

 Epidemiological studies have revealed that exposure to PM2.5 is linked to liver cancer.
Epidemiological studies have revealed that exposure to PM2.5 is linked to liver cancer.
Abstract
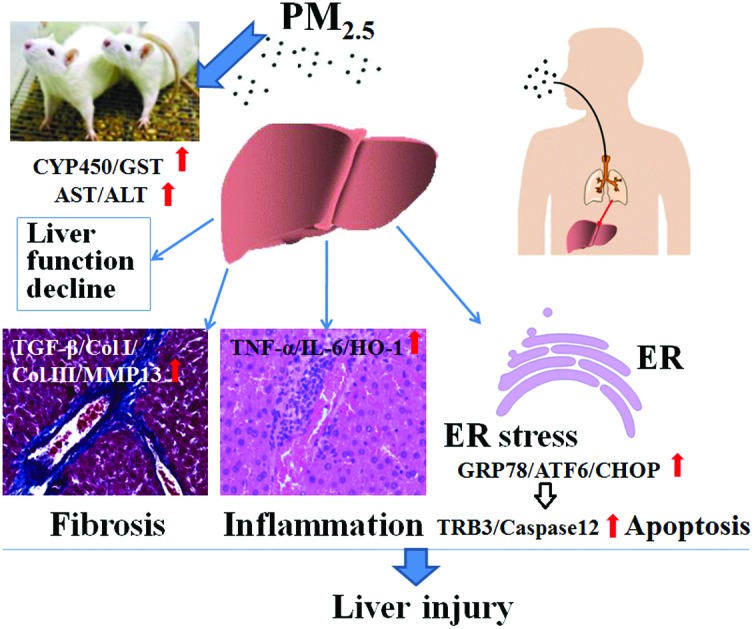
Epidemiological studies have revealed that exposure to PM2.5 is linked to liver cancer. However, the hepatic toxicity and relevant molecular mechanisms of PM2.5 have not yet been fully described. Herein, we report on our investigation of the fibrosis, inflammation, endoplasmic reticulum (ER) stress and apoptosis in the livers of rats, caused by exposure to PM2.5 during summer and winter in Taiyuan, China. Male SD rats were sub-chronically exposed to PM2.5 (in summer: 0.2, 0.6, 1.5 mg per kg of b.w.; in winter: 0.3, 1.5, 2.7 mg per kg of b.w.) via intratracheal instillation once every 3 days for 60 days. The results showed that exposure to high dosages of PM2.5 caused the following: (1) hepatic histopathological changes and liver function decline through elevating the activities of AST, ALT, CYP450 and GST; (2) triggered liver fibrosis, in which TGF-β1, Col I, Col III, and MMP13 mRNA and protein expression were significantly upregulated, and enhanced inflammation with the overexpression of TNF-α, IL-6 and HO-1 versus the control; (3) induced liver ER stress and cell apoptosis via activating the GRP78/ATF6/CHOP/TRB3/caspase 12 pathway. The data also indicated that the liver injury induced by winter PM2.5 in Taiyuan was more serious compared to that induced by summer PM2.5. This work provides new insight into the mechanisms of PM2.5-induced liver injury, and aids the understanding of the underlying mechanisms by which PM2.5 might affect liver diseases.
Introduction
Liver diseases are major global health problems. Cirrhosis has reportedly caused over a million deaths in 187 countries in 2010, and the death toll from liver cancers was estimated as 745 533 in 184 countries in 2012,1,2 in which socioeconomic development and environmental insults are the risk factors that play vital roles in hepatic pathogenesis.2,3
Fine particulate matter with an aerodynamic diameter ≤2.5 μm (PM2.5) is a ubiquitous atmospheric pollutant that is generated mainly from coal combustion, diesel engines, and biomass burning, etc. Alarmingly, it can deposit in pulmonary alveoli through respiration and endanger the lung. The PM2.5 surface may absorb large amounts of hazardous chemicals such as heavy metals and polycyclic aromatic hydrocarbons (PAHs). Through blood circulation, water-soluble fractions or insoluble nanoparticles of PM2.5 can reach other extra-pulmonary organs,4 particularly the liver, which is an important organ for the detoxification of exogenous chemicals.
Many epidemiological studies have revealed that inhalation exposure to airborne PM2.5 is linked to pulmonary diseases, cardiovascular disease and stroke.5,6 Notably, particulate matter has been classified as carcinogenic to humans (Group 1) by the International Agency for Research on Cancer.7,8 There is therefore growing concern about its toxicological roles and systemic effects on human health. Along with many research efforts on the adverse effects of PM2.5 on the cardiorespiratory and nervous systems,5–8 some epidemiological studies have recently confirmed that ambient PM2.5 exposure increases the incidence and mortality of liver cancer,9,10 and other reports have demonstrated that PM2.5 may be a significant risk factor for non-alcoholic fatty liver disease (NAFLD) progression.11,12 Experimental studies have indicated that PM2.5 could affect liver energy metabolism in mice by inhibiting the tricarboxylic acid cycle and stimulating glycolysis, reduce cell survival rates, and induce lipid accumulation and oxidative stress in hepatocytes.13,14 More importantly, Zheng et al. discovered that PM2.5 had a direct adverse health effect on the liver and triggered hepatic fibrosis in a murine model,15 and PM2.5-induced inflammatory responses promoted collagen deposition in the liver by activating the transforming growth factor-β1 (TGF-β1) signaling pathway.15 This is a breakthrough in the studies of PM2.5-induced liver toxicity, since hepatic fibrosis is a pathological condition characterized by the accumulation of the extracellular matrix (ECM) proteins that occur in most types of chronic liver diseases such as NAFLD and hepatocellular carcinoma (HCC).16,17 The work of Zheng et al. also indicated that the PM2.5 pollutant is an independent risk factor for liver fibrosis.15 Thus, the studies on PM2.5-triggered hepatic fibrosis are very significant in terms of identifying new health risk factors and understanding the pathogenesis of liver diseases. However, the current studies on PM2.5 on liver injury or fibrosis are very limited and need further investigation.
The imbalance between fibrogenesis and fibrolysis in the liver may directly influence the process of fibrosis, in which some special fibrotic-related genes exert a critical role in the pathogenesis of liver fibrosis.18–20 TGF-β1 is the major pro-fibrotic growth factor that stimulates the production of ECM proteins, mostly collagen (Col) Type I and III.19 Matrix metalloproteinase-13 (MMP-13) mainly hydrolyzes ECM and degrades collagen, and MMP13 expression may be markedly upregulated in rat liver fibrosis in order to reduce ECM.20 Nevertheless, the changes in TGF-β1, collagen and MMP13 gene expression in rat livers after PM2.5 sub-chronic exposure have been largely uninvestigated.
Inflammatory responses are very important in regulating liver pathological conditions induced by endogenous and exogenous substances. In the pathological process, the liver may produce pro-inflammatory cytokines such as tumor necrosis factor-α (TNF-α) and interlukine-6 (IL-6), which can modulate inflammatory responses.21 Heme oxygenase-1 (HO-1) is an essential enzyme presented in multiple mammalian tissues including the liver, and the induction of HO-1 may fulfill its protective function against inflammatory process and oxidative damage in the liver.22 It is proven that chronic unresolved inflammation is associated with persistent hepatic injury, leading to sequential development of fibrosis, cirrhosis, and eventually HCC.23 In this study, we focus on whether liver fibrosis appears in the rats after subchronic exposure to PM2.5, and meanwhile observed the inflammatory responses and the changes in TNF-α, IL-6 and HO-1 levels in rat livers to understand their relevance to the liver fibrosis.
The endoplasmic reticulum (ER) stress is an intracellular stress response induced by the accumulation of unfolded or misfolded proteins. It is a double-edged sword, that is, it can maintain cellular homeostasis and protect cells from adverse stress as far as possible, whereas the persistent ER stress will evoke cell apoptosis.24,25 Some biomarkers related to ER stress such as glucose-regulated protein (GRP) 78, activating transcription factor 6 (ATF6) and C/-EBP homologous protein (CHOP) contribute a lot to regulating the ER stress process. Previous studies have shown that ER stress may promote fibrogenesis, inflammation and apoptosis in the liver,26–30 in which activation of the ER stress-mediated GRP78 and CHOP/tribbles homolog 3 (TRB3)/caspase 12 could be the important mechanism responsible for the liver injury. Accordingly, we investigated the ER stress and apoptosis responses incurred by PM2.5 and the related molecular mechanisms.
Taiyuan is a typical northern city of China with the characteristics of the coal industry. The main problem of the atmospheric environment in Taiyuan is the soot pollution that usually appears in the heating period of winter, and accordingly, PM2.5 pollution is relatively serious.31–33 In order to clarify the liver injury responses of PM2.5 subchronic exposure in the typical pollution area in China, in the present study, liver tissue pathology, liver function enzyme changes, fibrosis, inflammation and ER stress were investigated in male rats exposed to PM2.5 in the winter and summer in Taiyuan (for 60 days). The following experiments were performed: (1) measuring the histopathological changes, collagen fiber distribution and cell apoptosis in livers of rats using the hematoxylin and eosin (HE) staining, Masson staining and the terminal dUTP nick-endlabelling (TUNEL) straining; (2) detecting the levels of aspartate aminotransferase (AST), alanine aminotransferase (ALT), albumin (ALB) and globulin (GLB) in serum as well as the activities of cytochrome P450 (CYP450) and glutathione S-transferase (GST) in the livers of rats; (3) analyzing the mRNA and protein expression of specific fibrosis related markers (TGF-β1, Col I, Col III and MMP13), inflammation-related markers (TNF-α, IL-6 and HO-1), ER stress markers (ATF6, GRP78 and CHOP) and apoptosis related genes (TRB3 and caspase 12) in rat livers using real-time reverse transcription-polymerase chain reaction (RT-PCR), enzyme linked immunosorbent assay (ELISA) and western blot exposed to PM2.5, respectively. We followed with interest the risks of liver disease occurrence induced by PM2.5 and highlighted the toxicological mechanisms in PM2.5-induced fibrosis, inflammatory responses, ER stress and apoptosis in this study. The findings provide new insight into the mechanisms of PM2.5-induced liver injury, and are informative for understanding the underlying mechanisms by which PM2.5 might affect liver diseases.
Experimental
PM2.5 sample preparation
Atmospheric PM2.5 samples were collected on quartz fiber filters during the summer and winter of 2012/2013 at Shanxi University in Taiyuan, China. PM2.5 concentrations were measured using a DustTrak™ II 8530 Aerosol Monitor (TSI Inc., USA). During sampling, the mean daily mass concentration of PM2.5 in June 2012 was 148 μg m–3, while the mean concentration of PM2.5 in winter from Dec. 2012 to Jan. 2013 was 336 μg m–3, in which PM2.5 concentrations were higher than the limit of the daily mass concentration of PM2.5 of 75 μg m–3, approved by the China National Ambient Quality Standard. During sampling, the exposure levels of heavy metals in PM2.5 were measured, in which the contents of Cu, Zn, Cd, Cr and Pb were 139, 528, 3, 132 and 232 ng m–3 in summer, while 279, 475, 3, 234 and 330 ng m–3 in winter, respectively.31 For PAHs in PM2.5, the contents of phenanthrene, anthracene, fluoranthene, and pyrene in winter were higher compared to summer.32 In particular, the levels of chrysene, benzo[a]anthracene, benzo[b]fluoranthene, benzo[k]fluoranthene and benzo[a]pyrene in PM2.5 in the winter in Taiyuan were much greater than those of the recommended safety standards.33
The PM2.5 physiological saline solutions were prepared as previously reported.34
Animal and treatment protocols
Male Sprague Dawley (SD) rats with body weight (b.w.) of 220–240 g were obtained from the Animal Center of the Academy of Military Medical Sciences (Beijing, China), and were fed in an animal house in the Institute of Environmental Science of Shanxi University (Taiyuan, China). Thirty-five rats were randomly divided into seven groups: the control group, groups given three different doses of summer PM2.5 (0.2, 0.6, 1.5 mg per kg b.w.) and groups given three different doses of winter PM2.5 (0.3, 1.5, 2.7 mg per kg b.w.) with five animals for each group. The rats in the PM2.5 groups were administered PM2.5 suspensions by intratracheal instillation every three days for 60 days. The animals were maintained in accordance with the guidelines of the Ministry of Health, People's Republic of China, Beijing, China, and the protocol was approved by the institutional ethical committee (IEC) of Shanxi University with permission (No. 201710010).
According to the China National Ambient Quality Standard for PM2.5, the mean mass concentrations of 35 and 75 μg m–3 are the annual and daily concentration limits of PM2.5 exposure levels. The mean mass concentrations of 500 and 700 μg m–3 are the orange and red alert criterion limits of haze PM2.5 in China. When the PM2.5 exposure levels were 35, 75, 350, 500 and 700 μg m–3, the PM2.5 instillation concentrations for three days to each rat would be estimated as 0.15, 0.30, 1.5, 2.2 and 3.0 mg per kg b.w., respectively, according to 200 mL min–1 of the respiratory volume limit of an adult rat. Combining the abovementioned contents with the daily mean concentrations of PM2.5 in Taiyuan during the summer and winter of 2012/2013, ranging from 49 to 544 μg m–3,31,32 the instillation concentrations of PM2.5 for summer were selected as 0.2, 0.6 and 1.5 mg per kg b.w., and 0.3, 1.5 and 2.7 mg per kg b.w. for winter in this study. The control rats were administered saline.
HE staining, Masson staining and TUNEL analysis
The rats were narcotized with sodium pentobarbital (80 mg kg–1, intraperitoneal injection) 24 h after the last intratracheal instillation. A piece of the liver from each rat was cut and fixed in 4% paraformaldehyde in phosphate buffer solution (PBS). Sections (5 μm thick) were cut from paraffin-embedded specimens of the liver tissues and used for the HE staining analysis and Masson trichrome staining analysis, which was performed using a special staining kit (Maixin Biocompany, Fujian, China). Light microscopic findings were graded semi-quantitatively from – (less collagen deposition), + (medium-grade fibrosis), to ++ (severe fibrosis change), according to the degrees of the hepatocyte fibrosis, in which the more the blue the collagen deposition observed was, the more serious was the degree of liver fibrosis. The sections were then subjected to TUNEL using a TUNEL kit (KeyGen Biotech Co., Ltd, Nanjing, China) for the detection of liver cell apoptosis. The numbers of TUNEL positive apoptotic cells in five random fields in each slide from liver tissues were counted and the apoptosis degree was estimated by counting the percentages of the TUNEL positive cells. The results were expressed as the fold increase compared to the TUNEL positive cell numbers in the control group, which was ascribed an arbitrary value of 1.
The rest of the liver was frozen in liquid nitrogen and then transferred to a refrigerator at –80 °C until analysis.
Biochemical assays
A sample of blood was taken from the caudal vein of the rat 24 h after the last treatment, and the serum sample was prepared. The rats were subjected to 24 h fasting before sacrifice. The levels of AST, ALT, ALB and GLB in the serum were detected using a Dimension RxL Max Chemistry System (Siemens AG, Munich, Germany). The liver tissue was homogenated with PBS (1 : 10, w/v) and centrifuged (3000 rpm, 4 °C, for 10 min), and the rat-liver homogenate supernatants were collected. The TNF-α, IL-6, HO-1, caspase 12 and TGF-β1 levels in the supernatants were detected using the corresponding ELISA kits (R&D Company, USA). The CYP450 levels were measured using a special ELISA kit from Beijing Fangcheng Biochemistry, China. In addition, GST activity in the supernatants was measured using a GST detection kit (Jiancheng Biochemistry, Nanjing, China). All the procedures were performed according to the manufacturer's instructions for special detection kits.
Real time quantitative RT-PCR
The frozen liver tissue samples were thawed for use in mRNA extraction and then the procedures of both RT of first-strand cDNA and quantitative PCR of the target genes and housekeeping gene (glyceraldehyde-3-phosphate dehydrogenase, GAPDH) were referenced as described previously.34 The mRNA expression levels were measured by using an iCycler iQ Real Time PCR Detection System (Bio-Rad, Richmond, CA, USA), and the SYBR Green I DNA binding dye was used. The GenBank accession numbers and primer information for the target genes and GAPDH are listed in Table 1. The primers were synthesized by Sangon Company, (Shanghai, China). The expression amounts of target genes were analyzed and assessed using the relative quantification method, following normalization using GAPDH.
Table 1. Primers used in real-time RT-PCR.
| Genes | Accession no. | Sequence |
|
| ATF6 | NM_001107196 | F | 5′-GCAGGTGTATTACGCTTCG-3′ |
| Products | 144bp | R | 5′-TTCGGTCTTGTGGTCTTGTT-3′ |
| GRP78 | NM_013083 | F | 5′-CCTGTTGCTGGACTCTGTGA-3′ |
| Products | 204bp | R | 5′-GAATACACCGACGCAGGAAT-3′ |
| CHOP | NM_001109986 | F | 5′-CCTCGCTCTCCAGATTCCA-3′ |
| Products | 147bp | R | 5′-CTCATTCTCCTGCTCCTTCTCC-3′ |
| TNF-α | NM_012675 | F | 5′-ACAGAAAGCATGATCCGAGA-3′ |
| Products | 148bp | R | 5′-TCAGTAGAGAGAAGAGCGTGG-3′ |
| IL-6 | NM_012589 | F | 5′-TCCTACCCCAACTTCCAATGCTC-3′ |
| Products | 79bp | R | 5′-TTGGATGGTCTTGGTCCTTAGCC-3′ |
| HO-1 | NM_012580 | F | 5′-GTCAAGCACAGGGTGACAGA-3′ |
| Products | 77bp | R | 5′-ATCACCTGCAGCTCCTCAAA-3′ |
| Col I | NM_053304 | F | 5′-ATCAGCCCAAACCCCAAGGAGA-3′ |
| Products | 128bp | R | 5′-CGCAGGAAGGTCAGCTGGATAG-3′ |
| Col III | NM_032085 | F | 5′-TGATGGGATCCAATGAGGGAGA-3′ |
| Products | 143bp | R | 5′-GAGTCTCATGGCCTTGCGTGTTT-3′ |
| MMP13 | NM_133530 | F | 5′-ATGCTGCATACGAGCATCCA-3′ |
| Products | 81bp | R | 5′-TGTCATACCCATTCAGGGCC-3′ |
| TGF-β1 | NM_021578 | F | 5′-TGAGTGGCTGTCTTTTGACG-3′ |
| Products | 146bp | R | 5′-TGGGACTGATCCCATTGATT-3′ |
| TRB3 | NM_144755 | F | 5′-TCAAGTTGCGTCGATTTGTCTTC-3′ |
| Products | 84bp | R | 5′-CAGTCATCACACAGGCATCCTC-3′ |
| Caspase12 | NM_130422 | F | 5′-GCTGCCAAGAGAACACATGA-3′ |
| Products | 169bp | R | 5′-GGTTCTCAGCTTTGCTCAGG-3′ |
| GAPDH | NM_017008 | F | 5′-ATGTATCCGTTGTGGATCTGAC-3′ |
| Products | 78bp | R | 5′-CCTGCTTCACCACCTTCTTG-3′ |
Western blotting
Total proteins for GRP78, ATF6, CHOP, Col I, Col III, MMP13, TRB3 and β-actin from frozen lung tissues were extracted with a protein extraction kit (Beyotime, Shanghai, China), and then protein concentrations were determined. After boiling the protein samples in the loading buffer, the protein expressions of the target genes were detected by western blot as the protocol in our lab.34 The rabbit polyclonal primary antibodies for ATF6 (ab203119, dilution ratio 1 : 100, Abcam, Cambridge, UK), GRP78 (cst3183s, 1 : 1000, Cell Signaling Technology, MA, USA), CHOP (cst5554, 1 : 1000, Cell Signaling Technology, MA, USA), MMP13 (sc-30073, 1 : 100, Santa Cruz, CA, USA), Col I (bs-10423R, 1 : 100, Bioss, Beijing, China), Col III (sc-28888, 1 : 100, Santa Cruz, CA, USA), TRB3 (sc-67122, 1 : 100, Santa Cruz, CA, USA) and β-actin (AB10024, 1 : 3000, Sangon, Shanghai, China) were incubated overnight at 4 °C. The infrared-labeled goat anti-rabbit secondary antibody at a dilution ratio of 1 : 20 000 was added to the membranes and incubated for 1.5 h at room temperature. The membranes were scanned and the protein band grayscale was quantified by the Odyssey Infrared Imaging System (Li-COR Biosciences, USA).
Statistical analysis
Data were expressed as means ± standard deviation and evaluated for statistical significance with a one-way analysis of variance (ANOVA) in SPSS Statistics. Post hoc tests were conducted to determine the difference between the groups, followed by a least significant difference (LSD) test. A level of P < 0.05 shows significant difference. The relationships between hepatotoxicity biomarkers and PM2.5 concentrations were assessed by using the correlation coefficient (r) method and a positive correlation is indicated by r > 0.8.
Results
Animal health status
No animals died during the experiment. The body weights of the animals in summer and winter PM2.5 groups were lower compared to the control, but no differences between the PM2.5 groups and the control group were observed. The body weight and the ratio of the liver weight to the whole animal body weight were also determined. We found that the body weight and liver weight/body weight ratio in PM2.5 exposure groups were not significant compared with the control (data not shown).
Liver histology
The histological changes in the livers after summer and winter PM2.5 exposure were measured by HE staining (Fig. 1). As expected, there were no pathological changes in the control animal livers. However, slight hyperemia and inflammatory cell infiltration were observed for the summer PM2.5 exposure group (0.6 and 1.5 mg kg–1), whereas moderate hyperemia and inflammatory cell infiltration were found for the winter PM2.5 exposure group (1.5 mg kg–1). In addition, we observed severe hyperemia and cellular vacuolar degeneration in the winter PM2.5 exposure group (2.7 mg kg–1). Therefore, higher dosages of PM2.5 in the summer or winter in Taiyuan could cause histopathological damage in the livers of male SD rats.
Fig. 1. HE staining results in the livers of rats. The red, blue and yellow arrows indicate the sites of hyperemia, inflammatory cell infiltration, and vacuolar degeneration cells. Magnification ×200.

Masson staining of the livers was performed to assess collagen distribution characterized by collagen fibrils, which were stained with a blue colour (Fig. 2). The control rats displayed a normal and very small distribution of collagen. Extensive collagen deposition was only evident in liver tissue from the summer PM2.5 exposure group (1.5 mg kg–1) and winter PM2.5 exposure group (1.5 and 2.7 mg kg–1). Besides, severe hyperemia and collagen deposition were found in livers of the winter PM2.5 exposure group (1.5 and 2.7 mg kg–1), and even some vacuolar degeneration cells appeared in the winter PM2.5 exposure group (2.7 mg kg–1). This suggested that higher dosages of PM2.5 in the summer or winter in Taiyaun could cause liver collagen accumulations and fibrosis responses.
Fig. 2. Masson staining results in the livers of rats. The red, blue and yellow arrows indicate the sites of hyperemia, collagen fibrils, and vacuolar degeneration cells, respectively. Degree of fibrosis: –, less collagen deposition; +: medium-grade fibrosis; ++: severe fibrosis change. ×200 magnification.

Levels of liver functional enzymes
In Table 2, the levels of AST and ALT in serum from rats were markedly increased when subjected to PM2.5 subchronic exposure at the higher doses of summer (0.6 and 1.5 mg per kg b.w.) or winter (1.5 and 2.7 mg per kg b.w.), relative to the control (P < 0.05). All the tested concentrations of PM2.5 obviously decreased ALB levels and the ratio of ALB/GLB in the serum from rats, in comparison to the control (P < 0.05). However, the increases in AST and ALT levels in the 0.2 and 0.3 mg per kg b.w. PM2.5 groups had little significant difference compared to the control group. The AST or ALT levels and PM2.5 concentrations had a positive correlation (r = 0.82–0.97), whereas the changes in ALB levels and the ratios of ALB/GLB with the elevation of PM2.5 concentration reflected a negative correlation (r = 0.79–0.84).
Table 2. Liver function parameters in the serum of rats treated with PM2.5.
| Groups (mg kg–1) | AST (U L–1) | ALT (U L–1) | ALB (g L–1) | Ratio of ALB/GLB |
| 0 | 100.4 ± 17.95 | 47.0 ± 4.53 | 11.18 ± 0.94 | 0.24 ± 0.02 |
| 0.2 | 134.8 ± 43.91 | 67.8 ± 19.79 | 9.36 ± 0.72** | 0.19 ± 0.02** |
| 0.6 | 167.4 ± 39.30* | 87.8 ± 28.87* | 8.78 ± 1.09** | 0.17 ± 0.03** |
| 1.5 | 190.0 ± 59.15** | 89.6 ± 19.81** | 8.36 ± 0.71** | 0.16 ± 0.02** |
| r | 0.92 | 0.82 | 0.80 | 0.79 |
| 0.3 | 131.2 ± 24.54 | 68.0 ± 20.06 | 9.20 ± 1.1** | 0.18 ± 0.02** |
| 1.5 | 170.2 ± 31.11* | 76.0 ± 27.72** | 8.66 ± 0.40** | 0.17 ± 0.01** |
| 2.7 | 199.0 ± 69.98** | 92.6 ± 25.65** | 8.06 ± 1.03** | 0.16 ± 0.02** |
| r | 0.97 | 0.93 | 0.84 | 0.83 |
In Fig. 3, PM2.5 the doses for summer (1.5 mg per kg b.w.) and winter (2.7 mg per kg b.w.) significantly increased the levels of CYP450 in the livers of rats in comparison to the control (P < 0.05 or P < 0.01), while no statistic alterations of CYP450 levels in the livers exposed to summer 0.2 and 0.6 mg per kg b.w. PM2.5 and winter 0.3 and 1.5 mg per kg b.w. PM2.5 were found, compared with the control. There was a significant positive correlation between summer or winter PM2.5 doses and CYP450 levels (r = 0.95 and 0.98). Besides, the changes in GST activities of PM2.5 treatment groups were greater compared to the control (P < 0.05 or P < 0.01) under the higher exposure levels (for summer: 0.6 and 1.5 mg per kg b.w. PM2.5; for winter: 1.5 and 2.7 mg per kg b.w. PM2.5). PM2.5 increased CYP450 or GST activities in a concentration-dependent relationship (r = 0.94–0.98).
Fig. 3. CYP450 and GST activities in rat livers treated with PM2.5. The values are mean ± SD from five individual samples. Using one-way ANOVA, compared with the control group, significant difference is indicated by *P < 0.05 and **P < 0.01.

Fibrosis-related gene expression induced by PM2.5 in rat livers
As shown in Table 3, the Col I, Col III and MMP13 mRNA and protein levels as well as TGF-β1 mRNA levels in the livers of rats exposed to PM2.5 at the higher dosages (for summer PM2.5: 1.5 mg per kg b.w., for winter PM2.5, 1.5 and 2.7 mg per kg b.w.) were markedly elevated compared to the control (P < 0.05 or P < 0.01). TGF-β1 protein levels in the livers of rats exposed to 1.5 mg per kg b.w. summer PM2.5 and 2.7 mg per kg b.w. winter PM2.5 were remarkably increased relative to the control (P < 0.05). The four gene expressions were not significantly changed in the livers of rats exposed to 0.2 mg per kg b.w. for summer PM2.5 and 0.3 mg per kg b.w. for winter PM2.5 (P > 0.05). PM2.5 caused an increase in mRNA or protein expression of the four genes above in a concentration-dependent manner (r = 0.79–0.99).
Table 3. Exposure to PM2.5 induces expression of fibrosis related molecules in the livers of rats in different groups.
| Groups (mg kg–1) | MMP13 | Col I | Col III | TGF-β1 |
| mRNA expressions in livers | ||||
| 0 | 1.00 ± 0.51 | 1.00 ± 0.31 | 1.00 ± 0.32 | 1.00 ± 0.25 |
| 0.2 | 1.15 ± 0.36 | 1.29 ± 0.46 | 1.27 ± 0.26 | 1.24 ± 0.23 |
| 0.6 | 1.54 ± 0.56 | 1.33 ± 0.43 | 1.48 ± 0.38 | 1.36 ± 0.27 |
| 1.5 | 2.21 ± 0.63** | 1.58 ± 0.45* | 1.66 ± 0.60* | 1.51 ± 0.29* |
| r | 0.99 | 0.92 | 0.92 | 0.91 |
| 0.3 | 1.64 ± 0.59 | 1.39 ± 0.20 | 1.50 ± 0.40 | 1.28 ± 0.44 |
| 1.5 | 2.16 ± 0.43** | 1.64 ± 0.57* | 1.69 ± 0.23** | 1.46 ± 0.40* |
| 2.7 | 2.33 ± 0.88** | 1.78 ± 0.30** | 1.71 ± 0.42** | 1.62 ± 0.40** |
| r | 0.90 | 0.91 | 0.79 | 0.93 |
| Groups | Protein expressions in livers |
|||
| 0 | 1.00 ± 0.15 | 1.00 ± 0.26 | 1.00 ± 0.26 | 53.88 ± 17.52 |
| 0.2 | 1.12 ± 0.21 | 1.46 ± 0.52 | 1.12 ± 0.16 | 57.19 ± 22.90 |
| 0.6 | 1.29 ± 0.31 | 1.73 ± 0.37 | 1.34 ± 0.55 | 77.58 ± 21.00 |
| 1.5 | 1.49 ± 0.25* | 1.92 ± 0.42* | 1.70 ± 0.43* | 81.62 ± 26.79* |
| r | 0.98 | 0.88 | 0.99 | 0.89 |
| 0.3 | 1.16 ± 0.13 | 1.63 ± 0.46 | 1.19 ± 0.39 | 56.24 ± 18.26 |
| 1.5 | 1.44 ± 0.28* | 1.84 ± 0.46* | 1.77 ± 0.43* | 67.37 ± 14.40 |
| 2.7 | 1.59 ± 0.17** | 2.0 ± 0.38* | 1.89 ± 0.31* | 86.80 ± 22.80* |
| r | 0.98 | 0.84 | 0.95 | 0.99 |

|
mRNA and protein expressions of TNF-α, IL-6 and HO-1
The inflammation occurred in the livers according to the histopathological observations as shown in Fig. 4. As seen in Fig. 4A, PM2.5 at higher doses (1.5 mg per kg b.w. for summer PM2.5, 1.5 and 2.7 mg per kg b.w. for winter PM2.5, respectively) significantly enhanced TNF-α, IL-6 and HO-1 mRNA expressions in the livers versus the control (P < 0.05 or P < 0.01), and such increases incurred by PM2.5 showed obvious positive dose-effect relationships (r = 0.89–0.95). As shown in Fig. 4B, protein levels of TNF-α (under 0.6 and 1.5 mg per kg b.w. for summer PM2.5, 1.5 and 2.7 mg kg–1 for winter PM2.5), IL-6 (under 1.5 mg per kg b.w. for summer PM2.5, three tested dosages of winter PM2.5) and HO-1 (under 1.5 mg per kg b.w. for summer PM2.5, 2.7 mg kg–1 winter PM2.5) were significantly promoted in a dose-dependent manner (r = 0.84–0.94).
Fig. 4. TNF-α, IL-6, HO-1 mRNA and protein levels in livers of rats treated with PM2.5. Mean expression of mRNA in each treated group is shown as an increase compared to the mean expression in the control group, which has been ascribed an arbitrary value of 1. The values are mean ± SD from five individual samples. Using one-way ANOVA, compared with the control group, significant difference is indicated by *P < 0.05 and **P < 0.01.

mRNA and protein expressions of ER stress and apoptosis related factors
On the basis of our data in Fig. 5A, 1.5 mg per kg b.w. for summer PM2.5 and 1.5 and 2.7 mg per kg b.w. for winter PM2.5 significantly induced mRNA expression of ATF6 (P < 0.05), and similarly increased gene expression of GRP78 and CHOP (P < 0.05 or P < 0.01) in the rat livers in comparison to the control. In Fig. 5B and C, ATF6 protein levels in the livers were obviously increased in the presence of PM2.5 at the dose of 2.7 mg per kg b.w. for winter PM2.5, relative to the control (P < 0.05), and GRP78 protein levels caused by 1.5 mg per kg b.w. summer PM2.5 and 2.7 mg per kg b.w. winter PM2.5 were significantly increased compared with the control (P < 0.05). As for CHOP, its over-expression of protein in the livers treated with PM2.5 at the concentrations of summer 1.5 mg per kg b.w. and winter 1.5 and 2.7 mg per kg b.w. had statistical significance versus the control (P < 0.05). Except for the above changes, no significant changes in the GRP78, ATF6 and CHOP gene expression were observed in the rats exposed to PM2.5 at lower concentrations, compared to that in the control (P > 0.05). The relationship between the gene expression of GRP78, ATF6 and CHOP and PM2.5 concentration had a positive correlation, with high r values ranging from 0.80 to 0.98.
Fig. 5. Exposure to PM2.5 induces expression of endoplasmic reticulum stress marker molecules in livers of rats in different groups. A and B: ATF6, GRP78 and CHOP expression of mRNA and protein; C: ATF6, GRP78 and CHOP protein bands. Mean expressions of mRNA and protein in each treated group are shown as increases compared to the mean expression in the control group, which has been ascribed an arbitrary value of 1. The values are mean ± SD from five individual samples. Using one-way ANOVA, compared with the control group, significant difference is indicated by *P < 0.05 and **P < 0.01.

As shown in Fig. 6A and B, summer PM2.5 (1.5 mg per kg b.w.) and winter PM2.5 (1.5 and 2.7 mg per kg b.w.) exposure induced liver cell apoptosis and increased the percentages of TUNEL positive cells in the livers. Further analysis showed that 1.5 mg per kg b.w. summer PM2.5 and 1.5 and 2.7 mg per kg b.w. winter PM2.5 significantly induced mRNA and protein expression of TRB3 and caspase 12 (P < 0.05 or P < 0.01) in the rat livers in comparison to the control (Fig. 6C and D).
Fig. 6. Effects of PM2.5 on liver cell apoptosis and gene expression of TRB3 and caspase 12 in different groups. (A) Liver cell apoptosis in the testis was detected by TUNEL. Arrows indicate TUNEL positive cells in the livers; ×200 magnification. (B) Increased folds of TUNEL positive cells in the livers in PM2.5-groups relative to the control. (C) and (D) TRB3 and caspase 12 mRNA and protein levels in livers of rats treated with PM2.5. Apoptosis change, mean expressions of mRNA and protein in each treated group are shown as increases, compared to the mean expression in the control group, which has been ascribed an arbitrary value of 1. The values are mean ± SD from five individual samples. Using one-way ANOVA, compared with the control group, significant difference is indicated by *P < 0.05 and **P < 0.01.

Discussion
The liver is an important and pivotal metabolism and detoxification organ and it also vulnerable to being affected and damaged by xenobiotics and drugs. It has been reported that PM2.5 may increase liver disease risks,9–12 but the understanding of the underlying mechanisms by which PM2.5 induces adverse health effects in the liver has been insufficient until now. In the current study, we have investigated the histopathology, changes in liver function enzyme and metabolic enzyme activity, and the responses of fibrosis, inflammation and ER stress as well as the alterations of the gene expression of hepatotoxicity biomarkers in the rats after sub-chronic exposure to summer and winter PM2.5 in Taiyuan of China, representing different compositions of PM2.5.
Firstly, the histopathology data showed that higher dosages of PM2.5 could cause liver damage in rats (Fig. 1), and Masson staining results indicated that excess collagen fibers were observed in the livers of the PM2.5 treated rats (Fig. 2). The results are consistent with the report by Zhang et al., which showed that the histopathological changes were more prevalent and severe in rat livers with the dose increasing after exposure to dosages of 1.8, 5.4, and 16.2 mg per kg b.w. PM2.5 suspensions for 30 days.35 In particular, under the same dosage of 1.5 mg per kg b.w. PM2.5, the liver injury caused by winter PM2.5 was more serious than summer PM2.5, which might be because the concentrations of harmful chemicals like heavy metals and PAHs in winter PM2.5 were higher than that of summer. Also, during the PM2.5 sampling in this study, the mean concentration of PM2.5 in the winter (336 μg m–3, comparable to the dosage of 1.5 mg per kg b.w. PM2.5) exceeded the average level of PM2.5 in the summer by almost 2.3 times (148 μg m–3, comparable to the dosage of 0.6 mg per kg b.w. PM2.5). Based on the HE and Masson staining results, 1.5 mg per kg b.w. PM2.5 in winter induced more severe liver tissue damage than that induced by 0.6 mg per kg b.w. PM2.5 in summer. Similarly, the mRNA or protein expressions of biomarkers of fibrosis, inflammation and ER stress in the livers incurred by 1.5 mg per kg b.w. PM2.5 in the winter were higher than that of 0.6 mg per kg b.w. PM2.5 in the summer. Taken together, PM2.5-caused liver injury in the winter was worse compared to that in the summer.
Along with the pathological injury, the alterations in liver function enzymes were measured in this study. In general, AST and ALT are very important proteins for the liver and are the representative indicators of liver damage. They can be released into the blood from the liver. When significant liver damage occurs, AST and ALT activities in serum are increased, leading to poor liver function.36 ALB and GLB are the major protein components in serum, which play an important role in inflammation. A low ALB/GLB ratio, meaning a decreased ALB level and increased GLB level, was reported to reflect the hepatic function decline and occurrence of chronic inflammation.37 In order to further indicate the biochemical mechanism of PM2.5-caused liver injury, the parameters for evaluating the liver function impairment in the serum were determined. The results showed that in the medium and high dosage of summer or winter PM2.5-treated groups, AST and ALT were significantly elevated compared with the control, and that values of AST (from 131.2–199.0 U L–1) were higher than those of ALT (from 67.8 to 92.6 U L–1). Also, in all the tested concentrations, ALB levels and ALB/GLB ratios were markedly reduced compared to the control. This suggests that PM2.5 caused liver function decline and inflammatory responses, which supported the report by Kim et al., in which PM2.5 exposure was associated with increased liver enzyme levels of AST and ALT in the blood of the elderly.38 In addition, the activation of CYP450 (phase I enzyme, participating in the metabolism of endogenous and exogenous substances including drugs, environmental compounds) and GST (phase II enzyme, catalyzing the conjugation of glutathione together with the intermediates of chemicals from the phase I reaction) partly means the excessive accumulation of harmful metabolites and the dysfunction of the liver metabolism.39 In the present study, higher concentrations of PM2.5 seriously induced CYP450 and GST in rat livers (Fig. 3), implying that PM2.5 may result in a metabolic disturbance of the liver via activating metabolic enzymes and further promoting the liver toxicity.
Secondly, liver fibrosis is the final stage of all chronic liver diseases. Therefore we specifically focused on and analyzed the correlation between fibrosis and the expressions of fibrosis-related genes (TGF-β1, Col I, Col III and MMP13) in the liver in the presence of PM2.5. The pathogenesis of fibrosis is involved in the inflammatory pathway and the growth factor signaling pathway mediated by TGF-β1,40 which is a pro-fibrogenic cytokine in hepatic fibrosis.41 The collagen fibers are localized and markedly increased around the tributaries of portal veins.41 Col I and Col III are two indicators of fibrosis and may enhance collagen matrix deposition in the liver, leading to fibrosis. TGF-β1 can stimulate matrix deposition and up-regulate Col I and Col III gene expression.42,43 In the present study, subchronic exposure to high dosages of PM2.5 had a pro-fibrosis effect via the regulation of the TGF-β1/Col I/Col III signaling pathway in the livers of rats, eventually impairing liver function. In contrast, MMP13 plays a protective role in the process of collagen degradation,20,44 and upregulation of MMP13 gene expression may elevate collagenase activity and enhance the collagen hydrolyzation in fibrotic livers of rats, consequently preventing liver fibrosis progression.44 As shown in Table 3, MMP13 mRNA and protein expressions in the livers were raised after 60 days exposure of PM2.5 at the higher dosages (1.5 and 2.7 mg per kg b.w.), implying that induction of MMP13 was a protective response against liver fibrosis upon PM2.5 exposure via hydrolyzing the collagen proteins. Although it is difficult to reconcile MMP13 high-expression with the pro-fibrosis gene (TGF-β1, Col I and Col III) over-expression in rat livers in this study (see Table 3), we noticed that PM2.5 provoked a significant increase in the collagen fiber formation in rat livers (Fig. 2). It may be speculated that under PM2.5 exposure conditions (1.5 and/or 2.7 mg per kg b.w.), the collagen fiber accumulation regulated by TGF-β1, Col I and Col III occupied a predominant position in the liver fibrotic response compared to the effect of MMP13, which reduced the fibrosis.
Thirdly, inflammation elicits a vital defense response to cellular or tissue injury. However, persistent inflammatory responses can lead to pathological injury related to the disease. When the liver cells are activated or damaged by exogenous harmful chemicals, pro-inflammatory cytokines are induced dramatically. If these cytokines are released excessively and persistently while the inflammation in the liver does not subside or heal during a short time, ultimately liver injury and liver tissue formation abnormity (i.e., fibrosis) would occur.45 It was proven that PM2.5 exposure was linked to HCC via chronic liver inflammation from a patient observational report.10 As we know, TNF-α and IL-6 are typical pro-inflammatory cytokines that serve a regulatory role in the pathogenesis of numerous diseases, including liver disease.21,46 Our study has demonstrated that exposure to PM2.5 for 60 days triggered inflammation responses (Fig. 1, 4 and Table 2) including hyperemia, inflammatory cell infiltration and the decrease of ALB levels, accompanied by the elevation of pro-inflammatory cytokines TNF-α and IL-6 levels in the liver, especially in the presence of high dosages of PM2.5 (1.5 and 2.7 mg per kg b.w.), suggesting that PM2.5 causes liver inflammatory injury, in which TNF-α and IL-6 high-expression promote the inflammatory responses. Additionally, MMP13 may mediate the release of inflammatory cytokines such as TNF-α,47 which implies that PM2.5-caused over-expression of MMP13 might promote the secretion of inflammatory cytokines. On the other hand, to investigate whether the liver inflammatory injury induced by PM2.5 was in relation to the anti-inflammatory factors, HO-1 activity in the livers of rats exposed to PM2.5 was measured. HO-1 is generally regarded as an antioxidant enzyme that catalyzes the oxidative degradation of heme to carbon monoxide and biliverdin, reducing reactive oxygen species formation from heme.22,48 Intriguingly, HO-1 is also proven to fulfill a key role in defense mechanisms against hepatic inflammation by inhibiting TNF-α gene expression,49 and carbon monoxide, which is yielded in the catalytic process of heme by HO-1 has anti-inflammatory properties.50 Our findings indicate that induction of HO-1 is an adaptive response to PM2.5 treatment, and it may play a protective role in the recovery of hepatocytes from inflammation injury. Nevertheless, based on the HE staining results (Fig. 1), inflammatory responses like hyperemia and inflammatory cell infiltration coexist in the liver of rats upon exposure to 1.5 and 2.7 mg per kg b.w. PM2.5. We speculate that the liver pathological injury induced by PM2.5 is mainly reflected as inflammation, while the pro-inflammatory effects of TNF-α and IL-6 are eventually dominant compared to the anti-inflammation effects of HO-1.
Finally, the ER is an organelle with important functions including intracellular calcium storage and synthesis, lipid biosynthesis, folding and post-translational modification of proteins. When the unfolded proteins accumulate in the ER lumen, ER stress response (also called unfolded protein response) happens, resulting in the impairment of ER functions. GRP78, ATF6 and CHOP are biomarkers of ER stress, and the up-regulations of these gene expressions are involved in enhancing the ER stress response.51–54 Upon activation in response to ER stress signals, the molecular chaperone GRP78 is activated and released from ER transmembrane sensors like ATF6. ATF6 is translocated to the Golgi and cleaved to an active fragment by the site 1 and site 2 proteases (S1P and S2P).52 The ATF6 fragment can then move to the nucleus where it would activate other functional molecules like CHOP,53 which is a stress-induced transcription factor that may be induced by ER stress and mediates apoptosis.54 It has been demonstrated that the ER stress response is a very complex regulation network,55 and it can exert an important self-defense role in the cellular protection against various exogenous stimuli. However, in conditions of prolonged and strong ER stress, along with over-expression of GRP78, ATF6, CHOP and other stress molecules, the signaling switches from a protective effect to a pro-apoptotic ER stress response, which is related to liver fibrosis and NAFLD.26,56 Laing et al. revealed that PM2.5 affected mouse liver cells by activating the PERK-eIF2α-CHOP pathway and stimulating the ER stress-mediated apoptosis.57 This viewpoint is partially in accordance with our study, in which PM2.5 activated the GRP78/ATF6 signaling pathway in the liver, and elevated the level of CHOP, further causing liver ER stress coupled with liver inflammatory responses and fibrosis. Moreover, TRB3 and caspase 12 are the important ER stress-inducible genes and are related to the ER stress-specific apoptosis pathway,58 in which CHOP may regulate and activate its downstream gene TRB3 and caspase 12, further activating the caspase cascade apoptotic pathway.59 Therefore, the gene expressions of CHOP, TRB3 and caspase 12 may reflect the degree of ER stress-mediated apoptosis. In this study, upregulation of ATF6, GRP78 and CHOP in livers of PM2.5 exposure groups indicated that the GRP78/CHOP pathway mediated ER stress activation, while the high expressions of TRB3 and caspase 12 protein suggested the apoptosis occurrence through the ER stress in response to PM2.5. Combined with the current TUNEL experimental results, it indicated that PM2.5 activated the GRP78/CHOP/TRB3/caspase 12 signaling pathway and further caused ER stress and apoptosis in the liver.
Taken together, identifying the molecular events by which PM2.5 induces fibrosis, inflammation, ER stress responses and apoptosis should aid in the understanding of air PM2.5 pollution-associated liver injury and related diseases. However, we cannot prove whether there is a direct relationship between fibrosis (or inflammation) and ER stress in the liver under PM2.5 subchronic exposure conditions. More in-depth work is still needed to investigate the molecular mechanisms of PM2.5 on ER stress-mediated inflammatory responses and fibrosis in the liver.
Conclusions
This study suggests that PM2.5 sub-chronic exposure causes hepatic pathological injury, fibrosis, inflammation, ER stress and apoptosis in rats, which might finally contribute to liver diseases. The liver injury induced by winter PM2.5 in Taiyuan was more serious than that induced by summer PM2.5.
Three possible mechanisms are proposed to explain PM2.5-induced liver injury: (1) PM2.5 caused hepatic pathological injury in the rats by significantly activating AST, ALT, CYP450 and GST and reducing the ratio of ALB/GLB, leading to liver function decline. The injury effects on the liver induced by winter PM2.5 were greater than that of summer PM2.5; (2) PM2.5-triggered rat liver fibrosis was mediated by upregulation of TGF-β1, Col I, Col III and MMP13 gene expressions, while it enhanced inflammation of the liver though elevating TNF-α, IL-6 and HO-1 levels; (3) PM2.5 activated the GRP78/ATF6/CHOP/TRB3/caspase 12 pathway, resulting in ER stress and apoptosis in the liver. Our findings are important in providing a new understanding to explain the promoted effects of PM2.5 on human liver diseases. At the same time, we realize that there is more work to be done. (1) It is necessary to explore the regulatory mechanisms of inflammation and ER stress on liver fibrosis caused by PM2.5. (2) More attention should be paid to the contributions of heavy metals or organic matter on PM2.5 liver toxicity.
Conflicts of interest
There are no conflicts to declare.
Acknowledgments
This research was supported by the National Natural Science Foundation of China (No. 91543202 and 21575084), HKBU Strategic Development Fund (No. 15-1012-P04), and 100 talents program of Shanxi Province of China.
References
- Mokdad A. A., Lopez A. D., Shahraz S., Lozano R., Mokdad A. H., Stanaway J., Murray C. J., Naghavi M. BMC Med. 2014;12:145. doi: 10.1186/s12916-014-0145-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong M. C., Jiang J. Y., Goggins W. B., Liang M., Fang Y., Fung F. D., Leung C., Wang H. H., Wong G. L., Wong V. W. Sci. Rep. 2017;7:45846. doi: 10.1038/srep45846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung B. K., Karlsen T. H. Dig. Dis. 2017;35:323–333. doi: 10.1159/000456583. [DOI] [PubMed] [Google Scholar]
- Kim J. W., Park S., Lim C. W., Lee K., Kim B. Toxicol. Res. 2014;30:65–70. doi: 10.5487/TR.2014.30.2.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pun V. C., Kazemiparkouhi F., Manjourides J., Suh H. H. Am. J. Epidemiol. 2017;186:961–969. doi: 10.1093/aje/kwx166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheers H., Jacobs L., Casas L., Nemery B., Nawrot T. S. Stroke. 2015;46:3058–3066. doi: 10.1161/STROKEAHA.115.009913. [DOI] [PubMed] [Google Scholar]
- IARC, Monographs on the evaluation of carcinogenic risks to humans, 2016, vol. 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loomis D., Grosse Y., Lauby-Secretan B., El Ghissassi F., Bouvard V., Benbrahim-Tallaa L., Guha N., Baan R., Mattock H. and Straif K., International Agency for Research on Cancer Monograph Working Group IARC, The carcinogenicity of outdoor air pollution, 2013, pp. 1262–1263. [DOI] [PubMed] [Google Scholar]
- Pedersen M., Andersen Z. J., Stafoggia M., Weinmayr G., Galassi C., Sørensen M., Eriksen K. T., Tjønneland A., Loft S., Jaensch A. Environ. Res. 2017;154:226–233. doi: 10.1016/j.envres.2017.01.006. [DOI] [PubMed] [Google Scholar]
- Pan W. C., Wu C. D., Chen M. J., Huang Y. T., Chen C. J., Su H. J., Yang H. I. J. Natl. Cancer Inst. 2016;108:djv341. doi: 10.1093/jnci/djv341. [DOI] [PubMed] [Google Scholar]
- Tarantino G., Capone D., Finelli C. World J. Gastroenterol. 2013;19:3951–3956. doi: 10.3748/wjg.v19.i25.3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan H. H., Fiel M. I., Sun Q., Guo J., Gordon R. E., Chen L. C., Friedman S. L., Odin J. A., Allina J. J. Immunotoxicol. 2009;6:266–275. doi: 10.3109/15476910903241704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S. N., Sang N. Acta Sci. Circumstantiae. 2017;37:3207–3212. [Google Scholar]
- Li M., Xie J. J., Wu T., Wang Y., Liu F. F., Wang F., Li Y. China Mod. Med. 2016:99–11. [Google Scholar]
- Zheng Z., Zhang X., Wang J., Dandekar A., Kim H., Qiu Y., Xu X., Cui Y., Wang A., Chen L. C. J. Hepatol. 2015;63:1397–1404. doi: 10.1016/j.jhep.2015.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M. Korean J. Gastroenterol. 2017;69:341–347. doi: 10.4166/kjg.2017.69.6.341. [DOI] [PubMed] [Google Scholar]
- Ding Y., Wu Z., Wei Y., Shu L., Peng Y. J. Cancer Res. Clin. Oncol. 2017;143:821–834. doi: 10.1007/s00432-017-2364-z. [DOI] [PubMed] [Google Scholar]
- Cong M., Iwaisako K., Jiang C., Kisseleva T. Int. J. Hepatol. 2012;2012:158547. doi: 10.1155/2012/158547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutroneo K. R., White S. L., Phan S. H., Ehrlich H. P. J. Cell Physiol. 2007;211:585–589. doi: 10.1002/jcp.20972. [DOI] [PubMed] [Google Scholar]
- Hironaka K., Sakaida I., Matsumura Y., Kaino S., Miyamoto K., Okita K. Biochem. Biophys. Res. Commun. 2000;267:290–295. doi: 10.1006/bbrc.1999.1910. [DOI] [PubMed] [Google Scholar]
- Streetz K., Wüstefeld T., Klein C., Manns M., Trautwein C. Cell. Mol. Biol. 2001;47:661–673. [PubMed] [Google Scholar]
- Chi X., Yao W., Xia H., Jin Y., Li X., Cai J., Hei Z. Oxid. Med. Cell. Longevity. 2015;2015:986075. doi: 10.1155/2015/986075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon O. S., Choi S. H., Kim J. H. Korean J. Gastroenterol. 2015;66:320–324. doi: 10.4166/kjg.2015.66.6.320. [DOI] [PubMed] [Google Scholar]
- Salminen A., Kauppinen A., Hyttinen J. M., Toropainen E., Kaarniranta K. Mol. Med. 2010;16:535–542. doi: 10.2119/molmed.2010.00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C., Bailly-Maitre B., Reed J. C. J. Clin. Invest. 2005;115:2656–2664. doi: 10.1172/JCI26373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iracheta-Vellve A., Petrasek J., Gyongyosi B., Satishchandran A., Lowe P., Kodys K., Catalano D., Calenda C. D., Kurt-Jones E. A., Fitzgerald K. A. J. Biol. Chem. 2016;291:26794–26805. doi: 10.1074/jbc.M116.736991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang T., Wang L., Li X., Song J., Wu X., Zhou S. Mol. Med. Rep. 2015;11:2941–2946. doi: 10.3892/mmr.2014.3020. [DOI] [PubMed] [Google Scholar]
- Kim H. M., Lee E. S., Lee B. R., Yadav D., Kim Y. M., Ko H. J., Park K. S., Lee E. Y., Chung C. H. PLoS One. 2015;10:e0120711. doi: 10.1371/journal.pone.0120711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohoka N., Yoshii S., Hattori T., Onozaki K., Hayashi H. EMBO J. 2005;24:1243–1255. doi: 10.1038/sj.emboj.7600596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou X., Qu Z., Fang Y., Shi X., Ji Y. Mol. Med. Rep. 2017;15:331–338. doi: 10.3892/mmr.2016.6016. [DOI] [PubMed] [Google Scholar]
- Lv J. L., Li M., Xie J. F., Di Z. D., Zhao L. J., Liu R. Q. Environ. Sci. Technol. 2016;39:126–131. [Google Scholar]
- Jia X. H., (dissertation), Shanxi University, Taiyuan, 2013. [Google Scholar]
- Li R. J., Kou X. J., Geng H., Dong C., Cai Z. W. Chin. Chem. Lett. 2014;25:663–666. [Google Scholar]
- Li R., Kou X., Geng H., Xie J., Tian J., Cai Z., Dong C. J. Hazard. Mater. 2015;287:392–401. doi: 10.1016/j.jhazmat.2015.02.006. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Hu H., Shi Y., Yang X., Cao L., Wu J., Asweto C. O., Feng L., Duan J., Sun Z. Sci. Total Environ. 2017;589:212–221. doi: 10.1016/j.scitotenv.2017.02.149. [DOI] [PubMed] [Google Scholar]
- Giannini E., Botta F., Fasoli A., Ceppa P., Risso D., Lantieri P. B., Celle G., Testa R. Dig. Dis Sci. 1999;44:1249–1253. doi: 10.1023/a:1026609231094. [DOI] [PubMed] [Google Scholar]
- Deng Y., Pang Q., Miao R.-C., Chen W., Zhou Y. Y., Bi J. B., Liu S. S., Zhang J. Y., Qu K., Liu C. OncoTargets Ther. 2016;9:5317. doi: 10.2147/OTT.S109736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K. N., Lee H., Kim J. H., Jung K., Lim Y. H., Hong Y. C. J. Prev. Med. Public Health. 2015;48:151–169. doi: 10.3961/jpmph.15.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura Y., Miyamoto M., Murakami A., Ohigashi H., Osawa T., Uchida K. Biochem. Biophys. Res. Commun. 2003;302:593–600. doi: 10.1016/s0006-291x(03)00219-5. [DOI] [PubMed] [Google Scholar]
- Sadasivan S. K., Siddaraju N., Khan K. M., Vasamsetti B., Kumar N. R., Haridas V., Reddy M. B., Baggavalli S., Oommen A. M., Rao R. P. Fibrog. Tissue Repair. 2015;8:1. doi: 10.1186/s13069-014-0017-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan X., Zhang Q., Li S., Lv Y., Su H., Jiang H., Hao Z. PLoS One. 2013;8:e82190. doi: 10.1371/journal.pone.0082190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gooch J. L., Gorin Y., Zhang B. X., Abboud H. E. J. Biol. Chem. 2004;279:15561–15570. doi: 10.1074/jbc.M308759200. [DOI] [PubMed] [Google Scholar]
- Lang Q., Liu Q., Xu N., Qian K. L., Qi J. H., Sun Y. C., Xiao L., Shi X. F. Biochem. Biophys. Res. Commun. 2011;409:448–453. doi: 10.1016/j.bbrc.2011.05.023. [DOI] [PubMed] [Google Scholar]
- Abe H., Kamimura K., Kobayashi Y., Ohtsuka M., Miura H., Ohashi R., Yokoo T., Kanefuji T., Suda T., Tsuchida M. Mol. Ther.–Nucleic Acids. 2016;5:e276. doi: 10.1038/mtna.2015.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuman M. G. Alcohol Res. Health. 2003;27:307–316. [PMC free article] [PubMed] [Google Scholar]
- Kawaratani H., Moriya K., Namisaki T., Uejima M., Kitade M., Takeda K., Okura Y., Kaji K., Takaya H., Nishimura N. Int. J. Mol. Med. 2017;40:263–270. doi: 10.3892/ijmm.2017.3015. [DOI] [PubMed] [Google Scholar]
- Yang Z., Zhang Y., Wang L. PLoS One. 2013;8:e65256. doi: 10.1371/journal.pone.0065256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T., Morita K., Akagi R., Sassa S. Curr. Med. Chem. 2004;11:1545–1561. doi: 10.2174/0929867043365080. [DOI] [PubMed] [Google Scholar]
- Nakahira K., Takahashi T., Shimizu H., Maeshima K., Uehara K., Fujii H., Nakatsuka H., Yokoyama M., Akagi R., Morita K. Biochem. Pharmacol. 2003;66:1091–1105. doi: 10.1016/s0006-2952(03)00444-1. [DOI] [PubMed] [Google Scholar]
- Otterbein L. E., Bach F. H., Alam J., Soares M., Lu H. T., Wysk M., Davis R. J., Flavell R. A., Choi A. M. Nat. Med. 2000;6:422–428. doi: 10.1038/74680. [DOI] [PubMed] [Google Scholar]
- Namba T., Ishihara T., Tanaka K.-i., Hoshino T., Mizushima T. Biochem. Biophys. Res. Commun. 2007;355:543–548. doi: 10.1016/j.bbrc.2007.02.004. [DOI] [PubMed] [Google Scholar]
- Tang X., Liang X., Li M., Guo T., Duan N., Wang Y., Rong G., Yang L., Zhang S., Zhang J. Mol. Cell. Biochem. 2015;407:197. doi: 10.1007/s11010-015-2469-0. [DOI] [PubMed] [Google Scholar]
- Shah A., Kumar A. Oncotarget. 2016;7:46100–46119. doi: 10.18632/oncotarget.10025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishitoh H. J. Biochem. 2012;151:217–219. doi: 10.1093/jb/mvr143. [DOI] [PubMed] [Google Scholar]
- Velasco G. Am. J. Physiol.: Cell Physiol. 2010;299:727–728. doi: 10.1152/ajpcell.00271.2010. [DOI] [PubMed] [Google Scholar]
- He L., Yuan F. H., Chen T., Huang Q., Wang Y., Liu Z. G. J. Huazhong Univ. Sci. Technol., Med. Sci. 2017;2:217–225. doi: 10.1007/s11596-017-1718-8. [DOI] [PubMed] [Google Scholar]
- Laing S., Wang G., Briazova T., Zhang C., Wang A., Zheng Z., Gow A., Chen A. F., Rajagopalan S., Chen L. C. Am. J. Physiol.: Cell Physiol. 2010;299:C736–C749. doi: 10.1152/ajpcell.00529.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K., Kaufman R. J. Neurology. 2006;66(2 Suppl 1):S102–S109. doi: 10.1212/01.wnl.0000192306.98198.ec. [DOI] [PubMed] [Google Scholar]
- Rao R. V., Peel A., Logvinova A., del R. G., Hermel E., Yokota T., Goldsmith P. C., Ellerby L. M., Ellerby H. M., Bredesen D. E. FEBS Lett. 2002;514:122–128. doi: 10.1016/s0014-5793(02)02289-5. [DOI] [PMC free article] [PubMed] [Google Scholar]