

 (5R)-5-Hydroxytriptolide (LLDT-8), a novel triptolide derivative, will proceed to phase II clinical trials for the treatment of rheumatoid arthritis and cancer.
(5R)-5-Hydroxytriptolide (LLDT-8), a novel triptolide derivative, will proceed to phase II clinical trials for the treatment of rheumatoid arthritis and cancer.
Abstract
(5R)-5-Hydroxytriptolide (LLDT-8), a novel triptolide derivative, will proceed to phase II clinical trials for the treatment of rheumatoid arthritis and cancer. However, the selection of disease and patients is largely limited by the testis toxicity, yet toxicity mechanisms are still poorly understood. In this study, LLDT-8 dose and time-dependently decreased the testes weight, germinal cell layers and induced abnormal spermatid development. Analysis of the germ cell-specific marker showed that spermatocytes were more sensitive to LLDT-8, which was confirmed by the in vitro sensitivity assay with spermatocyte-like GC-2spd and sertoli-like TM4 cells. In GC-2spd, LLDT-8 induced G1/S arrest and apoptosis. MAPK activity screening identified that TGF-β activated kinase 1 (Tak1) is critical in LLDT-8 induced apoptosis. LLDT-8 reduced the Tak1 protein and dephosphorylated Tak1 at Ser412 in GC-2spd and the testes, but not in TM4. RNAi mediated depletion or pharmacologic inhibition of Tak1 induced apoptosis in GC-2spd. Meanwhile, activating Tak1 rescued up to 50% of the GC-2spd cells from the apoptosis induced by LLDT-8. Altogether, our study firstly revealed the important role of Tak1 in the survival of spermatocytes, and dephosphorylation of Tak1 at Ser412 may contribute to the spermatocyte-specific testis toxicity induced by LLDT-8.
Introduction
Triptolide has unique anti-inflammatory, immunosuppressive and anticancer activities.1–4 Owing to its severe toxicity and poor water solubility, multiple triptolide analogues have been developed and evaluated for their safety and efficacy in clinical trials.5–9 However, none of these clinical trials have proceeded into phase II. The frequent, severe toxicity that occurred in the gastrointestinal tract, skin, liver, kidney and reproductive system prevented triptolide and its derivatives from further clinical trials7,8,10
(5R)-5-Hydroxytriptolide (LLDT-8) is a novel triptolide derivative, has potent immunosuppressive, anti-inflammatory and anticancer activities, and now is ready to enter phase II clinical trials in China for the treatment of rheumatoid arthritis and cancer.11–14 LLDT-8 suppresses the activation of T and B cells, reduces the production of the Th1 type cytokines (IFN-γ, IL-2) and inflammatory cytokines (TNF-α, IL-16), and inhibits NF-kB activation triggered by lipopolysaccharides.11,15 LLDT-8 also displays potent anticancer activity via its transcription inhibition.14 Unlike the systemic toxicity induced by trip- tolide, LLDT-8 only induced testicular injury in rodents (unpublished data in our group), while the testis toxicity largely limited the selection of disease and patients in the ongoing clinical trials of LLDT-8 and the mechanisms remain unknown.
TGF-β activated kinase 1(TAK1, MAP3K7) is a central regulator of cell death and is activated through a diverse set of intra- and extracellular stimuli, which controls cell viability and inflammation through activating NF-kB, mitogen-activated protein kinases and receptor interacting protein kinase.16–18 TAK1 activation correlates with phosphorylation of Thr184, Thr187 and Ser192 in its activation loop.19,20 Recently, Ser412 is also found to be required for its activation.21–24 However, the role of TAK1 in the testis is still unknown.
Here, we report that inhibition of Tak1 and its Ser412 phosphorylation repressed the survival of spermatocyte-like cells. LLDT-8 induced apoptosis via dephosphorylation of Tak1 at Ser412, which may contribute to the spermatocyte specific testis toxicity of LLDT-8.
Materials and methods
Materials
(5R)-5-Hydroxytriptolide (99.9%) was kindly provided by Professor Yuanchao Li (Shanghai Institute of Materia Medica, Shanghai, China). All other chemicals were commercially available and purchased as reagent grade from Sigma-Aldrich (St Louis, USA). The following antibodies were used for western blotting: Tak1, phosphor-Tak1 (Ser412), phosphor-Tak1 (Thr184/187), JNK, phosphor-JNK, P38, phosphor-P38, ERK1/2 and phosphor-ERK1/2 (Cell signaling), RNA polymerase II (Millipore), and β-actin (Santa Cruz).
Ethics statement, animal care and study design
Animal-use protocols were approved by the Institutional Animal Care and Use Committee of the Shanghai Institute of Materia Medica (Shanghai, China), IACUC No. 2014-07-RJ-88. C57BL/6 mice (9–12weeks old) were used in this study. All animals were maintained under controlled temperature with a light/dark cycle. Food and water were provided ad libitum.
Male C57BL/6 mice (9 weeks old, 22–24 g body weight) were used in the studies of toxicity of (5R)-5-hydroxytriptolide (LLDT-8) (0.5 and 1.0 mg kg–1). LLDT-8 was consecutively administered by gavage for 30 days. The control group received saline. Mice were sacrificed on the 15th, 21st and 30th day post-administration and the blood, liver, kidney, spleen, testis and epididymis were collected. The main lobe of the liver, kidney, spleen and epididymis were fixed in 10% neutral buffered formalin for histological examination, the left testicle was fixed in Davidson's buffer for 16 hours followed by 10% neutral buffered formalin. The tissue sections were stained with hematoxylin and eosin (H&E). The remaining tissues were stored at –80 °C for RNA and protein extraction.
Serum levels of blood urea nitrogen (BUN), creatine (Cre), alanine aminotransferase (ALT), aspartate transaminase (AST) and other indexes were determined using an automatic Hitachi Clinical Analyzer Model 7080 (Hitachi High-Technologies Corporation, Tokyo, Japan).
Cell culture and proliferation assay
Mouse TM4 (sertoli cells, ATCC number: CRL-1715), and GC-2spd (spermatocytes, ATCC number: CRL-2196) were purchased from ATCC (Manassas, VA, USA). These cells were grown under a 5% CO2 atmosphere at 37 °C. TM4 was cultured in DMEM supplemented with 5% horse serum and 2.5% fetal bovine serum. GC-2spd was cultured in DMEM supplemented with 10% fetal bovine serum. Cell viability was determined using the MTT assay. Cell number count was performed with the Vi-Cell XR cell counter (Beckman Coulter).
Western blotting
After drug treatment, the cells were washed twice with ice-cold phosphate-buffered saline (PBS; 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, and 1.8 mM KH2PO4, pH 7.4) and lysed in sodium dodecyl sulfate (SDS) sample buffer. Cell lysates, containing equal amounts of protein, were separated by SDS–PAGE and transferred to polyvinylidine difluoride membranes. After blocking in 5% nonfat milk in TBST (Tris-buffered saline containing 0.1% Tween-20, pH 7.6), the membranes were incubated with the appropriate primary antibodies at 4 °C overnight and then exposed to secondary antibodies for 1 h at room temperature. Immunoreactive proteins were visualized using the enhanced chemiluminescence system (Millipore, Shanghai, China).
RNA interference
The following sequence for TAK1–siRNA was designed and synthesized by GenePharma (Shanghai, China): 5′-GGCGCCUGAAGUAUUUGAATT. The scrambled siRNA sequence was 5′-AUUGUCAGACUGACGUCGU. Before transfection, 8 × 104 GC-2spd cells were seeded in a 6-well plate for 16 h. For transient transfections, 4 μl of Lipofectamine 2000 (Invitrogen) were mixed with 250 μl of Opti-MEM (Invitrogen) and incubated for 5 min at room temperature. Additionally, 2.5 μl of siRNA duplex (20 μM) were mixed with 250 μl of Opti-MEM. Then the Lipofectamine-containing mix was added dropwise to the siRNA containing mix and incubated at room temperature for another 20 min. The Lipofectamine/siRNA mix were added to GC-2spd cells and incubated for 4 h before addition of medium with serum (10%) overnight. After 72 h-transfection, GC-2spd cells were harvested for protein extraction, cell number count and apoptosis assay.
Construction of lentiviral vectors for Tak1 overexpression
Based on the published murine Tak1 (NM_009316.1), a forward primer with an XbaI and a reverse primer with an EcoRI restriction site were synthesized for the cloning of Tak1: forward 5′-CTAGTCTAGAGCCGCCACCATGTCGCAGGTCCTGAACTTCG and reverse 5′-CCGGAATTCTCATGAAGTGCCTTGTCGTTTCTGCTG. PrimeStar High Fidelity enzyme (Takara, Dalian, China) was used to perform PCR on cDNA from murine testis. Agarose gel electrophoresis revealed a single DNA product of about the expected size (1740 bp), which was used for cloning into the eukaryotic expression vector pcDNA3.1(–). The entire Tak1–cDNA insert was sequenced. By using this expression plasmid as a template, constitutively active Tak1 (CA-Tak1, lacking the 22 N-terminal amino acids as described by Yamaguchi et al.25) was cloned into pCDH vector, a lentiviral vector. The lentiviral particles were generated by calcium-phosphate using a three-plasmid system in 293 cells. After 48 h transfection, the supernatant was collected and filtered by using 0.45 μM filters and stored at –80 °C for infection. Before infection, 1 × 105 GC-2spd cells were seeded in 6 cm dishes. After 16 h, the supernatant containing lentiviral particles were added and puromycin (1.2 μg mL–1) was used for the selection and maintenance of GC-2spd cells with stably expressed CA-Tak1.
Flow cytometry
Cell cycle distribution was measured using a FACScan flow cytometer (BD Biosciences, San Jose, CA, USA) after standard fixation and propidium iodide staining and analyzed with CellQuest and ModFit LT3.0 software. In GC-2spd and TM4 cells, the Annexin V-FITC and propidium iodide dual labeling kit was used (Dojindo, Shanghai, China) according to the manufacturer's instructions. In GC-2spd cells with stably expressed CA-Tak1, the Annexin V-PE/7-AAD dual labeling kit was used (BD Pharmingen).
Quantitative real-time PCR (qPCR)
Total RNA from mouse testis was extracted using the UNIQ-10 total RNA isolation kit (Sangon Biotech, Shanghai, China). The remaining genomic DNA in the total RNA was digested using RNase-free DNase I (Fermentas, Burlington, Canada). DNase-digested RNA was reverse transcribed into cDNA using the PrimeScript RT reagent kit (TaKaRa, Dalian, China). DNase-digested RNA without reverse transcription was used as a negative control. QPCR was carried out using the SYBR Premix Ex Taq (TaKaRa) with primers listed in the ESI.† QPCR cycle parameters were: 95 °C for 10 s, followed by 40 cycles of 95 °C for 5 s, and 60 °C for 30 s with a melting-curve analysis at the end of the protocol. The amplification process was performed on a Rotor Gene Q PCR system (QIAGEN, Shanghai, China) and data were analyzed by the 2–ΔΔCt methods using the Sequence Detection Software.
Primers for QPCR
GATA binding protein 1 (gata1, NM_008089):
Forward: 5-CAGGTTTCTTTTCCTCTGGG-3, reverse: 5-AAAGGACTGGGAAAGTCAGC-3
Phosphoglycerate kinase-2 (pgk2, NM_031190):
Forward: 5-CTGTTGCTGATGAGCTCAAG-3, reverse: 5-ACTCCGACCATAGAACTGTG-3
Zbtb16 zinc finger and BTB domain containing 16 (Plzf, NM_001033324):
Forward: 5-TGAGATCCTCTTCCACCGAA-3, reverse: 5-GTAGGACTCATGGCTGAGAGA-3
Deleted in azoospermia-like (Dazl, NM_010021):
Forward: 5-CCTCCA ACCATGATGAATCC-3, reverse: 5-TCTGTATGCTTCGGTCCACA-3
Heat shock protein a2 (Hspa2, NM_008301):
Forward: 5-CATCATCAATGAGCCCACAG-3, reverse: 5-TCTTGTGTTTGCGCTTGAAC-3
Protamine 1 (Prm1, NM_013637):
Forward: 5-ATGCTGCCGCAGCAAAAGCA-3, reverse: 5-CACCTTATGGTGTATGAGCG-3
Gapdh (NM_008084):
Forward: 5-GGCTACACTGAGGACCAGGTT-3, reverse: 5-TGCTGTAGCCGTATTCATTGTC-3
Tak1 (NM_009316.1):
Forward: 5-GGGCTGTTCATAATGGCACT-3, reverse: 5-GAGTTGCTCTGCCCTTCATC-3
Immunohistochemistry
Paraffin-embedded sections (5 μm) of testes were mounted together on a gelatin-coated glass slide and dried overnight at 37 °C. The sections were dewaxed in xylene and hydrated in a graded series of alcohols. The sections were boiled three times for 10 min each in 0.01 M sodium citrate using a microwave oven. The sections were then incubated in 0.35% H2O2 in PBS for 10 min. Blocking occurred in 5% BSA (Sigma, St Louis, MO)/5% goat serum (Dingguo, Shanghai, China). The slides were then incubated with rabbit polyclonal antibodies against γ-H2AX diluted 1 : 50 in PBS including 1% BSA in a humidified chamber overnight at 4 °C. Incubation with the secondary biotinylated goat anti-rabbit IgGs (Santa Cruz Biotechnology, Santa Cruz, CA) diluted 1 : 100 in PBS including 1% BSA was performed in a humidified chamber for 60 min at room temperature. The horseradish peroxidase avidin–biotin complex reaction was performed according to the manufacturer's protocol (Dingguo, Shanghai, China). The bound antibodies were visualized using 0.3 μg μl–1 3,3′-diaminobenzidine (DAB; Sigma) in PBS, to which 0.03% H2O2 was added. The sections were counterstained with hematoxylin.
Statistical analysis
Statistical analysis was performed using two-tailed Student's t tests or one-way analysis of variation (one way anova) with the post hoc test method. A probability of <0.05 was considered to be statistically significant.
Results
LLDT-8 induced spermatocyte-specific testis toxicity in C57BL/6 mice
The dosage of LLDT-8 (0.5, 1 mg kg–1) (Fig. 1) in this study was the same as that of LLDT-8 for the treatment of cancer and rheumatoid arthritis in animal models.26 LLDT-8 (0.5, 1.0 mg kg–1) caused dose and time-dependent decrease of the testis weight (Fig. 2A and S1A, B†). As shown in Fig. S1D,† the testes in the ‘Saline’ group had normal spermatid development. In the ‘LLDT-8’ group, the testes showed reduced germinal cell layers (Fig. 2E2, E3 and F1–F3, star), severe vacuolar degeneration (Fig. 2F2 and F3, asterisk) and abnormal development of spermatids (Fig. 2E1–E3 and F1–F3, arrow). At the end of administration of LLDT-8 (1.0 mg kg–1, 30 day), the seminiferous tubules had lost the normal morphology; only a few spermatogonia or primary spermatocytes could be observed (Fig. 2F3). On the other hand, LLDT-8 did not induce any abnormality in other tissues including the epididymis, liver, kidney, spleen and blood circulation (Fig. S1C, S2 and S3†).
Fig. 1. The structure of LLDT-8.
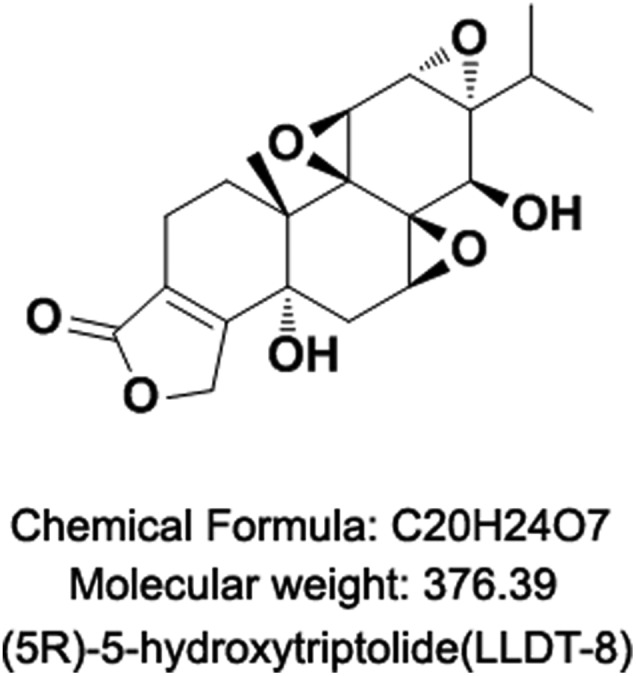
Fig. 2. LLDT-8 induced spermatocyte-specific testis toxicity in C57BL/6 mice. Mice (n = 3–5) were weighed before sacrificing on the 15th, 21st and 30th day post-administration of LLDT-8 or saline. The testes were collected and weighed. (A) Testis relative weight (testis absolute weight vs. body weight) is shown here; body weight and testis weight are shown in Fig. S2.† (B–D) The transcript levels of markers for spermatocytes were detected by qPCR. Dazl: a marker for type B spermatogonia and primary spermatocytes; Pgk2: a marker for meiotic spermatocytes; Hspa2: a marker for post-meiotic spermatocytes and spermatids. (E and F) The left testicle of each mouse was stained with H&E (×40); the ‘Saline’ group is shown in Fig. S2.† The ‘LLDT-8’ groups are shown here. Star: reduction of germinal layers; asterisk: vacuolar degeneration; arrow: abnormally developed spermatids. (G–H) The γ-H2AX level in mice testes is shown. The γ-H2AX level indicates the number of pachytene spermatocytes. The data are shown as mean ± SD; significant difference was determined by one way Anova, N = 3, *P < 0.05, **P < 0.01, ***P < 0.001 vs. the saline group.

To further elucidate the mechanisms of LLDT-8-induced testis toxicity, we investigated transcript levels of germ cell-specific differentiation markers.27 Gata1 (a marker for sertoli cells) and Plzf (a marker for germ stem cell and spermatogonial differentiation) were normally expressed in the testes (Fig. S1E and F†),28 while Dazl (a marker for Type B spermatogonia and primary spermatocytes),29,30 Pgk2 (a meiotic spermatocyte marker)31,32 and Hspa2 (a marker for post-meiotic spermatocytes and spermatids)33 were decreased from the 15th or 21st day (Fig. 2B–D). Prm1 (a spermatid-specific marker)34 was rapidly decreased from the 15th day (Fig. S1G†).
The phosphorylation of histone H2AX at serine 139 (γ-H2AX) is widely used as a marker for DNA double-strand breaks (DSBs). In the testes, γ-H2AX has an essential role in spermatogenesis. One prominent feature of the initiation of meiosis is the formation of genome-wide DSBs which are a prerequisite of homologous recombination. DSBs are specifically marked by γ-H2AX, which is distributed globally at the leptotene and zygotene stages but concentrated in the asynapsed XY body at the pachytene and diplotene stages. Therefore, the γ-H2AX level can well reflect the meiotic progress in spermatocytes, such as synapsis, DSB formation or meiotic recombination.35,36 In this study, the protein level and distribution of γ-H2AX were examined in the testes. In this study, the protein level of γ-H2AX was reduced by LLDT-8 from the 21st day (Fig. 2G and H). The distribution of γ-H2AX was also dramatically changed by LLDT-8. 1.0 mg kg–1 of LLDT-8 time-dependently decreased the number of γ-H2AX-positive germ cells. At the end of exposure, the γ-H2AX-postive germ cells almost completely disappeared (Fig. S4†). The disappeared γ-H2AX-postive germ cells strongly indicated the disrupted meiotic progress in the spermatocytes and the loss of various stages of the spermatocytes during the treatment of LLDT-8.
Using spermatocyte-like cell line GC-2spd and sertoli like cell line TM4, we also found that GC-2spd was more sensitive to LLDT-8 than TM4 (Fig. 3A). Taken together, all the above data strongly indicated that spermatocytes are the main target of LLDT-8 in the testes.
Fig. 3. LLDT-8 induced G1/S phase arrest and cellular apoptosis in GC-2spd. (A) GC-2spd and TM4 cells were exposed to various doses of LLDT-8 for 48 h to calculate IC50. (B) After incubation with LLDT-8 for 24 h, GC-2spd cells were stained with 5 μl Annexin V-FITC and 10μl PI (20 μg ml–1) and analyzed by flow cytometry for apoptosis. Representative results are shown in Fig. S4.† (C, D) After incubation with LLDT-8 for 24 h, GC-2spd cells were stained with 10 μl PI (20 μg ml–1) and analyzed by flow cytometry for the cell cycle. Significant difference was determined by the two-tailed Student's t tests or one way Anova, n = 3, mean ± SD, **P < 0.01, ***P < 0.001 vs. the saline group.

LLDT-8 induced G1/S phase arrest and cellular apoptosis in GC-2spd
GC-2spd, a spermatocyte-like cell line was used to investigate the effects of LLDT-8. Cell cycle progression and apoptosis were examined by flow cytometry. After 24 h-exposure, LLDT-8 (500 nM) resulted in apoptotic cell death in over 10% GC-2spd (Fig. 3B). LLDT-8 at 250 nM increased the percentage of GC-2spd cells in the S phase, while LLDT-8 at 500 nM increased the percentage of GC-2spd cells in the G1 phase (Fig. 3C and D).
LLDT-8 rapidly dephosphorylated Tak1 at Ser412 in GC-2spd but not in TM4
Tak1 is known to control inflammation through activating NF-kB and mitogen-activated protein kinases and LLDT-8 strongly inhibits the activation of NF-kB.15,16 In this study, several MAPKs were affected by LLDT-8 (Fig. S5†), and Tak1 was selected due to its potential role in LLDT-8-induced apoptosis. LLDT-8 rapidly dephosphorylated Tak1 at Ser412 from 8 h, and decreased Tak1 protein only at 24 h (Fig. 4A–C). Consistently, LLDT-8 also reduced Tak1 protein and Ser412 phosphorylation in the testes (Fig. 4D and E). Notably, LLDT-8 with the same concentration did not affect Tak1 of TM4, and failed to induce apoptosis in TM4 (Fig. 5). We also detected the transcription level of Tak1 in GC2-spd cells, and found that LLDT-8 decreases Tak1 mRNA at 16 h but not at 8 and 24 h (Fig. S6B†).
Fig. 4. LLDT-8 dephosphorylated Tak1 at Ser412 in GC-2spd and the testes. (A) After treatment with LLDT-8 (250, 500 nM) for 8 h, 16 h, and 24 h, the protein bands of TAK1/p-TAK1 (Ser412, Thr184/187) of GC-2spd were detected by western blotting. (B, C) Line graphs showed data from representative experiments, which were normalized to β-actin. (D) After administration with saline or LLDT-8 (0.5, 1 mg kg–1) for 15 days, the protein levels of Tak1 and p-Tak1 (Ser412, Thr184/187) in the testes were detected by western blotting. Significant difference was determined by the two-tailed Student's t tests or one way Anova, n = 3, mean ± SD., $$ (P < 0.01) indicates the significance of Tak1 between LLDT-8 and control; **(P < 0.01), ***(P < 0.001) indicates the significance of p-Tak1(Ser412) between LLDT-8 and control; ##(P < 0.01) indicates the significance between Tak1 and p-Tak1(Ser412).

Fig. 5. LLDT-8 had no effect on the dephosphorylation of Tak1 at Ser412 and cellular apoptosis in sertoli like cell line TM4. TM4 cells were exposed to LLDT-8 (250, 500 nM) for 24 h, then apoptosis was analyzed by flow cytometry. (A–C) LLDT-8 did not affect the expression or phosphorylation of Tak1 in TM4, (D) and failed to induce apoptosis in TM4. Histograms show data from representative experiments (n = 3, mean ± SD).

Role of Tak1 in the survival of GC-2spd and LLDT-8 induced apoptosis
The function of Tak1 in the testes is still unknown. Knockdown of Tak1 by siRNA in GC-2spd obviously inhibited the growth and induced apoptosis (Fig. 6A–C and S6A†). Similarly, the Tak1 inhibitor, 5Z-7-oxozeaenol (5Z), decreased the expression and Ser412 phosphorylation of Tak1, also inhibited the proliferation of GC-2spd and resulted in apoptotic cell death (Fig. 6D–F).
Fig. 6. Tak1 inhibition promoted apoptosis in GC-2spd. (A–C) GC-2spd cells were transfected with Tak1 siRNAs for 72 h. (A) The expression of Tak1 and p-Tak1 (Ser412) proteins was detected by western blotting; cell number (B) of GC-2spd was assessed by cell count; the apoptosis of GC-2spd (C) was analyzed by flow cytometry. Representative results are shown in Fig. S8.† (D–F) GC-2spd cells were incubated with 5Z-7-oxozeaenol (5Z) for 24 h. Cell number (E) of GC-2spd was assessed by cell count; the apoptosis of GC-2spd (F) was analyzed by flow cytometry.

To clarify the role of Tak1 in LLDT-8 induced apoptosis, constitutively activated Tak1 (CA-Tak1), lacking the 22 N-terminal amino acids was stably overexpressed in GC-2spd.25 CA-Tak1 enhanced Tak1 phosphorylation at Ser412, attenuated the cytotoxicity of LLDT-8 in GC-2spd and rescued up to 50% GC-2spd from the apoptosis induced by LLDT-8 of high concentration (1, 3 μM) (Fig. 7). Here, the concentration of LLDT-8 was increased to 1–3 μM, because LLDT-8 at 250–500 nM did not induce significantly apoptosis in GC-2spd cells with a stably transfected pCDH-empty vector or pCDH-CA-Tak1 vector (data not shown). This tolerance of GC-2spd to LLDT-8 may be related to the puromycin-induced resistance during the construction of stable cell line.37–39
Fig. 7. Constitutively activated Tak1 attenuated LLDT-8 induced cell death in GC-2spd. (A–C) GC-2spd cells with stably expressed CATAK1 (A) were treated with LLDT-8 for 24 h or 48 h. Cell viability (B) was measured using the MTT assay (48 h); after treatment with LLDT-8 (1000, 3000 nM), the apoptosis of GC-2spd (C) was analyzed by flow cytometry (24 h). Representative results are shown in Fig. S8.† Significant difference was determined by one way Anova, n = 3, mean ± SD. Si-NC: negative control. *P < 0.05, **P < 0.01, ***P < 0.001.

Discussion
In this study, we find that LLDT-8 specifically induces spermatocyte damage in vivo and in vitro through the inhibition of Tak1 Ser412 phosphorylation (Fig. 2–7). The initial cells in spermatogenesis are germ stem cells or spermatogonia, which divide mitotically into two types of spermatogonia. Type A spermatogonia serve for the renewal of germ stem cells, and type B spermatogonia yield primary spermatocytes by mitosis. The primary spermatocyte divides meiotically into two secondary spermatocytes (Meiosis I), which divide into four spermatids.40 In this study, markers for pre-meiotic (Dazl), meiotic (Pgk2), pachytene (γ-H2AX) and post-meiotic (Hspa2) spermatocytes all decreased from the 15th or 21st day, indicating severe spermatocyte injuries. Although the spermatid injuries seem to be more severe (Fig. S1G†), considering the fact that spermatids are derived from spermatocytes, we suggested that spermatocytes should be the main target of LLDT-8.
The role of TAK1 in the testis is still unknown. Here, the siRNA or inhibitor of Tak1 greatly promoted apoptosis in GC-2spd (Fig. 6), indicating the potential role of Tak1 in spermatogenesis. Both Thr184/187 and Ser412 phosphorylation are important for TAK1 kinase activity.22,23 In this study, Ser412 phosphorylation in mice seemed to be greatly higher than Thr184/187 phosphorylation (Fig. 4D and 7A), implying that Tak1 Ser412 phosphorylation may be more important in mice.
LLDT-8 dephosphorylated Tak1 in GC-2spd and induced apoptosis (Fig. 3, 4, 6, and 7). However, only 50% of cellular apoptosis was prevented by overexpressed CA-Tak1 (Fig. 7). A previous study has shown that LLDT-8 promotes apoptosis via transcription inhibition.14 Here, LLDT-8 also reduced RNA polymerase II in GC-2spd (Fig. S7†), which may lead to the incomplete recovery of CA-Tak1.
In this study, G1 phase arrest was induced by LLDT-8 at 500 nM, and S phase arrest was induced by LLDT-8 at 250 nM (Fig. 3C and D), which was consistent with a previous study, LLDT-8 at 100–300 nM induced S phase arrest.14 In the G1 phase, the cell synthesizes mRNA and proteins in preparation for DNA synthesis in the S phase, so the G1 phase arrest by LLDT-8 (500 nM) may be related to its stronger inhibitory effect on transcription.41–43
In our previous report, we confirm that testis is the most sensitive target to triptolide.44 Compared to triptolide, LLDT-8 displayed low toxicity to most of the tissues but not the testis. Testicular injury seems to be the most common adverse reaction to LLDT-8 and other triptolide derivatives. Tak1 and other factors may be involved in the strong sensitivity of testis to triptolide and its derivatives. More work is needed to clarify the potential mechanisms, which will be useful to develop a triptolide derivative with a better safety profile.
Our study firstly found the important role of Tak1 in the survival of spermatocytes, and the dephosphorylation of Tak1 at Ser412 may contribute to spermatocyte-specific testis toxicity induced by LLDT-8. Our data will be helpful for the further clinical development of LLDT-8 and the development of triptolide derivatives.
Conflict of interest
None.
Supplementary Material
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81102496), the National Key Technologies R&D Program (2012ZX09302-003, 2012ZX09301-001-006) and the Natural Science Foundation of Jiangsu Province (Grants No. BK2012358). We thank Prof. YC. Li for the kind provision of high purity (5R)-5-hydroxytriptolide (99.9%); we thank Prof. YZ. Wang for comments on this work.
Footnotes
†Electronic supplementary information (ESI) available. See DOI: 10.1039/c5tx00409h
References
- Leuenroth S. J., Okuhara D., Shotwell J. D., Markowitz G. S., Yu Z., Somlo S., Crews C. M. Proc. Natl. Acad. Sci. U. S. A. 2007;104:4389–4394. doi: 10.1073/pnas.0700499104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mujumdar N., Mackenzie T. N., Dudeja V., Chugh R., Antonoff M. B., Borja-Cacho D., Sangwan V., Dawra R., Vickers S. M., Saluja A. K. Gastroenterol. 2010;139:598–608. doi: 10.1053/j.gastro.2010.04.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titov D. V., Gilman B., He Q. L., Bhat S., Low W. K., Dang Y., Smeaton M., Demain A. L., Miller P. S., Kugel J. F., Goodrich J. A., Liu J. O. Nat. Chem. Biol. 2011;7:182–188. doi: 10.1038/nchembio.522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y., Zhang Y., Li L., Feng X., Ding S., Zheng W., Li J., Shen P. Chem. Biol. 2014;21:246–256. doi: 10.1016/j.chembiol.2013.12.009. [DOI] [PubMed] [Google Scholar]
- Kitzen J. J., de Jonge M. J., Lamers C. H., Eskens F. A., van der Biessen D., van Doorn L., Ter Steeg J., Brandely M., Puozzo C., Verweij J. Eur. J. Cancer. 2009;45:1764–1772. doi: 10.1016/j.ejca.2009.01.026. [DOI] [PubMed] [Google Scholar]
- Liu J., Chen X., Zhang Y., Miao H., Liu K., Li L., Zhong D. Anal. Chim. Acta. 2011;689:69–76. doi: 10.1016/j.aca.2011.01.016. [DOI] [PubMed] [Google Scholar]
- Harousseau A. P. J. L. D. H., Michallet M. and Brandely M., presented in part at the 13th Congress of the European Hematology Association Copenhagen Denmark, June 12–15, 2008.
- Xin M. J., Cui S. H., Liu S. A., Sun H. C., Li F., Sun J. B., Luo B. Hepatobiliary Pancreatic Dis. Int. 2010;9:312–318. [PubMed] [Google Scholar]
- Chen D. P., Ma Y. Y., Wang X. Q., Yu S. Q., Li L., Dai B., Mao Z. G., Liu H. C., Liu S. Y., Mei C. L. Am. J. Kidney Dis. 2014;63:1070–1072. doi: 10.1053/j.ajkd.2014.01.418. [DOI] [PubMed] [Google Scholar]
- Xiong J., Wang H., Guo G. M., Wang S. Z., He L. Q., Chen H. F., Wu J. PLoS One. 2011:6. doi: 10.1371/journal.pone.0020751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y. F., Zhu Y. N., Ni J., Zhong X. G., Tang W., Zhou R., Zhou Y., Dong J. R., He P. L., Wan H., Li Y. C., Yang Y. F., Zuo J. P. J. Neuroimmunol. 2006;175:142–151. doi: 10.1016/j.jneuroim.2006.03.011. [DOI] [PubMed] [Google Scholar]
- Tang W., Zhou R., Yang Y., Li Y. C., Yang Y. F., Zuo J. P. Transplantation. 2006;81:927–933. doi: 10.1097/01.tp.0000203299.39843.d2. [DOI] [PubMed] [Google Scholar]
- Ren Y. X., Zhou R., Tang W., Wang W. H., Li Y. C., Yang Y. F., Zuo J. P. Acta Pharmacol. Sin. 2007;28:518–525. doi: 10.1111/j.1745-7254.2007.00524.x. [DOI] [PubMed] [Google Scholar]
- Wang L., Xu Y., Fu L., Li Y., Lou L. Cancer Lett. 2012;324:75–82. doi: 10.1016/j.canlet.2012.05.004. [DOI] [PubMed] [Google Scholar]
- Tang W., Zuo J. P. Acta Pharmacol. Sin. 2012;33:1112–1118. doi: 10.1038/aps.2012.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuadrado I., Cidre F., Herranz S., Estevez-Braun A., de las Heras B., Hortelano S. Toxicol. Appl. Pharmacol. 2012;258:109–117. doi: 10.1016/j.taap.2011.10.013. [DOI] [PubMed] [Google Scholar]
- Mihaly S. R., Ninomiya-Tsuji J., Morioka S. Cell Death Differ. 2014;21:1667–1676. doi: 10.1038/cdd.2014.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morioka S., Broglie P., Omori E., Ikeda Y., Takaesu G., Matsumoto K., Ninomiya-Tsuji J. J. Cell Biol. 2014;204:607–623. doi: 10.1083/jcb.201305070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ajibade A. A., Wang H. Y., Wang R. F. Trends Immunol. 2013;34:307–316. doi: 10.1016/j.it.2013.03.007. [DOI] [PubMed] [Google Scholar]
- Bosman H. S. M. C., Jaques J., Brouwers-Vos A. Z., Quax W. J., Schuringa J. J., Vellenga E. Blood. 2014;124:3130–3140. doi: 10.1182/blood-2014-04-569780. [DOI] [PubMed] [Google Scholar]
- Cai P. C., Shi L., Liu V. W., Tang H. W., Liu I. J., Leung T. H., Chan K. K., Yam J. W., Yao K. M., Ngan H. Y., Chan D. W. OncoTargets Ther. 2014;5:7549–7562. doi: 10.18632/oncotarget.2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu M. D., Ouyang C., Lin W. L., Zhang T., Cao X. T., Xia Z. P., Wang X. J. J. Immunol. 2014;192:2846–2856. doi: 10.4049/jimmunol.1302537. [DOI] [PubMed] [Google Scholar]
- Singh A., Sweeney M. F., Yu M., Burger A., Greninger P., Benes C., Haber D. A., Settleman J. Cell. 2012;148:639–650. doi: 10.1016/j.cell.2011.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inokuchi-Shimizu S., Park E. J., Roh Y. S., Yang L., Zhang B., Song J. Y., Liang S., Pimienta M., Taniguchi K., Wu X. F., Asahina K., Lagakos W., Mackey M. R., Akira S., Ellisman M. H., Sears D. D., Olefsky J. M., Karin M., Brenner D. A., Seki E. J. Clin. Invest. 2014;124:3566–3578. doi: 10.1172/JCI74068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi K., Shirakabe T., Shibuya H., Irie K., Oishi I., Ueno N., Taniguchi T., Nishida E., Matsumoto K. Science. 1995;270:2008–2011. doi: 10.1126/science.270.5244.2008. [DOI] [PubMed] [Google Scholar]
- Zhou R., Zhang F., He P. L., Zhou W. L., Wu Q. L., Xu J. Y., Zhou Y., Tang W., Li X. Y., Yang Y. F., Li Y. C., Zuo J. P. Int. Immunopharmacol. 2005;5:1895–1903. doi: 10.1016/j.intimp.2005.06.009. [DOI] [PubMed] [Google Scholar]
- Sasaki T., Marcon E., McQuire T., Arai Y., Moens P. B., Okada H. J. Cell Biol. 2008;182:449–458. doi: 10.1083/jcb.200802113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buaas F. W., Kirsh A. L., Sharma M., McLean D. J., Morris J. L., Griswold M. D., de Rooij D. G., Braun R. E. Nat. Genet. 2004;36:647–652. doi: 10.1038/ng1366. [DOI] [PubMed] [Google Scholar]
- Kim B., Cooke H. J., Rhee K. Development. 2012;139:568–578. doi: 10.1242/dev.075846. [DOI] [PubMed] [Google Scholar]
- Schrans-Stassen B. H. G. J., Saunders P. T. K., Cooke H. J., de Rooij D. G. Biol. Reprod. 2001;65:771–776. doi: 10.1095/biolreprod65.3.771. [DOI] [PubMed] [Google Scholar]
- Yoshioka H., Geyer C. B., Hornecker J. L., Patel K. T., McCarrey J. R. Mol. Cell. Biol. 2007;27:7871–7885. doi: 10.1128/MCB.00990-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boer P. H., Adra C. N., Lau Y. F., Mcburney M. W. Mol. Cell. Biol. 1987;7:3107–3112. doi: 10.1128/mcb.7.9.3107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inselman A. L., Nakamura N., Brown P. R., Willis W. D., Goulding E. H., Eddy E. M. Genesis. 2010;48:114–120. doi: 10.1002/dvg.20588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleene K. C., Distel R. J., Hecht N. B. Dev. Biol. 1984;105:71–79. doi: 10.1016/0012-1606(84)90262-8. [DOI] [PubMed] [Google Scholar]
- Fernandez-Capetillo O., Mahadevaiah S. K., Celeste A., Romanienko P. J., Camerini-Otero R. D., Bonner W. M., Manova K., Burgoyne P., Nussenzweig A. Dev. Cell. 2003;4:497–508. doi: 10.1016/s1534-5807(03)00093-5. [DOI] [PubMed] [Google Scholar]
- Cordelli E., Paris L. Methods Mol. Biol. 2013;1044:293–310. doi: 10.1007/978-1-62703-529-3_15. [DOI] [PubMed] [Google Scholar]
- Cass C. E. J. Cell. Physiol. 1972;79:139–146. doi: 10.1002/jcp.1040790116. [DOI] [PubMed] [Google Scholar]
- Demeuse P., Fragner P., Leroy-Noury C., Mercier C., Payen L., Fardel O., Couraud P. O., Roux F. J. Neurochem. 2004;88:23–31. doi: 10.1046/j.1471-4159.2003.02071.x. [DOI] [PubMed] [Google Scholar]
- Nelson E. J., Hinkle P. M. Endocrinology. 1992;130:3246–3256. doi: 10.1210/endo.130.6.1350759. [DOI] [PubMed] [Google Scholar]
- Rolland A. D., Jegou B., Pineau C. Adv. Exp. Med. Biol. 2008;636:16–41. doi: 10.1007/978-0-387-09597-4_2. [DOI] [PubMed] [Google Scholar]
- Cao T. M., Hua F. Y., Xu C. M., Han B. S., Dong H., Zuo L., Wang X., Yang Y., Pan H. Z., Zhang Z. N. Ann. Hematol. 2006;85:434–442. doi: 10.1007/s00277-005-0046-4. [DOI] [PubMed] [Google Scholar]
- Liu X., Yang J. M., Zhang S. S., Liu X. Y., Liu D. X. BMC Cancer. 2010;10:684. doi: 10.1186/1471-2407-10-684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y. M., Pyo H. Cancer Biother.Radiopharm. 2013;28:138–145. doi: 10.1089/cbr.2012.1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C. Z., Xing G. Z., Maeda K., Wu C. Y., Gong L. K., Sugiyama Y., Qi X. M., Ren J., Wang G. J. Toxicol. Res. 2015;4:1260–1268. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.