

 Organic solvent-induced neurodegeneration is a severe public health problem which has no effective prevention measures yet.
Organic solvent-induced neurodegeneration is a severe public health problem which has no effective prevention measures yet.
Abstract
Organic solvent-induced neurodegeneration is a severe public health problem which has no effective prevention measures yet. Cystamine stands as a promising neuroprotective agent against many degenerative diseases. In the present study, we investigated the possible protective effects of cystamine against 2,5-hexanedione (2,5-HD) induced peripheral neuropathy. Chronic exposure to 2,5-HD (300 mg kg–1, 6 times per week for 6 weeks) resulted in obvious peripheral nerve damage shown as the elevation of gait scores and the increase of latency in an accelerating rota-rod test. Cystamine (30 mg kg–1 and 60 mg kg–1) co-treatment obviously ameliorated 2,5-HD-induced impairments of the peripheral nervous system. To decipher the underlying mechanisms, we investigated the effects of cystamine on the regulation of brain-derived neurotrophic factor (BDNF) and heat shock protein-70 (Hsp70) expression and the PI3K/Akt signaling pathway. The results revealed that cystamine up-regulated the protein levels of BDNF and Hsp70, accompanied by the activation of the PI3K/Akt pathway in the spinal cord, which might account for the protection of cystamine against 2,5-HD-induced neuropathy.
Introduction
n-Hexane is an important aliphatic compound widely used in industry to make glue, paints, varnishes, printing inks and shoe manufacturing. Occupational and experimental exposure to n-hexane may induce serious impairment of the nervous system. Historically, this type of nerve damage has been classified as a central–peripheral distal axonopathy and has been presumed to be responsible for the associated neurological defects.1,2 Metabolism studies have suggested that the neurotoxicity of n-hexane is mediated by the ultimate toxic metabolite, 2,5-hexanedione (2,5-HD).3 The morphological hallmark of 2,5-HD-induced neuropathy has been traditionally considered as swollen axons containing aggregations of neurofilaments (NFs). Pathologically, NFs accumulate in the distal axon and form massive axonal swellings leading to retraction of myelin from the Ranvier nodes.4,5 Although many hypotheses including the inhibition of energy metabolism, NF cross linking and impaired axonal transport, have been proposed; however, the molecular mechanism for 2,5-HD-induced axonopathy remains to be elucidated.6,7 As there are few prevention and control measures to be proposed in occupational n-hexane intoxication, there is great interest in developing drugs that can protect against n-hexane-induced axonal degeneration.
Cystamine (Cys) is a linear aliphatic diamine composed of a disulfide bridge. It has been demonstrated that cystamine and its metabolite cysteamine exhibited protective effects in several models of neurodegenerative diseases including Huntington's disease, Parkinson's disease and amyotrophic lateral sclerosis (ALS).8 The exact mechanism by which cystamine exerts its neuroprotective effects is controversial. It can inhibit caspase-3 and transglutaminase activity, act as an antioxidant, stimulate brain-derived neurotrophic factor (BDNF) secretion, and upregulate heat shock proteins (HSPs).9,10 These potentially involved mechanisms are also related to the pathway of toxicant-induced neuropathy. Therefore, cystamine might serve as a novel therapeutic agent for n-hexane-induced neuropathy.
In this study, we showed direct evidence that cystamine treatment ameliorated the impaired behavior and reduced the axonal degeneration in rats exposed to 2,5-hexanedione, which might be related to the increased expression of BDNF and the activation of the PI3K/Akt signaling pathway.
Materials and methods
Chemicals and reagents
2,5-Hexanedione, cystamine dihydrochloride and a primary antibody of BDNF were purchased from Sigma-Aldrich, Inc. (St Louis, MO, USA). Antibodies against total Akt and p-Akt were obtained from ImmunoWay Biotechnology Company (Plano, TX, USA). Antibodies against PI3K and p-PI3K were provided by Cell Signaling Technology Inc. (Beverly, MA, USA). The BCA™ protein assay kit was obtained from Pierce Biotechnology (Rockford, IL, USA). All other chemicals were of the highest grade commercially available.
Animal treatment
All animal experimental procedures and protocols of the study were approved by the Ethics Committee for Animal Experiments of Shandong University Institute of Preventive Medicine (permit number: 20111231) in accord with the NIH Guide for Care and Use of Laboratory Animals. Adult male Wistar rats, weighing 220–240 g, were provided by the Experimental Animal Center of Shandong University. Drinking water and commercial animal feed were available ad libitum. The animal room was maintained at approximately 22 °C and 50% relative humidity with a 12 h light/dark cycle.
After the acclimatization period, the animals were randomly divided into 5 groups (n = 10), that is, the control group, 2,5-HD group, cystamine group (60 mg per kg bw), and 2 Cys/2,5-HD groups (30 and 60 mg per kg bw). The rats in the 2,5-HD group and Cys/2,5-HD groups were treated with 300 mg per kg bw 2,5-HD orally. The animals in other two groups received equal volumes of the vehicle. Cys was administered to the rats in the Cys group and Cys/2,5-HD groups by intraperitoneal injection, while animals in the other two groups received equal volumes of sterile saline. All groups of animals were treated for continuous 6 weeks (6 times per week).
Behavior assessment
2,5-HD-induced neurological deficiency was detected and quantified using gait scores and rota-rod every week. To measure gait abnormalities, rats were placed in a clear plexiglass box (90 × 90 cm) and were observed for 3 min, and a gait score was assigned from 1 to 4 where 1 = a normal, unaffected gait; 2 = a slightly abnormal gait (hindlimbs show uncoordinated placement, exaggerated or overcompensated movements, or are splayed slightly, walks on tiptoes); 3 = moderately abnormal gait (obvious movement abnormalities characterized by markedly splaying hindlimbs, ataxia, swaying, rocking, lurching, stumbling); 4 = severely abnormal gait (flat foot walk, hindlegs flat on the surface, crawling, or unable to support weight). A trained, blinded observer who was not involved in animal care or 2,5-HD administration performed the behavioral evaluation.
The accelerating rota-rod was performed according to the method described by Monville et al.11 Firstly, rats were habituated with a training session on the rota-rod (ZS-ROM, Beijing Zhongshidichuang Technology and Development Co., Ltd, Beijing, China) set at a constant speed of 8 rpm until they achieved a criterion of remaining on the rotating spindle for 60 seconds. During the tests, rats were placed on a rod that accelerated smoothly from 4 to 40 rpm over a period of 200 seconds. The length of time that each animal stayed on the rod was recorded as the latency to fall, registered automatically by a trip switch. Animals receive three trials per week; the average of the three trials is presented weekly.
Tissue preparation, electrophoresis and immunoblotting
The animals were sacrificed by decapitation after 6 weeks of 2,5-HD treatment. The spinal cords were dissected from animals of each group on ice, snap-frozen in liquid nitrogen and then stored at –80 °C until analyzed. The samples were homogenized in ice-cold RIPA lysis buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, 1 mM Na3VO4, 5 mM NaF, 50 mM Tris-HCl) supplemented with 1 mM PMSF and 1% cocktail protein protease inhibitors. After a 20 min centrifugation at 12 000g, the supernatant was collected and the protein concentration was determined using the BCA™ protein assay kit. Equal aliquots of samples were mixed with loading buffer, and then heated at 100 °C for 10 min. The pre-treated protein samples (about 20–50 μg) were separated by 10% or 15% SDS/polyacrylamide gel electrophoresis, transferred to a polyvinylidene fluoride (PVDF) membrane (Millipore, USA). Blots were blocked with 5% skim milk in TBST for 1 h at room temperature. Then the membrane was incubated with the primary antibody diluted in 0.5% BSA overnight at 4 °C. After washing off the unbound primary antibodies with TBST, the membranes were incubated with appropriate horseradish peroxidase (HRP)-conjugated antibodies for 2 h at room temperature. The membranes were washed and then detected by using an enhanced chemiluminescence kit. The immunoreactive bands of proteins were scanned with an Agfa Duoscan T1200 scanner, and the relative optical densities (IDO) of the bands were quantified using the Kodak Imaging Program and Image-Pro Plus software (Eastman Kodak Company, New Haven, CT, USA).
Statistical analysis
The data of the gait score and accelerating rota-rod were presented as mean ± SEM and analyzed by two-way repeated-measures analysis of variance followed by Dunnett's multiple comparison test to determine the significance of the results. For all other data, the data were expressed as mean ± SD and one-way ANOVA coupled with Dunnett's post hoc analysis was adopted. The level of statistical significance was set at P < 0.05.
Results
Effects of cystamine on 2,5-HD-induced neurobehavioral changes
Gait changes
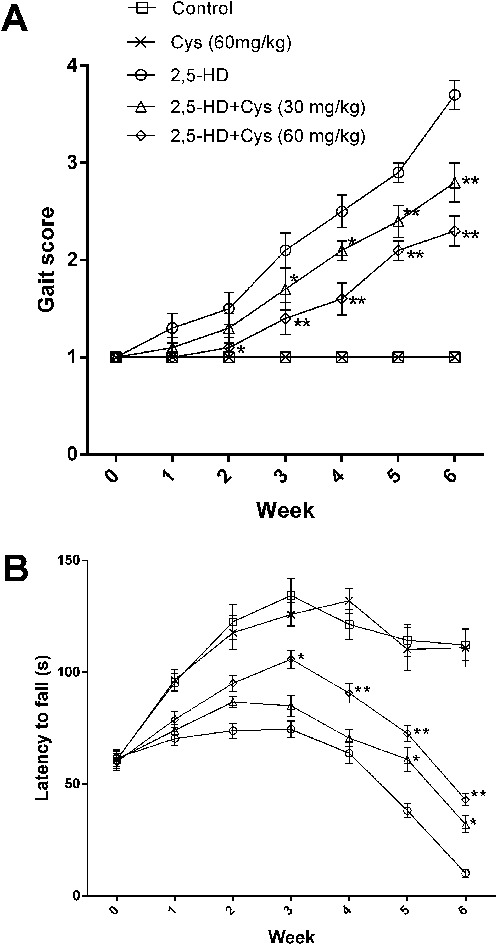
Exposure to 2,5-HD resulted in significant characteristic neurotoxic symptoms including ataxia, tip-toe walking and hindlimb weakness. As shown in Fig. 1A, all rats in the control group showed normal gait (gait score: 1.00 ± 0.00), while the rats in the 2,5-HD group showed obvious motor deficits (gait score: 3.70 ± 0.48) at the end of the 6 weeks. Interestingly, cystamine co-treatment profoundly suppressed 2,5-HD-induced motor deficits. Compared with the rats in the 2,5-HD group, the cystamine co-treated animals were able to walk normally although they moved slowly than the rats in the control group. The gait scores of the rats in 30 and 60 mg kg–1 co-treatment groups were increased to 2.80 ± 0.63 and 2.3 ± 0.48, respectively, with significant differences compared to that of the 2,5-HD group (P < 0.01).
Fig. 1. Effects of cystamine on the neurobehavioral changes of rats treated with 2,5-HD. (a) shows gait changes. (b) shows the performance of the accelerating rota-rod. *p < 0.05, **p < 0.01, compared with the 2,5-HD group.

Accelerating rota-rod
The performance of rats on the rota-rod is shown in Fig. 1B. The performance of the rats in the control and cystamine groups rose steadily over the whole test period. The latency to fall of the 2,5-HD intoxicated rats was significantly decreased compared to control rats since the 1st week (P < 0.01). Compared with the rats in the 2,5-HD group, the duration of stay was significantly prolonged since the 5th week in the low dose cystamine group and the 2nd week in the high dose cystamine group, respectively. In the last week of test, the rats in the 2,5-HD group could not stay on the rod; however, the rats in the cystamine group were able to stay on the rod for 32.0 ± 11.8 s and 43.0 ± 8.5 s in the lower cystamine group and higher cystamine group, respectively.
Cystamine co-treatment increased the protein levels of BDNF
The effect of cystamine on growth factor expression is certainly among its most explicit and significant mechanisms of action.8 Therefore, we investigated the protein levels of BDNF. As shown in Fig. 2, compared with that of the control rats, the mature-BDNF protein level in the spinal cord of 2,5-HD treated rats was decreased by 37.6% (P < 0.01). However, cystamine co-treatment significantly increased the protein levels of the precursor and the mature form of BDNF.
Fig. 2. Cystamine co-treatment significantly increased the protein levels of the precursor and mature BDNF. Total protein samples were prepared using RIPA buffer, and the protein levels of the precursor and mature BDNF were detected by western blotting. (a) Representative western blot bands; (b) quantitative data analyses. Data are presented as mean ± SD from at least 3 independent experiments, and expressed as the percentage of the control. **p < 0.01, compared with the control group; ##p < 0.01, compared with the 2,5-HD group.

Cystamine co-treatment led to the activation of the PI3K/Akt signaling pathway
To investigate whether the protective effect of cystamine was related to the PI3K/Akt pathway, the protein levels of p-PI3K, PI3K, p-Akt and Akt in the spinal cord were detected. As shown in Fig. 3, compared with those of the control group rats, the ratio of p-PI3K/t-PI3K in the rats of the 2,5-HD group was decreased by 34.7% (P < 0.01), while the ratio of p-Akt/t-Akt was decreased by 32.2% (P < 0.01). Compared with those of the 2,5-HD group rats, the ratio of p-PI3K/t-PI3K and p-Akt/t-Akt in the cystamine and co-treated group was significantly increased.
Fig. 3. Cystamine co-treatment led to the increased phosphorylation and activation of PI3K and Akt. Total protein samples were prepared using RIPA buffer, and the protein levels of p-PI3K, PI3K, p-Akt and Akt were detected by western blotting. (a) Representative western blot bands; (b) quantitative data analyses. Data are presented as mean ± SD from at least 3 independent experiments, and expressed as the percentage of the control. *p < 0.05, compared with the control group; ##p < 0.01, compared with the 2,5-HD group.

Cystamine co-treatment increased the Hsp70 protein levels in the spinal cord
Hsp-70 is one of the downstream substrates of the PI3K/Akt pathway, and has been demonstrated to be involved in the neuroprotective effect of cystamine.12 Therefore, we detected the protein levels of Hsp70. As shown in Fig. 4, the protein level of Hsp70 was not significantly affected by 2,5-HD compared with the control group. Cystamine treated alone significantly increased Hsp70 expression. Compared with the 2,5-HD group, the protein levels of Hsp70 in the cystamine co-treated group were increased by 34.1% and 54.7% (P < 0.01), respectively.
Fig. 4. Cystamine co-treatment significantly increased the protein levels of Hsp70. Total protein samples were prepared by using RIPA buffer, and the protein levels of Hsp70 were detected by western blotting. (a) Representative western blot bands; (b) quantitative data analyses. Data are presented as mean ± SD from at least 3 independent experiments, and expressed as the percentage of the control. #p < 0.05, ##p < 0.01, compared with the 2,5-HD group.

Discussion
In this study, we evaluated the protective effects of cystamine, a potential neuroprotective agent, on 2,5-HD induced peripheral neuropathy. The rats were treated with or without cystamine and exposed to 2,5-HD for 6 weeks. The results showed that cystamine significantly attenuated 2,5-HD induced neuropathy which was illustrated by the decrease of gait scores and long latency to fall during the rota-rod test.
To explore the underlying mechanisms for the protective effects of cystamine, we investigated the changes of BDNF and Hsp70, the explicit and significant targets of cystamine action, and the PI3K/AKT signal pathway. A decrease in the neurotrophic factor levels is generally observed in patients suffering from neurodegenerative diseases.13,14 A previous study had demonstrated that the endogenous neurotrophic factor levels could be decreased after 2,5-HD treatment.15 The present study also found that the protein levels of mature BDNF were significantly decreased in the spinal cord of 2,5-HD exposed rats, while cystamine co-treatment significantly reversed the decline of the precursor and the mature form of the BDNF protein levels. Of the neurotrophins, BDNF appears to be the most potent in promoting the survival of motor neurons.16 It was reported that BDNF could prevent neuronal death, enhance neuronal activity, and promote axon growth.17,18 Therefore, high levels of BDNF induced by cystamine might improve the resistance of the nervous system to 2,5-HD toxicity and promote the regeneration of axons.
The phosphatidylinositol 3 kinase (PI3K)/Akt pathway is known to play important roles in numerous cellular functions such as cell growth, proliferation, differentiation, motility, survival, and intracellular trafficking.19 And this pathway is particularly important for mediating neuronal survival under a wide variety of circumstances and has been found to be sufficient and, in some cases, necessary for the trophic-factor-induced cell survival of several neuronal cell types.20 The present study revealed that chronic 2,5-HD exposure could lead to a decrease of the protein level of p-PI3K and p-Akt, which was consistent with a previous report.21 It has been reported that cystamine could activate the PI3K/Akt pathway.22 The current study also found that cystamine co-treatment significantly increased the p-PI3K/PI3K and p-Akt/Akt ratio. As an in vitro study had demonstrated that cystamine treatment could activate the PI3K/Akt pathway through the BDNF/TrkB (BDNF cell surface receptor) signaling,23 it may be possible that cystamine-induced elevation of the BDNF levels led to the activation of the TrkB receptor, and then activated the intracellular PI3K/Akt signaling pathways to exert its neuroprotective effects.
It is well documented that heat-shock proteins (HSPs) are involved in protecting the nervous system from a variety of insults such as stroke, epilepsy, and cerebralischemia.24 The overexpression of HSPs could reduce nervous system damage and promote restoration of the injury. HSPs can exert chaperone functions which can prevent abnormal protein folding or aggregation. This might decline the amount of NF accumulation induced by 2,5-HD and delay the progression of the neuropathy. This is supposed by the demonstration that Hsp70 plays a role in preventing the formation of NF aggregation induced by acrylamide.25 Thus, agents which could promote the endogenous protein levels of HSPs might protect neuropathy induced by 2,5-HD. In this study, the protein levels of Hsp70 were slightly increased in the rats of the 2,5-HD group, which might be a compensatory response. Interestingly, cystamine treatment led to a significant increase of the Hsp70 protein level, which was consistent with previous studies.10,26 These results suggested that neuronal heat shock responses may underlie the cystamine neuroprotective action against 2,5-HD neuropathy as well.
To date, it is realized that a diversity of mechanisms are involved in exogenous induced neurodegenerative diseases. The use of multi-effect agents would represent the most efficacious strategy to treat neurodegenerative diseases. The use of cystamine capable of increasing endogenous BDNF and activating PI3K/Akt signaling, along with potential actions such as inhibiting oxidative stress, reducing apoptosis and transglutaminase inhibition, would benefit neurodegeneration recovery and prevention. Several lines of evidence have emerged recently to support the beneficial properties of cystamine and cysteamine in the context of neurodegenerative diseases. However, the current study firstly proved that cystamine can also prevent exogenous compound-induced peripheral neuropathy.
In summary, this study provided convincing evidence that cystamine could attenuate behavioral deficiency induced by 2,5-HD. The results indicated that the neuroprotective activity of cystamine might be related to the increase in the BDNF and Hsp70 expression, and the activation of the PI3K/Akt signaling pathway.
Conflict of interest
The authors declare that they have no conflict of interest.
Acknowledgments
This research was supported by grants from the National Natural Science Foundation of China (81373044, to K. X.).
References
- Spencer P. S., Schaumburg H. H. J. Neuropathol. Exp. Neurol. 1977;36:300–320. doi: 10.1097/00005072-197703000-00006. [DOI] [PubMed] [Google Scholar]
- LoPachin R. M., DeCaprio A. P. Toxicol. Appl. Pharmacol. 2004;199:20–34. doi: 10.1016/j.taap.2004.03.008. [DOI] [PubMed] [Google Scholar]
- Spencer P. S., Bischoff M. C., Schaumburg H. H. Toxicol. Appl. Pharmacol. 1978;44:17–28. doi: 10.1016/0041-008x(78)90280-6. [DOI] [PubMed] [Google Scholar]
- Lehning E. J., Jortner B. S., Fox J. H. Toxicol. Appl. Pharmacol. 2000;165:127–140. doi: 10.1006/taap.2000.8937. [DOI] [PubMed] [Google Scholar]
- Graham D. G., Anthony D. C., Boekelheide K. Neurobehav. Toxicol. Teratol. 1982;4:629–634. [PubMed] [Google Scholar]
- Spencer P. S., Sabri M. I., Schaumburg H. H. Ann. Neurol. 1979;5:501–507. doi: 10.1002/ana.410050602. [DOI] [PubMed] [Google Scholar]
- Lopachin R. M., Decaprio A. P. Toxicol. Sci. 2005;86:214–225. doi: 10.1093/toxsci/kfi197. [DOI] [PubMed] [Google Scholar]
- Gibrat C., Cicchetti F. Prog. Neuropsychopharmacol. Biol. Psychiatry. 2011;35:380–389. doi: 10.1016/j.pnpbp.2010.11.023. [DOI] [PubMed] [Google Scholar]
- Hsu T. C., Chen Y. C., Lai W. X. Eur. J. Pharmacol. 2008;591:307–314. doi: 10.1016/j.ejphar.2008.06.078. [DOI] [PubMed] [Google Scholar]
- Borrell-Pages M., Canals J. M., Cordelieres F. P. J. Clin. Invest. 2006;116:1410–1424. doi: 10.1172/JCI27607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monville C., Torres E. M., Dunnett S. B. J. Neurosci. Methods. 2006;158:219–223. doi: 10.1016/j.jneumeth.2006.06.001. [DOI] [PubMed] [Google Scholar]
- Chatterjee M., Andrulis M., Stuhmer T. Haematologica. 2013;98:1132–1141. doi: 10.3324/haematol.2012.066175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuccato C., Cattaneo E. Nat. Rev. Neurol. 2009;5:311–322. doi: 10.1038/nrneurol.2009.54. [DOI] [PubMed] [Google Scholar]
- Allen S. J., Watson J. J., Shoemark D. K. Pharmacol. Ther. 2013;138:155–175. doi: 10.1016/j.pharmthera.2013.01.004. [DOI] [PubMed] [Google Scholar]
- Qiang L., Qing-jun L., Hua-wei D. J. Hyg. Res. 2005;3:006. [Google Scholar]
- Apfel S. C. Brain Pathol. 1999;9:393–413. doi: 10.1111/j.1750-3639.1999.tb00234.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lykissas M. G., Batistatou A. K., Charalabopoulos K. A. Curr. Neurovasc. Res. 2007;4:143–151. doi: 10.2174/156720207780637216. [DOI] [PubMed] [Google Scholar]
- Henderson C. E., Camu W., Mettling C. Nature. 1993;363:266–270. doi: 10.1038/363266a0. [DOI] [PubMed] [Google Scholar]
- Zeng T., Zhang C. L., Song F. Y. Toxicology. 2012;296:56–66. doi: 10.1016/j.tox.2012.03.005. [DOI] [PubMed] [Google Scholar]
- Brunet A., Datta S. R., Greenberg M. E. Curr. Opin. Neurobiol. 2001;11:297–305. doi: 10.1016/s0959-4388(00)00211-7. [DOI] [PubMed] [Google Scholar]
- Wang Q., Sun G., Gao C. Sci. Rep. 2016;6:34715. doi: 10.1038/srep34715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillai A., Veeranan-Karmegam R., Dhandapani K. M. J. Neurochem. 2008;107:941–951. doi: 10.1111/j.1471-4159.2008.05665.x. [DOI] [PubMed] [Google Scholar]
- Yao R. Q., Qi D. S., Yu H. L. Neurochem. Res. 2012;37:2777–2786. doi: 10.1007/s11064-012-0871-5. [DOI] [PubMed] [Google Scholar]
- Wyttenbach A. and Arrigo A. P., in Heat shock proteins in neural cells, Springer, 2009, pp. 81–99. [Google Scholar]
- Gold B. G., Voda J., Yu X. Exp. Neurol. 2004;187:160–170. doi: 10.1016/j.expneurol.2004.01.005. [DOI] [PubMed] [Google Scholar]
- Dong Z., Wolfer D. P., Lipp H.-P. Mol. Ther. 2005;11:80–88. doi: 10.1016/j.ymthe.2004.09.007. [DOI] [PubMed] [Google Scholar]