been shown, using ultrasound, to induce both peri-sinusoidal and perivascular fibrosis in humans. Vinyl chloride is metabolized and detoxified in the liver by CYP2E1, aldehyde dehydrogenase 2 (ALDH2) and glutathione S-transferase theta 1 (GSTT1) enzymes. The genetic polymorphism of CYP2E1 might be associated with the observed idiosyncratic response seen in patients exposed to vinyl chloride.24 In experimental animal models, carbon tetrachloride has been used as a model compound to induce liver damage and produce a similar hepatic fibrosis to that seen in humans. Carbon tetrachloride, like vinyl chloride is also metabolized in the liver by CYP2E1 resulting in the generation of reactive metabolites that induce oxidative damage and lead to hepatocyte cell death.25 The resulting hepatocyte necrosis leads to the activation of Kupffer cells and the release of cytokines. This promotes the activation of hepatic stellate cells leading to remodeling of extracellular matrix and fibrosis.26 Thioacetamide has also been widely used as a model chemical to induce fibrosis in rodents. Mice treated by 100 mg kg–1 Thioacetamide 3 times per week for 8 weeks developed hepatic fibrosis accompanied with inflammation and centrilobular necrosis.27 In rats, administration of thioacetamide also produced fibrotic livers and hepatocyte damage.28 While not completely understood, the thioacetamide induced hepatic fibrosis in rodents has been associated with increased oxidative damage and lipid peroxidation.22
Evidence on the induction of hepatic fibrosis by other chemical agents is limited. Nevirapine, an inhibitor of HIV replication, appears to exacerbate the already present fibrosis in patients with hepatitis C.29 Nevirapine treatment alone did not induce liver toxicity in rats. However, when co-treated with d-galactosamine which produces liver damage similar to viral hepatitis in humans, Nevirapine activated hepatic stellate cells and induced bridging fibrosis in rats after only eight days.29 While the mechanism of the Nevirapine effects on fibrosis are not fully understood, apoptotic cell death pathways, toll-like receptors, and maturation of dendritic cells have been suggested as target pathways for the effect. The appearance of liver fibrosis is an important determinant in the morbidity and mortality of patients with NASH and ASH mortality.20,21 While several chemical agents have been identified as either inducing fibrosis or amplifying the already present hepatic fibrosis, this area of research requires further study. In particular, on how the fibrosis induction and modulation occurs with chemical exposure in fatty liver disease.
Genetic predisposition
Genetic predisposition has also been identified as a potential risk factor that correlates with non-alcohol fatty liver disease susceptibility between individuals. Romeo et al. identified a single nucleotide polymorphism in the patatin-like phospholipase domain-containing 3 (PNPLA3) that changes a highly conserved codon 148 from isoleucine to methionine and is significantly correlated with intra-hepatic fat content.30 Variants in PNPLA3 have been partially explained the susceptibility of non-alcohol fatty liver disease among different ethnic groups, in which Hispanics (G allele frequency: 0.49%) tend to have a higher risk to develop hepatic steatosis whereas African American (G allele frequency: 0.17%) is more protective from non-alcohol fatty liver disease. While the mechanism underlying PNPLA3148M associated hepatic steatosis is not well understood, it appears to modify three pathways: (1) increasing the biosynthesis of triglycerides in the cell due to elevated acetyltransferase activity;31 (2) reducing the lipolytic ability of the hepatocyte;32 (3) and depleting of TAG long-chain polyunsaturated fatty acids.33 In addition, PNPLA3148M appears to modify the profibrogenic capability of hepatic stellate cells (which are involved in the fibrosis seen in NASH).34 An additional polymorphism, TM6SF2, has also been identified that appears to confer susceptibility to non-alcohol fatty liver disease.35 TM6SF2 results in an increased hepatic susceptibility to steatosis due to impaired functioning of apolipoprotein B and VLDL secretion.
Diet
The role of diet has been well documented in the initiation and progression of non-alcohol fatty liver disease. Hepatocytes absorb fatty acids proportional to the fatty acid concentrations in the blood. An overabundance of saturated fats, sugars and cholesterol in the diet may lead to the steatosis in the liver. Increased lipid in the hepatocytes has been linked to an increased production of reactive oxygen species, mitochondrial dysfunction, and apoptosis.36 In addition, excess storage of saturated fat in white adipose tissue can lead to the hypertrophy and hyperplasia of adipocytes, which in turn recruit macrophages and release proinflammatory cytokines into circulation and liver.37 Besides saturated fats, excess fructose consumption has also been linked to the pathogenesis of non-alcohol fatty liver disease through an increase in lipogenesis and an inhibition of fatty acid β-oxidation. Fructose is converted to fructose-1-phosphate, a precursor for fatty acids synthesis in hepatocytes.38 Fructose appears to contribute to the changes involved in the progression from simple steatosis to NASH by inducing oxidative stress, causing mitochondrial dysfunction and ER stress.39 In addition, fructose consumption and fibrosis has also been noted in patients with non-alcohol fatty liver disease.40 It is important to also note the role of increase calories in the induction of steatosis. In particular, a hyper caloric diet along with high fat is an important contributor to the NASH in Western Cultures.
Excessive carbohydrates in the diet can activate sterol regulatory element-binding protein-1c (SREBP-1c) and carbohydrate response element-binding protein (ChREBP) in the liver, increasing de novo fatty acids synthesis.41 When obesity develops, the overexpression of tumor necrosis factor α (TNFα) in the adipose tissue inhibits phosphorylation of insulin receptor substrates, impairs insulin-mediated suppression of hormone-sensitive lipase, and increases the release of LCFA into circulation.42 Mitochondrial and peroxisomal β-oxidation is the major pathway of fatty acid catabolism in hepatocytes. Carnitine palmonitoyl transferase 1 (CPT1) and peroxisome proliferator-activated receptor α (PPARα) play important roles in the process. CPT1 catalyzes the transfer of acyl group from cytoplasm into mitochondria, where β-oxidation normally carried out. PPARα regulates the expression of many genes associated with fatty acids transport, oxidation, and synthesis. PPARα null mice develop hepatic steatosis spontaneously and the administration of PPARα activators reverses methionine choline-deficient diet (MCD) induced NASH and fibrosis in mice.43,44 Lastly, the accumulated fatty acids in the liver are esterified into triglycerides and exported into circulation by VLDL. The biogenesis of VLDL is controlled by mitochondrial triglyceride transfer (MTT). Studies have shown that drugs can modify MTT function that in turn will lead to hepatic steatosis.45,46
Rodent models for non-alcohol fatty liver disease
Several rodent models for non-alcohol fatty liver disease have been described that allow for the further investigation of the cellular and molecular changes that occur during the progression from steatosis to cirrhosis. Given the strong association between diet and fatty liver disease in humans, rodent models for the initiation and progression of non-alcohol fatty liver disease have for the most part been diet based. A summary of some of the model diets that have been used for the induction of steatosis and NASH are shown in Table 1. One diet, a methionine choline-deficient diet reproduces the steatosis, inflammation and fibrosis seen in human liver in mice.47 The lack of methionine and choline in the rodent diet leads to the impaired biosynthesis and secretion of VLDL. This results in the rapid accumulation of lipids in hepatocytes resulting in toxicity, oxidative stress, and inflammation in the liver. However, the methionine choline-deficient diet does not replicate the metabolic profile seen in human non-alcohol fatty liver disease.48 In patients with non-alcohol fatty liver disease, the metabolic syndrome (obesity and dyslipidemia) is prevalent. However, in the methionine choline-deficient diet model, rodents exhibited a decrease body weight, the absence of insulin resistance, and a decreased triglyceride in the circulation. A modification of the methionine choline-deficient diet using a choline deficient high fat diet (CD-HFD), while not physiological, has been shown to better mimic the liver changes seen in humans with non-alcoholic liver disease in mice.49
Table 1. Dietary models of steatosis and steatohepatitis in rodents.
| Models | Kcal from fat in diet | Added sugars in water | Treatment duration (week) | Obesity | Insulin resistance | Steatosis | Lobular inflammation | Hepatocytes ballooning | Perisinusoidal fibrosis | HCC | Ref. |
| MCD | 20% | – | Up to 52 | – | – | + | + | + | + | – | 48 |
| CD-HFD | NP | – | 48 | + | NP | + | + | + | + | + | 49 |
| HFD | 60% | – | Up to 50 | + | + | + | + | – | + | – | 50 |
| HFTFD | 58% | + | 16 | + | + | + | + | + | + | – | 58 |
| ALIOS | 45% | + | Up to 16 | + | + | + | + | + | – | – | 54 |
| HFD32 | 64% | – | 60 | + | + | + | + | – | + | + | 50 |
| AMLN | 40% | + | Up to 30 | + | + | + | + | + | + | – | 56 |
| WD | 42% | – | Up to 60 | + | + | + | + | – | + | + | 52 |
| WD | 42% | + | Up to 52 | + | + | + | + | + | + | + | 58 |
A second dietary approach in rodent models of nonalcoholic fatty liver disease utilizes a high fat diet. In these models, between 30% to 85% of the calories are derived from fat.50 The high fat diets produce obesity, insulin resistance, dyslipidemia, and hepatic steatosis. The development of steatohepatitis and fibrosis with these diets depends on the composition of the high fat diet, the test species, and the duration of time on diet. In an effort to replicate the human diet, a high fat diet commonly referred to as the western diet, with 42% of calories from fat, 43% of calories from carbohydrates, and 0.15% cholesterol by weight comparable with that of unhealthy human diet in western countries has been developed. The western diet model has seen increase use in studying NASH.51–53
Additional studies have shown that dietary cholesterol and fructose may promote the development of NASH by inducing insulin resistance, oxidative stress, and inflammation.54 Mice fed with a higher level of cholesterol and fructose in the diet exhibited clinically relevant characteristics of non-alcohol fatty liver disease/NASH seen in humans.55,56 With the Western diet, simple hepatic steatosis in mice occurs as early as 6 weeks and progresses to NASH and fibrosis in 20–30 weeks.52,57 With the supplementation of sugar water, Asgharpour et al. were able to observe hepatocellular neoplasms in mice fed with western diet at 52 weeks.58 The metabolic features, histopathology, as well as transcriptomic patterns in the western diet treated mouse liver matches with that seen in human non-alcohol fatty liver disease. Thus, there is strong support that the western diet used in rodent models best approximates the physiopathology and etiology of the spectrum of liver damages induced by non-alcohol fatty liver disease in humans.
Chemical induced non-alcohol fatty liver disease
Drugs
Besides dietary factors, the formation of hepatic steatosis and its progression to NASH has also been linked to the pharmaceuticals and environmental chemicals.1,4,59,60 Pharmaceuticals drug induced steatosis is one of the more common manifestations of drug induced liver injuries (Table 2). Chronic treatment with amiodarone has produced steatosis in patients through impaired mitochondrial function and inhibition of lipid oxidation.61 Similarly, long term use of valproic acid, an antiepileptic drug, is associated with fatty liver.62 Valproic acid induced steatosis appears to correlate with the induction of metabolic disorders leading to fatty acid retention in human liver. Chronic exposure to valproic acid has been shown to increase hepatic steatosis in mice on a high fat diet by impairing mitochondrial β-oxidation.63 Other drugs such as tamoxifen, tetracycline, methotrexate, and corticosteroids have also been associated with steatosis in the human liver.4,64–67
Table 2. Drugs induced steatosis and steatohepatitis.
| Chemicals | Hepatic effects | Potential mechanism | Ref. |
| Tamoxifen | Steatosis/steatohepatitis | Increase fatty acids biosynthesis | 65 |
| Amiodarone | Steatosis/steatohepatitis | Inhibit mitochondrial fatty acid oxidation | 55 |
| Valproic acid | Steatosis | Induce metabolic disorders; impair mitochondrial functions | 57 |
| Tetracyclines a | Steatosis | Upregulate lipogenic genes | 61 |
| Methotrexate | Steatosis/steatohepatitis | Induce oxidative stress | 63 and 64 |
| Corticosteroids | Steatosis | Inhibit mitochondrial fatty acid oxidation; increase de novo fatty acids synthesis | 55 |
aTetracyclines compounds, including tetracycline, doxycycline and minocycline, may induce acute microvesicular steatosis.
Non-pharmaceutical chemicals
Given the confirmed role for the induction of steatosis and non-alcohol fatty liver disease by selective pharmaceuticals, recent efforts have also focused on the possible linkage environmental chemical exposure and fatty liver changes. Despite this, few studies have been done to show a causal effect of chemicals on NASH. Recently, Cave and colleagues have proposed novel nomenclature to describe the spectrum of toxicant-associated fatty liver diseases (TAFLD) including steatosis, steatohepatitis (TASH).3 The use of the terms TAFLD and TASH were proposed to highlight the unique differences between the mechanisms underlying the drug-induced liver injury and hepatotoxicity induced by industrial toxicants compared with dietary induced NASH. Many classes of industrial chemicals, including halogenated hydrocarbons, volatile organic mixtures, POPs, pesticides, and some nitro-organic compounds, have been associated with TAFLD.3 The specific mechanisms of TAFLD/TASH appear in many cases to be chemical-specific. While insulin resistance, proinflammatory cytokines, and apoptotic cell death have been reported in TAFLD/TASH,68 modulation of nuclear receptors by these environmental and industrial chemicals may also be critical in the onset of steatosis, the progression to steatohepatitis, and the development of fibrosis and hepatocellular carcinoma. The term TASH was first used to describe vinyl chloride induce liver disease in workers. Studies from the Cave group showed that vinyl chloride exposure resulted in impairment of mitochondrial beta oxidation leading to an increased sensitivity of the liver to steatohepatitis from high fat diet.68 They have proposed that the mitochondrial dysfunction seen in the initial hit with vinyl chloride may be due to the formation of protein adducts from vinyl chloride metabolites. Metabolomics data from vinyl chloride workers showed increased serum lipid peroxides and altered acyl carnitines consistent with incomplete hepatic lipid peroxidation was also apparent.69 In addition, a potential role for PPAR receptors was suggested.
Efforts have been made to investigate the mechanisms of environmental chemical induced steatosis and non-alcohol fatty liver disease using rodent models. The Japanese Toxicogenonics Project (TG-GATEs) has included steatosis as an endpoint. In the evaluation of 131 chemicals Using TG-GATEs at multiple doses and time points, 17 chemicals in this database caused steatosis formation in the rat liver.70 While the underlying mechanism of steatosis induced by each compound may be different, transcriptomic analysis showed genes involved in glucose metabolism, lipid biosynthesis and transportation are modified by the majority of these chemicals.71 Kaiser et al.72 identified 16 environmental chemicals that have mechanistic support to induce fatty liver in rodents. Mitochondrial malfunction, impaired fatty acid exportation, increased cytokine production, and insulin resistance were suggested as possible mechanisms for the development of steatosis by these chemicals. Interestingly, most of these chemicals share a similar molecular structure with a high degree of chlorination. More recently using two large databases; the Toxicological Reference Database (ToxRefDB) and the Chemical Effects in Biological Systems (CEBS) with the key words fatty change, fatty necrosis, Oil red O-positive staining, steatosis, and lipid deposition, Al-Eryani et al. identified 123 chemicals associated with liver steatosis.73
Questions remain concerning the potential causal relationship between chemical exposure and the development of non-alcohol fatty liver disease. Treviño and Katz74 in a recent review suggested a possible linkage between endocrine disruptors and fatty liver diseases in human via modulation of xenobiotic nuclear receptors. In addition, the role of these nuclear receptors has been suggested in the pathogenesis of non-alcohol fatty liver disease.75 Activation of nuclear receptors following xenobiotic exposure results in the induction of phase I and phase II drug-metabolism enzymes.76,77 Specifically, the peroxisome proliferator activated receptors (PPARs), the pregnane X receptors (PXR), the constitutive androstane receptor (CAR), the liver X receptor (LXR), and the farnesoid X receptor (FXR), and acyl hydrocarbon receptor (AHR) have been shown to participate in non-alcohol fatty liver disease induction.75,78 These same receptors are activated by xenobiotics in the formation of liver tumors in rodents.79 The following section reviewed selective nuclear receptor mediated chemicals that have been associated with steatosis and steatohepatitis (Table 3).
Table 3. Selective nuclear receptor mediated chemicals and their effect on NAFLD.
| Chemicals | Nuclear receptor | Effects on NAFLD | Ref. |
| Perfluoroalkyl acids (PFOA, PFOS, PFNA, etc.) | PPARα, CAR, PXR | Steatosis, inflammation, cell proliferation | 84–87 |
| Phalates(DEHP) | PPARα, PPARγ | Steatosis, inflammation, oxidative stress, cell proliferation | 94–97 |
| Dioxins(TCDD) | AhR | Steatosis, inflammation, cell proliferation | 98–102 |
| Polychlorinated biphenyls | CAR, PXR, AhR | Steatosis, inflammation | 105–108 |
| Cyclodienes | CAR, PXR | Steatosis | 109–111 |
| Trichloroethylene and perchloroethylene | PPARα, PPARγ | Steatosis, inflammation, cell cycle | 113, 117 and 118 |
| Vinyl chloride | PPARα | Steatosis, inflammation, cell death | 69 |
Selected chemicals
Perfluoralkyl acids
Perfluoralkyl acids (PFAAs) such as perfluorooctanoic acid (PFOA), perfluorooctane sulfonate (PFOS), and perfluorononanoic acid (PFNA) are persistent and widely distributed environmental contaminants.80 Exposure to PFAAs has been attributed to a wide range of adverse effects in human and animal studies.80,81 Mechanistically, PFAAs have been shown to activate PPARα which appears to be linked to many of the adverse effects in the rodent liver with exposure.82,83 The activation of PPARα has been shown to upregulate are critical in hepatic lipid oxidation genes such as CYP4A, acylCoA oxidase 1 (ACOX1), and CPT-1A. The activation of PPARα also enhances the clearance of lipids in the rodent liver.
PFAAs have been shown to cause micro- and macro-vesicular steatosis in the hepatocytes.84–87 PFOS was found to induce fatty liver in mice in a dose and time dependent manner via the upregulation of fatty acid translocase and inhibition of mitochondrial β-oxidation.88 PFOA also exaggerated high fat diet induced hepatotoxicity as well as lipid accumulation in the liver.89 In a recent study, short term administration of PFAAs (PFOA, PFNA, and PFHxS) was found to induce hepatic steatosis in mice in a PPARα dependent way.87 The molecular mechanism underlying the correlation between PFAAs and lipid accumulation is still unresolved. The activation of PPARα leads to an increase of fatty acid oxidation genes in the lipogenic pathway such as CD36, fatty acids synthase, and SREBP-1. An imbalance of this pathway may lead to either an accumulation of fat or oxidation of fatty acids in hepatocytes.87 In contrast to the PFAAs, other PPARα activators such as Wy-14643, do not induce hepatic steatosis. However, the PFAAs also activate other xenobiotic nuclear receptors besides PPARα such as CAR and PXR.90,91 Therefore, the lipogenic effects in the liver following PFAA exposure may possibly be linked to the cross-talk between PPARα and CAR/PXR. Administration of Wy-14643, a potent PPARα agonist, prevented the hepatic steatosis and liver injury in the mouse from dietary administration of a high fat diet.92 Similarly treatment with the hypolipidemic drug, clofibrate (a PPARα agonist) also decreased high fat diet induced hepatic steatosis and liver inflammation in mice.93
Di (2-ethylhexyl) phthalate (DEHP)
Another PPARα activator, di(2-ethylhexyl) phthalate (DEHP), is a common phthalate ester that is widely used as a plasticizer.94 DEHP induces liver tumors in rodents through the activation of PPARα.95 Meanwhile, DEHP and its metabolites may also interact PPARγ and in turn affect the regulation of energy metabolism in the liver.96 DEHP feeding to rats exacerbated the liver effects of concomitantly administered high fat diet to induce non-alcohol fatty liver disease.97 Similar to the PFAA compounds, DEHP administration induced a dose-dependent modification of gene expression and resulting proteins in both the lipogenesis and lipolysis pathways. Thus, the activation of liver PPARα activation by environmental chemicals remains unclear if this is a beneficial or detrimental effect on the induction and progression of non-alcohol fatty liver disease.
2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD)
Exposure to dioxins, specifically, 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), is associated with abnormal lipid metabolism and diabetes in humans.98 TCDD is metabolized through acyl hydrocarbon receptor (AhR) mediate enzymes. The disruption of the normal function of AhR may lead to the hepatic steatosis and inflammation.99 In transgenic mice with constitutively activated AhR, hepatic steatosis developed spontaneously. This was considered to be related to the upregulation of CD36.100 Short tern administration of TCDD to wild type female mice also produced an increased hepatic steatosis and lipid composition.101 AhR activation has been shown to also suppress the normal functions of PPARα dependent pathways, resulting in increased insulin resistance and lipid accumulation.98,102
Polychlorinated biphenyls (PCBs)
Polychlorinated biphenyls (PCBs) are polyhalogenated aromatic hydrocarbons that persistent in environment.103 Epidemiological studies have associated PCBs exposure with obesity, insulin resistance and non-alcohol fatty liver disease.104,105 PCB126 which possesses dioxin-like AhR activation capability induced fatty change in the liver in female Sprague-Dawley rats. Similarly, PCB126 induced an accumulation of triglycerides in primary cultured hepatocytes through the upregulation of SREBP-1c and microsomal triglyceride transfer protein.106 PCB126 (similar to TCDD) inhibited PPARα activation and expression of the PPARα downstream genes Acox1 and hydroxy-3-methylglutaryl-CoA synthase 2 (Hmgcs2) in rat liver resulting in lipid accumulation in the hepatocytes.107
Non TCDD- like PCBs that have limited AhR activity (e.g. PCB153, Aroclor 1260) activate the CAR/PXR pathway and inhibit the progression of non-alcohol fatty liver disease. PCB153 treatment alone produced minimal effects on rodent hepatic steatosis but a combination of PCB153 and co administered high fat diet significantly impaired fatty acid β-oxidation and upregulated lipogenic genes.108 These observations were attributed to the modulation and activation of the nuclear receptors AhR, CAR/PXR by the high fat diet which may make the liver more susceptible to chemically induced toxicities. In contrast to PCB153, Aroclor 1260 increased hepatic inflammation in western diet induced hepatic steatosis in mice.53 In this study, hepatic Cyp2b10 and Cyp3a11 were increased as were an elevation of proinflammatory cytokines release.
Cyclodienes
The cyclodienes are organochlorine insecticides that were widely used but are now banned by EPA due to the adverse effects on human and environmental health. Chronic feeding of cyclodienes to rodents resulted in hepatic steatosis. For example, liver fatty changes were seen in female rats fed the cyclodiene, Chlordecone, for 2 years.109 Chlordane induced hepatic steatosis in mice after prolonged dietary treatment.110 Both Chlordane and Chlordecone were found to be activators of CAR/PXR.111,112 Although not fully understood, the modulation of PXR induced by Chlordecone may partially contribution to the observed fatty changes in the liver through the alterations of cholesterol homeostasis and lipoprotein metabolism.112
Tri and tetrachloroethylene
Trichloroethylene (TCE) and Tetrachloroethylene (PERC) are widely used organic solvents and have been reported to induce hepatotoxicity including liver tumors in mice following oral or dermal exposure.113 TCE activation of PPARα has been suggested to be responsible for the formation of liver tumors in mice.114 The association between TCE and fatty changes in the liver has been reported.3 Interestingly, Ramdhan et al. showed that TCE increased liver triglyceride levels and steatosis in Pparα-null and humanized PPARα mice, but not in mPPARα mice.115 TCE treatment also significantly increased the expression of PPARγ in Pparα-null and hPPARα mice and but not in mPPARα mice. PPARγ regulates genes in the lipogenesis pathway in the liver, which may account for the observed increase in triglyceride accumulation with TCE treatment.115 PERC has also been shown to be a PPARα activator116 and has been associated with altered hepatic lipid metabolism. Increased hepatic triglycerides along with a decrease in serum triglycerides and an induction of Cyp4a10 and Acox1 were seen in mice treated with PERC for 24 hours.117 More recently, non-alcohol fatty liver disease was found to be a susceptibility factor of PERC induced liver effects.118
Xenobiotic nuclear receptors
The alteration of drug metabolism enzymes in non-alcohol fatty liver disease is an emerging topic. Studies have shown that both phase I and phase II metabolic enzymes are modulated in models of non-alcohol fatty liver disease.119 While the mechanism underlying these changes has not been well understood, activation or downregulation of the xenobiotic nuclear receptors has been suggested to be important in regulating the expression of drug metabolism and transport enzymes. Work in our lab showed that nuclear receptors were modulated in the mouse model of the high fat diet induced hepatic steatosis (Table 4). The nuclear receptors modified by the high fat diet are the same receptors involved in drug and other xenobiotic metabolism and activation.120
Table 4. Gene expression analysis of xenobiotic nuclear receptors and their target genes in a mouse model of NAFLD a .
| Genes | Fold change over control by qPCR | Fold change over control by RNA-seq b |
| Pparα | 2.27 | 1.35 |
| Pparγ | 2.24 | 1.72 |
| Pparδ/β | –3.85 | –2.02 |
| Cyp4a10 | 2.58 | 1.25 |
| Car (Nr1i3) | 1.48 | 1.72 |
| Pxr (Nr1i2) | 1.56 | 1.23 |
| Cyp2b10 | 21.92 | 3.27 |
| Cyp3a11 | 4.20 | 1.83 |
| Ahr | 2.30 | 1.42 |
| Cyp1a1 | 1.76 | 1.36 |
| Cyp1a2 | –1.69 | –1.86 |
| Lxrα | –1.02 | –1.11 |
| Abcg5 | 4.08 | 2.42 |
| Abcg8 | 3.58 | 2.06 |
| Fxr | 1.14 | 1.12 |
| Abcb4 | 2.12 | 1.89 |
| Abcb11 | 1.11 | –1.36 |
aFatty and normal liver samples were generated from C57BL/6 mice fed with western diet or low-fat control diet for 16 weeks, respectively.
bDifferential expression analysis was done using DESeq-2 package in R. Bold: Statistically different from controls (for qPCR analysis: N = 4–6; student two-tailed t-test, p < 0.05; for RNA-seq: N = 4; fold change >1.5; adjusted p < 0.05).
PPARs
The PPAR family of nuclear receptors consists of three members; PPARα, PPARβ/δ, and PPARγ, with PPARα the predominant subtype in hepatocytes.121 PPARα regulates numerous signaling pathways involving in lipid metabolism in hepatocytes. For example, activation of PPARα increases carnitine palmitoyltransferase 1A (CPT-1A), the rate limiting enzyme in the mitochondrial β-oxidation.122 The uptake of free fatty acids from circulation to hepatocytes is tightly mediated by CD36, which is also upregulated by PPARα. Other lipid metabolism pathways mediated by PPARα includes: peroxisomal β-oxidation, acyl-CoA formation and hydrolysis, fatty acid biosynthesis, lipoprotein transportation and metabolism, cholesterol transportation, and ketogenesis.123
In patients with NASH, the mRNA level of PPARα is negatively correlated with the severity of steatosis and insulin resistance.124 A decreased in PPARα may result in an impaired function of mitochondrial and peroxisomal β-oxidation, which further leads to the accumulation of fatty acids in the hepatocytes. Elevation of PPARα gene expression correlated with improved liver histopathology of these patients. Thus, the activation of PPARα has been proposed to be a therapeutic target in non-alcohol fatty liver disease. In animal models, PPARα is activated by chronic high fat diet feeding.125–127 PPARα is considered a nutritional sensor that allows the adaption based on the rate of lipid catabolism or biosynthesis.128 Thus, the increased long chain fatty acids influx to hepatocytes may lead to the adaptive activation of PPARα.129 C57BL/6N mice fed by western diet for 12 weeks had a significant higher level of hepatic phosphatidylcholine, which has been demonstrated as the natural ligand of PPARα.130,131
The other two members PPARγ and PPARβ/δ are also present in the liver. PPARγ responses to fatty acids, eicosanoids, and various anti-diabetics drugs and regulate lipid homeostasis mainly in the muscles and adipose tissues.132 In a hepatic steatosis mouse model, PPARγ mRNA showed a significant increase in the liver.133,134 This increase may contribute to the abnormal accumulation of triglycerides in the liver, while protecting other tissues from insulin resistance. Mice with liver specific Pparγ-null showed a reduced level of hepatic steatosis but hyperlipidemia and muscle insulin resistance.132 PPARβ/δ is a critical regulator of glucose and lipoprotein metabolism in the liver.135 Information on the role of hepatic PPARβ/δ rodent models of non-alcohol fatty liver disease is limited. However, some studies have shown that activating PPARβ/δ reduces steatosis, inflammation, and liver fibrosis in the mouse liver,136–138 suggesting the expression and activity of PPARβ/δ may be impaired in non-alcohol fatty liver disease. In contrast to the rodent models of steatosis, Francque and colleagues reported no changes in gene expression of PPARγ and PPARβ/δ in patients with steatosis and steatohepatitis.124
CAR/PXR
A role for CAR/PXR in energy metabolism has been demonstrated.139 PXR can regulate lipid metabolism in the liver through activation of lipogenic genes (such as CD36 and Stearoyl-CoA desaturase-1 (SCD1)) and induce triglyceride accumulation. PXR can also suppress PPARα function, which further impairs β-oxidation in mitochondria. Reports on the role of CAR in mediating lipid homeostasis are less clear. CAR activation has been associated with the induction of Insig-1, which inhibits the translocation of SREBP-1, an important protein in fatty acids biosynthesis.140 In contrast, CAR may compete with PPARα for the binding of 3-hydroxyacyl-CoA dehydrogenase and thus inhibit β-oxidation.139 Moreover, the crosstalk between CAR and PXR makes it more difficult to investigate the functions of CAR alone in the lipid metabolism. CAR/PXR regulates the expression of CYP3A, which has been reported to be altered in both human and rodent non-alcohol fatty liver disease. A pilot study conducted by Kolwankar et al. suggested that hepatic CYP3A4 activity was inversely associated with the severity of steatosis in patients.141 Subsequently, Fisher et al. found no change in the CYP3A4 mRNA level in human fatty liver samples, although a marginal decrease of protein level of CYP3A4 was seen.142 More recently, biopsies of patients compared to healthy controls.143 CYP3A mRNA and protein level was decreased in high-fat diet fed rats.144,145 In the mouse, high fat diet induced changes to mRNA and enzyme activity of Cyp3a11 showed inconsistent results. Ning & Jeong using a western diet (∼42% fat) reported on a significant increase in Cyp3a11 mRNA and activity level in mice.146 Kirpich et al. using a “pure” high fat diet (∼60% fat) reported on the suppression of Cyp3a11 gene expression.147 Besides high fat content, the western diet model also contains high levels cholesterol. Previous study showed that high cholesterol diet significantly increased hepatic Cyp3a11 level in wild-type but not Pxr-null mice, suggesting that dietary cholesterol is a ligand of PXR.148 In a study from our laboratory using the western diet (Table 4), Pxr and Cyp3a11 mRNA level in the liver of mice were significantly higher than that in controls. Therefore, the dietary content of cholesterol may influence the activation of PXR and more work is needed to clarify the modulation of PXR and its downstream Cyp3a11 induction in high fat diet models for fatty liver disease.
CYP2B is the prototypical marker of CAR/PXR activation in the liver. Limited studies have reported the linkage between non-alcohol fatty liver disease and CYP2B6 expression in humans.119 Concordantly, there is also no robust evidence on the modification of Cyp2b activity in rat models of non-alcohol fatty liver disease.149,150 In mice, the induction of Cyp2b10 gene expression and enzymatic activity is inconsistent among studies. Similar with previous findings, our data suggested Cyp2b10 was marginally induced by western diet feeding (Table 5). Interestingly, the trend of Cyp2b10 and Cyp3a11 modulation tends to be comparable within a single study,147,151 indicating the cross-talk between CAR and PXR upon the effects of dietary components.152
Table 5. Ingenuity pathway analysis predicts hepatic xenobiotic nuclear receptor activation in a mouse model of NAFLD a .
| Upstream nuclear receptor | Predicted activity | Activation z-score | p-Value |
| PPARα | +++ | 4.159 | 1.08 × 10–50 |
| PPARγ | + | 1.342 | 5.73 × 10–15 |
| PPARδ | +++ | 2.815 | 9.53 × 10–15 |
| CAR | + | 0.972 | 1.71 × 10–13 |
| PXR | +++ | 3.153 | 1.13 × 10–19 |
| AHR | - | -1.740 | 3.67 × 10–16 |
| LXR | + | 0.886 | 8.89 × 10–9 |
| FXR | + | 0.943 | 1.41 × 10–10 |
aSteatoic and normal liver samples were generated from C57BL/6 mice fed with western diet for 16 weeks. RNA-seq (4 samples per group) was done to profile the whole transcriptome of these samples. Differential expression analysis was done using DESeq-2 package in R. Ingenuity pathway analysis was done to predict the activation of xenobiotic nuclear receptors based on differential expression analysis results. Activation z-score and p-value were calculated by the build-in algorithms in IPA.
AHR
As a major functional member in CYP1A subfamily, CYP1A2 constitutes about 15% of total hepatic CYP enzymes.153 It is responsible for the metabolism of a variety of drugs including antipsychotics, antihistamines, β-blockers, cyclooxygenase-2 inhibitors.154 CYP1A2 also is important in the biotransformation of a number of environmental toxicants including aflatoxin B1 and aromatic/heterocyclic amines.155 The inhibition of CYP1A2 in fatty liver has been consistently shown in human studies. Fisher and colleagues reported a significant decrease of CYP1A2 protein levels as well as microsomal activity with non-alcohol fatty liver disease progression.142 In cirrhotic livers, the CYP1A2 protein levels decreased to 50% of that of non-cirrhotic liver.156 In rodents, high fat diets also consistently decrease Cyp1a2 mRNA and protein level.146,147 However, the alteration of hepatic Cyp1a2 in mice on the MCD diet or with transgenic mouse models of non-alcohol fatty liver disease has been inconsistent.119 AHR is a major upstream inducer of CYP1A2. Activation of AhR by TCDD caused hepatic steatosis in rodents, likely due to the upregulation of CD36.100 The mRNA level of Cyp1a2 was also significantly increased with the activation of AhR. Recently, treatment with α-naphthoflavone (an antagonist of AhR) prevented high fat western diet induced obesity and fatty liver in male and female C57BL/6 mice.157 While the mRNA or protein level of Cyp1a2 was not reported in this study, the authors suggested that Cyp1b1 (also mediated by AhR) may be critical in modulation the lipid metabolism in non-alcohol fatty liver disease. These studies suggest that AhR activation (and the resulting increased CYP1A2 expression) is linked to the modulation of hepatic steatosis however more work is needed to understand the observed paradox.
LXR/FXR
LXR has two isoforms LXRα and LXRβ, with LXRα predominantly expressed in the liver.158 LXR is associated with non-alcohol fatty liver disease because (1) it controls the expression of lipogenic genes such as SREBP-1 and SCD-1, and thus promotes fatty acids biosynthesis; (2) LXR is a regulates cholesterol excretion of the liver via Abcg5/8; (3) LXR upregulates CYP7A1 and facilitate cholesterol degradation in bile acids.159 In humans, LXR expression has been correlated with the degree of hepatic fat deposition, as well as with hepatic inflammation and fibrosis.160 In rat fed with HFD, the elevated levels of Lxrα and Lxrβ as well as their target genes Abca1 and Abcg5 were seen at the stage of hepatic steatosis.161 Concordantly, increased Lxrα mRNA level has been observed in mice fed with high fat high sucrose diet as early as 2 weeks.162 In high fat high fructose induced non-alcohol fatty liver disease in mice, Abca1 was also increased after prolonged treatment.163 Taken together, evidence strongly support that LXR is activated during the progression of non-alcohol fatty liver disease.
FXR plays critical roles in the bile acids and cholesterol metabolism. Both conjugated and unconjugated forms of bile acids are the endogenous ligands of FXR.164 It should be noted that FXR is expressed in stellate cells in human and rodent livers. The activation of FXR not only suppresses obesity and hepatic steatosis but also inhibits the formation of extracellular matrix in rodent models.165,166 Currently, limited study reported FXR and its downstream ATP-binding cassette transporters in dietary models of NAFLD in rodents. In rat, high fat diet seemed to significantly increase the expression of Fxr as well as Abcb11 and Abcb4.161
Gene expression analysis of hepatic nuclear receptors
Recent studies in our laboratory have examined the changes in nuclear receptor in a mouse model with high fat diet treatment (Table 4).120 In this study male C57BL/6 were placed randomly into two groups consisting of a control diet (13.5% calories from fat) or a high fat diet (42% calories from fat). Animals were allowed food and water ad libitum and mice from each group were sampled after 16 weeks on dietary treatment. Histopathology and receptor activation analysis was performed as well as measurements for steatosis. In this model, sequential hepatic steatosis (8 weeks onwards) was seen along with increased hepatocytes DNA synthesis, lobular inflammation (16–52 weeks), and fibrosis (24–52 weeks).120 The expression of PPARα, CAR/PXR, AHR, LXR, FXR and their target genes were measured in the livers of mice fed with control and high fat diet for 16 weeks (Table 4). We observed the activation of PPARα, CAR/PXR, and LXR in mice with hepatic steatosis. Consistent with previous results, AHR mediated Cyp1a2 was significantly downregulated in high fat diet mice compared to controls. In addition, moderate induction of FXR mediated Abcb4 was seen, suggesting a modification of bile acid biosynthesis and transport. Ingenuity pathway analysis based on RNA-seq results confirmed the modulation of nuclear receptors in mice with NAFLD compared to controls (Table 5).
Conclusions
Fatty liver disease involves the accumulation of hepatocellular fat from either alcoholic or non-alcoholic derived steatosis. Non-alcoholic fatty liver disease is a major cause of chronic liver disease in humans and is the predominantly the result of an increased supply of fat to the liver from dietary sources. A number of drugs and other chemicals have been shown to induce steatosis and subsequently steato fibrosis. Many of the same hepatic pathways, including metabolism, inflammation, and cell growth that are targeted by xenobiotic treatment are also modified in fatty liver disease. Based on the published literature and our preliminary results (Tables 4 and 5) it is apparent that nuclear receptor activation is definitely modify during the progression of the steatosis to steatohepatitis. Thus, the presence of fatty liver disease prior to exposure to drugs and other chemicals may modify the usual pattern of adaptive and adverse cellular and molecular effects of these xenobiotics on the liver. Conversely, exposure to xenobiotics may modify the progression and pathology of the non-alcoholic fatty liver disease resulting in either an enhancement or reduction of the toxicopathologic effects. Understanding the way that fatty liver changes modify hepatic pathways, in particular those dependent on nuclear receptor activation, are important in understanding how the presence of the fatty changes may modify the potential human risk of a drug or environmental chemical.
Conflicts of interest
There are no conflicts to declare for this manuscript.
References
- Joshi-Barve S., Kirpich I., Cave M. C., Marsano L. S., McClain C. J. Cell. Mol. Gastroenterol. Hepatol. 2015;1:356–367. doi: 10.1016/j.jcmgh.2015.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinella M. E., Sanyal A. J. Nat. Rev. Gastroenterol. Hepatol. 2016;13:196–205. doi: 10.1038/nrgastro.2016.3. [DOI] [PubMed] [Google Scholar]
- Wahlang B., Beier J. I., Clair H. B., Bellis-Jones H. J., Falkner K. C., McClain C. J., Cave M. C. Toxicol. Pathol. 2013;41:343–360. doi: 10.1177/0192623312468517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinowich L., Shibolet O. BioMed Res. Int. 2015;2015:168905. doi: 10.1155/2015/168905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day C. P., James O. F. Gastroenterology. 1998;114:842–845. doi: 10.1016/s0016-5085(98)70599-2. [DOI] [PubMed] [Google Scholar]
- Takaki A., Kawai D., Yamamoto K. Int. J. Mol. Sci. 2013;14:20704–20728. doi: 10.3390/ijms141020704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzzetti E., Pinzani M., Tsochatzis E. A. Metabolism. 2016;65:1038–1048. doi: 10.1016/j.metabol.2015.12.012. [DOI] [PubMed] [Google Scholar]
- Arrese M., Cabrera D., Kalergis A. M., Feldstein A. E. Dig. Dis. Sci. 2016;61:1294–1303. doi: 10.1007/s10620-016-4049-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung C., Rivera L., Furness J. B., Angus P. W. Nat. Rev. Gastroenterol. Hepatol. 2016;13:412–425. doi: 10.1038/nrgastro.2016.85. [DOI] [PubMed] [Google Scholar]
- Bashiardes S., Shapiro H., Rozin S., Shibolet O., Elinav E. Mol. Metab. 2016;5:782–794. doi: 10.1016/j.molmet.2016.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaki A., Kawai D., Yamamoto K. Int. J. Mol. Sci. 2013;14:20704–20728. doi: 10.3390/ijms141020704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baffy G., Brunt E. M., Caldwell S. H. J. Hepatol. 2012;56:1384–1391. doi: 10.1016/j.jhep.2011.10.027. [DOI] [PubMed] [Google Scholar]
- Park E. J., Lee J. H., Yu G. Y., He G., Ali S. R., Holzer R. G., Osterreicher C. H., Takahashi H., Karin M. Cell. 2010;140:197–208. doi: 10.1016/j.cell.2009.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts R. A., Ganey P. E., Ju C., Kamendulis L. M., Rusyn I., Klaunig J. E. Toxicol. Sci. 2006;96:2–15. doi: 10.1093/toxsci/kfl173. [DOI] [PubMed] [Google Scholar]
- Angulo P., Alba L. M., Petrovic L. M., Adams L. A., Lindor K. D., Jensen M. D. J. Hepatol. 2004;41:943–949. doi: 10.1016/j.jhep.2004.08.020. [DOI] [PubMed] [Google Scholar]
- Aleffi S., Petrai I., Bertolani C., Parola M., Colombatto S., Novo E., Vizzutti F., Anania F. A., Milani S., Rombouts K. Hepatology. 2005;42:1339–1348. doi: 10.1002/hep.20965. [DOI] [PubMed] [Google Scholar]
- Thakur V., Pritchard M. T., McMullen M. R., Nagy L. E. Am. J. Physiol.: Gastrointest. Liver Physiol. 2006;290:G998–1007. doi: 10.1152/ajpgi.00553.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaser S., Moschen A., Cayon A., Kaser A., Crespo J., Pons-Romero F., Ebenbichler C. F., Patsch J. R., Tilg H. Gut. 2005;54:117–121. doi: 10.1136/gut.2003.037010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunt E. M., Kleiner D. E., Wilson L. A., Belt P., Neuschwander-Tetri B. A. Hepatology. 2011;53:810–820. [Google Scholar]
- Angulo P., Kleiner D. E., Dam-Larsen S., Adams L. A., Bjornsson E. S., Charatcharoenwitthaya P., Mills P. R., Keach J. C., Lafferty H. D., Stahler A., Haflidadottir S., Bendtsen F. Gastroenterology. 2015;149:389–397. doi: 10.1053/j.gastro.2015.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younossi Z. M., Stepanova M., Rafiq N., Henry L., Loomba R., Makhlouf H., Goodman Z. Hepatol. Commun. 2017;1:421–428. doi: 10.1002/hep4.1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanguas S. C., Cogliati B., Willebrords J., Maes M., Colle I., van den Bossche B., de Oliveira C., Andraus W., Alves V. A. F., Leclercq I., Vinken M. Arch. Toxicol. 2016;90:1025–1048. doi: 10.1007/s00204-015-1543-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao T. J., Wang J. D., Yang P. M., Yang P. C., Cheng T. J. J. Occup. Environ. Med. 2004;46:962–966. doi: 10.1097/01.jom.0000137722.66767.38. [DOI] [PubMed] [Google Scholar]
- Hsieh H. I., Chen P. C., Wong R. H., Wang J. D., Yang P. M., Cheng T. J. Toxicology. 2007;239:34–44. doi: 10.1016/j.tox.2007.06.089. [DOI] [PubMed] [Google Scholar]
- Weber L. W., Boll M., Stampfl A. Crit. Rev. Toxicol. 2003;33:105–136. doi: 10.1080/713611034. [DOI] [PubMed] [Google Scholar]
- Iwaisako K., Jiang C., Zhang M., Cong M., Moore-Morris T. J., Park T. J., Liu X., Xu J., Wang P., Paik Y. H., Meng F., Asagiri M., Murray L. A., Hofmann A. F., Iida T., Glass C. K., Brenner D. A., Kisseleva T. Proc. Natl. Acad. Sci. U. S. A. 2014;111:E3297–E3305. doi: 10.1073/pnas.1400062111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen I. S., Chen Y. C., Chou C. H., Chuang R. F., Sheen L. Y., Chiu C. H. J. Sci. Food Agric. 2012;92:1441–1447. doi: 10.1002/jsfa.4723. [DOI] [PubMed] [Google Scholar]
- Qin D., Nie Y., Wen Z. Iran. J. Basic Med. Sci. 2014;17:879–885. [PMC free article] [PubMed] [Google Scholar]
- Brown H. R., Castellino S., Groseclose M. R., Elangbam C. S., Mellon-Kusibab K., Yoon L. W., Gates L. D., Krull D. L., Cariello N. F., Arrington-Brown L., Tillman T., Fowler S., Shah V., Bailey D., Miller R. T. Toxicol. Pathol. 2016;44:112–131. doi: 10.1177/0192623315617033. [DOI] [PubMed] [Google Scholar]
- Romeo S., Kozlitina J., Xing C., Pertsemlidis A., Cox D., Pennacchio L. A., Boerwinkle E., Cohen J. C., Hobbs H. H. Nat. Genet. 2008;40:1461–1465. doi: 10.1038/ng.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari M., Schoiswohl G., Chitraju C., Paar M., Cornaciu I., Rangrez A. Y., Wongsiriroj N., Nagy H. M., Ivanova P. T., Scott S. A., Knittelfelder O., Rechberger G. N., Birner-Gruenberger R., Eder S., Brown H. A., Haemmerle G., Oberer M., Lass A., Kershaw E. E., Zimmermann R., Zechner R. Cell Metab. 2012;15:691–702. doi: 10.1016/j.cmet.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J. W., Yang H., Mitchell G. A. Hepatology. 2016;63:676–677. doi: 10.1002/hep.27943. [DOI] [PubMed] [Google Scholar]
- Li J. Z., Huang Y., Karaman R., Ivanova P. T., Brown H. A., Roddy T., Castro-Perez J., Cohen J. C., Hobbs H. H. J. Clin. Invest. 2012;122:4130–4144. doi: 10.1172/JCI65179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruschi F. V., Claudel T., Tardelli M., Caligiuri A., Stulnig T. M., Marra F., Trauner M. Hepatology. 2017;65:1875–1890. doi: 10.1002/hep.29041. [DOI] [PubMed] [Google Scholar]
- Kozlitina J., Smagris E., Stender S., Nordestgaard B. G., Zhou H. H., Tybjærg-Hansen A., Vogt T. F., Hobbs H. H., Cohen J. C. Nat. Genet. 2014;46:352–356. doi: 10.1038/ng.2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado M. V., Ravasco P., Jesus L., Marques-Vidal P., Oliveira C. R., Proença T., Baldeiras I., Camilo M. E., Cortez-Pinto H. Scand. J. Gastroenterol. 2008;43:95–102. doi: 10.1080/00365520701559003. [DOI] [PubMed] [Google Scholar]
- Funaki M. J. Med. Invest. 2009;56:88–92. doi: 10.2152/jmi.56.88. [DOI] [PubMed] [Google Scholar]
- Ouyang X., Cirillo P., Sautin Y., McCall S., Bruchette J. L., Diehl A. M., Johnson R. J., Abdelmalek M. F. J. Hepatol. 2008;48:993–999. doi: 10.1016/j.jhep.2008.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jegatheesan P., De Bandt J. P. Nutrients. 2017;9:230. doi: 10.3390/nu9030230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdelmalek M. F., Suzuki A., Guy C., Unalp-Arida A., Colvin R., Johnson R. J., Diehl A. M. Hepatology. 2010;51:1961–1971. [Google Scholar]
- Dentin R., Girard J., Postic C. Biochimie. 2005;87:81–86. doi: 10.1016/j.biochi.2004.11.008. [DOI] [PubMed] [Google Scholar]
- Anstee Q. M., Goldin R. D. Int. J. Exp. Pathol. 2006;87:1–16. doi: 10.1111/j.0959-9673.2006.00465.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambasiva Rao M., Reddy J. K. Hepatology. 2004;40:783–786. doi: 10.1002/hep.20453. [DOI] [PubMed] [Google Scholar]
- Rao M., Papreddy K., Musunuri S., Okonkwo A. In Vivo. 2001;16:145–152. [PubMed] [Google Scholar]
- Chen Z., Newberry E. P., Norris J. Y., Xie Y., Luo J., Kennedy S. M., Davidson N. O. J. Lipid Res. 2008;49:2013–2022. doi: 10.1194/jlr.M800240-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berriot-Varoqueaux N., Aggerbeck L., Samson-Bouma M.-E., Wetterau J. Annu. Rev. Nutr. 2000;20:663–697. doi: 10.1146/annurev.nutr.20.1.663. [DOI] [PubMed] [Google Scholar]
- Marcolin E., Forgiarini L. F., Tieppo J., Dias A. S., Rodrigues de Freitas L. A., Marroni N. P. Arq. Gastroenterol. 2011;48:72–79. doi: 10.1590/s0004-28032011000100015. [DOI] [PubMed] [Google Scholar]
- Itagaki H., Shimizu K., Morikawa S., Ogawa K., Ezaki T. Int. J. Clin. Exp. Pathol. 2013;6:2683–2696. [PMC free article] [PubMed] [Google Scholar]
- Wolf M. J., Adili A., Piotrowitz K., Abdullah Z., Boege Y., Stemmer K., Ringelhan M., Simonavicius N., Egger M., Wohlleber D., Lorentzen A., Einer C., Schulz S., Clavel T., Protzer U., Thiele C., Zischka H., Moch H., Tschop M., Tumanov A. V., Haller D., Unger K., Karin M., Kopf M., Knolle P., Weber A., Heikenwalder M. Cancer Cell. 2014;26:549–564. doi: 10.1016/j.ccell.2014.09.003. [DOI] [PubMed] [Google Scholar]
- Takahashi Y., Soejima Y., Fukusato T. World J. Gastroenterol. 2012;18:2300–2308. doi: 10.3748/wjg.v18.i19.2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon L. J., Flask C. A., Papouchado B. G., Feldstein A. E., Nagy L. E. PLoS One. 2013;8:10. doi: 10.1371/journal.pone.0056100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanSaun M. N., Lee I. K., Washington M. K., Matrisian L., Gorden D. L. Am. J. Pathol. 2009;175:355–364. doi: 10.2353/ajpath.2009.080703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlang B., Song M., Beier J. I., Cameron Falkner K., Al-Eryani L., Clair H. B., Prough R. A., Osborne T. S., Malarkey D. E., Christopher States J., Cave M. C. Toxicol. Appl. Pharmacol. 2014;279:380–390. doi: 10.1016/j.taap.2014.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetri L. H., Basaranoglu M., Brunt E. M., Yerian L. M., Neuschwander-Tetri B. A. Am. J. Physiol.: Gastrointest. Liver Physiol. 2008;295:G987–G995. doi: 10.1152/ajpgi.90272.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlton M., Krishnan A., Viker K., Sanderson S., Cazanave S., McConico A., Masuoko H., Gores G. Am. J. Physiol.: Gastrointest. Liver Physiol. 2015;308:G159–G159. doi: 10.1152/ajpgi.00145.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapper J. R., Hendricks M. D., Gu G., Wittmer C., Dolman C. S., Herich J., Athanacio J., Villescaz C., Ghosh S. S., Heilig J. S., Lowe C., Roth J. D. Am. J. Physiol.: Gastrointest. Liver Physiol. 2013;305:G483–G495. doi: 10.1152/ajpgi.00079.2013. [DOI] [PubMed] [Google Scholar]
- Li Z. Z., Berk M., McIntyre T. M., Feldstein A. E. J. Biol. Chem. 2009;284:5637–5644. doi: 10.1074/jbc.M807616200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asgharpour A., Cazanave S. C., Pacana T., Seneshaw M., Vincent R., Banini B. A., Kumar D. P., Daita K., Min H.-K., Mirshahi F. J. Hepatol. 2016;65:579–588. doi: 10.1016/j.jhep.2016.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Eryani L., Wahlang B., Falkner K., Guardiola J., Clair H., Prough R., Cave M. Toxicol. Pathol. 2015;43:482–497. doi: 10.1177/0192623314549960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heindel J. J., Blumberg B., Cave M., Machtinger R., Mantovani A., Mendez M. A., Nadal A., Palanza P., Panzica G., Sargis R. Reprod. Toxicol. 2017;68:3–33. doi: 10.1016/j.reprotox.2016.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kneeman J. M., Misdraji J., Corey K. E. Ther. Adv. Gastroenterol. 2012;5:199–207. doi: 10.1177/1756283X11430859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farinelli E., Giampaoli D., Cenciarini A., Cercado E., Verrotti A. World J. Hepatol. 2015;7:1251–1257. doi: 10.4254/wjh.v7.i9.1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L. F., Liu L. S., Chu X. M., Xie H., Cao L. J., Guo C., A J. Y., Cao B., Li M. J., Wang G. J., Hao H. P. Acta Pharmacol. Sin. 2014;35:363–372. doi: 10.1038/aps.2013.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antherieu S., Rogue A., Fromenty B., Guillouzo A., Robin M. A. Hepatology. 2011;53:1895–1905. doi: 10.1002/hep.24290. [DOI] [PubMed] [Google Scholar]
- López-Riera M., Conde I., Tolosa L., Zaragoza Á., Castell J. V., Gómez-Lechón M. J., Jover R. Front. Pharmacol. 2017;8:3. doi: 10.3389/fphar.2017.00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakthiswary R., Chan G. Y. L., Koh E. T., Leong K. P., Thong B. Y. H. Sci. World J. 2014;2014:823763. doi: 10.1155/2014/823763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan H.-J., Chang H.-T., Lee C.-H. J. Formosan Med. Assoc. 2016;115:411–417. doi: 10.1016/j.jfma.2015.05.006. [DOI] [PubMed] [Google Scholar]
- Cave M., Falkner K. C., Ray M., Joshi-Barve S., Brock G., Khan R., Bon Homme M., McClain C. J. Hepatology. 2010;51:474–481. doi: 10.1002/hep.23321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guardiola J. J., Beier J. I., Falkner K. C., Wheeler B., McClain C. J., Cave M. Toxicol. Appl. Pharmacol. 2016;313:47–56. doi: 10.1016/j.taap.2016.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igarashi Y., Nakatsu N., Yamashita T., Ono A., Ohno Y., Urushidani T., Yamada H. Nucleic Acids Res. 2014;43:D921–D927. doi: 10.1093/nar/gku955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahini N., Selvaraj S., Borlak J. PLoS One. 2014;9:e114085. doi: 10.1371/journal.pone.0114085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser J. P., Lipscomb J. C., Wesselkamper S. C. Int. J. Toxicol. 2012;31:551–563. doi: 10.1177/1091581812466418. [DOI] [PubMed] [Google Scholar]
- Al-Eryani L., Wahlang B., Falkner K., Guardiola J. J., Clair H., Prough R., Cave M. Toxicol. Pathol. 2015;43:482–497. doi: 10.1177/0192623314549960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treviño L. S., Katz T. A. Endocrinology. 2017;159:20–31. doi: 10.1210/en.2017-00887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cave M. C., Clair H. B., Hardesty J. E., Falkner K. C., Feng W., Clark B. J., Sidey J., Shi H., Aqel B. A., McClain C. J., Prough R. A. Biochim. Biophys. Acta. 2016;1859:1083–1099. doi: 10.1016/j.bbagrm.2016.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handschin C., Meyer U. A. Pharmacol. Rev. 2003;55:649–673. doi: 10.1124/pr.55.4.2. [DOI] [PubMed] [Google Scholar]
- Prakash C., Zuniga B., Song C. S., Jiang S., Cropper J., Park S., Chatterjee B. Nucl. Recept. Res. 2015;2:101178. doi: 10.11131/2015/101178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. H., Wada T., Febbraio M., He J., Matsubara T., Lee M. J., Gonzalez F. J., Xie W. Gastroenterology. 2010;139:653–663. doi: 10.1053/j.gastro.2010.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corton J. C., Cunningham M. L., Hummer B. T., Lau C., Meek B., Peters J. M., Popp J. A., Rhomberg L., Seed J., Klaunig J. E. Crit. Rev. Toxicol. 2014;44:1–49. doi: 10.3109/10408444.2013.835784. [DOI] [PubMed] [Google Scholar]
- Lau C., Anitole K., Hodes C., Lai D., Pfahles-Hutchens A., Seed J. Toxicol. Sci. 2007;99:366–394. doi: 10.1093/toxsci/kfm128. [DOI] [PubMed] [Google Scholar]
- Chang E. T., Adami H. O., Boffetta P., Cole P., Starr T. B., Mandel J. S. Crit. Rev. Toxicol. 2014;44(Suppl 1):1–81. doi: 10.3109/10408444.2014.905767. [DOI] [PubMed] [Google Scholar]
- Klaunig J. E., Babich M. A., Baetcke K. P., Cook J. C., Corton J. C., David R. M., DeLuca J. G., Lai D. Y., McKee R. H., Peters J. M. CRC Crit. Rev. Toxicol. 2003;33:655–780. doi: 10.1080/713608372. [DOI] [PubMed] [Google Scholar]
- Takacs M. L., Abbott B. D. Toxicol. Sci. 2007;95:108–117. doi: 10.1093/toxsci/kfl135. [DOI] [PubMed] [Google Scholar]
- Haughom B., Spydevold Ø. Biochim. Biophys. Acta, Lipids Lipid Metab. 1992;1128:65–72. doi: 10.1016/0005-2760(92)90258-w. [DOI] [PubMed] [Google Scholar]
- Kudo N., Kawashima Y. Toxicol. Appl. Pharmacol. 1997;145:285–293. doi: 10.1006/taap.1997.8186. [DOI] [PubMed] [Google Scholar]
- Martin M. T., Brennan R. J., Hu W., Ayanoglu E., Lau C., Ren H., Wood C. R., Corton J. C., Kavlock R. J., Dix D. J. Toxicol. Sci. 2007;97:595–613. doi: 10.1093/toxsci/kfm065. [DOI] [PubMed] [Google Scholar]
- Das K. P., Wood C. R., Lin M. T., Starkov A. A., Lau C., Wallace K. B., Corton J. C., Abbott B. D. Toxicology. 2017;378:37–52. doi: 10.1016/j.tox.2016.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan H., Zhao Y., Wei X., Hui K., Giesy J., Wong C. K. Biochim. Biophys. Acta, Gen. Subj. 2012;1820:1092–1101. doi: 10.1016/j.bbagen.2012.03.010. [DOI] [PubMed] [Google Scholar]
- Tan X., Xie G., Sun X., Li Q., Zhong W., Qiao P., Sun X., Jia W., Zhou Z. PLoS One. 2013;8:e61409. doi: 10.1371/journal.pone.0061409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjork J., Butenhoff J., Wallace K. Toxicology. 2011;288:8–17. doi: 10.1016/j.tox.2011.06.012. [DOI] [PubMed] [Google Scholar]
- Elcombe C. R., Elcombe B. M., Foster J. R., Farrar D. G., Jung R., Chang S. C., Kennedy G. L., Butenhoff J. L. Arch. Toxicol. 2010;84:787–798. doi: 10.1007/s00204-010-0572-2. [DOI] [PubMed] [Google Scholar]
- Ip E., Farrell G., Hall P., Robertson G., Leclercq I. Hepatology. 2004;39:1286–1296. doi: 10.1002/hep.20170. [DOI] [PubMed] [Google Scholar]
- Lalloyer F., Wouters K., Baron M., Caron S., Vallez E., Vanhoutte J., Baugé E., Shiri-Sverdlov R., Hofker M., Staels B. Arterioscler., Thromb., Vasc. Biol. 2011;31:1573–1579. doi: 10.1161/ATVBAHA.110.220525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latini G. Clin. Chim. Acta. 2005;361:20–29. doi: 10.1016/j.cccn.2005.05.003. [DOI] [PubMed] [Google Scholar]
- Rusyn I., Peters J. M., Cunningham M. L. Crit. Rev. Toxicol. 2006;36:459–479. doi: 10.1080/10408440600779065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kambia N., Renault N., Dilly S., Farce A., Dine T., Gressier B., Luyckx M., Brunet C., Chavatte P. J. Enzyme Inhib. Med. Chem. 2008;23:611–616. doi: 10.1080/14756360802205059. [DOI] [PubMed] [Google Scholar]
- Chen H., Zhang W., Rui B. B., Yang S. M., Xu W. P., Wei W. Environ. Toxicol. Pharmacol. 2016;42:38–44. doi: 10.1016/j.etap.2015.12.016. [DOI] [PubMed] [Google Scholar]
- Remillard R. B., Bunce N. J. Environ. Health Perspect. 2002;110:853–858. doi: 10.1289/ehp.02110853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angrish M. M., Dominici C. Y., Zacharewski T. R. Toxicol. Sci. 2013;131:108–115. doi: 10.1093/toxsci/kfs277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. H., Wada T., Febbraio M., He J., Matsubara T., Lee M. J., Gonzalez F. J., Xie W. Gastroenterology. 2010;139:653–663. doi: 10.1053/j.gastro.2010.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angrish M. M., Jones A. D., Harkema J. R., Zacharewski T. R. Toxicol. Sci. 2011;124:299–310. doi: 10.1093/toxsci/kfr226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaban Z., El-Shazly S., Abdelhady S., Fattouh I., Muzandu K., Ishizuka M., Kimura K., Kazusaka A., Fujita S. J. Vet. Med. Sci. 2004;66:1377–1386. doi: 10.1292/jvms.66.1377. [DOI] [PubMed] [Google Scholar]
- Schecter A., Colacino J., Haffner D., Patel K., Opel M., Päpke O., Birnbaum L. Environ. Health Perspect. 2010;118:796. doi: 10.1289/ehp.0901347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D.-H., Lee I.-K., Song K., Steffes M., Toscano W., Baker B. A., Jacobs D. R. Diabetes Care. 2006;29:1638–1644. doi: 10.2337/dc06-0543. [DOI] [PubMed] [Google Scholar]
- Lee D.-H., Lee I.-K., Porta M., Steffes M., Jacobs D. Diabetologia. 2007;50:1841–1851. doi: 10.1007/s00125-007-0755-4. [DOI] [PubMed] [Google Scholar]
- Boucher M.-P., Lefebvre C., Chapados N. A. J. Diabetes Metab. Disord. 2015;14:88. doi: 10.1186/s40200-015-0218-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadupudi G. S., Klaren W. D., Olivier A. K., Klingelhutz A. J., Robertson L. W. Toxicol. Sci. 2016;149:98–110. doi: 10.1093/toxsci/kfv215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlang B., Falkner K. C., Gregory B., Ansert D., Young D., Conklin D. J., Bhatnagar A., McClain C. J., Cave M. J. Nutr. Biochem. 2013;24:1587–1595. doi: 10.1016/j.jnutbio.2013.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson P. S., Egle J. L., Hennigar G. R., Lane R. W., Borzelleca J. F. Toxicol. Appl. Pharmacol. 1979;48:29–41. doi: 10.1016/s0041-008x(79)80005-8. [DOI] [PubMed] [Google Scholar]
- Khasawinah A. M., Grutsch J. F. Regul. Toxicol. Pharmacol. 1989;10:244–254. doi: 10.1016/0273-2300(89)90051-2. [DOI] [PubMed] [Google Scholar]
- Ross J., Plummer S. M., Rode A., Scheer N., Bower C. C., Vogel O., Henderson C. J., Wolf C. R., Elcombe C. R. Toxicol. Sci. 2010;116:452–466. doi: 10.1093/toxsci/kfq118. [DOI] [PubMed] [Google Scholar]
- Lee J., Scheri R. C., Zhang Y., Curtis L. R. Toxicol. Appl. Pharmacol. 2008;233:193–202. doi: 10.1016/j.taap.2008.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray J. L., Kopec A. K., Joshi N., Cline-Fedewa H., Lash L. H., Williams K. J., Leung P. S., Gershwin M. E., Luyendyk J. P. Toxicol. Sci. 2017;156:428–437. doi: 10.1093/toxsci/kfw264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laughter A. R., Dunn C. S., Swanson C. L., Howroyd P., Cattley R. C., Corton J. C. Toxicology. 2004;203:83–98. doi: 10.1016/j.tox.2004.06.014. [DOI] [PubMed] [Google Scholar]
- Ramdhan D. H., Kamijima M., Wang D., Ito Y., Naito H., Yanagiba Y., Hayashi Y., Tanaka N., Aoyama T., Gonzalez F. J., Nakajima T. Environ. Health Perspect. 2010;118:1557–1563. doi: 10.1289/ehp.1001928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyton K. Z., Hogan K. A., Scott C. S., Cooper G. S., Bale A. S., Kopylev L., Barone S., Makris S. L., Glenn B., Subramaniam R. P., Gwinn M. R., Dzubow R. C., Chiu W. A. Environ. Health Perspect. 2014;122:325–334. doi: 10.1289/ehp.1307359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cichocki J. A., Furuya S., Venkatratnam A., McDonald T. J., Knap A. H., Wade T., Sweet S., Chiu W. A., Threadgill D. W., Rusyn I. Environ. Health Perspect. 2017;125:057006. doi: 10.1289/EHP788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cichocki J. A., Furuya S., Luo Y. S., Iwata Y., Konganti K., Chiu W. A., Threadgill D. W., Pogribny I. P., Rusyn I. Toxicol. Sci. 2017;159:102–113. doi: 10.1093/toxsci/kfx120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrell M. D., Cherrington N. J. Drug Metab. Rev. 2011;43:317–334. doi: 10.3109/03602532.2011.577781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaunig J. E., Li X. and Wang Z., Xenobiotic Nuclear Receptors Expression in High Fat Diet-Induced Non-Alcoholic Fatty Liver Disease: A Time-Course Study, in The Toxicologist, 2018, vol. 162, abstract no. 2051. [Google Scholar]
- Evans R. M., Mangelsdorf D. J. Cell. 2014;157:255–266. doi: 10.1016/j.cell.2014.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song S., Attia R. R., Connaughton S., Niesen M. I., Ness G. C., Elam M. B., Hori R. T., Cook G. A., Park E. A. Mol. Cell. Endocrinol. 2010;325:54–63. doi: 10.1016/j.mce.2010.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakhshandehroo M., Knoch B., Müller M., Kersten S. PPAR Res. 2010;2010 doi: 10.1155/2010/612089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francque S., Verrijken A., Caron S., Prawitt J., Paumelle R., Derudas B., Lefebvre P., Taskinen M.-R., Van Hul W., Mertens I. J. Hepatol. 2015;63:164–173. doi: 10.1016/j.jhep.2015.02.019. [DOI] [PubMed] [Google Scholar]
- Patsouris D., Reddy J. K., Müller M., Kersten S. Endocrinology. 2006;147:1508–1516. doi: 10.1210/en.2005-1132. [DOI] [PubMed] [Google Scholar]
- Zou Y., Du H., Yin M., Zhang L., Mao L., Xiao N., Ren G., Zhang C., Pan J. Mol. Cell. Biochem. 2009;323:195–205. doi: 10.1007/s11010-008-9982-3. [DOI] [PubMed] [Google Scholar]
- de Fourmestraux V., Neubauer H., Poussin C., Farmer P., Falquet L., Burcelin R., Delorenzi M., Thorens B. J. Biol. Chem. 2004;279:50743–50753. doi: 10.1074/jbc.M408014200. [DOI] [PubMed] [Google Scholar]
- Pawlak M., Lefebvre P., Staels B. J. Hepatol. 2015;62:720–733. doi: 10.1016/j.jhep.2014.10.039. [DOI] [PubMed] [Google Scholar]
- Varga T., Czimmerer Z., Nagy L. Biochim. Biophys. Acta. 2011;1812:1007–1022. doi: 10.1016/j.bbadis.2011.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarthy M. V., Lodhi I. J., Yin L., Malapaka R. R., Xu H. E., Turk J., Semenkovich C. F. Cell. 2009;138:476–488. doi: 10.1016/j.cell.2009.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmarchelier C., Dahlhoff C., Keller S., Sailer M., Jahreis G., Daniel H. BMC Genomics. 2012;13:84. doi: 10.1186/1471-2164-13-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavrilova O., Haluzik M., Matsusue K., Cutson J. J., Johnson L., Dietz K. R., Nicol C. J., Vinson C., Gonzalez F. J., Reitman M. L. J. Biol. Chem. 2003;278:34268–34276. doi: 10.1074/jbc.M300043200. [DOI] [PubMed] [Google Scholar]
- Inoue M., Ohtake T., Motomura W., Takahashi N., Hosoki Y., Miyoshi S., Suzuki Y., Saito H., Kohgo Y., Okumura T. Biochem. Biophys. Res. Commun. 2005;336:215–222. doi: 10.1016/j.bbrc.2005.08.070. [DOI] [PubMed] [Google Scholar]
- Souza-Mello V. World J. Hepatol. 2015;7:1012. doi: 10.4254/wjh.v7.i8.1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C. H., Olson P., Hevener A., Mehl I., Chong L. W., Olefsky J. M., Gonzalez F. J., Ham J., Kang H., Peters J. M., Evans R. M. Proc. Natl. Acad. Sci. U. S. A. 2006;103:3444–3449. doi: 10.1073/pnas.0511253103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staels B., Rubenstrunk A., Noel B., Rigou G., Delataille P., Millatt L. J., Baron M., Lucas A., Tailleux A., Hum D. W., Ratziu V., Cariou B., Hanf R. Hepatology. 2013;58:1941–1952. doi: 10.1002/hep.26461. [DOI] [PubMed] [Google Scholar]
- Iwaisako K., Haimerl M., Paik Y.-H., Taura K., Kodama Y., Sirlin C., Yu E., Ruth T. Y., Downes M., Evans R. M. Proc. Natl. Acad. Sci. U. S. A. 2012;109:E1369–E1376. doi: 10.1073/pnas.1202464109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagasawa T., Inada Y., Nakano S., Tamura T., Takahashi T., Maruyama K., Yamazaki Y., Kuroda J., Shibata N. Eur. J. Pharmacol. 2006;536:182–191. doi: 10.1016/j.ejphar.2006.02.028. [DOI] [PubMed] [Google Scholar]
- Wada T., Gao J., Xie W. Trends Endocrinol. Metab. 2009;20:273–279. doi: 10.1016/j.tem.2009.03.003. [DOI] [PubMed] [Google Scholar]
- Roth A., Looser R., Kaufmann M., Blattler S. M., Rencurel F., Huang W., Moore D. D., Meyer U. A. Mol. Pharmacol. 2008;73:1282–1289. doi: 10.1124/mol.107.041012. [DOI] [PubMed] [Google Scholar]
- Kolwankar D., Vuppalanchi R., Ethell B., Jones D. R., Wrighton S. A., Hall S. D., Chalasani N. Clin. Gastroenterol. Hepatol. 2007;5:388–393. doi: 10.1016/j.cgh.2006.12.021. [DOI] [PubMed] [Google Scholar]
- Fisher C. D., Lickteig A. J., Augustine L. M., Ranger-Moore J., Jackson J. P., Ferguson S. S., Cherrington N. J. Drug Metab. Dispos. 2009;37:2087–2094. doi: 10.1124/dmd.109.027466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolsey S. J., Mansell S. E., Kim R. B., Tirona R. G., Beaton M. D. Drug Metab. Dispos. 2015;43:1484–1490. doi: 10.1124/dmd.115.065979. [DOI] [PubMed] [Google Scholar]
- Hanagama M., Inoue H., Kamiya M., Shinone K., Nata M. Legal Med. 2008;10:177–184. doi: 10.1016/j.legalmed.2007.12.006. [DOI] [PubMed] [Google Scholar]
- Osabe M., Sugatani J., Fukuyama T., Ikushiro S., Ikari A., Miwa M. Drug Metab. Dispos. 2008;36:294–302. doi: 10.1124/dmd.107.017731. [DOI] [PubMed] [Google Scholar]
- Ning M., Jeong H. Drug Metab. Dispos. 2017;45:707–711. doi: 10.1124/dmd.117.075655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirpich I. A., Gobejishvili L. N., Homme M. B., Waigel S., Cave M., Arteel G., Barve S. S., McClain C. J., Deaciuc I. V. J. Nutr. Biochem. 2011;22:38–45. doi: 10.1016/j.jnutbio.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonoda J., Chong L. W., Downes M., Barish G. D., Coulter S., Liddle C., Lee C.-H., Evans R. M. Proc. Natl. Acad. Sci. U. S. A. 2005;102:2198–2203. doi: 10.1073/pnas.0409481102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugatani J., Wada T., Osabe M., Yamakawa K., Yoshinari K., Miwa M. Drug Metab. Dispos. 2006;34:1677–1687. doi: 10.1124/dmd.106.010645. [DOI] [PubMed] [Google Scholar]
- Cho S. J., Kim S. B., Cho H. J., Chong S., Chung S. J., Kang I. M., Lee J. I., Yoon I. S., Kim D. D. J. Agric. Food Chem. 2016;64:5598–5606. doi: 10.1021/acs.jafc.6b01663. [DOI] [PubMed] [Google Scholar]
- Ma Y., Liu D. PLoS One. 2012;7:e38734. doi: 10.1371/journal.pone.0038734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez J., Mota L., Baldwin W. Curr. Pharmacogenomics Pers. Med. 2009;7:81–105. doi: 10.2174/187569209788654005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H., Tompkins L. M. Curr. Drug Metab. 2008;9:598–610. doi: 10.2174/138920008785821710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanger U. M., Schwab M. Pharmacol. Ther. 2013;138:103–141. doi: 10.1016/j.pharmthera.2012.12.007. [DOI] [PubMed] [Google Scholar]
- Gunes A. and Dahl M.-L., Variation in CYP1A2 activity and its clinical implications: influence of environmental factors and genetic polymorphisms, 2008. [DOI] [PubMed] [Google Scholar]
- George J., Murray M., Byth K., Farrell G. C. Hepatology. 1995;21:120–128. [PubMed] [Google Scholar]
- Moyer B. J., Rojas I. Y., Kerley-Hamilton J. S., Nemani K. V., Trask H. W., Ringelberg C. S., Gimi B., Demidenko E., Tomlinson C. R. Nutr. Res. 2017;44:38–50. doi: 10.1016/j.nutres.2017.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teboul M., Enmark E., Li Q., Wikström A., Pelto-Huikko M., Gustafsson J.-A. Proc. Natl. Acad. Sci. U. S. A. 1995;92:2096–2100. doi: 10.1073/pnas.92.6.2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducheix S., Montagner A., Theodorou V., Ferrier L., Guillou H. Biochem. Pharmacol. 2013;86:96–105. doi: 10.1016/j.bcp.2013.03.016. [DOI] [PubMed] [Google Scholar]
- Ahn S. B., Jang K., Jun D. W., Lee B. H., Shin K. J. Dig. Dis. Sci. 2014;59:2975–2982. doi: 10.1007/s10620-014-3289-x. [DOI] [PubMed] [Google Scholar]
- Ghoneim R. H., Ngo Sock E. T., Lavoie J. M., Piquette-Miller M. Br. J. Nutr. 2015;113:507–516. doi: 10.1017/S0007114514003717. [DOI] [PubMed] [Google Scholar]
- Yang Z.-H., Miyahara H., Takeo J., Katayama M. Diabetol. Metab. Syndr. 2012;4:32. doi: 10.1186/1758-5996-4-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basciano H., Miller A. E., Naples M., Baker C., Kohen R., Xu E., Su Q., Allister E. M., Wheeler M. B., Adeli K. Am. J. Physiol.: Endocrinol. Metab. 2009;297:E462–E473. doi: 10.1152/ajpendo.90764.2008. [DOI] [PubMed] [Google Scholar]
- Wang Y.-D., Chen W.-D., Moore D. D., Huang W. Cell Res. 2008;18:1087. doi: 10.1038/cr.2008.289. [DOI] [PubMed] [Google Scholar]
- Ali A. H., Carey E. J., Lindor K. D. Ann. Transl. Med. 2015;3:5. doi: 10.3978/j.issn.2305-5839.2014.12.06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Wang Z., Klaunig J. E. Soc. Toxicol. 2017:1082. [Google Scholar]