

 Metabolomics is a useful tool for discovering biomarkers to predict the side effects of cancer therapy.
Metabolomics is a useful tool for discovering biomarkers to predict the side effects of cancer therapy.
Abstract
Cisplatin, which is an inorganic molecule containing a platinum ion, is an antineoplastic agent that has been used to treat various solid tumors. However, its side effects include nephrotoxicity, neurotoxicity, bone marrow toxicity, gastrointestinal toxicity, and ototoxicity, which can limit its use. In this study, nephrotoxicity was caused by the intraperitoneal injection of cisplatin into rats, and then metabolome analysis was performed using gas chromatography/mass spectrometry (GC/MS) and liquid chromatography/mass spectrometry (LC/MS) to find plasma metabolite biomarker candidates that would facilitate the early detection of cisplatin-induced nephrotoxicity. As a result, chronological changes were detected in the plasma levels of cysteine–cystine and 3-hydroxy-butyrate in the GC/MS-based metabolomics study. In the LC/MS-based metabolomics study, 3 acylcarnitines and a phosphatidylethanolamine with C18:2–C18:2 were identified as potential plasma biomarkers of cisplatin-induced nephrotoxicity. The plasma levels of these 6 metabolites altered significantly after the administration of cisplatin, and these alterations occurred quicker than the equivalent changes in the plasma levels of creatinine and blood urea nitrogen, which are usually used as indicators of renal dysfunction. These results indicate that the abovementioned metabolites might be reliable biomarkers that would allow the earlier detection of cisplatin-induced nephrotoxicity and that metabolomics is a useful tool for discovering biomarkers that could be used to predict the side effects of cancer therapy.
Introduction
Cisplatin, which is an inorganic molecule containing a platinum ion, is an antineoplastic agent that has been used to treat various solid tumors, including head and neck, lung, testicular, ovarian, and bladder cancer.1–3 Cisplatin binds to DNA, resulting in a platinum fragment cross-linking the two DNA strands within the double helix. This cross-linking triggers DNA damage, such as the production of defective DNA templates, DNA synthesis arrest, and the inhibition of DNA replication in cancer cells.4 However, its side effects, which include nephrotoxicity, neurotoxicity, bone marrow toxicity, gastrointestinal toxicity, and ototoxicity, can sometimes limit its use. Among these side effects, nephrotoxicity is a serious problem. Cisplatin is absorbed into renal tubular cells, and exposing tubular cells to cisplatin triggers complex signaling pathways including reactive oxygen species (ROS), p53, and mitogen-activated protein kinase pathways, leading to the promotion of cell death. Cisplatin also induces inflammatory responses, such as tumor necrosis factor-α production. In addition, cisplatin causes renal vascular injuries. These events can ultimately lead to acute renal failure,5,6 and approximately one-third of patients who receive cisplatin subsequently suffer renal dysfunction.7,8
Currently, serum creatinine and blood urea nitrogen (BUN) levels are used as indicators of renal function, but the serum levels of these markers only increase markedly after renal injuries have occurred. Therefore, better and more reliable biomarkers that would allow the early detection of nephrotoxicity are needed. Until now, various studies aimed at identifying novel biomarkers have been performed using genomic and proteomic approaches,9 and several candidate biomarkers, which are more sensitive than classical serum biomarkers, have been found.10–12 However, genomics provides extensive information regarding the genotype, and proteomics mainly helps to explain information based on genomic evidence. Therefore, it might be difficult to obtain detailed phenotypic information based on these approaches.
Metabolomics (or metabolome analysis) is the comprehensive study of chemical processes involving low molecular weight metabolites. Metabolites are the end products of cellular processes, so metabolomic data includes not only information derived from innate genetic variability, but also information based on post-birth environmental factors. Therefore, it has been recognized that metabolomics is useful for evaluating final phenotypes in detail. We have carried out many metabolomics-based studies in order to discover novel metabolite biomarkers and evaluate various diseases.13–19 In this study, biological samples from a nephrotoxicity animal (rat) model was subjected to gas chromatography/mass spectrometry (GC/MS)- and liquid chromatography/mass spectrometry (LC/MS)-based metabolome analyses to find metabolite biomarker candidates that would aid the early detection of cisplatin-induced nephrotoxicity. In the GC/MS analysis, various metabolites, such as organic acids, amino acids, sugars, and sugar alcohols, were evaluated. In the LC/MS analysis, lipid metabolites, such as lysophosphatidylcholines, phosphatidylcholines (PCs), lysophosphatidylethanolamines, phosphatidylethanolamines (PEs) fatty acids (FAs), and acylcarnitines (ACs), were evaluated. In searching for the biomarkers that contribute to early detection of cisplatin-induced nephrotoxicity, urine is often used. However, urine has the thickness, of which affects the concentrations of molecules in the urine, but blood has its very low possibility. Usually, the urine levels of molecules are corrected with the urine levels of creatinine/creatine. However, if the urine levels of creatinine/creatine are changed by cisplatin-induced nephrotoxicity, the creatinine/creatine correction is inadequate. Therefore, we selected blood but not urine in this study.
Methods
Study design and the animal model of cisplatin-induced nephrotoxicity
All animal treatments in this study were approved by the institutional animal care and use committee of the Sumika Technoservice Corporation (Osaka, Japan), and then were performed according to the animal experimentation regulations of the Sumika Technoservice Corporation. Six-week-old male Crl:CD (SD) rats (Japan Charles River Laboratories International; Yokohama, Japan) were used in this study. The animals were housed in a cage with a raised mesh base under constant environmental conditions (room temperature, 22–24 °C; relative humidity, 40%–70%) and a 12 h light–dark cycle (8:00/20:00). Prior to the experiment, food and water were provided ad libitum. The animals were randomly divided into three groups; the high-dose group (n = 13), low-dose group (n = 10), and untreated group (n = 10). The animals were given 10 mg per kg body weight (BW) of cisplatin intraperitoneally in the high-dose group, 5 mg per kg BW cisplatin in the low-dose group, and saline alone in the untreated group. Cisplatin was purchased from Nichi-Iko Pharmaceutical Corporation (Toyama, Japan), and it was diluted with saline obtained from Fuso Pharmaceutical Industries (Osaka, Japan). The administration concentrations of cisplatin in our study were decided according to previous reports,10,20 and these concentrations have been often applied into many articles. Blood samples were collected from the tail vein at 24 h, 48 h, and 96 h after the injection of cisplatin. Then, the rats were sacrificed at 96 h, and kidney samples were harvested. The blood samples for the metabolome analysis were centrifuged at 4 °C and 3000 rpm for 10 min to obtain plasma, and then the plasma was immediately frozen and stored at –80 °C. The kidney samples for the metabolome analysis were cut into small pieces (approximately 50 mg), which were immediately frozen and stored at –80 °C.
Biochemical examination and histopathology
The plasma levels of creatinine, BUN, total protein (TP), and albumin were measured by the Oriental Yeast Corporation (Tokyo, Japan). The kidney samples used for the histopathological evaluations were preserved in 10% buffered formaldehyde solution. Then, these samples were embedded in paraffin, cut into 5 μm slices, and stained with hematoxylin and eosin (H&E). The H&E-stained tissues were examined by microscopy in a blinded manner. The part of kidney samples was used to measure the level of Pt in the kidney tissue, and its level was measured by Sumika Chemical Analysis Service (Osaka Japan).
Sample preparation for the metabolome analysis
The plasma sample preparation for the GC/MS-based metabolome analysis was performed according to the method described in our previous study.21 In the metabolome analysis in which kidney samples were subjected to GC/MS, each 10 mg kidney sample was mixed with 1000 μL of mixed-solvent (MeOH : H2O : CHCl3 = 2.5 : 1 : 1) and 10 μL of 0.5 mg ml–1 2-isopropylmalic acid as an internal standard, before being homogenized using 2 beads and a crushing machine. The mixture was shaken at 1200 rpm for 30 min at 4 °C and was then centrifuged at 16 000 rpm for 3 min at 4 °C, before the upper layers were transferred into a clean tube, and 200 μL of distilled water were added. After being centrifuged at 15 000 rpm for 3 min at 4 °C, the resultant supernatant was lyophilized using a freeze dryer. The lyophilized samples were mixed with 20 mg ml–1 methoxyamine hydrochloride (Sigma-Aldrich, Tokyo, Japan) dissolved in pyridine and then shaken at 1200 rpm for 90 min at 30 °C. Next, N-methyl-N-trimethylsilyl-trifluoroacetamide (GL Science, Tokyo, Japan) was added for derivatization, and the mixture was incubated at 1200 rpm for 30 min at 37 °C. The mixture was centrifuged at 15 000 rpm for 5 min, and the resultant supernatant was subjected to GC/MS analysis.
The plasma sample preparation for the LC/MS-based metabolome analysis was performed according to the method described in our previous study.22 In the metabolome analysis in which kidney samples were subjected to LC/MS, each 5 mg kidney sample was homogenized with 225 μL of MeOH and 25 μL of PC 12:0–12:0 as an internal standard. After being centrifuged, 175 μL of the resultant kidney sample mixture were subjected to LC/MS analysis.
GC/MS and LC/MS metabolite profiling of plasma and kidney tissue
The GC/MS analysis was conducted using a GCMS-QP2010 Ultra (Shimadzu Co., Kyoto, Japan) with a fused silica capillary column (CP-SIL 8 CB low bleed/MS; inner diameter: 30 m × 0.25 mm, film thickness: 0.25 μm; Agilent Co., Palo Alto, CA) according to the method described in a previous report.18,21 The LC/MS analyses were carried out using a Nexera LC system (Shimadzu Co., Kyoto, Japan) with two LC-30AD pumps, a DGU-20A5 degasser, an SIL-30AC autosampler, a CTO-20AC column oven, and a CBM-20A control module, coupled to an LCMS-8040 triple quadrupole mass spectrometer (Shimadzu Co., Kyoto, Japan) according to the method described in a previous report.22,23 The peak height and area of each metabolite detected in the plasma samples during the GC/MS and LC/MS analyses were normalized to the peak height and area of the internal standard, respectively. The peak height and area of each metabolite detected in the kidney samples during the GC/MS and LC/MS analysis were normalized to the peak height and area of the internal standard, respectively, and the fresh weight of the kidney sample.
Statistical analysis
The metabolite data collected during the GC/MS and LC/MS analyses were compared between each treatment group and the untreated group at 24 h, 48 h, and 96 h after the injection of cisplatin, and the results were evaluated for statistical significance using Welch's t-test.
Results and discussion
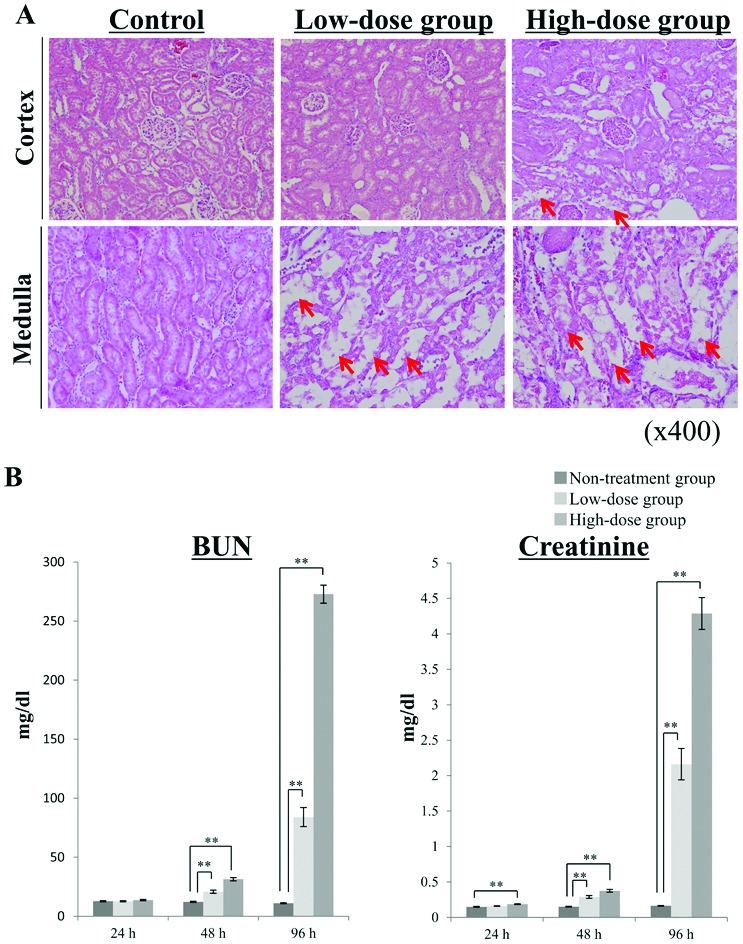
In our study, the rats were randomly divided into three groups; the high-dose group, which was intraperitoneally injected with 10 mg per kg BW cisplatin (n = 13); the low-dose group, which was intraperitoneally injected with 5 mg per kg BW cisplatin (n = 10), and the untreated group, which was intraperitoneally injected with saline alone (n = 10). The rats in the low-dose and untreated groups did not show any BW loss during the experiment, although the significant differences between the low-dose and untreated groups were observed (ESI Table 1†). In the high-dose group, the rats developed diarrhea, and their bodies became dirty with any significant changes in their BW, and 2 rats died at 96 h after the administration of cisplatin (data not shown). The dead rats were excluded from the experiment. Kidney samples were collected at 96 h after the administration of cisplatin, and then pathological evaluations were performed (Fig. 1A). H&E staining of the kidney cortex and medulla showed irregular tubular and cellular structures. This cisplatin-induced kidney damage was dependent on the injected dose. The level of Pt in the kidney tissues was measured to investigate whether cisplatin is located in the kidney. In the rats treated with 5 mg per kg BW cisplatin, the Pt level was 8.0 ± 1.1 μg g–1 tissue (mean ± S.E.). The Pt level of the rats treated with 10 mg per kg BW cisplatin was 16.2 ± 1.6 μg g–1 tissue (mean ± S.E.), and its dose-dependency could be confirmed. At 24 h, 48 h, and 96 h after the injection of cisplatin, the chronological changes in the rats’ plasma creatinine, BUN, TP, and albumin levels were evaluated (Fig. 1B, ESI Table 2†). The plasma levels of creatinine and BUN exhibited marked fold-changes (fold-change: >2) after both 48 and 96 h in the high-dose group and after 96 h in the low-dose group (P < 0.01). In the low-dose group, significant changes in plasma creatinine and BUN levels were detected after 48 h (P < 0.01), but the associated fold-change values were low. In the high-dose group, only the plasma creatinine level had changed significantly after 24 h (P < 0.01), although the associated fold-change value was low (fold-change: 1.25). These results suggest that plasma creatinine and BUN levels may not be useful for evaluating nephrotoxicity at 24 h after the administration of cisplatin, even in patients given high doses of the drug. In addition, these biochemical parameters are not very sensitive markers of nephrotoxicity because they only change after the kidneys have been damaged. Regarding TP, no significant differences between the low-dose and untreated groups were observed, and we could not confirm the significant difference at 24 h after the administration of cisplatin in the high-dose group (ESI Table 2†). At 24 h after the administration of cisplatin in the low-dose group, the plasma level of albumin was significantly increased, but the associated fold-change value was low (fold-change: 1.05). Therefore, in this study using a metabolomics-based approach we tried to identify more sensitive biomarker candidates that could be used to detect nephrotoxicity earlier than is possible with clinical biochemical parameters.
Fig. 1. Biochemical and pathological changes detected in plasma after the injection of cisplatin. Rats were divided into 3 groups (high-dose group, low-dose group, untreated group) and then were intraperitoneally administered cisplatin at a concentration of 10 (high-dose group) or 5 (low-dose group) mg per kg BW. The rats in the untreated group were administered saline as a vehicle control. (A) Pathological findings of the rats’ kidney tissues at 96 h after the injection of cisplatin. Kidney samples were collected at 96 h after the injection of cisplatin and then were immediately fixed in 10% formaldehyde. Histological sections (of the cortex and medulla) of the kidney samples were stained with hematoxylin and eosin (H&E). Each image shows representative results for each group. Red arrows exhibit the sites of remarkable renal tubular injury. (B) The chronological changes in the plasma levels of creatinine and BUN. Blood samples were collected at 24 h, 48 h, and 96 h after the administration of cisplatin, and the plasma levels of creatinine and BUN were measured. Data are shown as mean ± S.E. values, and double asterisks indicate statistical significance at P < 0.01 compared with the untreated group according to Welch's t test. n = 11 for the high-dose group, n = 10 for the low-dose group, and n = 10 for the untreated group; BUN, blood urea nitrogen.

We identified 80 metabolites in rat plasma samples using GC/MS (ESI Table 3A†). In both the high- and low-dose groups, the levels of 9 metabolites exhibited significant differences (P < 0.05) compared with the untreated group at 48 h after the injection of cisplatin. Among the 9 metabolites, 8 and 2 displayed significant changes in their levels at 24 h in the high-dose group and low-dose group, respectively. We also detected 205 metabolites in the rat plasma samples using LC/MS (ESI Table 3B†). In both treatment groups, 72 metabolites demonstrated significant differences (P < 0.05) in their levels compared with the untreated group at 48 h after the injection of cisplatin. In the high-dose group, among the 72 metabolites, 56 exhibited significant changes in their levels (P < 0.05) at 24 h. In the low-dose group, among the 72 metabolites, 48 exhibited significant changes in their levels (P < 0.05) at 24 h.
Furthermore, we identified 44 and 223 metabolites in rat kidney samples using GC/MS and LC/MS, respectively (ESI Tables 4A and 4B†). Among the 44 metabolites detected by GC/MS, 9 metabolites exhibited significant changes (P < 0.05) in their levels in both the high- and low-dose groups compared with the untreated group, and among the 223 metabolites detected by LC/MS, 76 metabolites displayed significant changes in their levels in both treatment groups (P < 0.05).
Our goal was to find biomarker candidates that could be used to detect cisplatin-induced nephrotoxicity earlier than is possible based on plasma creatinine BUN, TP and albumin levels. Therefore, we focused on the data obtained for the high-dose group at 24 h after the injection of cisplatin and used the following criteria to select biomarker candidates: statistical significance (P < 0.01) and a marked fold-change value (fold-change: >2 or <0.5). To analyze the data, we evaluated the changes in metabolite levels induced by the administration of cisplatin using volcano plots. A volcano plot is a type of scatterplot that is used to identify changes in large datasets composed of replicated data.24 The volcano plot of the GC/MS-based metabolite data obtained in the high-dose group at 24 h showed that 3-hydroxy-butyrate, cysteine–cystine, and tryptophan satisfied the abovementioned criteria (Fig. 2A). Among these 3 metabolites, 3-hydroxy-butyrate and cysteine–cystine also exhibited statistically significant changes in their plasma levels (P < 0.01) at 48 h after the administration of cisplatin in the low-dose group (Fig. 3A). As for the LC/MS-based metabolite data obtained at 24 h after the injection of cisplatin in the high-dose group, the volcano plot showed that the plasma levels of AC 14:0, AC 18:1, AC 18:2, and PE 18:2–18:2 displayed significant changes (P < 0.01) as well as large fold-changes (fold-change: <0.5 or >2) (Fig. 2B). These 4 metabolites also demonstrated significant changes in their plasma levels at 48 h after the injection of cisplatin in the low-dose group (P < 0.01) (Fig. 3B). In the control group, the plasma levels of cysteine–cystine and PE 18:2–18:2 were decreased, so cysteine–cystine and PE 18:2–18:2 may be the metabolites that show the interday variation. Furthermore, these 6 candidates showed statistically significant changes (P < 0.01) as well as large fold-changes (fold-change: <0.5 or >2) in their plasma levels at 48 h after the injection of cisplatin in the high-dose group (ESI Fig. 1A and 1B†). Taking these results together, the plasma levels of 3-hydroxy-butyrate, AC 14:0, AC 18:1, and AC 18:2 increased, and the plasma levels of cysteine–cystine and PE 18:2–18:2 decreased after the administration of cisplatin. In addition, the plasma level of 3-hydroxy-butyrate exhibited the greatest change at 48 h after the injection of cisplatin, whereas the changes in the remaining 6 metabolites were largest at 96 h. These findings indicate that the abovementioned plasma metabolites are candidate metabolite biomarkers that could be used to aid the early detection of cisplatin-induced nephrotoxicity.
Fig. 2. Candidate biomarkers of nephrotoxicity selected by volcano plot analysis of the plasma metabolite data obtained using GC/MS or LC/MS in the high-dose group at 24 h after the injection of cisplatin. Volcano plots were used to show the changes in the metabolite data obtained with GC/MS (A) or LC/MS (B) at 24 h after the administration of 10 mg per kg BW cisplatin (in the high-dose group). The volcano plots were constructed with P-values on the y-axis and fold-change values on the x-axis. The P-values (compared with the untreated group) were calculated using Welch's t test. The fold-change values represent ratios of the value for the relevant treatment group to the value for the untreated group. The arrows indicate the metabolites that exhibited statistically significant changes in their plasma levels at 24 h after the administration of 10 mg per kg BW cisplatin (in the high-dose group) or 48 h after the administration of 5 or 10 mg per kg BW cisplatin (in the low and high-dose groups, respectively) (Fig. 3). The horizontal dotted line represents P = 0.01, the vertical dotted line indicates a fold-change value of 2 or 0.5, and the gray zones represent P-values of <0.01 and fold-change values of >2 or <0.5. PE, phosphatidylethanolamine; AC, acylcarnitine.

Fig. 3. Chronological changes in the levels of representative plasma metabolites after the administration of cisplatin. In the volcano plot analysis of the high-dose group shown in Fig. 2, some candidate biomarkers of nephrotoxicity were selected. Among these candidates, 2 candidates (3-hydroxy-butyrate and cysteine–cystine) were selected based on the GC/MS analysis (A), and 4 metabolites (AC 14:0, AC 18:1, AC 18:2, and PE 18:2–18:2) were selected based on the LC/MS analysis (B). Data are shown as mean ± S.E. values. Single asterisks indicate statistical significance at P < 0.05, and double asterisks indicate statistical significance at P < 0.01 compared with the untreated group according to Welch's t test. n = 11 for the high-dose group, n = 10 for the low-dose group, and n = 10 for the untreated group; PE, phosphatidylethanolamine; AC, acylcarnitine.

Among these 6 candidates, cysteine–cystine, AC 18:1, AC 18:2, and PE 18:2–18:2 were detected in kidney tissue. In the low- and high-dose groups, the levels of cysteine–cystine, AC 18:2, and PE 18:2–18:2 exhibited significant changes (P < 0.05) compared with the control group (Fig. 4); i.e., the levels of these metabolites in kidney tissue decreased after the injection of cisplatin.
Fig. 4. Changes in the kidney tissue levels of the representative metabolites selected in the plasma analysis at 96 h after the injection of cisplatin. Among the 6 representative metabolites selected during the plasma analyses, the GC/MS analysis of kidney tissue detected 1 metabolite (cysteine–cystine), and the LC/MS analysis of kidney tissue detected 3 metabolites (AC 18:1, AC 18:2, and PE 18:2–18:2). The kidney tissue samples were obtained at 96 h after the administration of cisplatin. Data are represented as mean ± S.E. values, and double asterisks indicate statistical significance at P < 0.01 compared with the untreated group according to Welch's t test. n = 11 for the high-dose group, n = 10 for the low-dose group, and n = 10 for the untreated group; PE, phosphatidylethanolamine; AC, acylcarnitine.

The relationships between the plasma levels of the biomarker candidates and the plasma creatinine level at 96 h were evaluated using Spearman's rank correlation coefficient (ESI Tables 5A and 5B†), and the plasma levels of 6 candidates exhibited significant correlations with the plasma creatinine level at 96 h (P < 0.05).
GC/MS-based metabolomics analysis detected chronological changes in the levels of some metabolites in tests of rat plasma samples obtained at 24 h, 48 h, and 96 h after the injection of cisplatin. We selected biomarker candidates based on the detection of statistically significant changes (P < 0.01) and large fold-change values (fold-change: <0.5 or >2) after 24 h in the high-dose group. Among the identified metabolites, cysteine–cystine and 3-hydroxybutyrate satisfied the abovementioned criteria. In the LC/MS-based metabolomics analysis of rat plasma samples collected at 24 h, 48 h, and 96 h after the injection of cisplatin, it was found that the levels of some metabolites changed, and some ACs and PE 18:2–18:2 satisfied the criteria for novel biomarker candidates of cisplatin-induced nephrotoxicity.
Cysteine is a sulfur-containing nutritionally semi-essential amino acid, and cystine is an oxidized form of cysteine. Cysteine and cystine are found as a major thiol/disulfide redox couple in plasma. Extracellularly, cysteine mainly exists in the form of cystine because cysteine is rapidly oxidized to cystine in normoxic states. On the other hand, cysteine is the prevailing form of intracellular cysteine because of the strong reducing states found within cells.25 Previous studies have shown that an imbalance in the extracellular levels of cysteine/cystine is involved in oxidative stress and other pathological conditions.26,27 Furthermore, it is known that intracellular cysteine is catabolized to glutathione (GSH) and hydrogen sulfide (H2S).28 H2S has a cytoprotective effect against oxidative stress by normalizing the GSH level, scavenging ROS, and suppressing the intracellular Ca2+ concentrations of neurons, cardiovascular cells, and kidney cells.29–32 In this study, the total levels of cysteine and cystine in plasma and kidney tissue fell after the injection of cisplatin, although the individual levels of cysteine and cystine could not be evaluated. Therefore, it is suspected that cysteine and cystine are consumed by renal cells to protect them from the ROS stress induced by cisplatin, and our results might reflect this phenomenon.
3-Hydoroxy-butyrate is the predominant ketone body made from acetyl-CoA. It is mainly produced in the liver in the presence of a negative energy state. Acetyl-CoA cannot be transported via the blood to organs that need energy, so it is converted to 3-hydroxy-butyrate, which can be transported via the blood. After being transported to the organs, 3-hydroxy-butyrate rapidly diffuses through the peripheral tissues and then readily enters cells through their membranes, where it exhibits antioxidative activity.33 Exogenous 3-hydroxy-butyrate has been shown to have therapeutic effects on stress-related conditions, such as hemorrhagic shock, cerebral hypoxia, anoxia, and ischemia.34,35 Furthermore, exogenous 3-hydroxy-butyrate demonstrated protective effects against paraquat-induced oxidative stress in rats by suppressing lipid peroxidation (by increasing the concentration of GSH) and decreasing apoptosis (by regulating Bcl-2 and Bax).36 In the present study, the plasma level of 3-hydroxy-butyrate was elevated during the early phase after the injection of cisplatin. Therefore, it is suggested that 3-hydroxy-butyrate was produced after the injection of cisplatin to protect renal cells from cisplatin-induced nephrotoxicity.
ACs play an important role in β-oxidation, which is the catabolic process by which FAs are broken down in mitochondria to generate acetyl-CoA that enters the citric acid cycle. ACs are produced via the binding of carnitine to acyl-CoA, which is produced from FAs after FAs have been transported to the inner mitochondrial membrane. Regarding urinary excretion, it is known that the kidneys are the major organ responsible for the homeostasis of carnitines including ACs.37,38 In some previous studies, it was found that FAs compose a major source of metabolic fuel for energy production in renal tubular cells.39 It was also shown that cisplatin deactivates proximal tubule peroxisome proliferator-activated receptor-alpha (PPARα), which activates β-oxidation of FAs, and regulates mitochondrial functions in kidney tissue. Furthermore, the activation of PPARα could ameliorate the reductions in renal function brought about by cisplatin-induced nephrotoxicity.40 In our study, the plasma levels of most ACs were elevated at 24 h, 48 h, and 96 h after the injection of cisplatin. On the other hand, the levels of ACs in the rats’ kidney tissues were decreased at 96 h after the injection of cisplatin. These results suggest that the failure of β-oxidation in renal cells and the urinary excretion of ACs resulted in the accumulation of ACs in plasma, and it is considered that the ACs that accumulated in the rats’ kidney tissues were lost because of the destruction of kidney tissue. In addition, in patients that were treated with cisplatin the urinary excretion of carnitine and ACs was upregulated, and the urinary levels of these molecules were suggested to be a marker of proximal tubular damage.41 In our study, the plasma levels of ACs were increased by the injection of cisplatin, and the levels of ACs in the rats’ kidney tissue were decreased. These results indicate that cisplatin-induced side effects affect the levels of ACs in the body and that changes in AC levels might represent useful biomarkers of cisplatin's side effects.
PE 18:2–18:2 is one of the PE groups, which are a class of phospholipid found in biological membranes. In our study, other PEs did not show the same tendency. In addition, PCs, which are also a class of phospholipid found in biological membranes, did not exhibit the same tendency. Therefore, the alterations in the plasma level of PE 18:2–18:2 might explain some aspects of cisplatin-induced nephrotoxicity, although detailed investigations are needed to confirm this.
Several previous studies have attempted to identify metabolite biomarkers of cisplatin-induced nephrotoxicity in rats. In some of these studies, urine was used as the biomaterial,20,42 but it is difficult to use urine for such studies because of the interindividual differences in its density. In the study by Uehara et al.,43 plasma was used, as was the case in our study. However, only anion and cation metabolites were targeted in the study by Uehara et al., while we analyzed lipid metabolites. In addition, recently protein-based biomarkers have been reported for early detection of acute kidney injury. For example, the urinary level of Kim-1 was significantly higher at 24 h after the 5.5 mg per kg BW cisplatin administration in rats, which were injected with cisplatin once through their tail veins.44 In the rats that were once intraperitoneally-injected with cisplatin at 6 mg per kg BW, urinary levels of SBP1, NGAL and TIMP-1 were markedly elevated at 72 h following cisplatin treatment, although their changes could not be observed at 24 h.45 In our study, some of plasma biomarker candidates were significantly altered at 24 h after the intraperitoneal injection at 5 mg per kg BW. Therefore, our study might have identified reliable biomarker candidates that could be used to detect cisplatin-induced nephrotoxicity earlier than is possible with the biomarkers identified in other studies. However, metabolite biomarker candidates have some problems to be resolved. For example, alterations in the urinary level of Kim-1 were confirmed in the human study,46 so assessment of metabolite biomarker candidates in the human study is necessary, and moreover it is also important to elucidate why metabolite biomarker candidates are changed by nephrotoxicity, although the possible mechanisms were discussed in this paper.
In conclusion, cysteine–cystine and 3-hydroxy-butyrate (based on GC/MS analysis) and AC 14:0, AC 18:1, AC 18:2, and PE 18:2–18:2 (based on LC/MS analysis) were identified as candidate biomarkers that could be used to detect cisplatin-induced nephrotoxicity early. The conventional biomarkers, such as creatinine, BUN, TP and albumin, did not show the prominent differences at 24 h after the administration of cisplatin in the low-dose group, although the plasma level of albumin was significantly increased with very low fold change, and these results indicate that the metabolite candidates are effective as early detection markers compared with the conventional biomarkers. In our study, the pronounced tubular cell damage was observed (Fig. 1A), so the altered metabolite biomarker candidates may be due to the tubular cell damage, although we cannot deny the possibility that apoptosis, inflammation and necrosis, which are known to be closely related to cisplatin-induced nephrotoxicity, are not involved in the changed metabolite biomarker candidates. Our results demonstrate that metabolomics is useful for revealing the chronological plasma metabolite changes induced by cisplatin exposure and for identifying reliable biomarkers that can be used to detect cisplatin-induced nephrotoxicity early. The cancer patients receive multiple courses of cisplatin, so it is needed to perform the validation in the human study on the basis of our findings, but the side effects of cisplatin are the critical issue, so our study is important.
Source of funding
This study was supported in part by a Grant-in-Aid for Scientific Research (B) from the Japan Society for the Promotion of Science (JSPS) [M. Y.]; a Grant-in-Aid for Scientific Research (C) from the JSPS [S. N.]; and the AMED-CREST from the Japan Agency for Medical Research and Development (AMED) [S. N., T. A., and M. Y.].
Conflicts of interest
There are no conflicts of interest to declare.
Supplementary Material
Acknowledgments
We are deeply grateful to Makoto Suzuki and Takashi Kobayashi (Division of Gastroenterology, Department of Internal Medicine, Kobe University Graduate School of Medicine) for their technical assistances and valuable comments for our study.
Footnotes
†Electronic supplementary information (ESI) available. See DOI: 10.1039/c7tx00171a
References
- Delord J. P., Puozzo C., Leesne E., Bugat R. Anticancer Res. 2009;29:553–560. [PubMed] [Google Scholar]
- Johnson S. W. and O'Dwyer P. J., Pharmacology of cancer chemotherapy, in Cancer: Principles and practice of oncology, ed. V. T. De Vita Jr., S. Hellman and S. A. Rosenberg, Lippincott Williams & Wilkins, Philadelphia, 7th edn, 2005, pp. 344–358. [Google Scholar]
- Kodama A., Watanabe H., Tanaka R., Kondo M., Chuang V. T., Wu Q., Endo M., Ishima Y., Fukagawa M., Otagiri M., Maruyama T. Biochim. Biophys. Acta. 2014;1840:1152–1162. doi: 10.1016/j.bbagen.2013.12.007. [DOI] [PubMed] [Google Scholar]
- Wang D., Lippard S. J. Nat. Rev. Drug Discovery. 2005;4:307–320. doi: 10.1038/nrd1691. [DOI] [PubMed] [Google Scholar]
- Ozkok A., Edelstein C. L. BioMed Res. Int. 2014;2014:967826. doi: 10.1155/2014/967826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pabla N., Dong Z. Kidney Int. 2008;73:994–1007. doi: 10.1038/sj.ki.5002786. [DOI] [PubMed] [Google Scholar]
- Arany I., Safirstein R. L. Semin. Nephrol. 2003;23:460–464. doi: 10.1016/s0270-9295(03)00089-5. [DOI] [PubMed] [Google Scholar]
- Bajorin D. F., Bosl G. J., Alcock N. W., Niedzwiecki D., Gallina E., Shurgot B. Cancer Res. 1986;46:5969–5972. [PubMed] [Google Scholar]
- Hewitt S. M., Dear J., Star R. A. J. Am. Soc. Nephrol. 2004;15:1677–1689. doi: 10.1097/01.asn.0000129114.92265.32. [DOI] [PubMed] [Google Scholar]
- Amin R. P., Vickers A. E., Sistare F., Thompson K. L., Roman R. J., Lawton M., Kramer J., Hamadeh H. K., Collins J., Grissom S., Bennett L., Tucker C. J., Wild S., Kind C., Oreffo V., Davis 2nd J. W., Curtiss S., Naciff J. M., Cunningham M., Tennant R., Stevens J., Car B., Bertram T. A., Afshari C. A. Environ. Health Perspect. 2004;112:465–479. doi: 10.1289/ehp.6683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devarajan P. Contrib. Nephrol. 2008;160:1–16. doi: 10.1159/000125893. [DOI] [PubMed] [Google Scholar]
- Thukral S. K., Nordone P. J., Hu R., Sullivan L., Galambos E., Fitzpatrick V. D., Healy L., Bass M. B., Cosenza M. E., Afshari C. A. Toxicol. Pathol. 2005;33:343–355. doi: 10.1080/01926230590927230. [DOI] [PubMed] [Google Scholar]
- Yoshida M., Hatano N., Nishiumi S., Irino Y., Izumi Y., Takenawa T., Azuma T. J. Gastroenterol. 2012;47:9–20. doi: 10.1007/s00535-011-0493-8. [DOI] [PubMed] [Google Scholar]
- Nishiumi S., Shinohara M., Ikeda A., Yoshie T., Hatano N., Kakuyama S., Mizuno S., Sanuki T., Kutsumi H., Fukusaki E., Azuma T., Takenawa T., Yoshida M. Metabolomics. 2010;6:518–528. [Google Scholar]
- Ooi M., Nishiumi S., Yoshie T., Shiomi Y., Kohashi M., Fukunaga K., Nakamura S., Matsumoto T., Hatano N., Shinohara M., Irino Y., Takenawa T., Azuma T., Yoshida M. Inflammation Res. 2011;60:831–840. doi: 10.1007/s00011-011-0340-7. [DOI] [PubMed] [Google Scholar]
- Ikeda A., Nishiumi S., Shinohara M., Yoshie T., Hatano N., Okuno T., Bamba T., Fukusaki E., Takenawa T., Azuma T., Yoshida M. Biomed. Chromatogr. 2012;26:548–558. doi: 10.1002/bmc.1671. [DOI] [PubMed] [Google Scholar]
- Shiomi Y., Nishiumi S., Ooi M., Hatano N., Shinohara M., Yoshie T., Kondo Y., Furumatsu K., Shiomi H., Kutsumi H., Azuma T., Yoshida M. Inflamm. Bowel Dis. 2011;17:2261–2274. doi: 10.1002/ibd.21616. [DOI] [PubMed] [Google Scholar]
- Nishiumi S., Kobayashi T., Ikeda A., Yoshie T., Kibi M., Izumi Y., Okuno T., Hayashi N., Kawano S., Takenawa T., Azuma T., Yoshida M. PLoS One. 2012;7:e40459. doi: 10.1371/journal.pone.0040459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T., Nishiumi S., Ikeda A., Yoshie T., Sakai A., Matsubara A., Izumi Y., Tsumura H., Tsuda M., Nishisaki H., Hayashi N., Kawano S., Fujiwara Y., Minami H., Takenawa T., Azuma T., Yoshida M. Cancer Epidemiol., Biomarkers Prev. 2013;22:571–579. doi: 10.1158/1055-9965.EPI-12-1033. [DOI] [PubMed] [Google Scholar]
- Won A. J., Kim S., Kim Y. G., Kim K. B., Choi W. S., Kacew S., Kim K. S., Jung J. H., Lee B. M., Kim S., Kim H. S. Mol. BioSyst. 2016;12:133–144. doi: 10.1039/c5mb00492f. [DOI] [PubMed] [Google Scholar]
- Terashima Y., Nishiumi S., Minami A., Kawano Y., Hoshi N., Azuma T., Yoshida M. Arch. Biochem. Biophys. 2014;555–556:55–65. doi: 10.1016/j.abb.2014.05.013. [DOI] [PubMed] [Google Scholar]
- Sakai A., Suzuki M., Kobayashi T., Nishiumi S., Yamanaka K., Hirata Y., Nakagawa T., Azuma T., Yoshida M. Biomarkers Med. 2016;10:577–586. doi: 10.2217/bmm-2016-0020. [DOI] [PubMed] [Google Scholar]
- Matsubara A., Izumi Y., Nishiumi S., Suzuki M., Azuma T., Fukusaki E., Bamba T., Yoshida M. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2014;969:199–204. doi: 10.1016/j.jchromb.2014.08.013. [DOI] [PubMed] [Google Scholar]
- Cui X., Churchill G. A. Genome Biol. 2003;4:210. doi: 10.1186/gb-2003-4-4-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad M., Sato H. Amino Acids. 2012;42:231–246. doi: 10.1007/s00726-011-0867-5. [DOI] [PubMed] [Google Scholar]
- Kumar P., Maurya P. K. Rejuvenation Res. 2013;16:179–184. doi: 10.1089/rej.2012.1394. [DOI] [PubMed] [Google Scholar]
- Go Y. M., Jones D. P. Free Radical Biol. Med. 2011;50:495–509. doi: 10.1016/j.freeradbiomed.2010.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cresenzi C. L., Lee J. I., Stipanuk M. H. J. Nutr. 2003;133:2697–2702. doi: 10.1093/jn/133.9.2697. [DOI] [PubMed] [Google Scholar]
- Kimura Y., Kimura H. FASEB J. 2004;18:1165–1167. doi: 10.1096/fj.04-1815fje. [DOI] [PubMed] [Google Scholar]
- Kimura Y., Goto Y., Kimura H. Antioxid. Redox Signaling. 2010;12:1–13. doi: 10.1089/ars.2008.2282. [DOI] [PubMed] [Google Scholar]
- Elrod J. W., Calvert J. W., Morrison J., Doeller J. E., Kraus D. W., Tao L., Jiao X., Scalia R., Kiss L., Szabo C., Kimura H., Chow C. W., Lefer D. J. Proc. Natl. Acad. Sci. U. S. A. 2007;104:15560–15565. doi: 10.1073/pnas.0705891104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripatara P., Patel N. S., Collino M., Gallicchio M., Kieswich J., Castiglia S., Benetti E., Stewart K. N., Brown P. A., Yaqoob M. M., Fantozzi R., Thiemermann C. Lab. Invest. 2008;88:1038–1048. doi: 10.1038/labinvest.2008.73. [DOI] [PubMed] [Google Scholar]
- Robinson A. M., Williamson D. H. Physiol. Rev. 1980;60:143–187. doi: 10.1152/physrev.1980.60.1.143. [DOI] [PubMed] [Google Scholar]
- Katayama M., Hiraide A., Sugimoto H., Yoshioka T., Sugimoto T. JPEN, J. Parenter. Enteral Nutr. 1994;18:442–446. doi: 10.1177/0148607194018005442. [DOI] [PubMed] [Google Scholar]
- Suzuki M., Suzuki M., Sato K., Dohi S., Sato T., Matsuura A., Hiraide A. Jpn. J. Pharmacol. 2001;87:143–150. doi: 10.1254/jjp.87.143. [DOI] [PubMed] [Google Scholar]
- Wei T., Tian W., Liu F., Xie G. Biochem. Biophys. Res. Commun. 2014;447:666–671. doi: 10.1016/j.bbrc.2014.04.074. [DOI] [PubMed] [Google Scholar]
- Jacob C., Belleville F. Pathol. Biol. 1992;40:910–919. [PubMed] [Google Scholar]
- Bremer J. Physiol. Rev. 1983;63:1420–1480. doi: 10.1152/physrev.1983.63.4.1420. [DOI] [PubMed] [Google Scholar]
- Portilla D. Semin. Nephrol. 2003;23:432–438. doi: 10.1016/s0270-9295(03)00088-3. [DOI] [PubMed] [Google Scholar]
- Li S., Nagothu K. K., Desai V., Lee T., Branham W., Moland C., Megyesi J. K., Crew M. D., Portilla D. Kidney Int. 2009;76:1049–1062. doi: 10.1038/ki.2009.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haschke M., Vitins T., Lüde S., Todesco L., Novakova K., Herrmann R., Krähenbühl S. Nephrol., Dial., Transplant. 2010;25:426–433. doi: 10.1093/ndt/gfp456. [DOI] [PubMed] [Google Scholar]
- Boudonck K. J., Mitchell M. W., Német L., Keresztes L., Nyska A., Shinar D., Rosenstock M. Toxicol. Pathol. 2009;37:280–292. doi: 10.1177/0192623309332992. [DOI] [PubMed] [Google Scholar]
- Uehara T., Horinouchi A., Morikawa Y., Tonomura Y., Minami K., Ono A., Yamate J., Yamada H., Ohno Y., Urushidani T. J. Appl. Toxicol. 2014;34:1087–1095. doi: 10.1002/jat.2933. [DOI] [PubMed] [Google Scholar]
- Sinha V., Vence L. M., Salahudeen A. K. J. Invest. Med. 2013;61:564–568. doi: 10.2310/JIM.0b013e31828233a8. [DOI] [PubMed] [Google Scholar]
- Kim K. S., Yang H. Y., Song H., Kang Y. R., Kwon J., An J., Son J. Y., Kwack S. J., Kim Y. M., Bae O. N., Ahn M. Y., Lee J., Yoon S., Lee B. M., Kim H. S. J. Toxicol. Environ. Health, Part A. 2017:1–12. doi: 10.1080/15287394.2017.1299655. [DOI] [PubMed] [Google Scholar]
- George B., Wen X., Mercke N., Gomez M., O'Bryant C., Bowles D. W., Hu Y., Hogan S. L., Joy M. S., Aleksunes L. M. Clin. Pharmacol. Ther. 2017;101:510–518. doi: 10.1002/cpt.606. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.