

Abstract
Ionic transport in organometal halide perovskites is of vital importance because it dominates anomalous phenomena in perovskite solar cells, from hysteresis to switchable photovoltaic effects. However, excited state ionic transport under illumination has remained elusive, although it is essential for understanding the unusual light-induced effects (light-induced self-poling, photo-induced halide segregation and slow photoconductivity response) in organometal halide perovskites for optoelectronic applications. Here, we quantitatively demonstrate light-enhanced ionic transport in CH3NH3PbI3 over a wide temperature range of 17–295 K, which reveals a reduction in ionic transport activation energy by approximately a factor of five (from 0.82 to 0.15 eV) under illumination. The pure ionic conductance is obtained by separating it from the electronic contribution in cryogenic galvanostatic and voltage-current measurements. On the basis of these findings, we design a novel light-assisted method of catalyzing ionic interdiffusion between CH3NH3I and PbI2 stacking layers in sequential deposition perovskite synthesis. X-ray diffraction patterns indicate a significant reduction of PbI2 residue in the optimized CH3NH3PbI3 thin film produced via light-assisted sequential deposition, and the resulting solar cell efficiency is increased by over 100% (7.5%–15.7%) with little PbI2 residue. This new method enables fine control of the reaction depth in perovskite synthesis and, in turn, supports light-enhanced ionic transport.
Keywords: activation energy, ion migration, light, perovskite
Introduction
Electrical conduction in materials can be classified into two categories, electronic and ionic, depending on the conducting species. A mixed conductor is a material that possesses both electronic and ionic conductivity, and the recently emerging family of organometal halide perovskites, ABX3 (A: CH3NH3(MA)/NH2CH=NH(FA)/Cs, B: Pb/Sn, X: I/Cl/Br), has proven to be mixed conductors1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12. Ion migration has been reported to be one of the main mechanisms responsible for anomalous I-V hysteresis and poor stability in perovskite solar cells. For optoelectronic applications of these organometal halide perovskites, light-induced effects will inevitably occur in optoelectronic devices such as solar cells, light-emitting diodes (LEDs) and lasers13, 14, 15. Under working conditions and continuous light illumination, the long-term output of perovskite-based solar cells is reported to be unstable. LED applications also exhibit a decrease in efficiency after some period of operation. It is reasonable to speculate that these perovskites possess different properties in their excited state under photoexcitation, which deserve further investigation to gain a better understanding of the optoelectronic properties of perovskites, especially with regard to these unusual light-induced effects10, 16, 17, 18, 19, 20, 21, 22, 23. However, to date, excited state ion migration for perovskites under photoexcitation has rarely been reported in CH3NH3PbI3 (MAPbI3).
Herein, we demonstrate a systematic and quantitative study of light-dependent ionic transport in MAPbI3 film over a wide range of temperatures (17–295 K) and light intensities (0–20 mW cm−2), by means of combined cryogenic galvanostatic6, 24 and voltage-current measurements. Distinct from the band-like nature of the high-mobility electronic transport in this material, the ionic transport exhibits an obvious hopping mechanism with varying activation energies under different illumination intensities. The activation energy for ionic transport shows a significant decrease by approximately a factor of five (from 0.82 to 0.15 eV) as the light intensity increases from 0 to 20 mW cm−2. To the best of our knowledge, this result is the first demonstration of light-dependent ionic transport in organometal halide perovskites. This light-enhanced ion migration can explain the photo-induced giant dielectric constant in the low-frequency regime and the photo-instability in perovskite-based devices as well as the light-induced halide segregation in mixed perovskites. We further demonstrate that this property can be utilized to manipulate the synthesis kinetics of perovskites through light-assisted ion migration. The results yield an obvious increase in solar cell efficiency due to reduced interfacial PbI2. Our findings provide important fundamental insights into the excited state properties of perovskite film, which deserves future investigation in inorganic halide perovskites, even for the whole ionic crystal family.
Materials and methods
MAPbI3 was prepared from an MAI/PbAc2 (Sigma-Aldrich, Shanghai, China) (3:1) precursor solution (215 and 172 mg ml−1 for MAI and PbAc2, respectively) with an additional 4 μl of H3PO2 (Aladdin, Shanghai, China). Then, the solution was spin coated onto a plasma-cleaned FTO/TiO2 substrate. The film was sintered at 100 °C for 50 min in ambient air (~30% relative humidity). The hole transport material (HTM) was then spin coated onto the perovskite film at 3000 rpm for 40 s. The spin-coating formulation was prepared by dissolving 72.3 mg of 2,2',7,7'-tetrakis(N,N-p-dimethoxy-phenylamino)-9,9'-spirobifluorene (spiro-MeOTAD) purchased from Yingkou OPV Tech New Energy Co., Ltd. (Shanghai, China), 30 μl of 4-tert-butylpyridine (tBP) and 20 μl of a stock solution of 520 mg ml−1 lithium bis(trifluoromethylsulphonyl)imide (Li-TFSI) in acetonitrile in 1 ml of chlorobenzene. Finally, 90-nm-thick gold electrodes were deposited via thermal evaporation. The active electrode area was fixed at 9 mm2.
For the light-assisted method of sequential deposition used to prepare the perovskite, PbI2 in DMF at a concentration of 450 mg ml−1 was spin coated onto an FTO/TiO2 substrate (3000 rpm for 30 s), followed by drying at 90 °C for 10 min. An MAI/MACl (45/5 mg ml−1) mixed solution was then spin coated onto the prepared PbI2 film (4000 rpm for 30 s), followed by annealing at 110 °C for 10 min. Three batches of samples were prepared for comparison: annealing without light (0 min), annealing with 5 min of light exposure (5 min), and annealing with 10 min of light exposure (10 min). The light intensity was ~40 mW cm−2. All samples were treated with the same annealing temperature and duration (110 °C/10 min) and were then solvent annealed with DMF at 100 °C for 40 min. For XRD characterizations, these as-prepared samples were directly mounted on the sample stage. For the preparation of solar cell devices, these samples were covered with an electron-blocking layer of spiro-MeOTAD and a gold layer, as described above.
X-ray powder diagrams were recorded using an X’PertMPD PRO from PANalytical equipped with a ceramic tube (Cu anode, λ=1.54060 A), a secondary graphite (002) monochromator and an RTMS X’Celerator detector, operating in the Bragg-Brentano geometry. For a description of the steady-state photoluminescence (PL) spectra and time-resolved fluorescence spectra, which were recorded using a high-resolution streak camera system (Hamamatsu C10910), and the current-voltage measurements, please refer to reference 12. X-ray photoelectron spectra were measured using a PHI Quantera SXM system (Chigasaki, Kanagawa, Japan) under a vacuum of 1.0 × 10−7 Torr, after using an Ar+ gun at 2 kV/30 s to clean the sample surface.
For high-field electric poling experiments, four pairs of electrodes were prepared on a glass substrate using a hard silica template with four pairs of square apertures (with 50-μm gaps and 500-μm widths). The thickness of the gold electrodes was ~0.8 μm. After 30 s of oxygen plasma treatment to remove residual organic material to enhance the wetting properties of the plates, an MAPbI3 precursor solution (MAI/PbAc2 (Sigma-Aldrich) (3:1)) was spin coated onto the substrate, followed by annealing at 100 °C for 50 min in ambient air (30% relative humidity). In the high-field poling experiments, optical images under different illuminations were recorded in ambient air at room temperature (~300 K). In situ observations of the ion drift were performed by placing the samples under an optical microscope (Olympus BX51) coupled to a colored CMOS digital camera, model GCI-070103 (Daheng New Epoch Technology, Inc., Shanghai, China). The optical microscope was operating in the reflection mode with the sample illuminated from the bottom.
The current–voltage (I–V) and galvanostatic characteristics of the samples were obtained using an Agilent B2900 Series precision source/measure unit (Beijing, China). The time duration for each galvanostatic measurement was 0.1 s, and the applied currents were 0.02, 0.04, 0.2, 0.8 and 2 nA for 0, 0.05, 1, 5 and 20 mW cm−2, respectively, considering that the gradually increasing conductivity under stronger illumination would have resulted in very weak signals if the same current of 0.02 nA had been used. After these data were collected, we used the procedures described in the main text to extract the ionic and electronic conductances. Finally, the ideal formula below was used to obtain the conductivity: 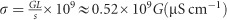 , where G is the conductance, s is the cross-sectional area, and L is the gap in the lateral device architecture.
, where G is the conductance, s is the cross-sectional area, and L is the gap in the lateral device architecture.
For cryogenic electrical experiments, we used a small silica template to prepare Au electrodes confined to the sample stage in the chamber, which left a gap on the perovskite film of 50 μm in width. The cryogenic experiments were conducted in a cryostation (Montana model C2) at temperatures ranging from 17 K to room temperature. The lateral device was directly mounted on the He-cooled cryostat with a temperature controller, in a high-vacuum box at 0.9 μTorr. The device was measured at increments of temperature from 17 to 295 K, with stabilization for more than 10 min at each temperature. The temperature increase was found to result in an increase of less than 2 °C in the temperature of the device under 20 mW cm−2 illumination (230V MI-150 Fiber Optic Illuminator).
Results and discussion
High-field poling behavior of the Au/MAPbI3/Au lateral structure
First, for macroscopic in situ detection of ionic motion under different illumination conditions, we performed high-field electrical poling experiments using an Au/MAPbI3/Au lateral device structure with a 50-μm gap filled with MAPbI3. Because a change in the contrast of optical images recorded under an optical microscope can be observed as a result of mobile ions under high-field poling1, 2, 25, a 100-V bias was applied to this device under three different light intensities (0, 5 and 20 mW cm−2). The dynamic process was recorded on video using a time accelerated mode (Supplementary Movie 1–3), nine snapshots of which are presented in Figure 1. Under dark conditions (Figure 1a), no contrast change of the perovskite film induced by ion migration was observed, whereas under illumination, a black line formed after 10 s of poling (Figure 1b). Moreover, many plane-dendrite structures formed under stronger illumination (Figure 1c), implying more severe ionic motion. Similar evidence of light-enhanced ionic motion was also observed in vacuum under high-field poling (Supplementary Fig. S1). As illustrated in Figure 1d, I− reduction and a subsequent I2 volatilization process occurred at the cathode under high-field poling, and MA+ could also move toward the anode, where it evaporated away in the form of CH3NH2, assisted by moisture and illumination. The gradual transformation25, 26 from MAPbI3 into PbI2 (Supplementary Fig. S2) was accompanied by MA+ migration from the cathode to the anode, which together created the observed threads in the perovskite film. This scenario was further confirmed by comparing the elemental distributions before and after high-field poling, which revealed no discernible ionic pile-up near the electrodes after high-field poling and the formation of many voids around these black threads (Supplementary Fig. S2).
Figure 1.

Optical dynamic images of perovskite film under electric poling and illumination of various intensities in ambient air at room temperature. (a) The film shows little change under dark conditions at a 100-V bias (2 V μm−1 high electric field). The scale bar represents 20 μm. (b) Under 5 mW cm−2 illumination, the film shows black threads at the cathode, with black dots expanding at the anode after 10 s of high-field poling. (c) Under 20 mW cm−2 illumination, the film shows black threads and plane-dendrite structures forming at the cathode and expanding from the cathode to the anode after 10 s of poling. (d) Schematic illustration of the migration of I− anions and MA+ cations through the film. The temperature of the perovskite film under light illumination was monitored with a precision of 1 °C by an infrared thermometer.
Characterization of light-enhanced ionic transport
With these macroscopic findings in mind, we proceeded to a quantitative evaluation of the change in the energy barrier for ion migration under a varying light intensity. Gold was used for the electrodes to guarantee ohmic contact because of the p-type nature of our perovskite film (Supplementary Fig. S3). The time-resolved PL spectrum confirmed the high intrinsic quality of the perovskite film, with a ~270 ns lifetime (Supplementary Fig. S3). To extract the ionic conductivity (σion), a current–voltage (I–V) scan was first performed to obtain the mixed conductivity σtotal in the perovskite film at a 50 V s−1 scan rate in the Au/MAPbI3/Au device. Here, the ionic accumulation effect was essentially negligible because the scanning period was much shorter than the relaxation time for ionic motion (Figure 2a and 2c). Then, we performed galvanostatic characterization with a sufficiently weak current to separate the pure electronic conductivity σe from the mixed conductivity σtotal, which is a standard technique in mixed conductor investigations24, 27, 28. When the current was switched on from 0 to 20 pA, the measured resistance immediately reached an initial value and then gradually increased to a stable value (Figure 2b). The ionic migration and accumulation are the rate-determining processes, whereas the equilibrium value was determined only by the electronic conductivity σe. As the schematic illustrations (Figure 2c–2e) show, initially, both electrons and ions contributed to the conductance, corresponding to the fast-scan I-V measurements and region I in Figure 2b. Subsequently, these mobile ions were gradually depleted because of ionic accumulation at the two sides of the film, resulting in fewer ions contributing to the conductance of the perovskite film (region II and Figure 2d). Finally, the conductance reached a stable value, with all mobile ions blocked at the boundary; then, only the electronic conductance remained (region III and Figure 2e). The double-layer capacitance effect was very weak2, 5 because of the small bias voltage applied in the measurements and the high resistance of the perovskite film. An 800-nm short-pass filter was used here to exclude the influence of phonons on the ionic motion under illumination. Finally, the ionic contribution was extracted from the mixed conductivity by subtracting the electronic conductance from the mixed conductance: σion =σtotal−σe.
Figure 2.

Characterization method for separating the ionic conductance from the mixed conductance in the perovskite film. (a) Voltage–current (V–I) measurements of the Au/perovskite/Au lateral device at 290 K under dark conditions at a 50 V s−1 scan rate. (b) Galvanostatic characterization of the lateral structure under dark conditions. A 20 pA current was switched on at 0.1 s. Region II corresponds to the gradual depletion of the mobile ions, and region III corresponds to the pure electronic conductance in the perovskite film. (c) Schematic illustration of the conductance contributions from both ions and electrons, without any ionic depletion in the perovskite film (Region I). (d) Schematic illustration of the gradual depletion of mobile ions as they accumulate at the two sides of film, leading to a reduced ionic contribution to the conductance of the perovskite film (Region II). (e) Schematic illustration showing only electrons contributing to the conductance after the mobile ions in the film have been totally depleted at a sufficiently weak current of 20 pA, corresponding to the application of a ~0.1-V bias to the 50-μm perovskite film (Region III).
Through the analysis described above, we extracted the pure electronic and ionic conductivities over a wide range of temperatures (17 to 295 K) and light intensities (0 to 20 mW cm−2) using the apparatus shown in Figure 3a, in which incident light was shed on the sample through an objective lens (numerical aperture (NA)=0.2). Figure 3b presents the results of our galvanostatic measurements at different temperatures, where the gradual increase in the resistance corresponds to the slow depletion of the ionic conductivity in the perovskite thin film. In addition, the relaxation times required to reach the steady-state resistance reflect the kinetic constant for ion migration at different temperatures from 100 to 295 K, which is denoted by k=1/τ. The ionic relaxation time is inversely correlated with temperature because of the reduced vibration frequency at lower temperature.
Figure 3.

Determination of electronic and ionic conductivities of perovskite film at various temperatures and various illumination intensities from 0 to 20 mW cm−2. (a) The apparatus used in our experiments, with a helium cooling system. Our lateral device was prepared on a silica substrate with Au electrodes and mounted on a sample stage with a cooling system. (b) Five typical galvanostatic curves (resistance vs time) recorded at different temperatures under a 20 pA current and 1 mW cm−2 illumination. The temperatures from bottom to top are 295, 240, 200, 150 and 100 K, respectively. (c) Electronic conductivities at various temperatures from 17 to 295 K as a function of 1000/T under various illumination intensities. The solid lines represent the fitted results for the electronic conductivity from 150 to 17 K. (d) Ionic conductivities at various temperatures from 17 to 295 K under various illumination intensities. (e) Ionic conductivity multiplied by temperature, σionT, as a function of 1000/T under various illumination intensities. (f) Zoomed-in view of the data in the dashed box in e. The dashed line indicates the phase transition temperature. Note that the ionic conductivity under 5 mW cm−2 illumination has been multiplied by 2 to distinguish the green dots from the purple dots in the plot.
The pure electronic resistance decreases exponentially with increasing temperature, and an obvious linear region corresponding to defect ionization appears above 150 K (Figure 3c). The corresponding ionization energies for these defects in the perovskite film are 92, 84, 73, 44 and 53 meV under illumination intensities of 0, 0.05, 1, 5 and 20 mW cm−2, respectively. The results indicate shallow-level defects in the sample. The reduction in the ionization energy level under illumination is attributed to a screening effect induced by the photoexcited carriers. We also observe a conductivity flip below 70 K under illumination. This behavior strongly implies an inverse power dependence of the mobility on the temperature, μ∝T−3/2, at temperatures ranging from 150 to 17 K10, 29, originating from the acoustic phonon scattering in perovskites. The corresponding electronic conductivity is described by the semi-empirical formula below:
where Nd is the total defect concentration, β is 1/kBT, kB is the Boltzmann constant, Ea is the activation energy for trap defects, a is a constant, I0 is the light intensity, and f(I0) is the carrier concentration generated via photoexcitation at an excitation power of I0. Without photoexcitation (f(I0)=0), the conductivity flip is very weak because of the extremely low carrier concentration near 100 K. However, under constant illumination, the second term dominates the conductivity near 100 K, where μ(T) manifests itself by photogenerated carriers. Using the above formula, the experimental electronic conductivity data can be well fitted from 150 to 20 K (solid lines in Figure 3c).
The temperature-dependent ionic conductivity is plotted in Figure 3d. Unlike the exponential law that governs the electronic conductivity with respect to 1000/T, ln(σion) shows a much less clear behavior in the activation region, especially under photoexcitation. Unlike the band-like transport observed for electrons, the formula that describes the hopping-like ionic transport depends on the energy barrier Ea27:
Through transformation, we obtain
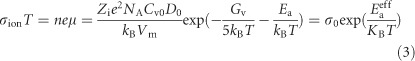 |
where Zi is the ionic charge, NA is Avogadro's constant, Cv is the concentration of intrinsic defects, kB is the Boltzmann constant, Vm is the molar volume of perovskite, D is the diffusion coefficient, Gv is the formation energy for vacancy defects, Ea is the activation energy for ionic diffusion, and Eaeff is defined to consider an excess vacancy formation energy in a vacancy-mediated mechanism. This formula shows many differences from the band-like electronic transport behavior and suggests that ln(σionT), rather than ln(σion), should be plotted versus 1000/T (Figure 3e). Linear regions from 140 to 295 K are observed in this plot, and the corresponding slopes markedly decrease with stronger illumination, reflecting the reduction in the activation energy for ionic transport. A zoomed-in view of the activation region (Figure 3f) further reveals two separate linear regions of the ionic conductance, Ea1 (T>250 K) and Ea2 (180<T<250 K), which are summarized in Table 1 and offer quantitative evidence for light-dependent ion migration.
Table 1. Summary of the activation energies extracted from the ionic/electronic conductivities under different light intensities for MAPbI3.
| Light intensity (mW cm-2) | Ea1 (T>250 K) (meV) | Ea2 (180<T<250 K) (meV) | Ea (electronic) (meV) |
|---|---|---|---|
| 0 | Null | 134 | 92 |
| 0.05 | 824 | 84 | 84 |
| 1 | 851 | 63 | 73 |
| 5 | 334 | Null | 44 |
| 20 | 144 | Null | 53 |
Note: The linear behavior from 180 to 250 K breaks down when the excitation power exceeds 5 mW cm−2 at the phase transition point, thus making it impossible to extract the corresponding activation energies at 5 and 20 mW cm−2 for the ionic conductance in the temperature range from 180 to 250 K.
The reported activation energies for I− and MA+ migration range from 0.1 to 1 eV8, 9, 10, 24. H+ migration is theoretically predicted to have an activation energy of 0.17 eV, which will decrease as a result of nuclear quantum tunneling7. The Ea1 values obtained in our measurements range from 0.82 to 0.14 eV and can thus be assigned to I- or MA+, whereas the Ea2 values are quite small, from 0.13 to 0.06 eV, and therefore should be assigned to H+. Further experimental evidence of proton migration via quantum tunneling is indicated by the crossover point of the kinetic constant near 100 K, where the effects of quantum tunneling and thermal hopping meet26. This behavior will be discussed in our future work. The generation of protons should be closely related to MA+. An H+ ion is attracted by the lone pair of nitrogen atom in CH3NH2, where H+ acts as a Lewis acid, leading to a charge transfer interaction; however, the resulting bond can be broken by thermal perturbation. For both I−/MA+ and H+, the activation energy shows a marked reduction as the illumination intensity increases (Table 1), consistent with the increased ionic conductivity in perovskites under stronger illumination. To the best of our knowledge, this result is the first demonstration of light-dependent ionic transport in organometal halide perovskites.
Unlike the electronic conductance10, 29, we observe that the ionic motion is influenced by phase transition (Supplementary Fig. S4), as indicated by a jump in the conductivity at ~190 K (Figure 3f). This phenomenon should be ascribed to the light-induced enhancement of the ion migration behavior caused by the reduced activation energy: the change in the energy barrier due to the phase transition results in a larger degree of influence on ionic transport with a smaller activation energy. In addition, the disappearance of the linear region corresponding to Ea1 (T>250 K) under dark condition may be related to a larger activation energy under dark condition, which should be observed at higher temperatures.
A structural transformation of the perovskite, a change in the valence of the ions or a weakened bond strength of the MA+ may explain the reduction in the migration barrier under illumination2, 7, 20, 21, 22. Theoretical calculations show that differently charged defects allow ionization-enhanced migration and that photoexcitation may modulate the defects’ charge, thereby influencing ionic transport7. Recent work has also revealed that polar molecules can greatly influence MA+ motion in perovskites because of the reduced bond strength between MA+ and adjacent I− cations2. Hence, illumination may also weaken the hydrogen bonding of MA+ by means of photo-induced carriers, thereby influencing the ionic motion in perovskites21, 30.
Light-induced phenomena in metal halide perovskites
Light-dependent ionic transport has important implications for the effects of light exposure on perovskite solar cells, such as the photo-induced halide segregation20, 31, the giant dielectric constant23, and the photo-instability of the long-term output16. We present a schematic diagram of the enhanced ion migration under illumination in Figure 4a. Under the same external field, the ionic accumulation is accelerated by light exposure because of the increase in the diffusion coefficient, 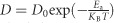 . Therefore, ionic transport becomes easier when the light intensity increases. The observed halide segregation behavior under illumination in mixed MAPb(IxBr1−x)3 can be understood as an effect of light-enhanced ionic transport. For MAPb(I0.5Br0.5)3, the initial PL peak at 640 nm splits into two peaks (~650 and 750 nm) under illumination (Figure 4b), accompanied by a monotonic increase in the PL intensity near 750 nm over time. These finding indicate that ion migration and redistribution occur when the sample is illuminated. However, this process is not fully reversible if the samples are stored under dark conditions for 60 min; only a slight blue shift is observed in the PL (Figure 4c). Phenomenologically, we speculate that light can induce a bonding transformation, thereby changing the Gibbs energy-concentration relation (Figure 4d), reducing the miscibility under light exposure. Meanwhile, because of the reduced energy barrier, ions move rapidly toward more stable sites to minimize the total Gibbs energy, causing the MAPb(I0.5Br0.5)3 to separate into MAPb(IxBr1−x)3 with various doping concentrations. Although the miscibility of the mixed perovskites returns to a high level after storage under dark conditions, Ea also immediately increases, thereby suppressing ionic motion; consequently, the perovskite tends to remain in a metastable state. The light-induced giant dielectric constant of this material is also related: since chemical capacitance can be induced by ionic motion in the low-frequency regime, a larger dielectric constant will result from the stronger ionic motion under illumination,
. Therefore, ionic transport becomes easier when the light intensity increases. The observed halide segregation behavior under illumination in mixed MAPb(IxBr1−x)3 can be understood as an effect of light-enhanced ionic transport. For MAPb(I0.5Br0.5)3, the initial PL peak at 640 nm splits into two peaks (~650 and 750 nm) under illumination (Figure 4b), accompanied by a monotonic increase in the PL intensity near 750 nm over time. These finding indicate that ion migration and redistribution occur when the sample is illuminated. However, this process is not fully reversible if the samples are stored under dark conditions for 60 min; only a slight blue shift is observed in the PL (Figure 4c). Phenomenologically, we speculate that light can induce a bonding transformation, thereby changing the Gibbs energy-concentration relation (Figure 4d), reducing the miscibility under light exposure. Meanwhile, because of the reduced energy barrier, ions move rapidly toward more stable sites to minimize the total Gibbs energy, causing the MAPb(I0.5Br0.5)3 to separate into MAPb(IxBr1−x)3 with various doping concentrations. Although the miscibility of the mixed perovskites returns to a high level after storage under dark conditions, Ea also immediately increases, thereby suppressing ionic motion; consequently, the perovskite tends to remain in a metastable state. The light-induced giant dielectric constant of this material is also related: since chemical capacitance can be induced by ionic motion in the low-frequency regime, a larger dielectric constant will result from the stronger ionic motion under illumination,  .
.
Figure 4.

Photo-induced effects in perovskite films and devices. (a) Schematic illustration of light-dependent ion migration in perovskite MAPbI3. (b) Photoluminescence spectra of the mixed perovskite MAPb(I0.5Br0.5)3 after various illumination durations. (c) Photoluminescence spectra of the mixed perovskite MAPb(I0.5Br0.5)3 after subsequent storage under dark conditions for 1 and 60 min. (d) Schematic plot of the Gibbs energy as a function of the doping concentration for MAPb(IxBr1−x)3 under dark and light conditions. β (I4/mcm) represents Br doping in the tetragonal MAPbI3 matrix, and α (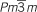 ) represents I doping in the cubic MAPbBr3 matrix. (e) Light-soaking stability tests under various illumination conditions. The devices were tested in an uncontaminated glove box at room temperature and were held at the maximum power point at 0.7 V, and the data were directly obtained from the steady-state values.
) represents I doping in the cubic MAPbBr3 matrix. (e) Light-soaking stability tests under various illumination conditions. The devices were tested in an uncontaminated glove box at room temperature and were held at the maximum power point at 0.7 V, and the data were directly obtained from the steady-state values.
Finally, we discuss the photo-instability observed in the stabilized power conversion efficiency (PCE) of perovskite solar cells in a glove box, which is among the greatest challenges hindering the commercialization of perovskite solar cells. It is well known that perovskite solar cells demonstrate worse stability under illumination than under dark conditions, even in the absence of decomposition induced by moisture or oxygen16. Moreover, this fundamental degradation mechanism is strongly related to the illumination intensity, as shown in Figure 4e. The steady-state PCE exhibits only a slight decrease under illumination at 0.5 or 4.5 mW cm−2, but it drops to ~70% of its initial value during the first 100 s when the light intensity is increased to 30 or 100 mW cm-2. We speculate that under strong illumination, more ions tend to migrate toward the two sides of the device because of the reduced energy barrier and that the charge transfer is consequently suppressed by these excess defects2, 8, 10, 13. Therefore, additional concerns regarding the stability of perovskite solar cells under illumination should be raised by the light-dependent ionic transport in MAPbI3.
Light-assisted sequential deposition
Considering that ionic interdiffusion is involved in the reaction between PbI2 and MAI stacking layers fabricated via sequential deposition, the limited ionic transport in the vertical direction usually results in excess PbI2 residue32. Microstructured PbI2 has been proposed to solve this problem in previous studies33, 34, 35. Herein, inspired by the observation of light-enhanced ion migration, we developed a novel light-assisted sequential deposition method to reduce excess PbI2. The procedures are illustrated in Figure 5a. After MAI is spin coated onto a prepared uniform film of PbI2, the subsequent annealing process is assisted by 40 mW cm−2 light exposure. Three batches of samples were produced for comparison, corresponding to 0, 5 and 10 min of light exposure during the annealing process (110 °C/10 min). The XRD patterns show evidence of a significant reduction in PbI2 residue (2θ=12.6°) in the case of light-assisted sequential deposition (Figure 5b). As a result, the corresponding device efficiency is increased from 7.5 to 15.7% with the reduction in PbI2 (Figure 5c). The series resistances Rs of the cells were derived from the intercepts of the linear fitting results for plots of −dV/dJ vs (JSC−J)−1, where J and Jsc are the current density and short-circuit current density, respectively36. The derived series resistance was found to decrease from 25.5 to 3.8 Ω cm2 (Figure 5d) as a result of light-assisted synthesis, indicating that the reduction in interfacial PbI2 enhances the charge transfer process. The steady-state performance at the maximum power point reveals an extremely slow response of the control device (0 min) when the light is switched on (Figure 5e). This slow process is consistent with the gradual improvement in the I–V measurements with increasing illumination duration observed for the control device (Figure 5f), in which stability is reached only after 4 min of light exposure. To detect the location of the residual PbI2, we used time-of-flight secondary-ion-mass spectrometry (ToF-SIMS) to obtain the chemical depth profiles for lead and iodide (Supplementary Fig. S5), which revealed that these substances are mainly located at the bottom of the film. Therefore, excess PbI2 at the bottom of the film will hinder charge transfer, and we speculate that, facilitated or activated by light, MA+/I− will slowly move and react with PbI2 under a chemical potential gradient. This slow reaction leads to the gradual improvement in charge transfer behavior observed here. Therefore, the results presented above further support the observation of light-enhanced ionic transport.
Figure 5.

Light-assisted sequential deposition. (a) Schematic illustration of light-assisted sequential deposition: during the annealing process, visible light is shed on the sample through an 800-nm short-pass filter. (b) XRD patterns of the as-prepared perovskite films without light exposure (0 min), with 5 min of light exposure (5 min) and with 10 min of light exposure (10 min) during an otherwise identical 10-min annealing process. (c) Current–voltage (I–V) measurements of solar cells fabricated using the corresponding films prepared with or without light exposure during the annealing process. (d) Plots of dV/dJ vs (JSC–J)−1 and the linear fit curves for these cells. (e) Steady-state performance of these devices at the maximum power point (0.55, 0.75 and 0.83 V represents for 0, 5 and 10 min of light exposure, respectively). (f) For the cell fabricated without light-assisted sequential deposition, the changes in the I–V curves during a continuous measurement under AM1.5 illumination. The values 0, 1, 2, 3 and 4 represent the light exposure durations, during which the cell was under open-circuit conditions.
Conclusions
Light-dependent ionic transport has been quantitatively demonstrated in perovskite film over a wide range of illumination intensities by separating the ionic conductivity from the mixed conductivity. Light exerts a significant influence on the ionic transport in MAPbI3 by reducing the activation energies for I−/MA+ and H+. This property of MAPbI3 has important implications for the photo-induced halide segregation, giant dielectric constant, and unstable behavior observed in perovskite films and devices. A light-assisted synthesis method is proposed to control the formation of PbI2 residues in perovskites to ensure better film quality by means of the light-enhanced ionic interdiffusion process. Further attention should be paid to the entire perovskite family, such as in NH2CH=NHPbI3 and CsPbI3, especially for inorganic system, to determine the role of organic component in light-induced effects. Our findings also imply a complicated interplay between light conditions and optoelectronic properties in excited state perovskite materials as a result of ion redistribution, thus making it urgent to investigate the microscopic mechanisms of these behaviors.
Acknowledgments
This work was supported by the National 973 Project (2013CB932602, MOST) of the Ministry of Science and Technology of China and the National Natural Science Foundation of China (NSFC51272007, 61571015, 11327902, 11234001, 91433102 and 51522201). QZ acknowledges the Beijing Nova Program (XX2013003) and the Program for New Century Excellent Talents in University of China.
Footnotes
Note: Supplementary Information for this article can be found on the Light: Science & Applications’ website(http://www.nature.com/lsa).
The authors declare no conflict of interest.
Supplementary Material
References
- Xiao ZG, Yuan YB, Shao YC, Wang Q, Dong QF et al. Giant switchable photovoltaic effect in organometal trihalide perovskite devices. Nat Mater 2015; 14: 193–198. [DOI] [PubMed] [Google Scholar]
- Leijtens T, Hoke ET, Grancini G, Slotcavage DJ, Eperon GE et al. Mapping electric field-induced switchable poling and structural degradation in hybrid lead halide perovskite thin films. Adv Energy Mater 2015; 5: 1500962. [Google Scholar]
- Azpiroz JM, Mosconi E, Bisquert J, de Angelis F. Defect migration in methylammonium lead iodide and its role in perovskite solar cell operation. Energy Environ Sci 2015; 8: 2118–2127. [Google Scholar]
- Leijtens T, Eperon GE, Noel NK, Habisreutinger SN, Petrozza A et al. Stability of metal halide perovskite solar cells. Adv Energy Mater 2015; 5: 1500963. [Google Scholar]
- Leijtens T, Srimath Kandada AR, Eperon GE, Grancini G, D'Innocenzo V et al. Modulating the electron-hole interaction in a hybrid lead halide perovskite with an electric field. J Am Chem Soc 2015; 137: 15451–15459. [DOI] [PubMed] [Google Scholar]
- Yang TY, Gregori G, Pellet N, Grätzel M, Maier J. The significance of ion conduction in a hybrid organic-inorganic lead-iodide-based perovskite photosensitizer. Angew Chem Int Ed Engl 2015; 54: 7905–7910. [DOI] [PubMed] [Google Scholar]
- Egger DA, Kronik L, Rappe AM. Theory of hydrogen migration in organic-inorganic halide perovskites. Angew Chem Int Ed Engl 2015; 54: 12437–12441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eames C, Frost JM, Barnes PRF, O'Regan BC, Walsh A et al. Ionic transport in hybrid lead iodide perovskite solar cells. Nat Commun 2015; 6: 7497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meloni S, Moehl T, Tress W, Franckevičius M, Saliba M et al. Ionic polarization-induced current-voltage hysteresis in CH3NH3PbX3 perovskite solar cells. Nat Commun 2016; 7: 10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner TM, Egger DA, Kronik L, Hodes G, Cahen D. Hybrid organic—inorganic perovskites: low-cost semiconductors with intriguing charge-transport properties. Nat Rev Mater 2016; 1: 15007. [Google Scholar]
- Xu JX, Buin A, Ip AH, Li W, Voznyy O et al. Perovskite-fullerene hybrid materials suppress hysteresis in planar diodes. Nat Commun 2015; 6: 7081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao YC, Zhou WK, Ma W, Meng S, Li H et al. Correlations between immobilizing ions and suppressing hysteresis in perovskite solar cells. ACS Energy Lett 2016; 1: 266–272. [Google Scholar]
- Cho H, Jeong SH, Park MH, Kim YH, Wolf C et al. Overcoming the electroluminescence efficiency limitations of perovskite light-emitting diodes. Science 2015; 350: 1222–1225. [DOI] [PubMed] [Google Scholar]
- Zhu HM, Fu YP, Meng F, Wu XX, Gong ZZ et al. Lead halide perovskite nanowire lasers with low lasing thresholds and high quality factors. Nat Mater 2015; 14: 636–642. [DOI] [PubMed] [Google Scholar]
- Wei HT, Fang YJ, Mulligan P, Chuirazzi W, Fang HH et al. Sensitive X-ray detectors made of methylammonium lead tribromide perovskite single crystals. Nat Photonics 2016; 10: 333–339. [Google Scholar]
- Zhao YC, Wei J, Li H, Yan Y, Zhou WK et al. A polymer scaffold for self-healing perovskite solar cells. Nat Commun 2016; 7: 10228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HS, Mora-Sero I, Gonzalez-Pedro V, Fabregat-Santiago F, Juarez-Perez EJ et al. Mechanism of carrier accumulation in perovskite thin-absorber solar cells. Nat Commun 2013; 4: 2242. [DOI] [PubMed] [Google Scholar]
- Deng YH, Xiao ZG, Huang JS. Light-induced self-poling effect on organometal trihalide perovskite solar cells for increased device efficiency and stability. Adv Energy Mater 2015; 5: 1500721. [Google Scholar]
- Lee JW, Kim DH, Kim HS, Seo SW, Cho SM et al. Formamidinium and cesium hybridization for photo- and moisture-stable perovskite solar cell. Adv Energy Mater 2015; 5: 1501310. [Google Scholar]
- Hoke ET, Slotcavage DJ, Dohner ER, Bowring AR, Karunadasa HI et al. Reversible photo-induced trap formation in mixed-halide hybrid perovskites for photovoltaics. Chem Sci 2015; 6: 613–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottesman R, Gouda L, Kalanoor BS, Haltzi E, Tirosh S et al. Photoinduced reversible structural transformations in free-standing CH3NH3PbI3 perovskite films. J Phys Chem Lett 2015; 6: 2332–2338. [DOI] [PubMed] [Google Scholar]
- Gottesman R, Haltzi E, Gouda L, Tirosh S, Bouhadana Y et al. Extremely slow photoconductivity response of CH3NH3PbI3 perovskites suggesting structural changes under working conditions. J Phys Chem Lett 2014; 5: 2662–2669. [DOI] [PubMed] [Google Scholar]
- Juarez-Perez EJ, Sanchez RS, Badia L, Garcia-Belmonte G, Kang YS et al. Photoinduced giant dielectric constant in lead halide perovskite solar cells. J Phys Chem Lett 2014; 5: 2390–2394. [DOI] [PubMed] [Google Scholar]
- Yokota I. On the theory of mixed conduction with special reference to the conduction in silver sulfide group semiconductors. J Physical Soc Japan 1961; 16: 2213–2223. [Google Scholar]
- Yuan YB, Chae J, Shao YC, Wang Q, Xiao ZG et al. Photovoltaic switching mechanism in lateral structure hybrid perovskite solar cells. Adv Energy Mater 2015; 5: 1500615. [Google Scholar]
- Yuan YB, Wang Q, Shao YC, Lu HD, Li T et al. Electric-field-driven reversible conversion between methylammonium lead triiodide perovskites and lead iodide at elevated temperatures. Adv Energy Mater 2016; 6: 1501803. [Google Scholar]
- Mizusaki J, Arai K, Fueki K. Ionic conduction of the perovskite-type halides. Solid State Ion 1983; 11: 203–211. [Google Scholar]
- Yokota I. On the electrical conductivity of cuprous sulfide: a diffusion theory. J Physical Soc Japan 1953; 8: 595–602. [Google Scholar]
- Oga H, Saeki A, Ogomi Y, Hayase S, Seki S. Improved understanding of the electronic and energetic landscapes of perovskite solar cells: high local charge carrier mobility, reduced recombination, and extremely shallow traps. J Am Chem Soc 2014; 136: 13818–13825. [DOI] [PubMed] [Google Scholar]
- Wu XJ, Yu H, Li LK, Wang F, Xu HH et al. Composition-dependent light-induced dipole moment change in organometal halide perovskites. J Phys Chem C 2015; 119: 1253–1259. [Google Scholar]
- McMeekin DP, Sadoughi G, Rehman W, Eperon GE, Saliba M et al. A mixed-cation lead mixed-halide perovskite absorber for tandem solar cells. Science 2016; 351: 151–155. [DOI] [PubMed] [Google Scholar]
- Xiao ZG, Bi C, Shao YC, Dong QF, Wang Q et al. Efficient, high yield perovskite photovoltaic devices grown by interdiffusion of solution-processed precursor stacking layers. Energy Environ Sci 2014; 7: 2619–2623. [Google Scholar]
- Zhang H, Mao J, He HX, Zhang D, Zhu HL et al. A smooth CH3NH3PbI3 film via a new approach for forming the PbI2 nanostructure together with strategically high CH3NH3I concentration for high efficient planar-heterojunction solar cells. Adv Energy Mater 2015; 5: 1501354. [Google Scholar]
- Liu TH, Hu Q, Wu J, Chen K, Zhao LC et al. Mesoporous PbI2 scaffold for high-performance planar heterojunction perovskite solar cells. Adv Energy Mater 2016; 6: 1501890. [Google Scholar]
- Wu CG, Chiang CH, Tseng ZL, Nazeeruddin MK, Hagfeldt A et al. High efficiency stable inverted perovskite solar cells without current hysteresis. Energy Environ Sci 2015; 8: 2725–2733. [Google Scholar]
- Shi JJ, Dong J, Lv ST, Xu YZ, Zhu LF et al. Hole-conductor-free perovskite organic lead iodide heterojunction thin-film solar cells: high efficiency and junction property. Appl Phys Lett 2014; 104: 063901. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.