

 Numerous studies have demonstrated that, upon maternal exposure, nano-TiO2 can cross the placental barrier, accumulate in offspring animals, and cause neurotoxicity.
Numerous studies have demonstrated that, upon maternal exposure, nano-TiO2 can cross the placental barrier, accumulate in offspring animals, and cause neurotoxicity.
Abstract
Background: Numerous studies have demonstrated that, upon maternal exposure, nano-TiO2 can cross the placental barrier, accumulate in offspring animals, and cause neurotoxicity. However, the neurotoxic mechanisms are not fully understood. The aim of this study is to determine the effects of nano-TiO2 on the dendritic outgrowth of hippocampal neurons and confirm the role of apoptosis and excessive autophagy in the neurotoxicity of offspring mice caused by nano-TiO2, as well as its molecular mechanisms. Methods: Pregnant mice were intragastrically administered 1, 2, or 3 mg per kg body weight nano-TiO2 consecutively from prenatal day 7 to postpartum day 21. The ultrastructure, mitochondrial membrane potential (MMP), levels of reactive oxygen species (ROS) and peroxides, and ATP contents, along with the expression of apoptosis- and autophagy-related factors, were investigated. Results: The dendritic length of hippocampal neurons was lower in the group treated with nano-TiO2 than in the control group. Apoptosis, excessive autophagy, and nano-TiO2 aggregation in hippocampal neurons resulted from maternal exposure to nano-TiO2. Maternal exposure to nano-TiO2 also resulted in the over-production of ROS, increases in malondialdehyde and protein carbonylation, reductions in MMP and ATP contents, up-regulation of apoptosis- or autophagy-related factors including histone H2AX at serine 139 (γH2AX), cytochrome C (Cyt C), caspase 3, phosphoinositide 3-kinase (PI3K3C), Beclin 1, c-Jun, LC3I, LC3II, JNK and p-JNK expression, and an increase of LC3II/LC3I, as well as down-regulation of Bcl-2 expression in hippocampal neurons of offspring mice. Conclusions: Maternal exposure to nano-TiO2 inhibited the dendritic outgrowth of hippocampal neurons. This effect is closely associated with excessive autophagy, which is related to severe oxidative stress and alterations in the expressions of apoptosis- and autophagy-related factors in the hippocampal neurons of offspring mice, due to maternal exposure to nano-TiO2.
Introduction
Due to their novel physicochemical properties and functions, nanomaterials are extremely attractive in various fields, including industry, medicine, agriculture and food. Among the many kinds of nanomaterials, nano-TiO2 is widely used in paints, wastewater treatment, cosmetics, food additives, sterilization, implanted biomaterials, and so on. However, the toxicity of nano-TiO2 to the nervous system attracts considerable attention because of the non-regeneration and susceptibility of neurons.1
Recent studies have shown that inhaled or injected nano-TiO2 can enter the systemic circulation and accumulate in various organs and tissues, causing adverse effects.2–10 In particular, nano-TiO2 can easily cross the blood–brain barrier (BBB) and central nervous system (CNS) via the olfactory pathway or by oral administration, damaging brain neurons and resulting in neurodegenerative diseases and the impairment of spatial recognition.11 Nano-TiO2 can also cross the placental barrier, leading to neurotoxicity in offspring animals.1 The body is in a stage of rapid cell growth and differentiation during the developmental stage. In particular, neurological development is a sensitive biological endpoint to nanomaterial exposure, and the ability of the body to remove poison is low.12,13 Therefore, we hypothesized that exposure to nano-TiO2 during pregnancy resulting from the use of nano-TiO2 in food additives, cosmetics, sunscreens,14–16 and orthopaedic and dental implants will pose a neurotoxicity risk to embryos and affect embryo or infant neural development.17,18 Many studies have unequivocally shown that nano-TiO2 administered during the prenatal period and transferred from pregnant mice to their offspring can damage the nervous system and impair long-term memory and spatial navigation.19–27 Moreover, nano-TiO2 can enter the cytoplasm or nucleus of rat primary hippocampal neurons, inducing apoptosis and inhibiting the growth of neuronal dendrites in vitro.28–30 The hippocampus, which is located in the infoldings of the cerebral cortex, is responsible for learning and long-term memory. Accordingly, we hypothesized that reductions in the long-term memory and spatial navigation of offspring due to maternal exposure to nano-TiO2 may be associated with the inhibition of hippocampal neuron development during embryo or infant development.
A neuron is a type of cell that has polarity, because axons and dendrites grow in different directions. As for hippocampal neurons, apical dendrites play a very important role in accepting information, basal dendrites are featured in the feedback loop, and axons determine the range of outgoing messages. Therefore, dendritic morphology and development are of great significance for signal transduction, signal processing, and feedback loop formation in neurons. However, neuron dendritic outgrowth may be related to autophagy since abnormal autophagy has been shown to seriously damage organism development and differentiation, accelerate the ageing process,31–35 and cause several neurodegenerative diseases.36 Numerous studies have reported that exposure to nano-TiO2 can induce autophagy in different types of cells, including human keratinocytes,37,38 normal lung cells,39–41 vascular endothelial cells, and podocytes.42,43 Whether the suppression of neuron dendritic growth caused by nano-TiO2 exposure involves autophagy is not yet clear.29,30
Therefore, in the present study, we examined the damage to hippocampal neurons and suppression of dendrite development caused by maternal exposure to nano-TiO2, along with the mechanisms, in developing offspring mice. Pregnant mice were consecutively administrated intragastrically with 1, 2, or 3 mg per kg body weight (BW) nano-TiO2 from prenatal day 7 to postpartum day 21. The ultrastructure, mitochondrial membrane potential (MMP), levels of reactive oxygen species (ROS) and peroxides, and ATP contents, as well as the expressions of apoptosis- and autophagy-related factors, were then determined in order to explore the developmental neurotoxicity and molecular mechanism of nano-TiO2 in the hippocampal neurons of offspring mice.
Materials and methods
Chemicals
Nanoparticulate TiO2 was prepared by the controlled hydrolysis of titanium tetrabutoxide. Details of the synthesis and characterization of nano-TiO2 are described in a previous report by our group.44 The nanoparticle characteristics were as follows: anatase phase; particle size = 6.5 nm; hydrodynamic diameter = mainly 294 nm; surface area = 174.8 m2 g–1; and zeta potential = 9.28 mV.44
Ethics approval
All animal experiments were conducted during the light/dark cycle and approved by the Animal Experimental Committee of Soochow University (Grant 2111270).
Animals and treatment
Female CD-1 (ICR) mice (20 ± 1.5 g) and male mice (23 ± 2 g) were purchased from the Animal Center of Soochow University (China). All mice were housed in stainless-steel cages in a ventilated animal room. The temperature of the housing facility was maintained at 24 °C ± 2 °C with a relative humidity of 60% ± 10% and a 12 h light/dark cycle. Distilled water and sterilized food were available ad libitum. Prior to dosing, the mice were acclimated to the environment for five days. Mice were kept in cages according to the ratio of 2 : 1 (♀ : ♂) at 18:00 every evening, and the vaginal suppository of female mice was checked the next morning at 06:00–08:00. Female mice were considered pregnant when the vaginal suppository was found (day 0). Pregnant mice were randomly divided into four subgroups (N = 6 per group), including a control group treated with 0.5% w/v hydroxypropylmethylcellulose (HPMC; Sigma-Aldrich, St Louis, MO, USA, Cat. #: 09963) and three experimental groups treated with 1, 2, or 3 mg per kg BW nano-TiO2 under SPF conditions. For appropriate dose selection, we consulted a World Health Organization report from 1969. According to the report, the LD50 of TiO2 for rats is >12 g per kg BW after oral administration. Dietary exposure to TiO2 in Western populations is 1–3 mg per kg BW per day on average for children under the age of 10 years and approximately 0.2 mg per kg BW per day of TiO2 for other age groups. Assuming that 36% of food-grade TiO2 is smaller than 100 nm in at least one dimension, this exposure limit decreases to approximately 0.1 mg of nanoscale TiO2/person each day.45,46 Animals received 1, 2, or 3 mg per kg BW nano-TiO2 orally using a gavage needle from prenatal day 7 to postnatal day (PND) 21. The same schedule was applied to control pregnant mice treated with 0.5% w/v HPMC. Thirty offspring mice in each group delivered spontaneously were reared with their respective dams until the time of initial experimentation at PND 7. Thirty pups in each group were separated from the mother at PND 21 and housed in cages in an isolated animal room under SPF conditions for 21 days. The room environment was set up at 24 °C ± 2 °C under 60% ± 10% relative humidity and a 12 h light/dark cycle.
All animal experiments were performed in accordance with the Guiding Principles in the Use of Animals in Toxicology.
Isolation and identification of hippocampal neurons
After PND 21, neurons were prepared from the hippocampal Cornus Ammon 1 (CA1) of offspring mice and treated with 0.25% trypsin (Gibco, New York, USA, Cat. #: 25200056) at 37 °C for 10 min. The cells were suspended in neurobasal medium (Gibco, New York, USA, Cat. #: 21103049) containing 2% B27 supplement (Gibco, New York, USA, Cat. #: 17504044) and 1% glutamine (Gibco, New York, USA, Cat. #: 25030081) and plated at a density of 1.0–5.0 × 105 cells per cm2 on poly-l-lysine-coated cell culture clusters in a humidified 5% CO2 atmosphere at 37 °C for 24 h. The reaction was then terminated by serum, and neurons were identified by immunocytochemistry with an antibody against neuron specific enolase (NSE, Thermo Fisher Scientific, USA), a marker for neurons. A culture with more than 95% neurons was used in the following experiments.
Observation of neuron ultrastructure
The primarily cultured hippocampal neurons from offspring mice were fixed in a fresh solution of 0.1 M sodium cacodylate buffer containing 2.5% glutaraldehyde for 2 h, washed 3 times with 0.1 M sodium cacodylate (pH 7.2–7.4), and fixed for 1 h in 1% osmium tetroxide at 4 °C. The specimens were dehydrated in a graded series of ethanol (30%, 50%, 70%, 80%, 90%, and 100%) and embedded in Epon 812. Ultrathin sections were stained with 0.5% aqueous uranyl acetate and lead citrate overnight and then observed using transmission electron microscopy (HITACHI H600, HITACHI, Japan). Dendrites were observed by scanning electron microscopy (QUANTA250, FEI Co., USA). For morphological classification, filopodia were defined as dendritic protrusions of 2–100 μm in length with pointed tips. Primary dendrite length was measured manual tracing using Image-Pro Plus 6.0 Image analysis software (Media Cybernetics, USA).47
MMP assay
MMP in the primarily cultured hippocampal neurons from offspring mice was measured using an MMP assay kit with 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetrathyl-benzimidazolyl-carbocyanine iodide (JC-1; Thermo Fisher Scientific, USA, Cat. #: T3168), using confocal laser scanning microscopy (Leica SP8, Bio-Rad, Hercules, CA, USA).48 JC-1 is a convenient voltage-sensitive probe to monitor MMP. JC-1 showed red fluorescence when J-aggregates formed in normal cells with high MMP; otherwise, it showed red fluorescence when JC-1 maintained its monomeric form in abnormal cells with low MMP. The relative proportions of red and green fluorescence were used to measure the ratio of mitochondrial depolarization.
Assay of titanium content
After postpartum day 21, the hippocampus tissues of offspring mice (N = 5 per group) were thawed. Samples (∼15 mg) were digested, and their titanium contents were determined using inductively coupled plasma-mass spectrometry (Thermo Elemental X7, Thermo Electron Co., Waltham, MA, USA).7 An indium concentration of 20 ng ml–1 was used as an internal standard, and the detection limit of Ti was 0.074 ng ml–1.
Assay of oxidative stress and ATP
ROS production and the levels of malondialdehyde (MDA) and protein carbonylation (PC) in the primarily cultured hippocampal neurons from offspring mice (N = 5 in each group) were assayed using commercial assay kits (Beijing Solarbio Science & Technology Co., Ltd, Beijing, China, Cat. #: CA1410 (ROS), Cat. #: BC0025 (MDA), Cat. #: BC1270 (PC)). ATP levels in the primarily cultured hippocampal neurons from offspring mice (N = 5 in each group) were measured using commercially available kits (Nanjing Jiancheng Bioengineering Institute, China, Cat. #: A095-2). All commercial assay kits were used according to the manufacturer's instructions.
Enzyme-linked immunosorbent assay (ELISA)
ELISA was performed using commercial kits (Shanghai Hushang Biotechnology Co., Ltd, Shanghai, China) to determine the levels of apoptosis- or autophagy-related factors such as phosphorylation of γH2AX (Cat. #: BPE35695m), Cyt C (Cat. #: BPE35771m), caspase 3 (Cat. #: BPE35623m), Bcl-2 (Cat. #: BPE35825m), PI3K3C (Cat. #: BPE35615m), Beclin l (Cat. #: BPE35988m) and c-Jun (Cat. #: BPE35289m) in the primarily cultured hippocampal neurons from offspring mice (N = 5 in each group). The manufacturer's instructions were followed. Absorbance was measured on a microplate reader at 450 nm (Varioskan Flash, Thermo Electron, Finland), and the concentrations were calculated from a standard curve for each sample.
Western blotting
The primarily cultured hippocampal neurons from offspring mice were harvested and resuspended in 0.1 mol L–1 pre-cooling Tris-buffered saline (TBS). Total proteins from the cell suspensions were extracted using cell lysis kits (Thermo Fisher Scientific, USA, Cat. #: 78510), centrifuged at 10 000g at 4 °C for 10 min and quantified using BCA protein assay kits (Thermo Fisher Scientific, USA, Cat. #: 23227). Protein samples were separated using sodium dodecyl sulphate–polyacrylamide gel electrophoresis and electroblotted onto polyvinylidene fluoride membranes (Bio-Rad Laboratories, Inc., USA, Cat. #: 162-0177). The membranes were first blocked with TBS containing 5% fat-free dry milk (5%BSA for P-JNK) for 1.5 h and then incubated overnight at 4 °C with primary antibodies: anti-LC3I (1 : 3000, Thermo Fisher Scientific, USA, Cat. #: PA5-22990), anti-LC3II (1 : 3000, Thermo Fisher Scientific, USA, Cat. #: PA1-16930), anti-JNK (1 : 2000, Thermo Fisher Scientific, USA, Cat. #: AHO1362), anti-p-JNK (1 : 2000, Thermo Fisher Scientific, USA, Cat. #: 700031) and anti-GAPDH (1 : 5000, Thermo Fisher Scientific, USA, Cat. #: AM4300). After washing three times with TBST (TBS + Tween 20), the membrane was incubated with HRP-conjugated secondary antibodies (Anti-rabbit, 1 : 10 000, Abcam Trading (Shanghai) Company Ltd, China, Cat. #: ab6721; Anti-mouse, 1 : 5000, Cell Signaling Technology, USA, Cat. #: 7076) for 1 h at room temperature, followed by detection using enhanced chemiluminescence (Thermo Fisher Scientific, Inc., IL, USA, Cat. #: 35055). Immunoreactive bands were visualized using X-ray films. For quantitative analysis, the bands were evaluated with Image J software and normalized for reference GAPDH density.
Statistical analysis
All data are expressed as mean ± standard deviation (SD). Statistical analysis was performed using SPSS 19.0 software (SPSS, Inc., Chicago, IL, USA), and statistical comparisons were made by one-way ANOVA followed by Dunnett's pair-wise multiple comparison t-test. Differences with P values of less than 0.05 were considered significant.
Results
Nano-TiO2 accumulation
To determine whether nano-TiO2 crossed the blood–fetal barrier to enter the brain of the offspring mice, we determined the Ti contents in the hippocampus. We found significant increases in Ti content in the hippocampus with increasing nano-TiO2 dose (Table 1, p < 0.05).
Table 1. Effect of prenatal exposure to nano-TiO2 on the titanium content of the hippocampus in offspring mice.
| Index | Nano-TiO2 (mg per kg BW) |
|||
| 0 | 1 | 2 | 3 | |
| Ti(ng ml–1) | 0.03 ± 0.001a | 15.82 ± 1.09b | 24.85 ± 1.73c | 38.86 ± 2.95d |
Evaluation of dendritic development
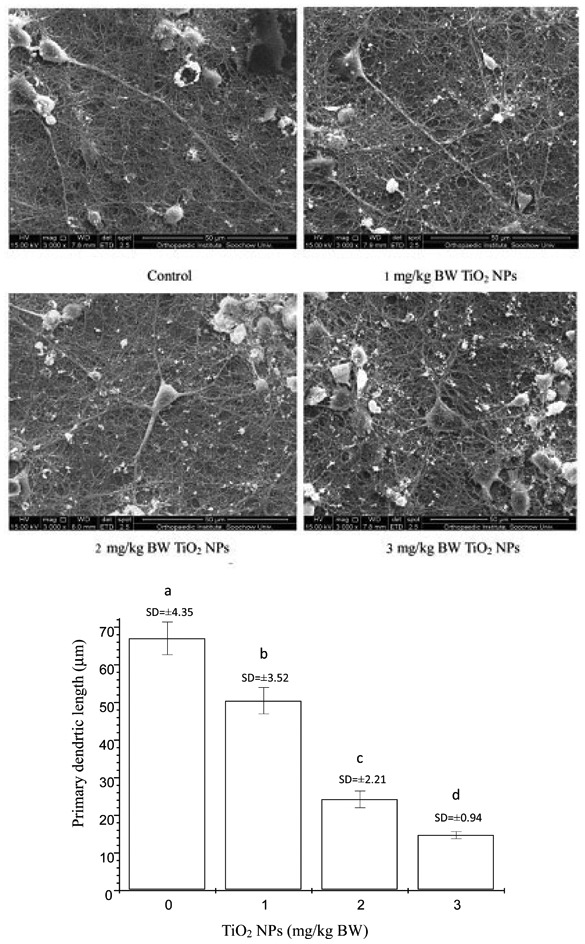
The effects of nano-TiO2 on dendritic outgrowth on offspring hippocampal neurons are shown in Fig. 1. Dendritic growth was significantly suppressed in the three nano-TiO2-treated groups. For nano-TiO2 doses of 1, 2 and 3 mg per kg BW, the primary dendrite length in hippocampal neurons was inhibited by 24.71%, 63.82%, and 77.99% compared to the control (p < 0.05), respectively.
Fig. 1. Neuronal morphometric changes of dendrites in the hippocampus CA1 area of offspring mice at postnatal day 21 following the exposure of pregnant mice to nano-TiO2. Letters b and a indicate significant difference between the groups treated with 1 mg per kg BW nano-TiO2 and the control (p < 0.05); letters c and b indicate significant difference between the groups treated with 2 and 1 mg per kg BW nano-TiO2 (p < 0.01); letters d and c indicate significant difference between the groups treated with 3 and 2 mg per kg BW nano-TiO2 (p < 0.05). Values represent mean ± SD (N = 5).

Observation of neuron ultrastructure
The ultrastructures of the hippocampal neurons are presented in Fig. 2. The hippocampal neurons of the control group contained nuclei with obvious nucleoli, homogeneous chromatin and intact lysosomes (Fig. 2). However, the ultrastructure in the nano-TiO2-treated groups indicated apoptosis, including significant mitochondrial swelling, carina disappearance, nucleus shrinkage, chromatin marginalization, anomalous nuclear membrane, and dilation of endoplasmic reticulum accompanied by the emergence of autolysosomes and the formation of vacuoles. Moreover, nano-TiO2 aggregation was observed in offspring hippocampal neurons (Fig. 2), indicating that nano-TiO2 can accumulate in offspring hippocampal neurons by crossing the placental barrier.
Fig. 2. Ultrastructures (5000×) of hippocampus neurons in offspring mice at postnatal day 21 following the exposure of pregnant mice to nano-TiO2. The control group exhibited round nuclei, obvious nucleoli, and the emergence of lysosomes. Yellow arrows indicate autophagosomes; red arrows indicate mitochondrial swelling; blue arrows indicate chromatin marginalization or nuclear pyknosis; green arrows indicate autophagic vacuoles; and blue ellipses indicate nano-TiO2 aggregation.

MMP changes
To confirm further effects of nano-TiO2 on mitochondrial injury within offspring hippocampal neurons, the changes in MMP of hippocampal neurons were assayed under confocal fluorescence microscopy (Fig. 3). The MMP values in the groups treated with 1, 2, and 3 mg per kg BW nano-TiO2 were significantly reduced by 17.14%, 36.57%, and 51.43% compared to the control (p < 0.05), respectively.
Fig. 3. Changes in mitochondrial membrane potential in offspring hippocampal neurons at postnatal day 21 following the exposure of pregnant mice to nano-TiO2. (a) Representative confocal images showing mitochondrial injury in the hippocampal neurons of the control and nano-TiO2-treated groups. (b) Levels of membrane potential, as indicated by the ratio of red to green fluorescence intensity, in the hippocampal neurons of the control and nano-TiO2-treated groups. Letters b and a indicate significant difference between the group treated with 1 mg per kg BW nano-TiO2 and the control (p < 0.05); letters c and b indicate significant difference between the groups treated with 2 and 1 mg per kg BW nano-TiO2 (p < 0.05); letters d and c indicate significant difference between the groups treated with 3 and 2 mg per kg BW nano-TiO2 (p < 0.05). Values represent mean ± SD (N = 5).

Oxidative stress and ATP content
To confirm the molecular mechanisms of nano-TiO2-induced damages to the hippocampal neurons of offspring mice, oxidative indices including ROS, MDA, and PC were examined. As shown in Fig. 4, the ROS, MDA, and PC levels were significantly elevated by 7.72%, 98.61%, and 98.78% for ROS; 17.37%, 57.09%, and 75.0% for MDA, and 78%, 193%, and 278% for PC (p < 0.05) in the groups treated with 1, 2, and 3 mg per kg BW nano-TiO2 compared to the control, respectively. To demonstrate the disruption of the mitochondrial electron transport chain (ETC) by nano-TiO2, we examined the mitochondrial energy (ATP) and ADP that derived from the hydrolysis of ATP (Fig. 4). The results showed that nano-TiO2 decreased the production of ATP, with reductions of 21.02%, 50.88%, and 59.85% in the groups treated with 1, 2 and 3 mg per kg BW nano-TiO2 compared to the control (p < 0.05), respectively. This suggests that nano-TiO2-induced ROS accumulation led to lipid and protein peroxidation in the hippocampal neurons of offspring mice, thus decreasing the production of ATP.
Fig. 4. Oxidative stress and ATP contents in offspring hippocampal neurons at postnatal day 21 following the exposure of pregnant mice to nano-TiO2. Letters b and a indicate significant difference between the groups treated with 2 or 3 mg per kg BW nano-TiO2 and the control or the group treated with 1 mg per kg BW nano-TiO2 (p < 0.05) in (a), respectively. Letters b and a indicate significant difference between the group treated with 1 mg per kg BW nano-TiO2 and the control (p < 0.05); letters c and b indicate significant difference between the groups treated with 2 and 1 mg per kg BW nano-TiO2 (p < 0.05); and letters d and c indicate significant difference between the groups treated with 3 and 2 mg per kg BW nano-TiO2 (p < 0.05) in (b), (c), and (d), respectively. Values represent mean ± SD (N = 5).

Expressions of apoptosis- and autophagy-related factors
To confirm the role of apoptosis- and autophagy-related factors in nano-TiO2-induced apoptosis and autophagy in the hippocampal neurons of offspring mice, the protein concentrations of apoptosis-related cytokines (γH2AX, Cyt C, caspase 3, and Bcl-2) and autophagy-related factors (PI3K3C, Beclin 1, and c-Jun) were detected by ELISA. As shown in Table 2, compared to the control, the expression of all apoptosis-related factors was significantly increased in the nano-TiO2-treated groups except for Bcl-2, in which it was dramatically decreased (p < 0.05). The expression of autophagy-related factors was significantly increased by nano-TiO2 (p < 0.05).
Table 2. Alterations in the expressions of apoptosis- and autophagy-related proteins in the hippocampal neurons of offspring mice at postnatal day 21 following the exposure of pregnant mice to nano-TiO2.
| Protein expression (ng mg–1 protein) | Nano-TiO2 (mg per kg BW) |
|||
| 0 | 1 | 2 | 3 | |
| rH2AX | 264.20 ± 41.23a | 302.14 ± 31.16b | 421.72 ± 14.12c | 502.41 ± 22.47d |
| Cytc | 34.10 ± 1.03a | 62.23 ± 2.77b | 156.87 ± 17.99c | 242.22 ± 15.89d |
| Caspase 3 | 24.25 ± 3.21a | 32.57 ± 1.55b | 51.75 ± 1.338c | 91.58 ± 3.03d |
| Bcl-2 | 17.19 ± 1.92a | 13.69 ± 1.77b | 8.34 ± 0.31c | 8.21 ± 1.60d |
| PI3K3C | 168.82 ± 13.37a | 948.95 ± 14.05b | 1044.22 ± 12.90c | 1129.31 ± 13.43c |
| Beclin 1 | 1313.33 ± 36.04a | 1517.01 ± 38.75b | 3873.67 ± 43.56c | 4135.34 ± 32.11d |
| c-Jun | 187.80 ± 35.70a | 258.47 ± 41.43b | 672.81 ± 10.32c | 973.42 ± 65.90d |
To further confirm the effects of nano-TiO2 on the expression of LC3I, LC3II, JNK, and p-JNK, the expression of these proteins was examined by western blot (Fig. 5). Nano-TiO2 markedly enhanced the expression of LC3II and phosphorylated JNK (p-JNK) in a dose-dependent manner (p < 0.05). The LC3II/LC3I and p-JNK/JNK ratios also markedly increased in a dose-dependent manner (p < 0.05).
Fig. 5. Changes in the expressions of LC3I, LC3II, JNK, and p-JNK in offspring hippocampal neurons at postnatal day 21 following the exposure of pregnant mice to nano-TiO2. Letters b and a indicate significant difference between the group treated with 1 mg per kg BW nano-TiO2 and the control (p < 0.05); letters c and b indicate significant difference between the groups treated with 2 and 1 mg per kg BW nano-TiO2 (p < 0.05); letters d and c indicate significant difference between the groups treated with 3 and 2 mg per kg BW nano-TiO2 (p < 0.05). Values represent mean ± SD (N = 5).

Discussion
Although recent studies have confirmed that maternal or prenatal exposure to nano-TiO2 can induce brain injury and decrease spatial recognition in offspring mice and rats,19–27 the mechanisms involving the development of hippocampal neurons in offspring are not well elucidated. In this study, the underlying mechanisms of neuronal injury and inhibition of dendritic outgrowth in offspring mice caused by maternal exposure to 1, 2, or 3 mg per kg BW nano-TiO2 from prenatal day 7 to postnatal day 21 were examined. The suppression of dendritic outgrowth (Fig. 1), and severe apoptosis (Fig. 2), excessive autophagy (Fig. 2) as well as nano-TiO2 accumulation in offspring hippocampal tissues (Table 1) or nano-TiO2 deposition in offspring hippocampal neurons (Fig. 2) were observed by nano-TiO2 in a dose-dependent manner, and were coupled with overproduction of ROS, peroxidation of lipid and protein, reduction in ATP production (Fig. 4), and alterations of apoptosis- or autophagy-related factor expression (Table 2 and Fig. 5). Recently, we demonstrated that exposure to nano-TiO2 could reduce dendritic length, the number of branches, and the spine density, and could also impair mitochondrial function in the developing neurons of primary cultured rat hippocampal neurons; however, the mechanism involving suppression of neuronal development is not well understood, due to nano-TiO2.29,30
Nano-TiO2 is known to have biological activity to produce ROS, which are phagocytized by neurons and microglia and then released.28 After exposure to nano-TiO2, ROS overproduction can impair the dynamic balance between oxidation and antioxidation in neurons, and ROS can further attack lipids, proteins and DNA, resulting in peroxidation in neurons, accompanied by increases in MDA and PC (Fig. 4). Mitochondria are known to be the sites of ATP synthesis by the electron transport chain (ETC), providing energy to the organism. However, ROS are also produced in this process. Thus, mitochondria are a major source of ROS production but are also a susceptible target of ROS.49,50 Excessive ROS production can increase the permeability of the mitochondrial membrane, decrease MMP (Fig. 3) and ATP synthesis (Fig. 4), induce the expression of the DNA-damaging marker γH2AX, and contribute to the release of Cyt C from the mitochondria to the cytoplasm, which leads to the up-regulation of caspase 3 and eventually to apoptosis. Moreover, mitochondrially-mediated apoptosis is also controlled by the Bcl-2 family; for example, the anti-apoptotic member Bcl-2 plays an important role in deciding the fate of cells.49–52 Our data show that nano-TiO2 can induce apoptosis (Fig. 2), which is accompanied by the up-regulation of γH2AX, Cyt C, caspase 3 and down-regulation of the Bcl-2 in a dose-dependent manner (Table 2). Our results are consistent with previous in vivo and in vitro findings.28,43 Mitoma et al. suggested that cell necrosis or apoptosis decreased dendritic outgrowth in immature neurons.53 Fukumitsu et al. indicated that oxidative stress induced a rapid drop in ATP levels throughout the entire Purkinje cell, although dendritic localization of mitochondria was required to maintain the local supply of ATP in growing dendrites; the authors thought that ATP generation by dendritic mitochondria may be critical for the normal growth of Purkinje cell dendrites.54 Accordingly, the suppression of dendritic outgrowth may be closely associated with the oxidative stress, low ATP levels, and apoptosis in offspring hippocampal neurons, caused by maternal exposure to nano-TiO2.
Autophagy is a general term for pathways by which cytoplasmic material is delivered to lysosomes for degradation. The autophagic machinery is considered to have evolved as a stress response that allows unicellular eukaryotic organisms to survive during harsh conditions.55–59 Autophagy can be rapidly enhanced when organelles sustain injuries (e.g., attack by ROS, nutrient starvation). Excessive autophagy not only leads to major diseases, including autoimmune diseases, cardiovascular diseases, nervous system diseases, and metabolic syndrome, but also inhibits the growth and development of cells and organisms and accelerates the aging process.31–36 In the present study, maternal exposure to nano-TiO2 led to severe inhibition of dendritic growth in offspring mice hippocampal neurons, which may be associated with the excessive autophagy of hippocampal neurons. Due to the excessive degradation of mitochondria via autophagy, ATP production was severely affected (Fig. 4), which contributed to the suppression of dendritic growth in offspring hippocampal neurons (Fig. 1).
Yoshimori et al. indicated that the formation of autophagic vacuoles is related to the conjugation process of Apg12 and that the modification of LC3. Apg12 can be activated by Apg7 and then conjugate with Apg5. The precursor of LC3 is processed into LC3I, which is also catalyzed by Apg7, and then converted to LC3II when the Apg12–Apg5–Apg16L complex is formed.60 The amount of LC3II is associated with the number of autophagosomes. Autophagy results in the increased conversion of LC3I to LC3II and the up-regulation of LC3 expression. Therefore, the alteration of LC3II expression can be regarded as an important indicator of autophagy. LC3II in autophagosomes can be degraded rapidly by lysosomal hydrolases when the autophagosomes fuse with lysosomes. Hence, the concentration of LC3II or the ratio of LC3II to LC3I is positively correlated with the number of autophagic vacuoles.61 Our data show that maternal exposure to nano-TiO2 not only induced the expression of LC3I and LC3II, but also increased the ratio of LC3II to LC3I (Fig. 5), leading to excessive autophagy (Fig. 2) in offspring hippocampal neurons.
As previously shown, the formation of autophagic vacuoles is closely associated with PI3K3C. PI3K3C phosphorylates phosphatidylinositol to produce phosphatidylinositol-3-P (PtdIns-3-P), recruits proteins containing –FYVE- or –PX- domains to form pre-autophagosomal membranes, and conjugates with Beclin 1 to form the PI3K3C–Beclin 1 complex, which plays an important role in autophagic response.62 Furthermore, a previous study revealed that the inhibitor of PI3K3C can inhibit the formation of autophagosomes; however, Bcl-2 can inhibit the Beclin-dependent autophagy to antagonize apoptosis.63 Our data show that maternal exposure to nano-TiO2 induced the expressions of PI3K3C and Beclin 1 but inhibited Bcl-2 expression (Table 2), thus resulting in autophagy. JNK has been suggested to promote autophagy by two distinct mechanisms.64 In one mechanism, activated JNK is translocated to the nucleus and promotes apoptosis by increasing the expressions of pro-apoptotic genes through the transactivation of c-Jun/AP1-dependent or p53/73 protein-dependent mechanisms. In the second mechanism, activated JNK is translocated to the mitochondria and phosphorylates the BH3-only family of Bcl-2 proteins to antagonize the antiapoptotic activity of Bcl-2 or Bcl-XL. JNK can also stimulate the release of Cyt C from the mitochondrial inner membrane through a Bid-Bax-dependent mechanism, promoting the formation of apoptosomes consisting of Cyt C, caspase 9 and Apaf-1.65 Our data show that maternal exposure to nano-TiO2 induced the expressions of c-Jun, p-JNK, LC3, Beclin 1 and Cyt C and attenuated Bcl-2 expression in offspring hippocampal neurons (Table 2 and Fig. 5), which was obviously associated with the activation of the JNK pathway. Lee et al. indicated that neuronal autophagy may play an important role in the structural refinement of neuritic outgrowth or regeneration, neural differentiation, synaptic growth, or synaptic plasticity; furthermore, the alteration of autophagy during neurodevelopment and synaptic plasticity might result in abnormal development and synaptic malfunction, thus inducing neurodevelopmental disorders.66 Indeed, the suppression of neuritic outgrowth in offspring hippocampal neurons induced by maternal exposure to nano-TiO2 may be closely related to excessive autophagy. However, their mechanisms may be very complex and need to be deeply explored in future studies.
Conclusions
The current study demonstrated that maternal exposure to nano-TiO2 can inhibit dendritic outgrowth in hippocampal neurons in offspring mice. This effect involved a diminished ATP level and the promotion of apoptosis and excessive autophagy. The apoptosis and excessive autophagy were related to severe oxidative stress and obvious alterations in the expressions of apoptosis- or autophagy-related factors due to maternal exposure to nano-TiO2. These findings contribute to the understanding of the mechanisms involved in the suppression of neuronal dendrite outgrowth along with learning and memory in offspring mice. The findings also illuminate the mechanisms of apoptosis and autophagy caused by maternal exposure to nano-TiO2.
Declaration
This submission complies with the Helsinki Declaration. None of the information has been published elsewhere. The submission contains no diagnostic testing or copyrighted material.
Author contributions
Yingjun Zhou, Fashui Hong and Yusheng Tian contributed to the design of the entire study; Xiangyu Zhao, Jie Hong and Yuguan Ze contributed to the experiments on animal treatment, histopathological examination, assay of MMP, oxidative stress, and ATP; Lei Sheng and Ling Wang contributed to the assay of protein expression; Fashui Hong contributed to the proof-reading of the paper.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (grant no. 31671033, 81473007, 81273036 and 30901218), the National Natural Science Foundation of Jiangsu Province (grant no. BK20161306), and the Top-notch Academic Programs Project of Jiangsu Higher Education Institutions (PPZY2015A018).
References
- Hong F. S., Yu X. H., Wu N., Zhang Y. Q. Toxicol. Res. 2017;6:115–133. doi: 10.1039/c6tx00338a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J. X., Zhou G. Q., Chen C. Y., Yu H. W., Wang T. C., Ma Y. M., Jia G., Gao Y. X., Li B., Sun J., Li Y. F., Jiao F., Zhao Y. L., Chai Z. F. Toxicol. Lett. 2007;168:176–185. doi: 10.1016/j.toxlet.2006.12.001. [DOI] [PubMed] [Google Scholar]
- Wang J. X., Liu Y., Jiao F., Lao F., Li W., Gu Y. Q., Li Y. F., Ge C. C., Zhou G. Q., Li B., Zhao Y. L., Chai Z. F., Chen C. Y. Toxicol. 2008;254:82–90. doi: 10.1016/j.tox.2008.09.014. [DOI] [PubMed] [Google Scholar]
- Wang J. X., Chen C. Y., Liu Y., Jiao F., Li W., Lao F., Li Y. F., Li B., Gu Y. Q., Ge C. C., Zhou G. Q., Gao Y. X., Zhao Y. L., Chai Z. F. Toxicol. Lett. 2008;183:72–80. doi: 10.1016/j.toxlet.2008.10.001. [DOI] [PubMed] [Google Scholar]
- Ma L. L., Liu J., Li N., Wang J., Duan Y. M., Yan J. Y., Liu H. T., Wang H., Hong F. S. Biomaterials. 2010;31:99–105. doi: 10.1016/j.biomaterials.2009.09.028. [DOI] [PubMed] [Google Scholar]
- Hu R. P., Gong X. L., Duan Y. M., Li N., Che Y., Cui Y. L., Zhou M., Liu C., Wang H., Hong F. S. Biomaterials. 2010;31:8043–8050. doi: 10.1016/j.biomaterials.2010.07.011. [DOI] [PubMed] [Google Scholar]
- Hu R. P., Zheng L., Zhang T., Cui Y. L., Gao G. D., Cheng Z., Chen J., Tang M., Hong F. S. J. Hazard. Mater. 2011;191:32–40. doi: 10.1016/j.jhazmat.2011.04.027. [DOI] [PubMed] [Google Scholar]
- Ze Y. G., Zheng L., Zhao X. Y., Gui S. X., Sang X. Z., Su J. J., Guan N., Zhu L. Y., Sheng L., Hu R. P., Cheng J., Cheng Z., Sun Q. Q., Wang L., Hong F. S. Chemosphere. 2013;92:1183–1189. doi: 10.1016/j.chemosphere.2013.01.094. [DOI] [PubMed] [Google Scholar]
- Ze Y. G., Hu R. P., Wang X. C., Li B., Su J. J., Sang X. Z., Guan N., Zhao X. Y., Gui S. X., Zhu L. Y., Cheng Z., Cheng J., Sheng L., Sun Q. Q., Wang L., Hong F. S. J. Biomed. Mater. Res., Part A. 2014;102A(2):470–478. doi: 10.1002/jbm.a.34705. [DOI] [PubMed] [Google Scholar]
- Su M. Y., Sheng L., Zhao X. Y., Wang L., Yu X. H., Hong J., Xu B. Q., Liu D., Jiang H., Ze X., Zhu Y. T., Long Y., Zhou J. L., Cui J. W., Li K., Ze Y. G., Hong F. S. Toxicol. Res. 2015;4:344–350. [Google Scholar]
- Song B., Liu J., Feng X. L., Wei L. M., Shao L. Q. Nanoscale Res. Lett. 2015;10:342. doi: 10.1186/s11671-015-1042-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson L. M., Diwan B. A., Fear N. T., Roman E. Environ. Health Perspect. 2000;108(Suppl. 3):573–594. doi: 10.1289/ehp.00108s3573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson L. M. Toxicol. Appl. Pharmacol. 2004;199(2):162–174. doi: 10.1016/j.taap.2004.02.008. [DOI] [PubMed] [Google Scholar]
- Powell J. J., Faria N., Thomas-McKay E., Pele L. C. J. Autoimmun. 2010;34:226–233. doi: 10.1016/j.jaut.2009.11.006. [DOI] [PubMed] [Google Scholar]
- Skocaj M., Filipic M., Petkovic J., Novak S. Radiol. Oncol. 2011;45:227–247. doi: 10.2478/v10019-011-0037-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir A., Westerhoff P., Fabricius L., Hristovski K., von Goetz N. Environ. Sci. Technol. 2012;46:2242–2250. doi: 10.1021/es204168d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster T. J., Kjiofor J. U. Biomaterials. 2004;25(19):4731–4739. doi: 10.1016/j.biomaterials.2003.12.002. [DOI] [PubMed] [Google Scholar]
- Choudhary S., Haberstroh K. M., Webster T. J. Tissue Eng. 2007;13(7):1421–1430. doi: 10.1089/ten.2006.0376. [DOI] [PubMed] [Google Scholar]
- Takeda K., Suzuki K. I., Ishihara A., Kubo-Irie M., Fujimoto R., Tabata M., Oshio S., Nihei T., Sugamata M. J. Health Sci. 2009;55(1):95–102. [Google Scholar]
- Shimizu M., Tainaka H., Oba T., Mizuo K., Umezawa M., Takeda K. Part. Fibre Toxicol. 2009;6(1):141–149. doi: 10.1186/1743-8977-6-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi Y., Shinkai Y., Mizuo K., Oshio S., Takeda K. J. Toxicol. Sci. 2010;35(5):749–756. doi: 10.2131/jts.35.749. [DOI] [PubMed] [Google Scholar]
- Yamashita K., Yoshioka Y., Higashisaka K., Mimura K., Morishita Y., Nozaki M., Yoshida T., Ogura T., Nabeshi H., Nagano K., Abe Y., Kamada H., Monobe Y., Imazawa T., Aoshima H., Shishido K., Kawai Y., Mayumi T., Tsunoda S., Itoh N., Yoshikawa T., Yanagihara I., Saito S., Tsutsumi Y. Nat. Nanotechnol. 2011;6(5):321–328. doi: 10.1038/nnano.2011.41. [DOI] [PubMed] [Google Scholar]
- Okada Y., Tachibana K., Yanagita S., Takeda K. J. Toxicol. Sci. 2013;38(3):363–370. doi: 10.2131/jts.38.363. [DOI] [PubMed] [Google Scholar]
- Mohammadipour A., Hosseini M., Fazel A., Haghir H., Rafatpanah H., Pourganji M., Bideskan A. E. Toxicol. Ind. Health. 2016;32(2):221–228. doi: 10.1177/0748233713498440. [DOI] [PubMed] [Google Scholar]
- Mohammadipour A., Fazel A., Haghir H., Motejaded F., Rafatpanah H., Zabihi H., Hosseini M., Bideskan A. E. Environ. Toxicol. Pharmacol. 2014;37(2):617–625. doi: 10.1016/j.etap.2014.01.014. [DOI] [PubMed] [Google Scholar]
- Cui Y. H., Chen X. Y., Zhu Z., Lei Y., Ma M. N., Cao R. J., Sun T. J., Xu J. L., Huo M. Y., Wen C. H., Che Y. Chemosphere. 2014;96:99–104. doi: 10.1016/j.chemosphere.2013.07.051. [DOI] [PubMed] [Google Scholar]
- Onoda A., Umezawa M., Takeda K., Ihara T., Sugamata M. PLoS One. 2014;9(4):e94336. doi: 10.1371/journal.pone.0094336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng L., Ze Y. G., Wang L., Yu X. H., Hong J., Zhao X. Y., Ze X., Liu D., Xu B. Q., Zhu Y. T., Long Y., Lin A. A., Zhang C., Zhao Y., Hong F. S. J. Biomed. Mater. Res., Part A. 2015;103A:1141–1149. doi: 10.1002/jbm.a.35263. [DOI] [PubMed] [Google Scholar]
- Hong F. S., Sheng L., Ze Y. G., Hong J., Zhou Y. J., Wang L., Yu X. H., Liu D., Xu B. Q., Ze X. Biomaterials. 2015;53(2):76–85. doi: 10.1016/j.biomaterials.2015.02.067. [DOI] [PubMed] [Google Scholar]
- Hong F. S., Ze Y. G., Zhou Y. M., Ye L. Q., Hong J., Yu X. H., Sheng L., Wang L. J. Biomed. Mater. Res., Part A. 2017;105A:2139–2149. doi: 10.1002/jbm.a.36073. [DOI] [PubMed] [Google Scholar]
- Wang G., Lu W. H., Li S., Gao L. R., Huang W. Q., Cui S. D., Wang X. Y., Lu D. X., Yang X. S. Chin. J. Pathophysiol. 2015;(10):1815. [Google Scholar]
- Rubinsztein D. C., Marino G., Kroemer G. Cell. 2011;146(5):682–695. doi: 10.1016/j.cell.2011.07.030. [DOI] [PubMed] [Google Scholar]
- Levine B., Mizushima N., Virgin H. W. Nature. 2011;469(7330):323–335. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meschini S., Condello M., Lista P., Arancia G. Curr. Cancer Drug Targets. 2011;11(3):357–379. doi: 10.2174/156800911794519707. [DOI] [PubMed] [Google Scholar]
- Xilouri M., Stefanis L. CNS Neurol. Disord.: Drug Targets. 2010;9(6):701–719. doi: 10.2174/187152710793237421. [DOI] [PubMed] [Google Scholar]
- Rubinsztein D. C., Codogno P., Levine B. Nat. Rev. Drug Discovery. 2012;11:709–730. doi: 10.1038/nrd3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y., Howe J. L., Yu Z., Leong D. T., Chu J. J. H., Loo J. S. C., Ng K. W. Small. 2013;9:387–392. doi: 10.1002/smll.201201363. [DOI] [PubMed] [Google Scholar]
- Lopes V. R., Loitto V., Audinot J. N., Bayat N., Gutleb A. C., Cristobal S. J. Nanobiotechnol. 2016;14:1. doi: 10.1186/s12951-016-0174-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern S. T., Adiseshaiah P. P., Crist R. M. Part. Fibre Toxicol. 2012;9:20. doi: 10.1186/1743-8977-9-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu K. N., Sung J. H., Lee S., Kim J. E., Kim S., Cho W. Y., Lee A. Y., Park S. J., Lim J., Park C. Food Chem. Toxicol. 2015;85:106–113. doi: 10.1016/j.fct.2015.08.001. [DOI] [PubMed] [Google Scholar]
- Yu K. N., Chang S. H., Park S. J., Lim J., Lee J., Yoon T. J., Kim J. S., Cho M. H. PLoS One. 2015;10:e0131208. doi: 10.1371/journal.pone.0131208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S. G., Newsome B., Hennig B. Toxicology. 2013;306:1–8. doi: 10.1016/j.tox.2013.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X. C., Yin H. Q., Li Z. G., Zhang T., Yang Z. Cell Biol. Toxicol. 2016;32:513–527. doi: 10.1007/s10565-016-9352-y. [DOI] [PubMed] [Google Scholar]
- Hu R. P., Zheng L., Zhang T., Cui Y. L., Gao G. D., Cheng Z., Chen J., Tang M., Hong F. S. J. Hazard. Mater. 2011;191(1–3):32–40. doi: 10.1016/j.jhazmat.2011.04.027. [DOI] [PubMed] [Google Scholar]
- National Institute for Occupational Safety and Health, Outlines Guidance on Handling Titanium Dioxide (TiO2), Atlanta, 2011. [Google Scholar]
- Baan R., Straif K., Grosse Y., Secretan B., El Ghissassi F., Cogliano V. Lancet Oncol. 2006;7:295–296. doi: 10.1016/s1470-2045(06)70651-9. [DOI] [PubMed] [Google Scholar]
- Xu X. H., Ye Y., Li T., Chen L., Tian D., Luo Q. Q., Lu M. Toxicol. Appl. Phamrmacol. 2010;249(2):188–196. doi: 10.1016/j.taap.2010.09.007. [DOI] [PubMed] [Google Scholar]
- Li X. B., Xu S. Q., Zhang Z. R., Hermann J. S. Chin. Sci. Bull. 2009;54(20):3830–3836. [Google Scholar]
- Simon H. U., Yehia A. H., Schaffer F. L. Apoptosis. 2000;5(5):415–418. [Google Scholar]
- Yao J. C., Jiang Z. Z., Duan W. G., Huang J., Zhang L., Hu L., He L., Li F., Xiao Y., Shu B., Liu C. Biol. Pharm. Bull. 2008;31(4):592–597. doi: 10.1248/bpb.31.592. [DOI] [PubMed] [Google Scholar]
- Kandasamy K., Srinivasula S. M., Alnemri E. S., Thompson C. B., Korsmeyer S. J., Bryant J. L., Srivastava R. K. Cancer Res. 2003;63(7):1712–1721. [PubMed] [Google Scholar]
- Suliman A., Lam A., Datta R., Srivastava R. K. Oncogene. 2001;20(17):2122–2133. doi: 10.1038/sj.onc.1204282. [DOI] [PubMed] [Google Scholar]
- Mitoma J., Ito M., Furuya S., Hirabayashi Y. J. Neurosci. Res. 1998;51(6):712–722. doi: 10.1002/(SICI)1097-4547(19980315)51:6<712::AID-JNR5>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Fukumitsu K., Fujishima K., Yoshimura A., Wu Y. K., Heuser J., Kengaku M. J. Neurosci. 2015;35:5707–5723. doi: 10.1523/JNEUROSCI.4115-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B., Kroemer G. Cell Death Differ. 2009;16(1):1–2. doi: 10.1038/cdd.2008.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinsztein D. C., Marino G., Kroemer G. Cell. 2011;146(5):682–695. doi: 10.1016/j.cell.2011.07.030. [DOI] [PubMed] [Google Scholar]
- Levine B., Mizushima N., Virgin H. W. Nature. 2011;469(7330):323–335. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meschini S., Condello M., Lista P., Arancia G. Curr. Cancer Drug Targets. 2011;11(3):357–379. doi: 10.2174/156800911794519707. [DOI] [PubMed] [Google Scholar]
- Xilouri M., Stefanis L. CNS Neurol. Disord.: Drug Targets. 2010;9(6):701–719. doi: 10.2174/187152710793237421. [DOI] [PubMed] [Google Scholar]
- Yoshimori T. Biochem. Biophys. Res. Commun. 2004;313(2):453–458. doi: 10.1016/j.bbrc.2003.07.023. [DOI] [PubMed] [Google Scholar]
- Crotzer V. L., Blum J. S. Proc. Natl. Acad. Sci. U. S. A. 2005;102(22):7779–7780. doi: 10.1073/pnas.0503088102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger S. B., Romero X., Ma C., Wang G. X., Faubion W. A., Liao G. X., Compeer E., Keszei M., Rameh L., Wang N. H., Boes M., Regueiro J. R., Reinecker H. C., Terhorst C. Nat. Immunol. 2010;11(10):920–7927. doi: 10.1038/ni.1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattingre S., Tassa A., Qu X., Garuti R., Liang X. H., Mizushima N., Packer M., Schneider M. D., Levine B. Cell. 2005;122(6):927–939. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- Jia G., Cheng G., Gangahar D. M., Agrawal D. K. Immunol. Cell Biol. 2006;84(5):448–454. doi: 10.1111/j.1440-1711.2006.01454.x. [DOI] [PubMed] [Google Scholar]
- Wei N., He H. B., Zhang C. C., Yuan D., Wang T. Chin. J. Clin. Pharm. Ther. 2013;18(7):807–812. [Google Scholar]
- Lee K. M., Hwang S. K., Lee J. A. Exp. Neurobiol. 2013;22(3):133–142. doi: 10.5607/en.2013.22.3.133. [DOI] [PMC free article] [PubMed] [Google Scholar]