

 The aim of the present study is to assess the toxic effect of dibutyl phthalate (DBP) and diethyl phthalate (DEP) on the freshwater fish Cyprinus carpio.
The aim of the present study is to assess the toxic effect of dibutyl phthalate (DBP) and diethyl phthalate (DEP) on the freshwater fish Cyprinus carpio.
Abstract
The aim of the present study is to assess the toxic effect of dibutyl phthalate (DBP) and diethyl phthalate (DEP) on the freshwater fish Cyprinus carpio. The median lethal concentrations of DBP and DEP for 96 h are found to be 35 and 53 mg L–1, respectively. Fish were exposed to 3.5 mg L–1 (Treatment I) and 1.75 mg L–1 (Treatment II) of DBP and 5.3 mg L–1 (Treatment I) and 2.65 mg L–1 (Treatment II) of DEP for a period of 35 days. The DBP and DEP exposed fish show a concentration based toxic effect on the selected parameters of this study. The hematological parameters, such as hemoglobin (Hb), hematocrit (Hct) and erythrocyte (RBC), were found to decrease in the DBP and DEP treated fish, whereas their leucocyte (WBC) count increased compared to that of the control groups. A biphasic response is noted in the erythrocyte indices, such as mean cellular volume (MCV), mean cellular hemoglobin (MCH) and mean cellular hemoglobin concentration (MCHC), throughout the study period. Exposure to DBP and DEP caused a significant (p < 0.05) decrease in sodium (Na+), potassium (K+), and chloride (Cl–) levels in the gill and brain of the fish throughout the study period when compared to that of their respective controls. The plasma protein level decreased in all the treatments, whereas the plasma glucose level significantly increased in the DBP and DEP exposed fish. Maximum inhibition of Na+/K+-ATPase activity was noticed in the gill and brain of the fish exposed to DBP and DEP. The cholinesterase (ChE) activity in the brain of the fish significantly decreased throughout the study period. A significant (p < 0.05) increase in glutamate oxaloacetate transaminase (GOT) and glutamic pyruvate transaminase (GPT) activity was noted in the fish exposed to both toxicants. The antioxidant enzymatic parameters such as superoxide dismutase (SOD) and catalase (CAT) activities were found to decrease in the gill and liver of the DBP and DEP treated fish, whereas a significant (p < 0.05) increase in lipid peroxidation (LPO) was observed. The above mentioned parameters could be used as potential biomarkers in clinical trials for the assessment of plasticizers. This study provides indispensable information towards future research on the effect of plasticizers on non-target organisms including humans.
Introduction
Daily, a wide range of chemicals are used for the enhancement of mankind, which increases parallel with the population growth. Plasticizers are manmade chemicals that are produced and utilized in high volume industrially and in the field of agriculture.1,2 For example, the concentration of plasticizers is up to 60% in fully flexible PVC products.3,4 Some of the commonly used plasticizers are phthalates, adipates, citrates, camphor and acelates, among which, phthalates are widely used in many industrial processes.5,6 Thus far, 23 different types of PAEs are commercially available, which are used as plasticizers, solvents and emulsifiers. Globally, their demand has gradually increased up to 6 million tonnes per year.7 Phthalates esters (PAEs) are synthetic chemicals which are industrially prepared from phthalic anhydride. Chemically, PAEs are the ortho-isomer of 1,2-benzenedicarboxylic acid with two side chains of either alkyl, benzyl, phenyl, cycloalkyl, or alkoxy groups.7,8 Furthermore, each PAE is categorized as low molecular or high molecular based on the carbon number of their alcohol.9
Although PAEs have wide favorable applications, a major concern is that they are not covalently bond to their products. This enhances the leaching activity of PAEs from products and their content in the environment.10 A measurable amount of PAEs has been detected in the air, water, soil, plastic wrapped items and human body fluids throughout the world.5,11 For example, in the U.S. retail market, 15 PAEs were detected (range from 0–95.2%) from 21 edible vegetable oil samples.5 The PAEs concentration was found to be very high in urban and rural fish eaters than urban and rural vegetarians.12 Cheng et al.10 reported the accumulation of PAEs in freshwater and marine water fish, which had values of <3.14 μg g–1 wet weight (ww) and <7.10 μg g–1 ww, respectively. PAEs have the potential to cause birth defects and reproductive anomalies and have hepatotoxic, teratogenic and carcinogenic effects on organisms.13–16 Recently, a novel report on the uptake of PAEs between the blood and nails of humans was estimated by Bui et al.6 using pharmacokinetic (PK) modelling from a Norwegian cohort. Hence, PAEs are considered as a priority chemical for scientists from a toxicological point of view. PAEs enter the aquatic environment through various point and non-point sources such as direct/indirect discharge, surface run-off, atmospheric deposition, and consumer products.17–19 PAEs were detected in surface marine water20–22 and freshwater sites21 and their concentration was found to be in the range of 0.1 to 100 μg g–1 in river sediments.23,24 These compounds can accumulate in the environment and their bioaccumulation rate is very high (range from 100 to 3000) in different organisms.25 Fish can absorb PAEs from water through gill respiration, ingestion of contaminated food particles and dermal exposure.26,27 As a result, these compounds pose a potential environmental threat to aquatic organisms.18,19 Previous literature indicated that PAEs can cause mutagenic effects in aquatic organisms4,28 and reproductive and developmental effects.29,30 Hence, the contamination of aquatic ecosystems with PAEs is a serious problem throughout the world.27,31
Among the PAEs, dibutyl phthalate (DBP) and diethyl phthalate (DEP) are ortho-short chain PAEs, which are used much more than the other forms of PAEs and are listed as Class 2 priority pollutants by the U.S. Environmental Protection Agency and Level 4 toxic chemical substances in Taiwan.32–34 DBP enters the environment through production, disposal in industrial and municipal landfills, waste incineration, and leaching from consumer products during use or after disposal35 and has been detected in various environmental matricies.36–38 Likewise, DEP, which is one of the low molecular weight phthalate esters, is used as a plasticizer in many products39–41 and enters the aquatic environment from industries and plastic and commercial products.42 These esters, which have been identified in all environmental compartments, also act as an endocrine disrupting chemicals.43 The impact of DBP and DEP on aquatic organisms has been reported by a few authors44–46 and are considered major contaminants in the aquatic environment.4
Even though plastic products are restricted for use in carry bags, food packaging materials and medical applications, DBP and DEP are extensively used in the manufacture of personal care products and incense sticks, as a perfume binder.6,47 In one of the major Indian rivers, the PAEs level detected was up to 1640 ng L–1 in water samples and 1438 ng g–1 dw in sediments.48 Among the PAEs, DBP and DEP were found to be the most predominant in all the samples with a contribution of 11% and 22% and a range between 0–372 ng L–1 and 36–520 ng L–1 in the water samples, respectively.
The presence of PAEs in water may reach the human body through ingestion, inhalation or dermal absorption.49 These low molecular weight PAEs do not accumulate in the body but are hydrolyzed to the corresponding monoesters and absorbed through phase I biotransformation.50–52 These monoester metabolites are oxidized in the body and conjugate with glucuronic acid in phase II biotransformation and are finally excreted through urine.50,53 The monomers could elicit ROS and cause an imbalance in the oxidative stress defence mechanism.54–56 This oxidative imbalance could lead to biochemical, physiological and pathological changes in a cell/organism.
To our knowledge, most of the previous literature illustrates the quantification of PAEs in the aquatic ecosystem. However, toxicological profiles, especially the aquatic toxicity of PAEs are scarce. Hence, we attempt to study the impact of DBP and DEP on the freshwater fish Cyprinus carpio using multiple biomarkers. Haematological, ionoregulation, biochemical, enzymological and antioxidant biomarkers are the routinely used biomarkers in fish which provide early warning signals in bio-monitoring.57–59 The fish Cyprinus carpio is a widespread freshwater fish in Indian rivers and also a cultivable fish singly or in combination with other major Indian carps.
Materials and methods
Chemicals and reagents
All chemicals were purchased from Loba Chemie Pvt. Ltd, Mumbai, India and were of analytical grade (99.99%) and did not contain additives or impurities that would affect the outcome of this study.
Animals and maintenance
Fingerlings of Cyprinus carpio (C. carpio) with an average length of 6.0–7.0 cm and weight of 8.0–9.0 g were acquired from the Tamil Nadu Fisheries Development Corporation, Aliyar, Tamil Nadu, India. Fingerlings cultured from the same brood stock were collected. After arriving at the laboratory, the fish were stocked in a large cement tank (1000 L capacity) for a minimum period of 30 days before the commencement of the experiment. During the acclimation period, the fish were fed ad libitum with rice bran and ground nut oil cake in dough form once in day before the water was replaced. Three quarters of the water was changed daily and the excess feed and fecal matter were removed. During acclimatization, the fish stock was maintained under natural photoperiods and ambient temperature.
All experiments were performed in compliance with relevant laws and guidelines of the Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA), with approval by the Ministry of Environment and Forests, Government of India (CPCSEA/CH/ORG/2006/2064).
Survival-to-mortality ratio at 96 h
Among the bioassay methods, the static bioassay method is the most convenient in laboratory conditions. Hence, the static bioassay method was chosen in the present investigation.60 The 96 h median lethal concentrations of DBP and DEP to the fish C. carpio was calculated via the probit analysis method.61 Separate glass tanks (20 L capacity) were used and different concentrations of DBP (5, 10, 15, 20, 25, 35 and 40 mg L–1) and DEP (5, 15, 25, 35, 45, 55 and 60 mg L–1) were added by removing the appropriate amount of water. 10 healthy fingerlings were added to each tank. A toxicant free control group was also maintained simultaneously. The fish were inspected at least after 24, 48, 72 and 96 hours. Dead fish were removed immediately. At the end of the 96 h exposure period, the survival and mortality of the fish in the toxicant exposed and control groups were recorded. Three replicates were also maintained.
Sublethal toxicity studies and sampling frequency
For sublethal studies, 3.5 mg L–1 (Treatment I) and 1.75 mg L–1 (Treatment II) of DBP and 5.3 mg L–1 (Treatment I) and 2.65 mg L–1 (Treatment II) of DEP were added in each glass aquarium (100 liter capacity). Then 50 fish were introduced to each glass aquarium. The water was changed daily in order to avoid the accumulation of fecal matter and excess feed and renewed with the toxicant. Positive and negative controls were also maintained. Additionally, suitable replicates were maintained. During the experimental period, the fish were fed ad libitum every day. The toxicity study was conducted for a period of 35 days with sampling on the 7, 14, 21, 28 and 35th day.
Preparation of samples and analytical procedures
Blood
Cardiac blood was collected from the DBP, DEP and control groups in plastic disposable syringes fitted with 26 gauge needles which were pre-chilled and coated with heparin (BeparineR heparin sodium, IP 1000 IU mL–1, an anticoagulant). Then the blood was expelled into separate heparinised plastic vials and kept immediately on ice. The whole blood was used for the analysis of Hb, Hct, RBCs and WBCs and the remaining blood samples were centrifuged at 93.9g, at 4 °C for 20 min to separate the plasma, which was used for the estimation of biochemical parameters (glucose and protein) and enzymes (GOT and GPT). A pooled blood sample was used for the determination of all the other parameters.
Organs/tissue
After drawing blood, the fish were washed with double distilled water and blotted dry with absorbent paper. The gills, brains and livers were collected separately for each group. 100 mg of each organ/tissue was homogenized with 1.0 mL of 0.1 M Tris-HCl buffer (pH 7.5) in a Teflon homogenizer and then centrifuged at 1000 rpm at 4 °C for 15 min. The supernatant was used for various analyses such as ionoregulation (Na+, K+ and Cl–), enzymes (GOT, GPT ChE and Na+/K+-ATPase) and antioxidants (SOD, CAT and LPO).
Hematological analysis
Hemoglobin content was estimated using the cyanmethemoglobin method of Drabkin.62 Hematocrit was estimated via the method by Nelson and Morris63 using an RM 12C micro centrifuge and a microhematocrit reader. Erythrocytes and leucocytes were counted using a haemocytometer.64 The erythrocyte indices, such as MCV, MCH and MCHC, were calculated using standard formulas.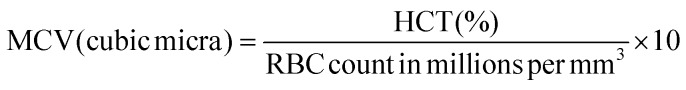
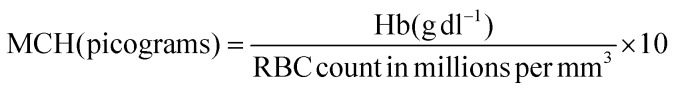
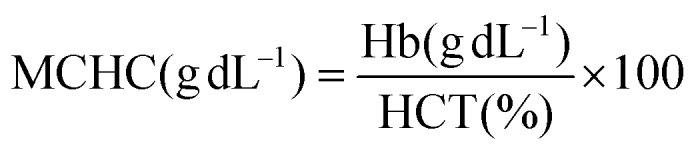
Electrolyte analysis
Sodium was estimated using the method of Maruna65 and Trinder.66
Step 1 Precipitation of sodium and protein: 1.0 mL of precipitating reagent was added to 0.02 mL of sample and 0.02 mL of standard solution was also added. The contents were mixed well, kept at room temperature for 5 min and centrifuged at 2500–3000 rpm for 2 min to obtain a clear supernatant.
Step 2 Colour development: 1.00 mL of acid reagent and 0.1 mL of color reagent were added to 0.02 mL of supernatant and mixed well. The mixture was allowed to stand at room temperature for 5 min and its optical density was measured within 15 min against distilled water using a UV Spectrophotometer at 530 nm and expressed as mmol L–1.
Potassium was estimated using the method of Sunderman and Sunderman67 and Terri and Sesin.68 1.0 mL potassium reagent was added to 0.02 mL of sample. Similarly, blank (0.02 mL of deionized water with 1.0 mL potassium reagent) and standard (0.02 mL of potassium standard solution with 1.0 mL potassium reagent) samples were also prepared. The contents were mixed well, allowed to stand for 5 min at room temperature and the absorbance measured within 15 min against deionized water using a UV Spectrophotometer at 630 nm and the level expressed as mmol L–1.
Chloride was estimated using a modified method of Schales and Schales69 and Schoenfeld and Lewellen.70 1 mL of chloride reagent was added to 0.01 mL. Similarly, standard (0.01 mL of chloride standard solution with 1 mL of chloride reagent) and blank (0.01 mL of distilled water with 1 mL of chloride reagent) samples were also prepared. The contents of the tubes were mixed well, kept for 2 min at room temperature and the optical density measured against distilled water using UV a spectrophotometer at 505 nm within 60 min of preparation which was expressed as mmol L–1.
Biochemical parameters
Plasma glucose was estimated using the O-toluidine method of Cooper and McDaniel.71 5 mL of O-toluidine colour reagent was added to 0.1 mL of sample. Similarly, 0.1 mL of distilled water with 5 mL of O-toluidine colour reagent and 0.1 mL of glucose standard solution with 5 mL of O-toluidine colour reagent were also prepared as blank and standard samples, respectively. The contents in the test tubes were mixed well, kept in a boiling water bath for 10 min, cooled under running tap water for 5 min and their optical density (O.D) measured against the blank at 630 nm within 30 minutes using a UV spectrophotometer and expressed as mg per 100 mL.
Plasma protein was estimated following the method of Lowry et al.72 0.90 mL of distilled water was added to 0.10 mL of sample. A blank was prepared with 1 mL of distilled water. A mixture of 5.0 mL of solution C [50 mL of solution A (2.00 g of sodium carbonate dissolved in 100.00 mL of 0.1 N NaOH) and 1 mL of solution B (500.00 mg of copper sulfate dissolved in 100.00 mL of 1% sodium potassium tartrate solution)] was added to both the test and blank samples, kept for 10 minutes at room temperature, then 0.5 mL of Folin phenol reagent was added and the optical density (O.D) was read after 15 min at 720 nm using a UV spectrophotometer. A standard was also prepared and the protein level was expressed as μg mL–1.
Enzymological analysis
GOT and GPT activity was determined following the method of IFCC.73 1.0 mL of working reagent was incubated at 37 °C for 1 min and 0.1 mL of sample was added. The contents were mixed well and the initial absorbance A0 was read after 1 min. The absorbance reading was repeated after 1, 2 and 3 min. The O.D values were measured against distilled water using a UV spectrophotometer at 340 nm. The mean absorbance change per minute (ΔA/min) was calculated and the unit was expressed as U L–1.
Na+/K+-ATPase activity was estimated according to the method of Shiosaka et al.74 0.3 mL of Tri-HCl buffer (pH7.5), 0.1 mL of 0.02 M ATP, 0.1 mL of 100 mM NaCl and 0.1 mL of KCl were mixed and 0.1 mL of sample was added. Similarly, 0.1 mL of distilled water with above solutions was also prepared. The contents of the tubes were mixed well, incubated in a water bath at 37 °C for 15 min and the reaction was terminated with 2.00 mL of 5% TCA. The tubes were kept at 4 °C for 30 min., centrifuged for 5 min at 500 rpm. Then 1 mL of ammonium molybdate and 0.4 mL of ANSA reagent were added to the supernatant and it was allowed to stand for 10 min at room temperature. The intensity of the blue colour that developed was read at 680 nm against a reagent blank a using UV spectrophotometer. The enzyme activity was expressed in terms of micrograms of inorganic phosphorous formed per gram of tissue.
The cholinesterase activity was measured using the method of Knedel and Bottger75 and Tietz.76 0.5 mL of sample was added to 1.0 mL of working reagent, mixed well and incubated at 37 °C for 30 s. A blank was also prepared with 1.5 mL of distilled water. The changes were measured using a UV spectrophotometer at 405 nm for 90 seconds with an interval of 30 seconds and the activity was expressed as U L–1.
Antioxidants analysis
SOD assays were estimated following the method of Marklund and Marklund.77 0.5 mL of sample was added to 0.25 mL of ice cold ethanol and 0.15 mL of ice cold chloroform in an Eppendorf tube. The contents were centrifuged for 15 min at 13 000 rpm and 0.5 mL of supernatant was separated and mixed with Tris buffer (2 mL) and the total volume made up to 4.5 mL by adding distilled water. Then 0.5 mL of freshly prepared pyrogallol was added. Simultaneously, a blank was also prepared by mixing 2 mL of Tris HCl and 2.5 mL of distilled water. A pyrogallol standard was prepared by adding 2 mL of Tris HCl, 2 mL of distilled water and 0.5 mL of pyrogallol. A brown colour developed in the freshly prepared pyrogallol solution as a result of oxidation. Immediately the O.D of the sample was measured using a UV-spectrophotometer at 470 nm for 3 min at an interval of 1 min each and expressed as IU g–1 protein.
CAT activity was estimated according to the method of Luck.78 0.04 mL of enzyme extract was added to 3.0 mL of H2O2–phosphate buffer and mixed thoroughly. The time required for a decrease in absorbance by 0.05 units was recorded at 240 nm using a UV-spectrophotometer. An enzyme solution containing H2O2–free phosphate buffer served as the control. One enzyme unit was calculated as the amount of enzyme required to decrease the absorbance at 240 nm by 0.05 units. CAT activity was expressed as μmol mg–1 protein.
LPO activity was estimated according to the method of Devasagayam and Tarachand.79 0.5 ml of Tris HCl buffer was added to 0.1 mL of sample and 0.15 mL of 10 mM KH2PO4 and 0.25 mL of distilled water added. The contents were incubated at 37 °C for 20 min with constant shaking. 1 mL of 10% TCA was added to stop the reaction. Then 0.75 mL of TBA was added and the tube was kept in a boiling water bath for 10 min after which a pink colour was observed. The contents were centrifuged at 5000 rpm for 10 min. The O.D of the mixture was measured at 532 nm using a UV-spectrophotometer and the unit was expressed in nmoles of MDA per g protein.
Statistical analysis
Statistical analysis was conducted individually for each sample and the mean value of five individual observations was taken for each parameter. All the values were expressed as means and analyzed by ANOVA, followed by a DMRT test using the SPSS software to determine the significant differences (p < 0.05) between the treatment and controls for each parameter.
Results and discussion
Animal exposure and preliminary observation
A series of behavioral changes were noted in the fish when exposed to both DBP and DEP. At the beginning of toxicant exposure, disruption of schooling behavior was observed, and then the fish stopped swimming and remained static in their position. Later, to avoid toxicant water, the fingerlings showed sign of erratic swimming and jumping activities. Additionally, hyper-excitability and rapid opercular movements were observed as surfacing and gulping of air. Their fins became very tough and stretched due to stretching of body muscles. Throughout their body, mucus secretion was found to be abundant. Later, the fish lost their balance, became lethargic, lost consciousness and settled at the bottom of the tank. Finally, their bellies turned upward, there was no opercular movement and they died. These changes were parallel with the increase in concentration of the toxicants. The observed behavioral changes were found to be more in the DEP treated fish.
It is clear that the load of DBP and DEP in the aquatic environment has increased dramatically in the last few decades. Thus, this study was performed to determine the potential effects of DBP and DEP on aquatic organisms at low concentrations. DBP and DEP are relatively water soluble compared to the other PAEs and result in chronic rather than acute effects in aquatic organisms.80 In general, lipophilic substances are absorbed by organic substances in the aquatic ecosystem and acclimate in sediment. This could be a potential way PAEs enter aquatic organisms.81 In the present study, the behavioral changes observed in both DBP and DEP exposed fish are a reflection of the adverse effects caused by PAEs, in which concentration dependent behavioral changes were also noticed. A similar behavioural response was also reported in Gammarus pulex exposed to DBP and C. carpio exposed to DEP.42,81 The observed behavioural changes such as rapid opercula movement, erratic swimming and loss of balance may be due to possible nervous disorder caused by the toxicants.82
In the present study, the 96 h LC50 was found to be 35 mg L–1 and 53 mg L–1 for the DBP and DEP exposed fish, respectively. Similar results were obtained in the study of Ghorpade et al.83 and Barse et al.42 The observed LC 50 value indicates that the toxicants (DBP and DEP) are moderately toxic to the fish C. carpio. The lethality occurred as a result of an abnormal oxygen supply and ion transport which affected physiological and biochemical processes. DBP and DEP are lipophilic substances which could accumulate in the gill region and prevent the normal influx and efflux of respiratory gases and ions. DBP and DEP are known to be peroxisomal proliferators (mediated by PPARα/β/γ) which cause uncoupled oxidative phosphorylation in the mitochondria.84,85 As uncouplers, they could withhold the process of energy production and ultimately create energy demand.86,87 This could lead to over activation of other energy producing sources and finally end in failure of the process and death of the organism. Furthermore the mortality of the fish may also result from oxidative stress.
Hematology
The responses of the hematological parameter of C. carpio fish exposed to sublethal concentrations of DBP and DEP for 35 days are presented in Table 1. All the hematological values are significant at the p < 0.05 level.
Table 1. Alterations in the hematological parameters of C. carpio exposed to PAEs for 35 days.
| Parameters/exposure period | Positive control | Negative control | DBP |
DEP |
||
| Treatment I (3.5 mg L–1) | Treatment II (1.75 mg L–1) | Treatment I (5.3 mg L–1) | Treatment II (2.65 mg L–1) | |||
| Hb (g dL –1 ) | ||||||
| 7 | 5.07a | 5.04a | 3.90a | 4.34a | 4.02a | 4.31a |
| 14 | 5.28a | 5.22a | 3.86b | 4.04ab | 3.97ab | 4.17ab |
| 21 | 5.16a | 5.06a | 3.77b | 3.95b | 3.9b | 4.11ab |
| 28 | 5.17a | 5.27a | 3.60b | 3.81b | 3.84b | 3.98b |
| 35 | 5.27a | 5.13a | 3.25b | 3.74b | 3.74b | 3.92b |
| Hct (%) | ||||||
| 7 | 15.14a | 14.52a | 11.01b | 12.78ab | 11.82ab | 12.6ab |
| 14 | 15.04ab | 15.42a | 11.3b | 11.18b | 11.68ab | 12.18ab |
| 21 | 15.3a | 14.96ab | 11.02c | 11.64c | 11.28c | 12.00bc |
| 28 | 15.22a | 15.50a | 10.40b | 11.38b | 11.04b | 11.82b |
| 35 | 15.38a | 15.40a | 9.40b | 10.92b | 10.92b | 11.46b |
| RBC (million per cu.mm of blood) | ||||||
| 7 | 2.06a | 2.15a | 1.40b | 1.82a | 1.80a | 2.01a |
| 14 | 2.12a | 2.07a | 1.13c | 1.64b | 1.73ab | 1.91ab |
| 21 | 2.03ab | 2.21a | 0.99d | 1.55c | 1.69bc | 1.82abc |
| 28 | 2.08a | 2.07a | 0.93c | 1.39b | 1.53b | 1.74ab |
| 35 | 2.27a | 2.14a | 0.87c | 1.20bc | 1.50b | 1.46b |
| WBCs (1000 cu.mm –1 of blood) | ||||||
| 7 | 25.45e | 25.30e | 47.44b | 35.39d | 48.99a | 40.68c |
| 14 | 26.10f | 27.08e | 59.05a | 40.51d | 50.30b | 45.02c |
| 21 | 27.80e | 25.82f | 75.17a | 61.90b | 53.84c | 48.59d |
| 28 | 27.05f | 28.18e | 88.36a | 62.68b | 57.82c | 52.64d |
| 35 | 29.31d | 29.64d | 93.36a | 70.98b | 60.75c | 58.73c |
| MCV (cubic micra) | ||||||
| 7 | 73.67a | 67.78ab | 77.38a | 69.38ab | 67.15ab | 62.42b |
| 14 | 70.99b | 75.97b | 98.62a | 67.30b | 66.68b | 63.68b |
| 21 | 76.46b | 68.54b | 109.10a | 74.35b | 66.10b | 66.76b |
| 28 | 73.85bc | 75.65bc | 111.15a | 81.22b | 71.82bc | 67.69c |
| 35 | 68.23c | 72.85c | 106.70a | 90.74b | 72.26c | 79.00c |
| MCH (picograms) | ||||||
| 7 | 24.68ab | 23.47b | 27.51a | 23.59b | 22.85b | 21.33b |
| 14 | 24.97b | 25.69b | 33.62a | 24.49b | 22.72b | 21.81b |
| 21 | 25.76b | 23.19b | 37.27a | 25.23b | 22.93b | 22.84b |
| 28 | 25.01bc | 25.74bc | 38.67a | 27.18b | 25.05bc | 22.89c |
| 35 | 23.35c | 24.23c | 36.81a | 31.09b | 24.75c | 27.02c |
| MCHC (g dL –1 ) | ||||||
| 7 | 33.51b | 34.76ab | 35.63a | 34.05ab | 34.04ab | 34.18ab |
| 14 | 35.20a | 33.84a | 34.10a | 37.50a | 34.12a | 34.23a |
| 21 | 33.70b | 33.84ab | 34.18ab | 33.95ab | 34.79a | 34.26ab |
| 28 | 33.91a | 34.02a | 34.80a | 33.48a | 34.91a | 33.85a |
| 35 | 34.23a | 33.29b | 34.48a | 34.28a | 34.33a | 34.20a |
In the present study the Hb, Hct and RBC values were found to decrease in the DBP and DEP exposed fish in both treatments compared to that in the respective control groups. In the DBP exposed fish a maximum mean value of 3.90 and 4.34 g dL–1 was noted at the end of the 7th day and a minimum value of 3.25 and 3.74 g dL–1 was noted at the end of the 35th day in Treatment I and II, respectively. A similar response pattern was also noticed in the DEP exposed fish in both treatments. There was no significant change in the positive control (acetone) group when compared to the negative control groups (toxicant and acetone free water). A significant (p < 0.05) reduction in Hct percentage was noted both in the DBP and DEP exposed fish in both treatments. In the DBP exposed fish a maximum percentage of 11.01% and 12.78% and a minimum percentage of 9.40% and 10.92% was noticed at the end of the 7th and 35th day in Treatment I and II, respectively. Similarly, in the DEP exposed fish the percentage of the Hct value was found to be 10.92% and 11.46% at the end of the 35th day in Treatment I and II, respectively.
The number of RBCs in the DBP and DEP treated groups (Treatment I and II) were found to decrease until the end of the 35th day of exposure when compare to that of the negative control groups. The decrease in the number of RBCs was found to be maximum at the end of the 7th day in both the treatments of the DBP and DEP exposed fish. As the exposure period extended the significant (p < 0.05) decrease in RBCs number was found to be minimum showing 0.87 and 1.20 million per cu.mm of blood in the DBP exposed fish and 1.50 and 1.46 million per cu.mm of blood in the DEP exposed fish at the end of the 35th day in both treatments. Among the treatments, Treatment II was found to be more toxic and, similarly, DEP was more toxic than DBP. The decreased haemoglobin concentration is an indication of hypochromic microcytic anemia and impaired oxygen delivery to the tissues, which could result in a slow metabolic rate and low energy production.88 Similarly, the decrease in the Hct value in the present study might have resulted from swelling of RBC due to DBP and DEP toxicity and effects. Furthermore, the decrease in Hct value is an indication of heamodilution due to erythrocyte sequestration.89 Swelling of RBC, spleen transfusion and fluid shifts may also lead to a decrease in Hct value.90 The reduction in Hb, RBC and PCV observed in Clarias gariepinus exposed to DEP indicated anemic condition of the fish or heamodilution due to erythrocyte sequestration or gill hemorrhage due to toxicant exposure.82 In the present study, a decrease in RBC count was also noted in both treatments, which indicates that the anaemic condition of the fish is due to DBP and DEP stress.
Compared with the control groups, the WBC count in both the DBP and DEP exposed fish was significantly (p < 0.05) increased for Treatment I and II. The significant increase in WBC count is directly proportional to the exposure period, which shows a maximum value of 93.36 and 70.98 1000 cu.mm–1 of blood in the DBP exposed fish and 60.75 and 58.73 1000 cu.mm–1 of blood in the DEP exposed fish at the end of the 35th day for Treatments I and II, respectively. Between the two treatments, Treatment I of both the DBP and DEP exposed groups was found to be more toxic. Similarly, among the toxicant groups, DEP was found to be more toxic. Alteration in the WBC level is an indication of activation of the immune response against stress (increased) and tissue damage (decreased).91 In the present study, the observed leucocytosis indicates stimulation of the immune system by the fish to protect themselves against DBP and DEP stress. The significant increase in WBC count in Oreochromis mossambicus exposed to DEP might have resulted from the hypersensitivity of leucocytes to DEP stress.92 Furthermore, entry of WBCs from the spleen into the blood circulation may also lead to an increase in WBC count under toxic conditions.93
When compared to the control groups, the MCV values were found to be increased throughout the study period in Treatment I of the DBP exposed fish. However, in Treatment II the MCV value was found to be increased after the 21st day showing a value of 74.35, 81.22 and 90.74 cubic micra when compared to the control groups on the 21st, 28th and 35th day, respectively. In the DEP treated groups, a significant (p < 0.05) decrease in MCV value was noted both in Treatment I and II (except on the 35th day). Upon comparison of the results DEP is found to be more toxic. As shown in Table 1, the MCH values increased in Treatment I and II (except on the 14th day) for the DBP exposed fish when compared to that in the normal control groups. However, in the DEP exposed groups the MCH values were found to decrease (except on the 35th day) both in Treatment I and II when compare to that of the control groups. The significant (p < 0.05) changes in the MCH value was found to be maximum in the DEP exposed groups. The MCHC values of the DBP exposed groups were found to increase in Treatment I throughout the study period (except on the14th day). However, in Treatment II a significant (p < 0.05) increase was noted at the end of the 7th and 35th day when compared to that in the control groups. In Treatment I of the DEP treated groups, the MCHC values increased (except on the 7th day) compared to that in the normal control. In Treatment II the MCHC values were found to increase on days 14 (34.23 g dL–1), 21 (34.26 g dL–1) and 35 (34.20 g dL–1) compared to that in the normal control group. Saleh and Marie94 stated that MCV, MCH and MCHC are blood indices and their alteration illustrates the anemic state of organisms. In our investigation, a biphasic condition of the MCV, MCH and MCHC values were recorded. An increase in MCV value is an indication of swelling of RBCs, whereas a decrease in MCV value indicates impaired oxygen uptake due to toxic stress. Similarly, an increase in immature RBCs in circulation may result in a decrease in MCH value.95,96 In contrast, an increase in MCH value might result from hypochromic microcystic anemia.97 The alteration in MCHC value may be due to congenital spherocytosis, as reported by Sobecka98 and Ramesh et al.58
Electrolytes
The responses of gill and brain Na+, K+ and Cl– levels of the DBP and DEP exposed fish are shown in Fig. 1–3. Both the gill and brain Na+, K+ and Cl– levels were found to be decreased significantly (p < 0.05) in Treatment I and II of the DBP and DEP exposed fish. A maximum reduction was observed at the end of the 35th day in both treatments. No significant changes in the level of Na+ were noted in the positive control when compared to the negative control. Generally, adrenaline-mediated stress response and osmoregulatory failure may decrease the gill electrolyte levels in fish.99,100 A decline in Na+, K+ and Cl– concentration in the brain of fish may be due to the increased urinary excretion of ions due to renal tubular dysfunction or reduced intestinal absorption.101,102 Previous studies indicate that PAEs affect the Na+/K+-pump and Ca2+/Mg2+-pump activities in abalone embryos.103 Inhibition of the Na+/K+ATPase enzymes results in the erratic entry of ions into the cell and as a result cell swelling could occur which finally leads to rupturing of the cell membrane.104 In the present investigation the significant decrease in electrolyte levels in the gill and brain of the fish might have resulted from the inhibition of Na+/K+ATPase activity due to DBP and DEP stress since significant inhibition of Na+/K+ATPase activity was observed. Osmoregulatory failure due to DBP and DEP stress may also lead to a decrease in electrolyte levels.
Fig. 1. Gill and brain sodium (mmol L–1) level of C. carpio exposed to DBP (3.5 mg L–1, Treatment I and 1.75 mg L–1, Treatment II) and DEP (5.3 mg L–1, Treatment I and 2.65 mg L–1, Treatment II) for 35 days. Each value represents the mean ± SD of 5 individuals. The same small letter on the different colored columns is not significantly different (p < 0.05).

Fig. 2. Gill and brain potassium (mmol L–1) level of C. carpio exposed to DBP (3.5 mg L–1, Treatment I and 1.75 mg L–1, Treatment II) and DEP (5.3 mg L–1, Treatment I and 2.65 mg L–1, Treatment II) for 35 days. Each value represents the mean ± SD of 5 individuals. The same small letter on the different colored columns is not significantly different (p < 0.05).

Fig. 3. Gill and brain chloride (mmol L–1) level of C. carpio exposed to DBP (3.5 mg L–1, Treatment I and 1.75 mg L–1, Treatment II) and DEP (5.3 mg L–1, Treatment I and 2.65 mg L–1, Treatment II) for 35 days. Each value represents the mean ± SD of 5 individuals. The same small letter on the different colored columns is not significantly different (p < 0.05).

Glucose and protein level
A duration dependent effect was noted in the level of glucose and protein in the DBP and DEP exposed fish in both treatments (Fig. 4 and 5). A maximum elevation of plasma glucose level was noted at the end of the 35th day in the DBP and DEP exposed fish with a value of 170.23 (Treatment I) and 165.18 (Treatment II) (mg per 100 mL) and 160.97 (Treatment I) and 155.21 (Treatment II) (mg per 100 ml), respectively. Similarly, a minimum elevation was noted at the end of 7th day in the DBP and DEP exposed fish with a value of 149.56 (Treatment I) and 143.95 (Treatment II) (mg per 100 mL) and 141.86 and 138.75 (Treatment II) (mg per 100 mL), respectively. Recently Mathieu-Denoncourt et al.105 reported that PAEs may interfere with the PPARα, PPARβ and PPARγ genes which control fatty acid degradation, fatty acid metabolism, and glucose levels. Oxidative phosphorylation is the biochemical process where a high amount of energy is produced by the transfer of electrons in the mitochondria. DBP and DEP cross the lipid membrane easily and are known to cause uncoupled oxidative phosphorylation in cellular organelles. Furthermore, an elevated blood glucose level is an indication of improper carbohydrate metabolism.106 In the present study, the elevation in the glucose level both in the DBP and DEP exposed fish might have resulted from the stimulation of glucocorticoids by DBP and DEP. In Treatment I and II of the DBP and DEP exposed fish the protein levels was found to be decreased significantly (p < 0.05) throughout the study period when compared to that of the control groups. In contrast to the glucose level, changes in the plasma protein level were found to be more in DEP exposed fish.
Fig. 4. Glucose (mg per 100 mL) level in the plasma of C. carpio exposed to DBP (3.5 mg L–1, Treatment I and 1.75 mg L–1, Treatment II) and DEP (5.3 mg L–1, Treatment I and 2.65 mg L–1, Treatment II) for 35 days. Each value represents the mean ± SD of 5 individuals. The same small letter on the different colored columns is not significantly different (p < 0.05).

Fig. 5. Protein (μg per mL) level in the plasma of C. carpio exposed to DBP (3.5 mg L–1, Treatment I and 1.75 mg L–1, Treatment II) and DEP (5.3 mg L–1, Treatment I and 2.65 mg L–1, Treatment II) for 35 days. Each value represents the mean ± SD of 5 individuals. The same small letter on the different colored columns is not significantly different (p < 0.05).

Fish under stress may also mobilize protein to meet the energy requirements needed to sustain increased physiological activity. In the present study, the higher energy demand during DBP and DEP exposure might have triggered an increase in protein catabolism, a process in which both blood and structural protein are converted into energy, thereby reducing the protein level. Furthermore, histopathological changes due to toxicant accumulation may lead to impaired functioning of organs which results in a reduction in protein level.107 Similar to the present investigation, a significant decrease in total protein was noted in E. fetida exposed to di-(2-ethylhexyl)phthalate (DEHP).108 The significant decrease in protein content may also be due to the induction of heat shock proteins (hsps).109 These proteins play a significant role in protein homeostasis110 and the cellular stress response within the cell111 and also help fish to cope with environmental changes.112,113 Hsp90 is active in supporting various components of the cytoskeleton114 and Hsp70 is known to support the repair and degradation of denatured proteins115 in fish. Expression of high levels of these proteins has been reported in fish exposed to toxicants.116,117
Enzyme activities
GOT
Plasma GOT activity was found to be increased throughout the study period both in Treatment I and II of the DBP and DEP exposed fish when compared to the control groups (Fig. 6). In the DBP treated group (Treatment I), a percentage increase of 50.09%, 52.26%, 56.16%, 59.32% and 62.86% was noted at the end of the 7, 14, 21, 28 and 35th day, respectively. Similarly, in Treatment II a percentage increase of 35.88%, 40.19%, 53.39%, 57.29% and 61.27% was noted at the end of the 7, 14, 21, 28 and 35th day, respectively. In the DEP exposed fish a similar trend was also noticed which showed a maximum percentage increase of 55.94% and 48.51% and minimum percentage increase of 36.38% and 31.65% at the end of the 35 and 7th day in Treatment I and II, respectively. Among the treated groups, the maximum percentage increase was observed in the DBP treated group.
Fig. 6. GOT (U L–1) level in the plasma and liver of C. carpio exposed to DBP (3.5 mg L–1, Treatment I and 1.75 mg L–1, Treatment II) and DEP (5.3 mg L–1, Treatment I and 2.65 mg L–1, Treatment II) for 35 days. Each value represents the mean ± SD of 5 individuals. The same small letter on the different colored columns is not significantly different (p < 0.05).

In the DBP exposed fish liver, GOT activity was significantly increased as the exposure extended, which showed a percentage increase of 96.17%, 99.63%, 105.85%, 118.66% and 125.44% and 88.40%, 99.07%, 102.32%, 113.41% and 114.26% at the end of the 7, 14, 21, 28 and 35th day in Treatment I and II, respectively (Fig. 6). Similarly, in the DEP exposed fish, the GOT activity was found to be increased throughout the exposure period, which showed a minimum percentage increase of 88.75% and 87.42% and maximum percent increase of 112.76% and 105.90% at the end of the 7 and 35th day in Treatment I and II, respectively.
GPT
Throughout the study period, GPT activity was found to be increased at all concentrations for both toxicants (Fig. 7). Moreover, the significant (p < 0.05) increase in GPT activity in the DBP exposed groups was directly proportional to the exposure period, which showed a minimum percentage increase of 26.14% and 23.37% and a maximum percentage increase of 48.30% and 41.47% at the end of the 7 and 35th day in Treatment I and II, respectively. A similar trend was also noticed in the DEP exposed fish which showed a minimum percentage increase of 20.27% and 18.10% and a maximum percentage increase of 36.97% and 32.20% at the end of the 7 and 35th day in Treatment I and II, respectively. Similar to the GPT activity in plasma, liver GPT activity was also found to be increased in the different concentrations of DBP and DEP exposed fish when compared to the control groups (Fig. 7). In the DBP exposed fish the maximum percentage increase of 92.75% was noted at the end of the 35th day in Treatment I. Similarly, in Treatment II, the maximum percent increase of 85.66% was noted at the end of the 35th day. In the DEP exposed fish, a maximum percent increase of 83.47% and 78.11% was observed at the end of the 35th day in Treatment I and II, respectively.
Fig. 7. GPT (U L–1) level in the plasma and liver of C. carpio exposed to DBP (3.5 mg L–1, Treatment I and 1.75 mg L–1, Treatment II) and DEP (5.3 mg L–1, DEP I and 2.65 mg L–1, DEP II) for 35 days. Each value represents the mean ± SD of 5 individuals. The same small letter on the different colored columns is not significantly different (p < 0.05).

PAEs as lipophilic substances easily penetrate the membrane and their affinity with lysosomal activity could alter enzymes. In the present study, the significant increase in GOT and GPT levels in the plasma of fish exposed to DBP and DEP indicates hepatic damage due to DBP and DEP accumulation which may in turn release these enzymes into bloodstream. Similarly, the observed increase in liver GOT and GPT activity indicates that the fish tried to mitigate the stress caused by DBP and DEP by an increased rate of metabolism. Ghorpade et al.83 and Kang et al.41 reported that the significant increase in transaminase activity in fish exposed to DEP indicates that DEP stimulates glutamate transaminase activity. This situation is an indication of high protein turnover and amino acid metabolism in PAEs exposed fish. Furthermore, disturbances in the Krebs cycle caused by DBP and DEP may be another possible reason for the observed increase in GOT and GPT activity.
Na+/K+-ATPase activity
Changes in the gill Na+/K+-ATPase activity in the DBP and DEP treated fish at different concentrations are presented in Fig. 8. In the DBP treated fish, a significant (p < 0.05) decrease in gill Na+/K+-ATPase activity was noted throughout the exposure period in both treatments when compared to the control groups. The significant inhibition of gill Na+/K+-ATPase activity was found to be maximum in Treatment II when compared to Treatment I (except at the end of the 35th day). A similar trend was also noted in the DEP exposed fish. Between the two toxicants, DEP was found to be more toxic when compare to the DEP exposed group. No inhibitions in the activity of the gill Na+/K+-ATPase were observed in the acetone treated groups throughout the study period. The enzyme activity was found to be significantly (p < 0.05) decreased in both the DBP and DEP exposed groups at all concentrations tested (Fig. 8). In the DBP exposed fish, the decrease in enzyme activity was indirectly proportional to the exposure period which showed a minimum percentage decrease of 11.12% and 15.67% at the end of the 35th day in Treatment I and II, respectively. Similarly, in the DEP exposed group a minimum percent decrease of 14.49% and 13.59% was also noted at the end of the 35th day in Treatment I and II, respectively. No significant changes were observed in the brain Na+/K+-ATPase activities between the positive and negative control groups.
Fig. 8. Na+/K+-ATPase activity in the gill and brain of C. carpio exposed to DBP (3.5 mg L–1, Treatment I and 1.75 mg L–1, Treatment II) and DEP (5.3 mg L–1, Treatment I and 2.65 mg L–1, Treatment II) for 35 days. Each value represents the mean ± SD of 5 individuals. The same small letter on the different colored columns is not significantly different (p < 0.05).

Inhibition of Na+/K+-ATPase activity in tissues indicates the inhibition of oxidative phosphorylation.54 Oruc et al.104 reported that the inhibition of the Na+/K+-ATPase in gill may result from toxicant-induced disturbances in the Na+/K+-ATPase pump. In the present study, the significant decrease in gill and brain Na+/K+-ATPase activity might have also resulted from the direct inhibition of the enzyme activity by DBP and DEP. The lowest amount of inhibition was recorded in the brain probably due to the presence of the blood brain barrier in the brain.118 The decline in brain ATPase activity in the fish Channa punctata may be due to LPO induced structural and functional alteration of the brain plasma membrane.119
Cholinesterase activity
A significant (p < 0.05) decrease in brain ChE activity was noted at all concentrations in both toxicant exposed groups (Fig. 9). In the DBP exposed groups, the decrease in brain ChE activity was found to be maximum in Treatment II compared to Treatment I. However, in the DEP exposed groups the decrease in brain ChE activity in both treatments was relatively similar. Between the two toxicants, DBP was found to be more toxic. No significant changes were observed in brain ChE activities between the positive and negative control groups.
Fig. 9. Brain cholinesterase activity of C. carpio exposed to DBP (3.5 mg L–1, Treatment I and 1.75 mg L–1, Treatment II) and DEP (5.3 mg L–1, Treatment I and 2.65 mg L–1, Treatment II) for 35 days. Each value represents the mean ± SD of 5 individuals. The same small letter on the different colored columns is not significantly different (p < 0.05).

Generally, PAEs accumulate in aquatic animals and cause adverse effects. In PAEs treated fish, neuronal genes (gap43, elavl3, gfap, mbp and ngn1) were highly expressed which indicates the neuro-toxicological effect of DBP and DEP.120 In the present study, the significant decrease in brain ChE activity might have resulted from DBP and DEP binding with a large number of functional sulfhydryl groups or the neurotoxic effect of these toxicants. A significant inhibition of the AChE activity in fish exposed to DEP indicates the lipophilic nature of DEP.82
Alterations of gill and liver antioxidant activity
SOD activity
A significant (p < 0.05) decrease in gill SOD activity was observed at all concentrations in both toxicant treated groups when compared to the control groups (Fig. 10). In the DBP treated groups, a percentage decrease of 0.076%, 0.063%, 0.059%, 0.054% and 0.049% was noted at the end of the 7, 14, 21, 28 and 35th day in Treatment I. In Treatment II, a maximum percentage decrease of 0.155% was noted at the end of the 7th day and a minimum percentage decrease of 0.078% was noted at the end of the 35th day. In the DEP treated groups, a maximum decrease (0.081% and 0.203%) and a minimum decrease (0.036% and 0.130%) were noted at the end of the 7 and 35th day, respectively. There was a significant decrease in the liver SOD activity in the fish exposed to DBP and DEP (Treatment I and II) throughout the study period (Fig. 10). In both the DBP and DEP exposed groups the decrease in liver SOD activity was indirectly proportional to the exposure period.
Fig. 10. SOD (IU g–1 protein) activity in the gill and liver of C. carpio exposed to DBP (3.5 mg L–1, Treatment I and 1.75 mg L–1, Treatment II) and DEP (5.3 mg L–1, Treatment I and 2.65 mg L–1, Treatment II) for 35 days. Each value represents the mean ± SD of 5 individuals. The same small letter on the different colored columns is not significantly different (p < 0.05).

Similar to our study, inhibited activities of SOD, CAT and glutathione peroxidase have been reported in the liver of the freshwater goldfish Carassius auratus injected intraperitoneally with 17 different PAEs.121 The alteration in the SOD activity indicates the compensatory response in stressed fish and its inhibitory action could be an indicator of damage in the antioxidant mechanisms.122 Low molecular weight compounds such as DBP and DEP may penetrate through the cell and generate a rapid response which leads to a decrease in SOD activity.123 Furthermore, an excess of H2O2 may reduce SOD activity, i.e., production of oxidants or ROS accumulation.124,125 Saglam et al.122 suggested that decreased SOD activity might be an indicator of damage in the antioxidant mechanisms caused by toxicants. In the present study, the decline in SOD activity also indicates damage in SOD protein due to the overproduction of ROS from DBP and DEP stress.
CAT activity
The catalase activity in the gill of Cyprinus carpio exposed to different concentrations of DBP and DEP are presented in Fig. 11. The gill CAT activities in Treatment I and II of the DBP and DEP exposed fish showed a significant decrease throughout the study period compared to that of the control groups. A maximum percentage decrease of 15.90% and 18.71% was noted at the end of the 7th day in Treatment I and II DBP treated groups. Similarly, in the DEP treated groups a maximum percent decrease of 18.80% and 18.86% was also noted at the end of the 7th day. Between the two toxicant groups, DEP was found to be the more toxic. The liver CAT activities in the DBP and DEP treated groups (Treatment I and Treatment II) were found to decrease significantly throughout the study period in relation to the control groups (Fig. 11). In the DBP exposed groups, a maximum percentage decrease of 21.59% and 24.65% and a minimum percentage decrease of 13.34% and 15.78% were noted at the end of the 7 and 35th day in Treatment I and II, respectively. Similarly, in the DEP treated group a maximum percentage decrease of 23.84% and 26.86% and a minimum percent decrease of 15.66% and 17.64% were noted at the end of the 7 and 35th day in Treatment I and II, respectively. Similar inhibition effects on SOD and CAT were found in Nile tilapia, Zebra fish and rodents exposed to PAEs.120,126,127 DBP and DEP at a given dose may increase ROS production resulting oxidative stress in fish. The inhibition of CAT activity results due to the accumulation or improper neutralization of H2O2 by SOD in the cell.124 Mankidy et al.128 noted the failure of gene expression of SOD and CAT resulted as oxidative stress caused by PAEs.
Fig. 11. CAT (μmol mg–1 protein) activity in the gill and liver of C. carpio exposed to DBP (3.5 mg L–1, Treatment I and 1.75 mg L–1, Treatment II) and DEP (5.3 mg L–1, Treatment I and 2.65 mg L–1, Treatment II) for 35 days. Each value represents the mean ± SD of 5 individuals. The same small letter on the different colored columns is not significantly different (p < 0.05).

LPO activity
During the exposure period LPO activity increased throughout the study period in both treatments (Fig. 12). The values were found to be statistically significant at 5% when compared to that of the control groups. In the DBP treated fish, a maximum percent increase of 21.72% and 20.04% was noted at the end of the 35th day in Treatment I and II, respectively. Similarly, in the DEP treated fish a maximum percent increase of 20.44% and 20.04% was also noted at the end of 35th day in Treatment I and II, respectively. Lipid peroxidation activity in the liver was found to gradually increase as the exposure period extended, which showed a maximum percent increase of 32.18% and 27.55% at the end of the 35th day in Treatment I and II, respectively, of the DBP exposed fish (Fig. 12). Similarly, in the DEP treated fish a maximum percent increase of 29.17% and 24.65% was also noted at the end of the 35th day in Treatment I and II, respectively. The statistical analysis reveals that all the values are significant at the 5% level. Similar to our study, a high production of LPO was noted in zebrafish embryos exposed to PAEs.128,129 Xu et al.120 also noted an elevation of ROS and MDA production in zebrafish embryos upon exposure to DBP and DEP, which reflects oxidative stress. In the present study, the observed elevation in LPO in the gill and liver of the fish exposed to DBP and DEP might have resulted from the oxidative stress caused by these toxicants at the given concentration. Furthermore, the poor response of the CAT enzyme to stressors could also elevate the LPO content in the organisms.130 The above alteration in antioxidant enzyme activities indicates that both DPB and DEP stimulate organ damage through oxidative stress via the generation of ROS, lipid peroxidation and disrupting cell function.131
Fig. 12. LPO (μmol mg–1 protein) activity in the gill and liver of C. carpio exposed to DBP (3.5 mg L–1, Treatment I and 1.75 mg L–1, Treatment II) and DEP (5.3 mg L–1, Treatment I and 2.65 mg L–1, Treatment II) for 35 days. Each value represents the mean ± SD of 5 individuals. The same small letter on the different colored columns is not significantly different (p < 0.05).

Conclusions
The median lethal concentration of this study reveals that DBP is more toxic than DEP. Both DBP and DEP at sublethal concentrations can cause adverse effects on fish. The data on hematological, electrolytes, biochemical and enzymological parameters are useful as baseline data for the control of PAEs contamination in the aquatic ecosystem. Furthermore, the alterations of antioxidant enzymes can be effectively used as biomarkers for the risk assessments of PAEs on aquatic organisms. However, further research is needed on the molecular mechanism and genotoxicity potential of these PAEs on aquatic organisms.
Conflict of interest
There are no conflicts of interest to declare.
Acknowledgments
The first/corresponding author thanks the funding agencies DST-SERB (NPDF-Ref. no.: PDf/2016/001215 dt. 05/07/2016), New Delhi, India and UGC (RFSMS-Ref. no.: NoF.4-7/2008(BSR)/11-49/2008(BSR) Dt. 16/09/2011) for providing fellowship.
References
- Sung H. H., Kao W. Y., Su Y. J. Aquat. Toxicol. 2003;64:25–37. doi: 10.1016/S0166-445X(03)00011-0. [DOI] [PubMed] [Google Scholar]
- Sun J., Wu X., Gan J. Environ. Sci. Technol. 2015;49:8471–8478. doi: 10.1021/acs.est.5b01233. [DOI] [PubMed] [Google Scholar]
- Kim W., Choi I., Jung Y., Lee J., Min S., Yoon C. Environ. Sci. Technol. 2013;47:12459–12468. doi: 10.1021/es4025996. [DOI] [PubMed] [Google Scholar]
- Zhang X., Chen Z. Langmuir. 2014;30:4933–4944. doi: 10.1021/la500476u. [DOI] [PubMed] [Google Scholar]
- Bi X., Pan X., Yuan S., Wang Q. J. Agric. Food Chem. 2013;61:9502–9509. doi: 10.1021/jf402576a. [DOI] [PubMed] [Google Scholar]
- Bui T. T., Alves A., Palm-Cousins A., Voorspoels S., Covaci A., Cousins I. T. Environ. Int. 2017;100:148–155. doi: 10.1016/j.envint.2017.01.007. [DOI] [PubMed] [Google Scholar]
- Wang Y., Zhang G., Wang L. J. Agric. Food Chem. 2015;63:75–84. doi: 10.1021/jf5046359. [DOI] [PubMed] [Google Scholar]
- Fatoki O. S., Ogunfowokan A. O. Environ. Int. 1993;19:619–623. doi: 10.1016/0160-4120(93)90314-8. [DOI] [Google Scholar]
- Chen B., Zhang L. Polym. Test. 2013;32:681–685. doi: 10.1016/j.polymertesting.2013.02.011. [DOI] [Google Scholar]
- Cheng Z., Nie X. P., Wang H. S., Wong M. H. Environ. Int. 2013;57–58:75–80. doi: 10.1016/j.envint.2013.04.005. [DOI] [PubMed] [Google Scholar]
- Zhang H., Zhang Z., Nakanishi T., Wan Y., Hiromori Y., Nagase H., Hu J. Chem. Res. Toxicol. 2015;28:1196–1204. doi: 10.1021/acs.chemrestox.5b00028. [DOI] [PubMed] [Google Scholar]
- Sepperumal U., Saminathan S. Int. J. Fish. Aquat. Stud. 2014;1:243–246. [Google Scholar]
- Kim E. J., Kim J. W., Lee S. K. Environ. Int. 2002;28:359–365. doi: 10.1016/S0160-4120(02)00058-2. [DOI] [PubMed] [Google Scholar]
- Bello U. M., Madekurozwa M. C., Groenewald H. B., Aire T. A., Arukwe A. Comp. Biochem. Physiol., Part C: Toxicol. Pharmacol. 2014;166:24–33. doi: 10.1016/j.cbpc.2014.06.005. [DOI] [PubMed] [Google Scholar]
- Mariana M., Feiteiro J., Verde I., Cairrao E. Environ. Int. 2016;94:758–776. doi: 10.1016/j.envint.2016.07.004. [DOI] [PubMed] [Google Scholar]
- Gao H. T., Xu R., Cao W. X., Qian L. L., Wang M., Lu L., Xu Q., Yu S. Q. Food Chem. Toxicol. 2017;101:94–104. doi: 10.1016/j.fct.2017.01.011. [DOI] [PubMed] [Google Scholar]
- Wang P., Wang S. L., Fan C. Q. Chemosphere. 2008;72:1567–1572. doi: 10.1016/j.chemosphere.2008.04.032. [DOI] [PubMed] [Google Scholar]
- Sun J. Q., Huang J., Zhang A. P., Liu W. P., Cheng W. W. J. Hazard. Mater. 2013;248–249:142–149. doi: 10.1016/j.jhazmat.2012.12.057. [DOI] [PubMed] [Google Scholar]
- Liu X. W., Shi J. H., Bo T., Zhang H., Wu W., Chen Q. C., Zhan X. M. Environ. Pollut. 2014;184:262–270. doi: 10.1016/j.envpol.2013.08.035. [DOI] [PubMed] [Google Scholar]
- Giam C. A., Chan H. S., Neff G. S., Atlas E. L. Science. 1978;199:419–421. [PubMed] [Google Scholar]
- Gledhill W. E., Kaley R. G., Adams W. J., Hicks O., Michael P. R., Saeger V. W., LeBlanc G. A. Environ. Sci.Technol. 1980;14:301–305. doi: 10.1021/es60163a001. [DOI] [PubMed] [Google Scholar]
- Fatoki O. S., Vernon F. Sci. Total Environ. 1990;95:227–232. [Google Scholar]
- Thuren A. Bull. Environ. Contam. Toxicol. 1986;36:33–40. doi: 10.1007/BF01623471. [DOI] [PubMed] [Google Scholar]
- Tan G. H. Bull. Environ. Contam. Toxicol. 1995;54:171–176. doi: 10.1007/BF00197427. [DOI] [PubMed] [Google Scholar]
- Li L., Zhu W., Chen L., Zhang P., Chen Z. J. Photochem. Photobiol., A. 2005;175:172–177. doi: 10.1016/j.jphotochem.2005.01.020. [DOI] [Google Scholar]
- European Commission, 1,2-Benzenedicarboxylic acid, di-C9-11-branched alkyl esters, C10-rich and di-“isodecyl” phthalate (DIDP), European Chemicals Bureau, 2003, vol. 36. [Google Scholar]
- Bhatia H., Kumar A., Ogino Y., Gregg A., Chapman J., McLaughlin M. J., Iguchi T. Aquat. Toxicol. 2014;149:103–115. doi: 10.1016/j.aquatox.2014.01.025. [DOI] [PubMed] [Google Scholar]
- Leitz J., Kuballa T., Rehm J., Lachenmeier D. W. PLoS One. 2009;4:1–7. doi: 10.1371/journal.pone.0008127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabjan E., Hulzebos E., Mennes W., Piersma A. H. Crit. Rev. Toxicol. 2006;36:695–726. doi: 10.1080/10408440600894914. [DOI] [PubMed] [Google Scholar]
- Lyche J. L., Gutleb A. C., Berman A., Eriksen G. S., Murk A. T., Ropstad E., Saunders M., Skaare J. U. J. Toxicol. Environ. Health, Part B. 2009;12:225–249. doi: 10.1080/10937400903094091. [DOI] [PubMed] [Google Scholar]
- Zeng F., Cui K. Y., Xie Z. Y., Wu L. N., Liu M., Li Y. J., Lin Y. J., Zhu F., Ma Z. L., Zeng Z. X. J. Hazard. Mater. 2009;169:719–725. doi: 10.1016/j.jhazmat.2009.04.006. [DOI] [PubMed] [Google Scholar]
- Huang M. Z., Ma Y. W., Wang Y., Wan J. Q., Zhang H. P. Bioresour. Technol. 2010;101:7767–7772. doi: 10.1016/j.biortech.2010.05.028. [DOI] [PubMed] [Google Scholar]
- Chi J., Cai X. Ecol. Eng. 2012;41:70–73. doi: 10.1016/j.ecoleng.2012.01.027. [DOI] [Google Scholar]
- Yang C. F., Wang C. C., Chen C. H. Int. Biodeterior. Biodegrad. 2014;95:55–60. doi: 10.1016/j.ibiod.2014.05.003. [DOI] [Google Scholar]
- Zeng Q., Wei C., Wu Y., Li K., Ding S., Yuan J., Yang X., Chen M. Food Chem. Toxicol. 2013;56:18–27. doi: 10.1016/j.fct.2013.01.045. [DOI] [PubMed] [Google Scholar]
- Xu X. R., Li H. B., Gu J. D. Int. Biodeterior. Biodegrad. 2005;55:9–15. doi: 10.1016/j.ibiod.2004.05.005. [DOI] [Google Scholar]
- Fang C. R., Long Y. Y., Shen D. S. Bioresour. Technol. 2009;100:5664–5670. doi: 10.1016/j.biortech.2009.06.039. [DOI] [PubMed] [Google Scholar]
- Bach C., Dauchy X., Chagnon M. C., Etienne S. Water Res. 2012;46:571–583. doi: 10.1016/j.watres.2011.11.062. [DOI] [PubMed] [Google Scholar]
- WHO (World Health Organization), Diethyl phthalate, 2003. http://www.inchem.org/documents/cicads/cicads/cicad52.htm. [Google Scholar]
- McCarroll N., Reassessment of the one exemption for the requirement of a tolerance for diethyl phthalate (CAS 84-66-2), USEPA, Washington, DC, 2006. [Google Scholar]
- Kang J. C., Jee J. H., Koo J. G., Keum Y. H., Jo S. G., Park K. H. Ecotoxicol. Environ. Saf. 2010;73:1449–1455. doi: 10.1016/j.ecoenv.2010.07.025. [DOI] [PubMed] [Google Scholar]
- Barse A. V., Chakrabarti T., Ghosh T. K., Pal A. K., Jadhao S. B. Pestic. Biochem. Physiol. 2007;88:36–42. doi: 10.1016/j.pestbp.2006.08.009. [DOI] [Google Scholar]
- Prasad B., Suresh S. Int. J. Environ. Sci. Dev. 2012;3:283–288. doi: 10.1016/j.apcbee.2012.03.004. [DOI] [Google Scholar]
- Chen W., Sung H. Aquat. Toxicol. 2005;74:160–171. doi: 10.1016/j.aquatox.2005.05.008. [DOI] [PubMed] [Google Scholar]
- Ortiz Zarragoitia M., Trant J. M., Cajaravillet M. P. Environ. Toxicol. Chem. 2006;25:2394–2404. doi: 10.1897/05-456R.1. [DOI] [PubMed] [Google Scholar]
- Mapuskar K., Pereira C., Vaman Rao C. Pestic. Biochem. Physiol. 2007;87:156–163. [Google Scholar]
- Sonde V., D'Souza A., Tarapore R., Pereira L., Khare M. P., Sinkar P., Krishnan S., Rao C. V. Toxicology. 2000;147:23–31. doi: 10.1016/S0300-483X(00)00164-5. [DOI] [PubMed] [Google Scholar]
- Selvaraj K. K., Sundaramoorthy G., Ravichandran P. K., Girijan G. K., Sampath S., Ramaswamy B. R. Environ. Geochem. Health. 2015;37:83–96. doi: 10.1007/s10653-014-9632-5. [DOI] [PubMed] [Google Scholar]
- Wittassek M., Koch H. M., Angerer J., Brüning T. Mol. Nutr. Food Res. 2011;55(1):7–31. doi: 10.1002/mnfr.201000121. [DOI] [PubMed] [Google Scholar]
- Albro P. W., Corbett J. T., Schroeder J. L., Jordan S., Matthews H. B. Environ. Health Perspect. 1982;45:19–25. doi: 10.1289/ehp.824519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albro P. W., Lavenhar S. R. Drug Metab. Rev. 1989;21:13–34. doi: 10.3109/03602538909029953. [DOI] [PubMed] [Google Scholar]
- Anderson W. A., Castle L., Scotter M. J., Massey R. C., Springall C. Food Addit. Contam. 2001;18(12):1068–1074. doi: 10.1080/02652030110050113. [DOI] [PubMed] [Google Scholar]
- Dirven H. A., van der Broek P. H., Jongeneelen F. J. Int. Arch. Occup. Environ. Health. 1993;64(8):555–560. doi: 10.1007/BF00517700. [DOI] [PubMed] [Google Scholar]
- Siva Prasada Rao K., Ramana Rao K. V. Toxicol. Lett. 1984;20:53–57. doi: 10.1016/0378-4274(84)90182-6. [DOI] [PubMed] [Google Scholar]
- Ferguson K. K., McElrath T. F., Chen Y. H., Mukherjee B., Meeker J. D. Environ. Health Perspect. 2015;123:210–216. doi: 10.1289/ehp.1307996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C. Y., Chen P. C., Hsieh C. J., Chen C. Y., Hu A., Sung F. C., Lee H. L., Su T. C. Sci. Rep. 2017;7:44318. doi: 10.1038/srep44318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poopal R. K., Ramesh M., Dinesh B. J. Trace Elem. Med. Biol. 2013;27(1):70–75. doi: 10.1016/j.jtemb.2012.06.002. [DOI] [PubMed] [Google Scholar]
- Ramesh M., Sankaran M., Veera-Gowtham V., Poopal R. K. Chem.-Biol. Interact. 2014;207:67–73. doi: 10.1016/j.cbi.2013.10.018. [DOI] [PubMed] [Google Scholar]
- Abhijith B. D., Ramesh M., Poopal R. K. J. Basic Appl. Zool. 2016;77:31–40. doi: 10.1016/j.jobaz.2015.11.002. [DOI] [Google Scholar]
- APHA, (American Public Health Association), Standard Methods for the Examination of Water and Wastewater, 1998, p. 1193. [Google Scholar]
- Finney D. J., Statistical method in biological assay, Griffin, London, 1978, p. 508. [Google Scholar]
- Drabkin D. L., J. Biol. Chem., 1946, 164 , 703 –723 , . http://www.jbc.org/content/164/2/703 . [PubMed] [Google Scholar]
- Nelson D. A. and Morris M. W., Basic methodology: hematology and coagulation, 1989, pp. 578–625.
- Rusia V. and Sood S. K., Routine haematological test, 1992.
- Maruna R. F. L. Clin. Chim. Acta. 1958;2:581–585. doi: 10.1016/0009-8981(57)90064-5. [DOI] [PubMed] [Google Scholar]
- Trinder P. Analyst. 1951;76:596–599. doi: 10.1039/AN9517600596. [DOI] [Google Scholar]
- Sunderman F. W., Sunderman F. W. Am. J. Clin. Pathol. 1959;29:95. [Google Scholar]
- Terri A. E., Sesin P. G. Am. J. Clin. Pathol. 1958;29:86–90. doi: 10.1093/ajcp/29.1_ts.86. [DOI] [PubMed] [Google Scholar]
- Schales O., Schales S. S., J. Biol. Chem., 1941, 140 , 879 –884 , . http://www.jbc.org/content/140/3/879 . [Google Scholar]
- Schoenfeld F. G., Lewellen C. J., Clin. Chem., 1964, 10 , 533 , . http://clinchem.aaccjnls.org/content/clinchem/10/6/533.full.pdf . [PubMed] [Google Scholar]
- Cooper G. R., McDaniel V. Stand. Methods Clin. Chem. 1970:159. [Google Scholar]
- Lowry O. H., Rosebrough N. J., Farr A. L., Randall R. J., J. Biol. Chem., 1951, 193 , 265 –275 , . http://www.jbc.org/content/193/1/265 . [PubMed] [Google Scholar]
- International Federation of Clinical Chemistry (IFCC) J. Clin. Chem. Clin. Biochem. 1986;24:481. [Google Scholar]
- Shiosaka T., Okuda H., Fujii S. Biochim. Biophys. Acta. 1971;246:171–183. doi: 10.1016/0005-2787(71)90125-0. [DOI] [PubMed] [Google Scholar]
- Knedel M., Bottger R. Klin. Wschr. 1967;45:325–327. doi: 10.1007/BF01747115. [DOI] [PubMed] [Google Scholar]
- Tietz N. W., Clinical guide to laboratory test, W.B. Saunders Co, 1990, p. 118. [Google Scholar]
- Marklund S., Marklund G. Eur. J. Biochem. 1974;47:469–474. doi: 10.1111/j.1432-1033.1974.tb03714.x. [DOI] [PubMed] [Google Scholar]
- Luck H., Methods in enzymatic analysis, Bergmeyer Academic press, 1974. [Google Scholar]
- Devasagayam T. P., Tarachand U. Biochem. Biophys. Res. Commun. 1987;145:134–138. doi: 10.1016/0006-291X(87)91297-6. [DOI] [PubMed] [Google Scholar]
- Oehlmann J., Schulte-Oehlmann U., Kloas W., Jagnytsch O., Lutz I., Kusk K. O., Wollenberger L., Santos E. M., Paull G. C., Van Look K. J. W., Tyler C. R. Philos. Trans. R. Soc. London, Ser. B. 2009;364:2047–2062. doi: 10.1098/rstb.2008.0242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thuren A., Woin P. Bull. Environ. Contam. Toxicol. 1991;46:159–166. doi: 10.1007/BF01688270. [DOI] [PubMed] [Google Scholar]
- Obiezue R. N., Ikele C. B., Mgbenka B. O., Okoye I. C., Attamah G. N., Uchendu C., Ezeamachi E., Onyia C. Q. Afr. J. Biotechnol. 2014;13:884–896. doi: 10.5897/AJB2013.13210. [DOI] [Google Scholar]
- Ghorpade N., Mehta V., Khare M., Sinkar P., Krishnan S., Rao C. V. Ecotoxicol. Environ. Saf. 2002;53:255–258. doi: 10.1006/eesa.2002.2212. [DOI] [PubMed] [Google Scholar]
- Latruffe N., Vamecq J. Biochimie. 1997;79:81–94. doi: 10.1016/S0300-9084(97)81496-4. [DOI] [PubMed] [Google Scholar]
- Corton J. C., Lapinskas P. J. Toxicol. Sci. 2005;83:4–17. doi: 10.1093/toxsci/kfi011. [DOI] [PubMed] [Google Scholar]
- Kessler R. J., Tyson C. A., Green D. E. Proc. Natl. Acad. Sci. U. S. A. 1976;73(9):3141–3145. doi: 10.1073/pnas.73.9.3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terada H. Environ. Health Perspect. 1990;87:213–218. doi: 10.1289/ehp.9087213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad F., Ali S. S., Shakoori A. R. Folia Biol. 1995;43:151–159. [Google Scholar]
- O'Connor D., Fromm P. Bull. Environ. Contam. Toxicol. 1975;14:406–411. doi: 10.1007/BF01721843. [DOI] [PubMed] [Google Scholar]
- Wells R. M. G., Weber R. E. J. Fish Biol. London. 1991;28:53–66. doi: 10.1111/j.1095-8649.1991.tb03090.x. [DOI] [Google Scholar]
- Witeska M. Electron. J. Ichthyol. 2005;1:35. [Google Scholar]
- Sepperumal U., Saminathan S. Eur. J. Zool. Res. 2013;2:55–59. [Google Scholar]
- Barcellos L. J. G., Kreutz L. C., Souza C., Rodrigues L. B., Fioreze I., Quevedo R. M., Cericato L., Soso A. B., Fagundes M., Conrad J., de Almeida Lacerda L., Terra S. Aquaculture. 2004;237:229–236. doi: 10.1016/j.aquaculture.2004.03.026. [DOI] [Google Scholar]
- Saleh Y. S., Marie M. A. Mar. Pollut. Bull. 2016;106:308–322. doi: 10.1016/j.marpolbul.2016.03.030. [DOI] [PubMed] [Google Scholar]
- Vutukuru S. S. Int. J. Environ. Res. Public Health. 2005;2:456–462. doi: 10.3390/ijerph2005030010. [DOI] [PubMed] [Google Scholar]
- Kavitha C., Malarvizhi A., Senthil Kumaran S., Ramesh M. Food Chem. Toxicol. 2010;48:2848–2854. doi: 10.1016/j.fct.2010.07.017. [DOI] [PubMed] [Google Scholar]
- Jastrzebska B. E., Protasowicki M. Acta Ichthyol. Piscatoria. 2005;35:29–38. [Google Scholar]
- Sobecka E. Acta Ichthyol. Piscatoria. 2001;31:127–143. [Google Scholar]
- Sathya V., Ramesh M., Poopal R. K., Dinesh B. Fish Shellfish Immunol. 2012;32:862–868. doi: 10.1016/j.fsi.2012.02.014. [DOI] [PubMed] [Google Scholar]
- Erhunmwunse N. O., Ainerua M. O. IOSR J. Environ. Sci., Toxicol. Food Technol. 2014;8:27–29. doi: 10.9790/2402-08152729. [DOI] [Google Scholar]
- Larsson A., Bengtsson B. E., Haux C. Aquat. Toxicol. 1981;1:19–35. doi: 10.1016/0166-445X(81)90004-7. [DOI] [Google Scholar]
- Kara Y., Wilkie M. P. Aquat. Toxicol. 2015;161:176–188. doi: 10.1016/j.aquatox.2015.01.032. [DOI] [PubMed] [Google Scholar]
- Zhou J., Cai Z. H., Xing K. Z. Environ. Pollut. 2011;159:1114–1122. doi: 10.1016/j.envpol.2011.02.016. [DOI] [PubMed] [Google Scholar]
- Oruc E. O., Uner N., Tamer L. Bull. Environ. Contam. Toxicol. 2002;69:271–277. doi: 10.1007/s00128-002-0057-y. [DOI] [PubMed] [Google Scholar]
- Mathieu-Denoncourt J., Wallace S. J., de Solla S. R., Langlois V. S. Gen. Comp. Endocrinol. 2015;219:74–88. doi: 10.1016/j.ygcen.2014.11.003. [DOI] [PubMed] [Google Scholar]
- Firat O., Kargin F. Arch. Environ. Contam. Toxicol. 2010;58:151–157. doi: 10.1007/s00244-009-9344-5. [DOI] [PubMed] [Google Scholar]
- Blahova J., Modra H., Sevcikova M., Marsalek P., Zelnickova L., Skoric M., Svobodova Z. BioMed Res. Int. 2014:1–8. doi: 10.1155/2014/980948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma T., Zhou W., Chen L., Wu L., Christie P., Zhang H., Luo Y. PLoS One. 2017;12:e0173957. doi: 10.1371/journal.pone.0173957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theodorakis N. G., Morimoto R. I. Mol. Cell. Biol. 1987;7(12):4357–4368. doi: 10.1128/mcb.7.12.4357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heikkila J. J. Comp. Biochem. Physiol., Part A: Mol. Integr. Physiol. 2017;203:179–192. doi: 10.1016/j.cbpa.2016.09.011. [DOI] [PubMed] [Google Scholar]
- Feder M. E., Hofmann G. E. Annu. Rev. Physiol. 1999;61:243–282. doi: 10.1146/annurev.physiol.61.1.243. [DOI] [PubMed] [Google Scholar]
- Basu N., Todgham A. E., Ackerman P. A., Bibeau M. R., Nakano K., Schulte P. M., Iwama G. K. Gene. 2002;295(2):173–183. doi: 10.1016/S0378-1119(02)00687-X. [DOI] [PubMed] [Google Scholar]
- Kayhan F. E., Duman B. S. Turk. J. Fish. Aquat. Sci. 2010;10:287–293. doi: 10.4194/trjfas.2010.0218. [DOI] [Google Scholar]
- Hahn G. M., Shiu E. C., Goldstein L., Li G. C. Cancer Res. 1985;45:4138–4143. [PubMed] [Google Scholar]
- Kiang J. G., Tsokos G. C. Pharmacol. Ther. 1998;80(2):183–201. doi: 10.1016/S0163-7258(98)00028-X. [DOI] [PubMed] [Google Scholar]
- Girilal M., Krishnakumar V., Poornima P., Mohammed Fayaz A., Kalaichelvan P. T. Chemosphere. 2015;139:461–468. doi: 10.1016/j.chemosphere.2015.08.005. [DOI] [PubMed] [Google Scholar]
- Zhao P., Guo Y., Zhang W., Chai H., Xing H., Xing M. Chemosphere. 2017;166:238–245. doi: 10.1016/j.chemosphere.2016.09.060. [DOI] [PubMed] [Google Scholar]
- Afsar S., Govind B., Jaiswal D. P., J. Exp. Sci., 2012, 3 , 01 –03 , . ISSN: 2218-1768 . [Google Scholar]
- Tabassum H., Afjal M. A., Khan J., Raisuddin S., Parvez S. Ecol. Indic. 2015;58:11–417. doi: 10.1016/j.ecolind.2015.06.008. [DOI] [Google Scholar]
- Xu H., Shao X., Zhang Z., Zou Y., Wu X., Yang L. Ecotoxicol. Environ. Saf. 2013;93:39–44. doi: 10.1016/j.ecoenv.2013.03.038. [DOI] [PubMed] [Google Scholar]
- Zheng Q., Feng M., Dai Y. Environ. Toxicol. Pharmacol. 2013;36:741–749. doi: 10.1016/j.etap.2013.07.008. [DOI] [PubMed] [Google Scholar]
- Saglam D., Atli G., Dogan Z., Baysoy E., Gurler C., Eroglu A., Canli M. Turk. J. Fish. Aquat. Sci. 2014;14:43–52. doi: 10.4194/1303-2712-v14_1_06. [DOI] [Google Scholar]
- Qu R., Feng M., Sun P., Wang Z. Environ. Toxicol. 2014;10:1125–1134. doi: 10.1002/tox.21985. [DOI] [PubMed] [Google Scholar]
- Vutukuru S. S., Chintada S., Madhavi R. K., Venkateswara Rao J., Anjaneyulu Y. Fish Physiol. Biochem. 2006;32:221–229. doi: 10.1007/s10695-006-9004-x. [DOI] [Google Scholar]
- Li Y., Yang J., Li S., Zhang J., Zheng J., Hou W., Zhao H., Guo Y., Liu X., Dou K., Situ Z., Yao L. J. Biol. Chem. 2011;286:32289–32299. doi: 10.1074/jbc.M111.247825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erkekoglu P., Giray B., Rachidi W., Hininger-Favier I., Roussel A. M., Favier A., Hincal F. Environ. Toxicol. 2011;29:98–107. doi: 10.1002/tox.20776. [DOI] [PubMed] [Google Scholar]
- Abu Zeid E. H., Khalil A. S. A. Am. J. Anim. Vet. Sci. 2014;9:269–276. doi: 10.3844/ajavsp.2014.269.276. [DOI] [Google Scholar]
- Mankidy R., Wisemana S., Maa H., Giesy J. P. Toxicol. Lett. 2013;217:50–58. doi: 10.1016/j.toxlet.2012.11.025. [DOI] [PubMed] [Google Scholar]
- Zhao X., Gao Y., Qi M. Ecotoxicology. 2014;23:626–632. doi: 10.1007/s10646-014-1194-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Menezes C. C., Loro V. L., da Fonseca M. B., Cattaneo R., Pretto A., dos Santos Miron D., Santi A. Pestic. Biochem. Physiol. 2011;100:145–150. doi: 10.1016/j.pestbp.2011.03.002. [DOI] [Google Scholar]
- Asghari M. H., Saeidnia S., Abdollahi M. Int. J. Pharmacol. 2015;11:95–105. [Google Scholar]