

SUMMARY
Altered glycolysis is a hallmark of diseases including diabetes and cancer. Despite intensive study of the contributions of individual glycolytic enzymes, systems-level analyses of flux control through glycolysis remain limited. Here, we overexpress in two mammalian cell lines the individual enzymes catalyzing each of the 12 steps linking extracellular glucose to excreted lactate, and find substantial flux control at four steps: glucose import, hexokinase, phosphofructokinase, and lactate export (and not at any steps of lower glycolysis). The four flux-controlling steps are specifically up-regulated by the Ras oncogene: optogenetic Ras activation rapidly induces the transcription of isozymes catalyzing these four steps and enhances glycolysis. At least one isozyme catalyzing each of these four steps is consistently elevated in human tumors. Thus, in the studied contexts, flux control in glycolysis is concentrated in four key enzymatic steps. Up-regulation of these steps in tumors likely underlies the Warburg effect.
ETOC BLURB
Here, we perform a systematic analysis of glycolytic flux control in mammalian cells. We identify four key flux-controlling steps: Glucose import and phosphorylation, fructose- 1,6-bisphosphate production and lactate export. In contrast, enzyme steps in lower glycolysis do not control pathway flux. Activation of glycolysis in cancer and immune cells is associated with enhanced expression of enzymes catalyzing these four key fluxcontrolling steps.
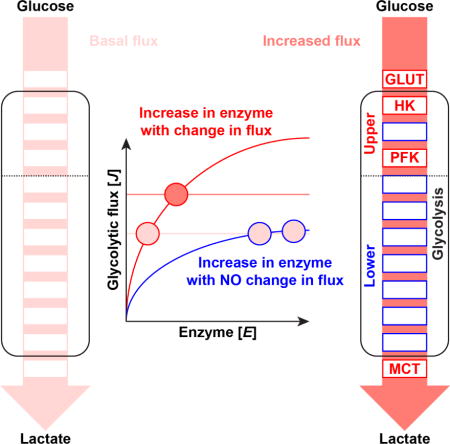
INTRODUCTION
Glycolysis provides cellular energy and metabolic precursors for biomass production. Seminal studies performed over the past hundred years have elucidated the mechanism and regulation of the ten enzymatic steps of glycolysis, which together catalyze the breakdown of glucose into two molecules of pyruvate. These ten enzymatic steps together with the two transport events at the plasma membrane (i.e., the uptake of glucose via glucose transporters and the excretion of lactate via monocarboxylate transporters) constitute the 12 potential steps for controlling glycolytic flux.
Control of glycolytic rate plays an important role in mammalian physiology, contributing to circulating glucose homeostasis and providing ATP and/or biomass building blocks in contexts such as cell proliferation, immune activation and angiogenesis (Buck et al., 2017; De Bock et al., 2013; Everts et al., 2014; Yu et al., 2017). Dysregulated glucose metabolism is a hallmark of diseases including diabetes and cancer.
Cancer cells extensively ferment glucose even in the presence of adequate oxygen (Warburg, 1956). While initially attributed by Warburg to defective mitochondria, it is now clear that most cancer cells have functional mitochondria that account for much of their ATP production (DeBerardinis and Chandel, 2016; Fan et al., 2014; Vander Heiden and DeBerardinis, 2017; Zong et al., 2016; Zu and Guppy, 2004). Accordingly, we use the term Warburg effect to refer to rapid aerobic glycolysis in cancer cells, irrespective of their utilization of oxidative phosphorylation. It has been argued that the Warburg effect promotes tumor growth by satisfying cancer cells’ high demand for both energy and central carbon metabolites for biosynthesis (Liberti and Locasale, 2016). The Warburg effect can be triggered both by oncogenic mutations (e.g., in Ras, PI3K/Akt, c-Myc) and by environmental cues (e.g., growth factors) (Gaglio et al., 2011; Hay, 2016; Vander Heiden et al., 2009; Hu et al., 2016; Lunt and Vander Heiden, 2011; Shim et al., 1997; Yu et al., 2017).
Consistent with their high utilization of glycolysis, cancers and other proliferating cells exhibit increased expression of many glycolytic enzymes (Vander Heiden et al., 2009). High expression of the glucose transporters GLUT1 and GLUT3 is associated with augmented glucose uptake and oncogenic growth (Birsoy et al., 2014; Onodera et al., 2014; Yun et al., 2009). Elevated activities of hexokinase and phosphofructokinase favor tumor initiation, immune cell activation, and angiogenesis (De Bock et al., 2013; Everts et al., 2014; Patra et al., 2013; Ros and Schulze, 2013; Webb et al., 2015; Yi et al., 2012; Ying et al., 2012; Yu et al., 2017). Aldolase A (ALDOA) has been shown to boost glycolysis upon PI3K/Akt signaling (Hu et al., 2016). When upper glycolysis is activated in cancer cells, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) has been reported to become a rate-limiting pathway step (Shestov et al., 2014). The importance of the final enzyme involved in pyruvate production, pyruvate kinase, to glycolytic flux control remains controversial. Earlier studies advocated for the PKM2 isoform as a key driver of the Warburg effect, but recent evidence suggests that the situation is more complex (Bluemlein et al., 2011; Christofk et al., 2008; Dayton et al., 2016). Finally, lactate dehydrogenase A (LDHA) has been implicated in c-Myc mediated transformation (Shim et al., 1997). Thus, nearly every enzyme linking glucose to lactate has been associated in some study with enhancing glycolytic flux.
Despite this extensive literature, a unified view of glycolytic flux control is lacking. In particular, the key glycolytic enzymes whose up-regulation is required for flux enhancement remains unclear. This in part likely reflects variation in flux control across cell lines and environmental conditions. It might also, however, reflect a failure to comprehensively experimentally probe the pathway. Specifically, to obtain a thorough understanding of flux control in any given biochemical pathway, it is valuable to systematically perturb each component to assess its contribution to overall pathway control. But previous experiments have largely focused on the role of individual glycolytic enzymes in isolation, and efforts to interrogate the entire pathway have primarily been theoretical and computational (Dai et al., 2016; Shestov et al., 2014).
Here we examine, in two mouse cell lines, the degree of flux control residing in each step of glycolysis by systematically overexpressing enzymes that catalyze every glycolytic step, and by subsequently analyzing pathway flux. We find that across all enzymes tested, only four steps show significant flux control: In both cell lines, flux is controlled by the two committed steps of upper glycolysis (i.e., hexokinase and phosphofructokinase, with the complication that excessive hexokinase leads to energy stress). In one cell line but not the other, glucose import and lactate export also control flux. In both cell lines, the enzymes of lower glycolysis did not control glycolytic flux. We further show that acute optogenetic stimulation of Ras is sufficient to enhance glycolytic flux and that Ras activates the transcription of genes encoding for the same four steps revealed in our overexpression experiments. Finally, analysis of expression data from solid tumors and paired normal tissue samples reveals a transcriptional signature in which gene isoforms from each of these four steps are strongly and consistently up-regulated across tumor types. While other enzymes may play additional regulatory roles in certain contexts, our findings show that glycolytic flux is controlled at a small number of pathway steps whose up-regulation underlies the Warburg effect.
RESULTS
Systematic enzyme overexpression can reveal glycolytic flux control
To systematically identify flux-controlling steps in glycolysis, we transiently overexpressed at least one human isozyme catalyzing every pathway step from import of extracellular glucose to secretion of lactate. Isozymes were chosen based on their high expression in glycolytic cells and human cancers (Hay, 2016). We tested multiple isozymes of glucose transporters, phosphofructokinases, and pyruvate kinases. In addition, we included fructose-1,6-bisphosphatase (FBP1), pyruvate dehydrogenase kinase 1 (PDK1), and the four isozymes of the phosphofructokinase 2 (PFK2) family, which phosphorylate fructose-6-phosphate to make fructose-2,6-bisphosphate, a key activator of the glycolytic enzyme phosphofructokinase 1 (PFK1). PFK2 isozymes, which are known as PFKFB1-4, are bifunctional and also contain phosphatase domains that can consume fructose-2,6-bisphosphate (Ros and Schulze, 2013). In total, we tested a set of 24 glycolysis-related genes. Green fluorescent protein (GFP) was used as a negative control.
Our initial experiments were performed in immortalized baby mouse kidney cells (iBMK), which were selected for their metabolic characterization (Fan et al., 2014) and high transfection efficiency. To minimize possible metabolic alterations induced by adaptation, glycolytic flux was measured within the first 30 h after transfection (Figure 1A & Figure S1A, B). Experiments were performed in both 25 mM glucose (common tissue culture concentration) and 5 mM glucose (typical circulating concentration in human).
Figure 1. Glycolytic flux is controlled by glucose import, fructose-1,6-bisphopshate production, and lactate export.
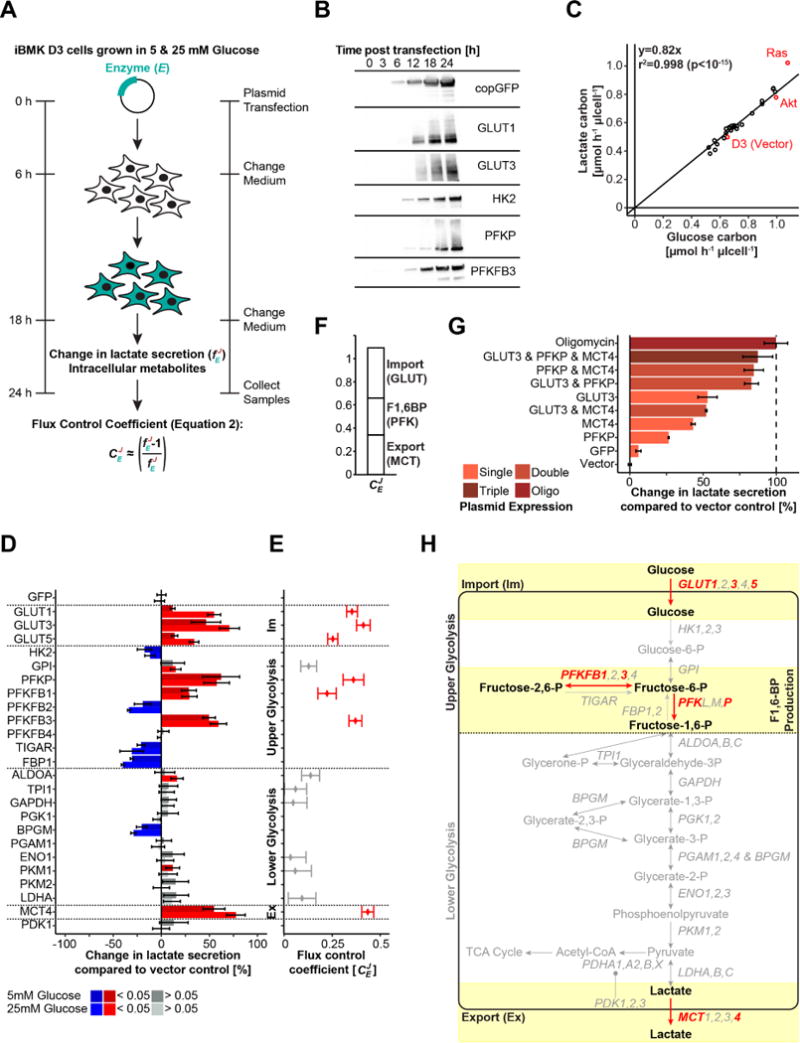
(A) Experimental setup used to determine glycolytic flux control. Enzymes were individually overexpressed in iBMK cells and change in lactate secretion (glycolytic flux) was assessed 24 h post transfection. (B) Western blot showing that the expression dynamics of different glycolytic enzymes is similar (see Figure S1 for additional enzymes). (C) Single glycolytic enzyme expression did not change the ratio of produced lactate to consumed glucose. Means of two biological replicates per condition are plotted. Parental, Akt, and Ras stably-expressing iBMK cells are colored in red. (D) Change in lactate secretion compared to vector control upon individual overexpression of glycolytic enzymes (in DMEM supplemented with 10% dFBS and 5 mM or 10 mM glucose as indicated) and (E) associated flux control coefficients according to the equation in A (for enzymes increasing glycolytic flux). Blue and red indicate statistically significant increases and decreases, respectively (q<0.05). Means and 95% confidence intervals (n≥6 biological replicates from at least two independent experiments) are plotted. (F) The sum of averaged flux control coefficients for glucose import (GLUT), FBP production (PFK), and lactate secretion (MCT) agrees with the summation theorem of metabolic control analysis. (G) Change in glycolytic flux upon combined overexpression of glucose transport (GLUT3), FBP production (PFKP), and lactate export (MCT4) in comparison to the glycolytic capacity measured upon oligomycin treatment. Data are means and 95% confidence intervals (n=3 biological replicates from one independent experiment; pTriple vs. Oligo>0.05; pTriple vs. GLUT3<0.01; pTriple vs. PFKP<0.01; pTriple vs. MCT4<0.02). (H) Cartoon highlighting in light yellow the experimentally determined glycolytic flux controlling steps. See also Figure S1.
Each of the tested constructs was well-expressed, with substantial accumulation of the recombinant protein by 18 h and corresponding five-fold or greater enhancement of the enzyme’s catalytic activity as measured in lysates (Figure 1B & Figure S1C, D, E). Overexpression of some individual glycolytic enzymes altered both glucose consumption and lactate secretion, but none significantly changed the ratio of glucose uptake to lactate secretion: cells excreted about 82% of incoming glucose carbon as lactate (Figure 1C & Figure S1F, G). Even greater conversion of glucose to lactate was observed in cells expressing oncogenic mutant Ras (H-RasV12G; Figure 1C), consistent with previous reports that oncogenic Ras inhibits pyruvate oxidation and enhances glycolytic flux (Fan et al., 2014; Gaglio et al., 2011).
Based on the nearly constant ratio of glucose uptake to lactate secretion across the cell lines, use of either metric leads to the same conclusions about glycolytic flux. Yet, measurements of lactate secretion are more precise (Figure S1F), as the fractional drop in extracellular glucose over 6 h is small and thus hard to measure. Accordingly, in all subsequent analyses, we use lactate secretion as a proxy for glycolytic flux. The uncertainty in the glucose uptake measurements was too large for us to determine whether the absolute flux of glucose carbon not being excreted as lactate tracks with glycolytic flux.
The extent to which a particular pathway step impacts glycolytic flux can be expressed as a flux control coefficient : the fractional change in glycolytic flux (J) induced by a given fractional change in enzyme catalytic activity (E) (Fell, 1998; Heinrich and Rapoport, 1974; Kacser and Burns, 1973),
| (Equation 1) |
Experimentally, measurement of based on Equation 1 is difficult. It requires quantitating small changes in both fluxes and enzyme activities. Fortunately, can be approximated by the fold-change in flux that occurs upon massively increasing the activity of a given enzymatic step (Small and Kacser, 1993a) (See Star Methods),
| (Equation 2) |
Note that dramatically increasing the activity of a given step does not correspondingly increase pathway flux, unless that is the sole rate-limiting pathway step, in which case . For steps that are not rate controlling, the pathway may not respond, and .
Flux is controlled by glucose import, phosphofructokinase, and lactate export
Of the 24 individually overexpressed genes, only seven significantly increased glycolytic flux in iBMK cells cultured in both high and low glucose media (Figure 1D, E): GLUT1, GLUT3, and GLUT5 (glucose transporters); MCT4 (lactate transporter); PFKP (phosphofructokinase; PFK1); and PFKFB1 and PFKFB3 (phosphofructo-2-kinase/fructose-2,6-biphosphatases; PFK2). The flux control coefficient for PFKP was comparable to previous literature values determined in other mammalian cells (Small and Kacser, 1993a; Torres et al., 1986). Our results are in agreement with literature pointing to the importance of glucose uptake and FBP production in controlling glycolytic flux (Birsoy et al., 2014; Onodera et al., 2014; Ros and Schulze, 2013; Webb et al., 2015; Yi et al., 2012; Ying et al., 2012; Yun et al., 2009). In contrast, no enzymes in lower glycolysis significantly impacted flux, including enzymes suggested in prior literature as inducers of the Warburg effect: ALDOA (aldolase), GAPDH, PGK (phosphoglycerate kinase), PKM1 and PKM2 (pyruvate kinases), and LDHA (lactate dehydrogenase). Nevertheless, we did observe significant flux control in the lactate export step, as overexpression of MCT4 led to a substantial increase in glycolytic flux (Figure 1C,D,E).
Certain genes, such as PKM2 and LDHA, have been classically associated with the Warburg effect but did not show any impact on glycolytic metabolism here. To rule out that these genes promote Warburg metabolism by suppressing oxidative phosphorylation or impairing glucose entry into the TCA cycle, we also examined oxygen consumption rate and contribution of glucose carbon to TCA intermediates. Although there was a trend towards higher oxygen consumption with GLUT3 over-expression, oxygen consumption rate did not vary significantly in response to overexpression of GLUT3, PFKP, GAPDH, PKM2, LDHA, or MCT4 (Figure S1H).
Glucose contribution to TCA was slightly but significantly decreased by PFKP overexpression but not affected by any other of these genes (Figure S1I). Thus, the GLUT, PFK, and MCT steps promote glycolytic flux, while most other steps neither drive glycolytic flux nor modulate glucose oxidation.
The summation theorem of metabolic control analysis states that the sum of the flux control coefficients in a linear pathway must equal 1 (Kacser and Burns, 1973). This theorem refers to the sum of flux control across the pathway steps, not across isozymes. In our case, we observe flux control of about 30% for glucose uptake, 30% for FBP production (either via expression of phosphofructokinase or its activators PFKFB1 or 3), and 40% for lactate export, together summing to approximately 100% (Figure 1F). Thus, our data are roughly consistent with the summation theorem.
We then tested the potential to further activate glycolytic flux in iBMK cells grown in 25 mM glucose by coexpressing GLUT3, PFKP, and MCT4 (Figure 1G). The ATP synthase inhibitor oligomycin was used as a positive control (Huijing and Slater, 1961; Lardy et al., 1958). Combined overexpression of GLUT3, PFKP, and MCT4 triggered a two-fold increase in glycolytic flux, which is comparable to oligomycin and significantly greater than any individual gene on its own (Figure 1G; pTriple vs. Oligo>0.05; pTriple vs. GLUT3<0.01; pTriple vs. PFKP<0.01; pTriple vs. MCT4<0.02). Similar increases in glycolytic flux were obtained with coexpression of either transporter with PFKP, but not the two transporters together. The roles for glucose import, phosphofructokinase, and lactate export in controlling iBMK cell glycolysis are highlighted in Figure 1H.
PFK2 isoforms exhibit divergent effects on glycolytic flux
Overexpression of five of the 24 tested genes significantly decreased glycolytic flux in both glucose conditions (Figure 1D). The reduction in glycolytic flux observed upon overexpression of fructose-1,6-bisphosphate phosphatase (FBP1) is consistent with its role in gluconeogenesis and inhibition of tumor progression (Li et al., 2014; Tejwani, 1983). We also captured the well-documented anti-glycolytic function of the TP53-induced glycolysis and apoptosis regulator (TIGAR) (Bensaad et al., 2006). In contrast, we were surprised to find that hexokinase 2 (HK2) overexpression decreased flux, an observation that results from energy stress based on its excessive overexpression, as described below.
Another anti-glycolytic gene was the phosphofructokinase 2 isozyme PFKFB2. This contrasted with PFKFB1 and 3, which as expected increased glycolysis, and PFKFB4 which had no discernable effect (Figure 1D). While PFK2 isoforms are generally considered pro-glycolytic, their impact on glycolysis depends on their balance of kinase and phosphatase activity. Recent in vitro biochemistry studies have assigned the highest kinase-to-phosphatase ratio to the PFKFB3 isoform (>700:1) (Ros and Schulze, 2013), which we found most strongly induced glycolytic flux. Existing biochemical data on the other three PFK2 isoforms, however, does not explain their divergent glycolytic flux phenotypes, as their in vitro kinase and phosphatase activities are similar. Our results highlight the functional diversity of PFK2 isoforms in cells, with the PFKFB2 isozyme acting to slow glycolysis.
Intracellular FBP concentration mirrors glycolytic flux
To explore potential connections between enzyme induction, flux, and metabolite levels, we measured changes in glycolytic intermediate levels by LC-MS. These metabolomics data confirmed the functional activities of the overexpressed enzymes (Figure 2A). For example, overexpression of PFKP led to a decrease in fructose-6-phosphate (F6P) and to a concomitant increase in FBP, whereas the opposite was observed for fructose bisphosphatase (FBP1). Increased ALDOA activity resulted in lower FBP but elevated dihydroxyacetone phosphate (DHAP). We observed that 2,3-bisphosphoglycerate (2,3BPG) accumulated in BPGM overexpressing cells, which likely explains their decreased glycolytic flux (Figures 1D & 2A). Our analysis also confirmed recently-reported side products of GAPDH and pyruvate kinase (Collard et al., 2016): we found that GAPDH overexpression induced erythronate, and PKM1 and especially PKM2 generated 2-phospholactate (Figure S2A, B). TIGAR overexpressing cells were characterized by a marked reduction of lower glycolytic intermediates, specifically 2,3BPG and phosphoenolpyruvate (PEP), but not FBP, and we did not detect any increase in hexose-phosphate species. These observations are consistent with recent evidence showing greater TIGAR phosphatase activity towards 2,3BPG and PEP than fructose-2,6-bisphosphate (Gerin et al., 2014). Collectively, these metabolomics data support the successful induction of the desired enzymatic activities via protein overexpression, including for enzymes that did not significantly impact glycolytic flux.
Figure 2. Intracellular FBP levels mirror glycolytic flux.
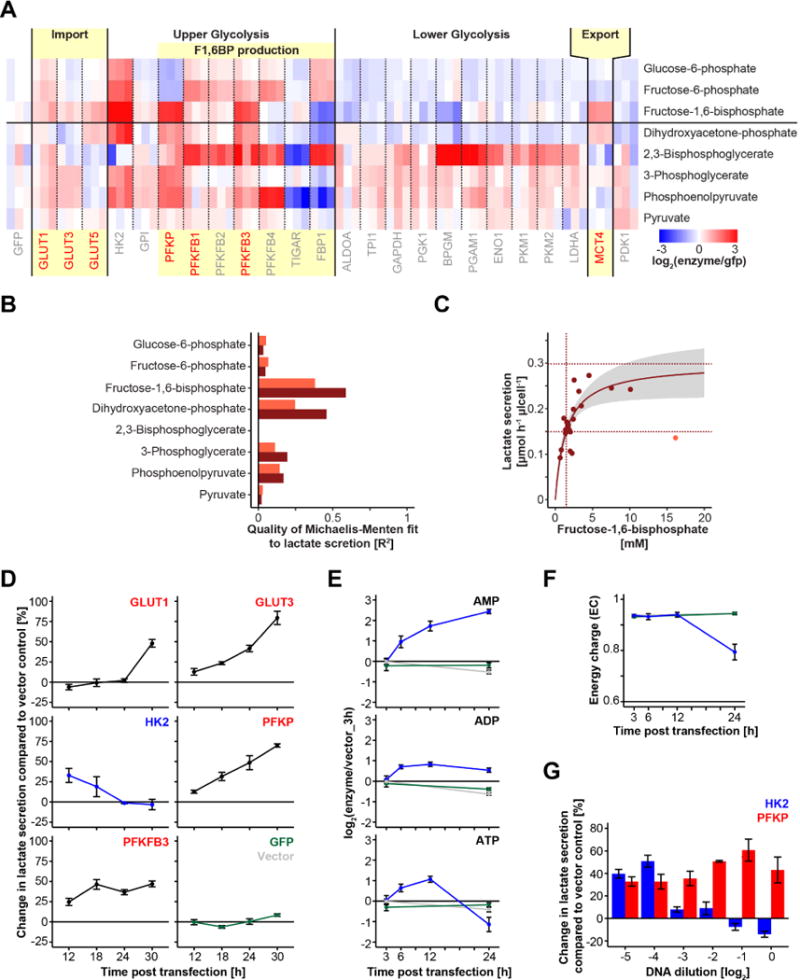
(A) Heat map summarizing changes in intracellular metabolite levels upon overexpression of glycolytic enzymes. Red and blue indicate increased and decreased intracellular metabolites, respectively. Light yellow colored boxes highlight the identified flux controlling steps from Figure 1. Glycolytic enzymes with positive glycolytic flux control are colored in red. (B) Quality of Michaelis-Menten fit of lactate secretion rate to the individual glycolytic intermediate concentration. Light brown shows R2 across all overexpressed constructs and dark brown represents R2 excluding HK2. R2 for fits were calculated by the residual sum of squares (RSS) and total sum of squares (TSS) with R2=1-RSS/TSS. (C) Absolute glycolytic flux as a function of FBP concentration follows Michaelis-Menten kinetics (p<10−4). Dark brown dotted lines indicate fitted Km (1.5 mM; p=0.002) and Vmax (0.3 μmol h−1 μl cell−1; p<10−8) values. Grey represents the 95% confidence intervals of the fitted function. Data from HK2 overexpressing cells is shown as the light brown dot and was omitted from the fitting. (D) Temporal dynamics of changes in lactate secretion upon single enzyme expression. (E) Adenosine phosphate levels upon HK2 overexpression (blue) compared to GFP (green) and vector control (grey) samples. (F) Energy charge upon HK2 overexpression. (G) Changes in lactate secretion as a function of DNA concentration. Means and 95% confidence intervals (n=3 biological replicates) are plotted in D, E, F and G. See also Figure S2.
In addition, our metabolite concentration data support a picture in which upper glycolytic enzymes control both glycolytic flux and metabolite levels. Overexpression of upper glycolytic enzymes caused strong metabolite changes across the entire pathway. In contrast, lower glycolytic enzymes tended to marginally increase lower glycolytic intermediates (with strong effects only on 2,3BPG) and to weakly deplete upper glycolytic intermediates (Figure 2A). Although the overexpressed glucose and lactate transporters generally had only weak effects on metabolite concentrations, they did elevate FBP and to a lesser extent DHAP, which is linked to FBP via the reversible aldolase reaction.
Consistent with prior literature in both microbes and mammalian cells (Kochanowski et al., 2013; Kotte et al., 2010; Peeters et al., 2017; Zhang et al., 2017), among the glycolytic intermediates, FBP and DHAP concentrations correlated most strongly with glycolytic flux (Figure 2B). AMP, ADP, ATP, and citrate are textbook regulators of glycolysis (Berg et al., 2002; Garland et al., 1963; Llorente et al., 1970; Newsholme et al., 1977), but none showed a comparable correlation with glycolytic flux (Figure S2C). With the exception of cells overexpressing toxic levels of hexokinase (see below), flux across the cell lines was generally well-explained by a Michaelis-Menten function of FBP concentration (p<10−4) (Figure 2C). The fitted Km value of 1.5 mM (p=0.002) is in the range of the basal FBP level in iBMK cells (1.8 mM), and the Vmax value of 0.3 μmol h−1 μl cell−1 (p<10−8) is two-fold higher than the basal glycolytic flux (0.15 μmol h−1 μl cell−1) and equal to the maximum glycolytic flux obtained upon oligomycin treatment or combined GLUT3, PFKP and MCT4 overexpression. Thus, glycolytic flux appears to be a saturable function of FBP levels.
Hexokinase is pro-glycolytic but toxic in excess
Hexokinase catalyzes an effectively irreversible step of glycolysis that has previously been shown to have significant flux control (Marín-Hernández et al., 2006). HK2 overexpression resulted in accumulation of most glycolytic intermediates, including FBP (Figure 2A). Accordingly, we were perplexed by the negative effect of HK2 overexpression on glycolytic flux (Figure 1D). To address this discrepancy, we measured the temporal dynamics of glycolytic flux in HK2 overexpressing cells, as well as in cells overexpressing GLUT1, GLUT3, PFKP, PFKFB3, and PKM2 (Figure 2D & Figure S2D). This analysis revealed that HK2 initially accelerates glycolytic flux, but suppresses it at later time points. No other enzyme showed a similar biphasic response. Thus, HK2 exerts positive glycolytic flux control, but over time also impairs glycolysis.
To obtain a better understanding of the underlying mechanism, we measured the temporal evolution of metabolite concentrations in HK2-overexpressing cells (Figure 2E & Figure S2E). We found that glycolytic intermediates accumulated monotonically. Adenosine monophosphate (AMP) and to a lesser extent adenosine diphosphate (ADP) also built up, suggestive of energy stress. ATP levels and adenylate energy charge dropped at the same time as glycolytic flux decreased (Figure 2D, F). Lower energy charge also manifested in depletion of creatine phosphate (Figure S2F). These metabolic alterations likely result from excessive HK2 enzyme activity detrimentally consuming ATP through overactive hexose phosphorylation, with downstream glycolysis unable to maintain pace. Cell growth and viability were, however, largely maintained over this time interval (Figure S2G). This HK2-induced energy stress is evocative of other instances where excessive ATP-driven phosphorylation leads to energy stress, e.g,. glucose-induced ATP depletion in yeast lacking the enzyme trehalose-6-phosphate synthase (TPS1) which acts in part as a brake on hexokinase (Van Aelst et al., 1993; Ernandes et al., 1998; González et al., 1992) and fructose-induced drainage of ATP in hepatocytes via ketohexokinase (Abdelmalek et al., 2012; Ishimoto et al., 2012; Johnson et al., 2013).
To prove that HK2 is fundamentally pro-glycolytic, but that excessive HK2 activity causes energy stress, we transfected iBMK cells with serial dilutions of plasmid DNA encoding HK2 or, for comparison, PFKP (Figure 2G). Both low and high copy numbers of PFKP had similar effects, increasing glycolytic flux by ~ 40%. In contrast, at the 30 h time point used in Figure 1, while concentrated HK2 decreased glycolytic flux, dilute HK2 increased glycolytic flux by ~ 40%. Glycolytic flux began to decrease as the introduced hexokinase activity exceeded endogenous activity by more than five-fold (Figure S2H). Thus, hexokinase exerts positive flux control in iBMK cells, but this was masked in our initial experiments involving massive overexpression, because excessive hexokinase activity causes ATP depletion.
Hexokinase and phosphofructokinase also control flux in NIH3T3 fibroblasts
To examine glycolytic flux control in another mouse cell line, we conducted single enzyme overexpression experiments in NIH3T3 mouse fibroblasts (3T3). Parental 3T3 cells were transduced by lentiviruses carrying a glycolytic gene of interest and lactate secretion was measured after 48 h. Overall, the results closely mirrored those observed in iBMK cells (Figure 3A & Figure S3A). Of the eight glycolytic reactions that did not substantially control flux in iBMK cells, none had a significant effect in 3T3 cells. Similar to iBMK cells, the hexokinase and phosphofructokinase steps exerted substantial flux control, with expression of the PFK1-activator PFKFB3 yielding a similar effect as PFKP itself. In contrast to iBMK cells, we did not observe increased flux in response to single overexpression of glucose or lactate transporters. Overall, our results suggest that hexokinase and phosphofructokinase are common flux-controlling enzymes in glycolysis, with other core glycolytic enzymes exerting limited flux control.
Figure 3. Glycolytic flux increase upon Ras activation is controlled by glucose import, glucose-6-phosphate production, and lactate export.
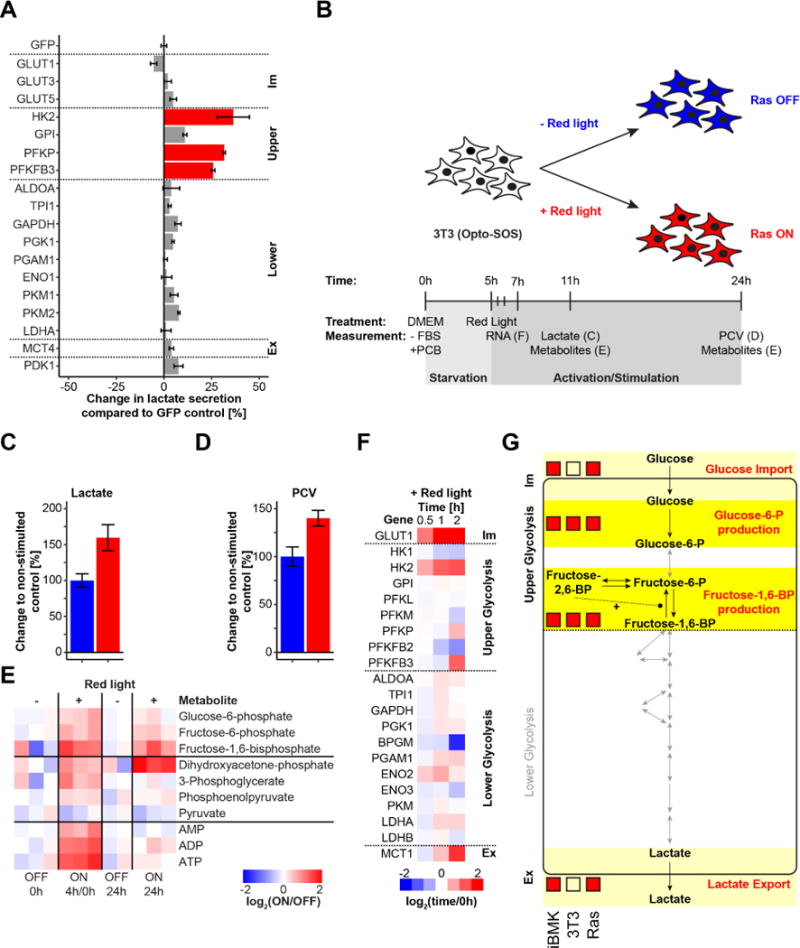
(A) Change in glycolytic flux in 3T3 cells upon individual overexpression of enzymes catalyzing every step in glycolysis. Means and 95% confidence intervals (n=3 biological replicates) are plotted. (B) Experimental setup to investigate acute metabolic perturbation in 3T3 cells upon optogenetic Ras activation. Changes in (C) lactate secretion at 6 h (n=12 biological replicates from two independent experiments; p<10−7) and (D) cell growth at 24 h (n=3 biological replicates; p<0.02) after red light exposure. Means and 95% confidence intervals are plotted. (E) Changes in intracellular metabolites 4 h and 24 h after activating red light exposure. The 4 h red light exposed time point was normalized to the mean of the 0 h time point; the stimulated (Ras ON) 24 h samples were normalized to the mean of the 24 h non-stimulated (Ras OFF) samples. (F) Gene expression dynamics of glycolytic enzymes (from RNAseq data) upon optogenetic Ras activation (Data was analyzed from a recent study (Wilson et al., 2017)). (G) Overview of results in iBMK and 3T3 cells. For the iBMK and 3T3 columns, red boxes indicate significant flux control based on enzyme overexpression experiments. For the Ras column, red boxes indicate significant transcriptional up-regulation upon acute optogenic Ras activation. See also Figure S3.
Acute Ras stimulation transcriptionally activates all four flux-controlling steps
The Ras/Erk pathway is a well-studied growth-promoting signal transduction pathway whose constituents are among the most frequently mutated genes in human cancers (Prior et al., 2012). Oncogenic mutations in the Ras/Erk pathway have been shown to enhance both uptake and utilization of glucose (Flier et al., 1987; Prior et al., 2012; Racker et al., 1985; Ying et al., 2012; Yun et al., 2009). We have previously observed that acute optogenetic stimulation of wildtype Ras is sufficient to drive 3T3 cell transcription and proliferation (Toettcher et al., 2013; Wilson et al., 2017). We wondered if this acute Ras-driven proliferation was associated with glycolytic enhancement, and, if so, whether the enhanced flux is driven by the flux-controlling reactions identified by our overexpression analysis.
To address these questions, we employed the optogenetic system (Opto-SOS) to activate Ras signaling activity in 3T3 cells (Goglia et al., 2017). Serum starved 3T3 cells expressing the light-activatable Opto-SOS system were stimulated with red light and glycolytic flux was measured at 6 h and cell growth after 24 h (Figure 3B). As expected, cell growth was increased by acute Ras signaling, and even at the 6 h time point, glycolytic flux was already enhanced (Figure 3C, D & Figure S3B).
To obtain a more thorough understanding of early metabolic perturbations upon Ras signaling, we measured glycolytic intermediates after 4 h and 24 h of activating light (Figure 3E). We observed a general increase in glycolytic intermediates and adenosine nucleotide levels at 4 h, similar to changes observed upon HK2 overexpression in iBMK cells (Figures 2A & 3E). FBP and DHAP were the only metabolites that remained elevated after 24 h of Ras activation. The two-fold elevation of FBP at 4 h was associated with a 1.5-fold increase in glycolytic flux, consistent with the relationship between FBP and glycolytic flux in iBMK cells (Figures 2B, C & 3C, E).
To explore potential mechanisms by which acute Ras activation might enhance glycolysis, we analyzed published RNAseq data collected from Opto-SOS 3T3 cells stimulated with activating red light over a 2 h time course (Wilson et al., 2017). Most gene expression changes were, as expected, in signaling pathways (Figure S3C). Among metabolic pathways, nucleotide biosynthesis was substantially increased, likely to support cell proliferation. In central carbon metabolism, four glycolytic genes were up-regulated more than two-fold at some time point. In contrast, such up-regulation was observed for only a single gene in the pentose phosphate pathway (ribose-5-phosphate isomerase A; RPIA) and none in the TCA cycle (Figure S3D). The more than two-fold up-regulated glycolytic genes were GLUT1, HK2, PFKFB3 and MCT1, corresponding to the four flux-controlling steps identified in the overexpression studies. PFKP was up to a lesser extent (Figure 3F). Induction of GLUT1 and HK2 was particularly strong and rapid. These acute changes differ from those resulting from chronic Ras activation, which also induces multiple lower glycolytic enzymes (Gaglio et al., 2011; Ying et al., 2012). In addition, we observed Ras-dependent decreases in expression of some glycolytic enzymes (BPGM and PFKFB2) which were “anti-glycolytic” in the single enzyme overexpression experiments (Figure 1D). We did not observe altered expression of any glycolytic genes with low flux control coefficients. Taken together, our results suggest that Ras controls a coordinated expression program to both up- and down-regulate glycolytic enzyme targets in a manner that increases glycolytic flux. Figure 3G summarizes the flux-controlling steps identified in iBMK and 3T3 cells together with the transcriptional targets induced by acute Ras signaling.
Solid tumors consistently overexpress genes for glucose transport, FBP production, and lactate export
Enhanced glycolysis was the first molecular phenotype assigned to cancer, and occurs in tumors originating from many different tissues and harboring distinct oncogenes and genomic alterations (Dang, 2012; Vander Heiden et al., 2009; Warburg, 1956). We wondered if the same four glycolytic flux-controlling steps identified in our cell culture data would also emerge in gene expression data across a variety of human tumors. To this end, we analyzed expression of glycolytic genes in 666 paired solid tumor tissue samples from The Cancer Genome Atlas (TCGA) (http://cancergenome.nih.gov/).
Based on KEGG database annotations, we curated a set of 40 glycolytic genes, covering all isozymes of glycolytic enzymes, including PFK2 isoforms and glucose and lactate transporters (Kanehisa and Goto, 2000; Kanehisa et al., 2016, 2017). Gene expression of tumor samples was normalized to the corresponding benign adjacent tissue. Unsupervised clustering of solely this limited set of glycolytic genes was sufficient to group tumor samples based on their tissue of origin (Figure 4A, B).
Figure 4. Increased glucose uptake, FBP production, and lactate export is a general hallmark of tumor metabolism.
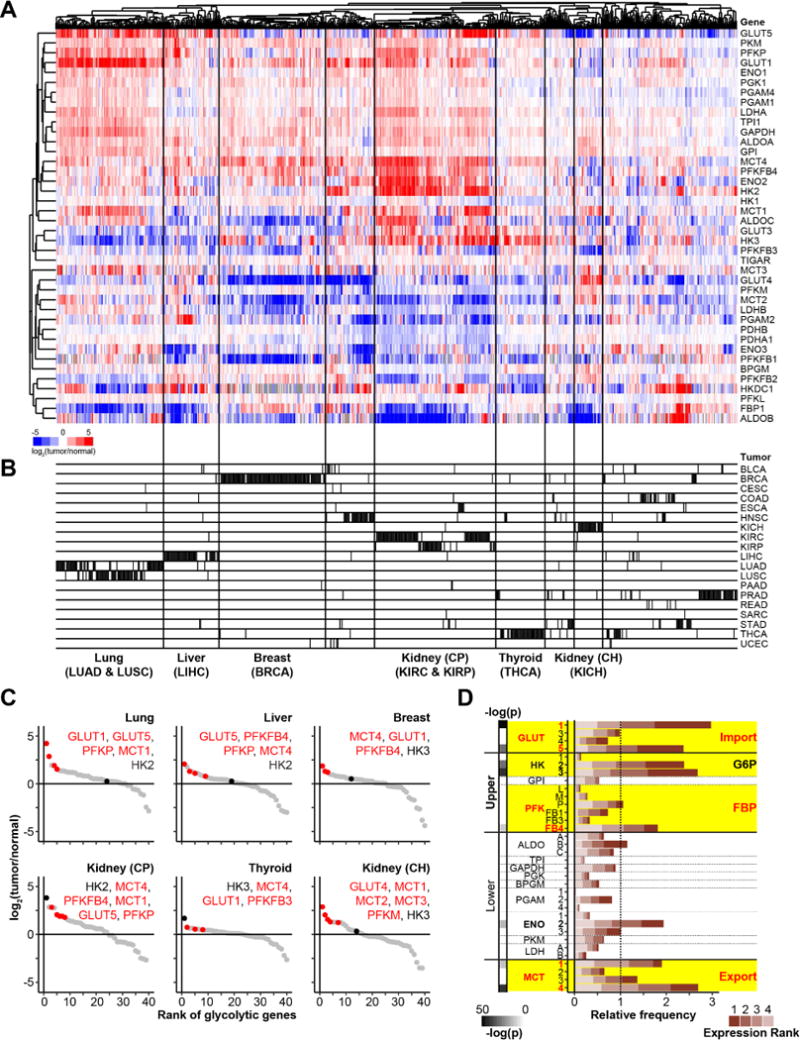
(A) Expression levels of glycolytic enzymes in solid tumor tissue samples compared to paired healthy tissue samples analyzed from TCGA data. (B) Clustering of tumor types based on glycolytic gene expression data only; Bladder Urothelial Carcinoma (BLCA), Breast invasive carcinoma (BRCA), Cervical squamous cell carcinoma and endocervical adenocarcinoma (CESC), Colon adenocarcinoma (COAD), Esophageal carcinoma (ESCA), Head and Neck squamous cell carcinoma (HNSC), Kidney Chromophobe (KICH), Kidney renal clear cell carcinoma (KIRC), Kidney renal papillary cell carcinoma (KIRP), Liver hepatocellular carcinoma (LIHC), Lung adenocarcinoma (LUAD), Lung squamous cell carcinoma (LUSC), Pancreatic adenocarcinoma (PAAD), Prostate adenocarcinoma (PRAD), Rectum adenocarcinoma (READ), Sarcoma (SARC), Stomach adenocarcinoma (STAD), Thyroid carcinoma (THCA), and Uterine Corpus Endometrial Carcinoma (UCEC). (C) Rank order of glycolytic genes from the most to the least expressed relative to benign adjacent tissue, on average, for different tumor types. Red represents top ranked gene isoform catalyzing glucose transport, FBP production and lactate export for each tumor type. Black indicates the highest expressed hexokinase isoform. (D) Relative frequencies with which gene isoforms across glycolysis are among the four most up-regulated glycolytic genes in an individual patient tumor. The expected frequency for each gene due to chance alone (78 occurances across 666 tumors) is defined as a relative frequency of 1. Statistically significant enrichment of a given gene isoform in the top four most up-regulated genes was calculated using a Fisher’s exact ttest with Bonferroni correction for multiple hypothesis testing (highlighted in bold). See also Figure S4, and Tables S2 and S3.
At least one isoform of every glycolytic step was generally up-regulated in each patient tumor (Figure 4A). In most cancer types, strong increases were seen in at least one isoform of GLUT, of PFK (including PFK2), and of MCT, representing three of the four key flux-controlling nodes (Figure 4C & Figure S4A). While no hexokinase isozyme was particularly strongly expressed in lung, liver or breast cancer, HK2 was the most strongly up-regulated glycolytic enzyme in kidney renal clear cell carcinoma (KIRC) and kidney renal papillary cell carcinoma (KIRP) tumors, and HK3 in thyroid tumors (THCA). The less consistent high up-regulation of HK expression may reflect the toxicity of excess HK activity.
To more systematically evaluate overexpression at different glycolytic steps, we ranked the glycolytic gene isoforms in each individual tumor specimen based on their extent of transcriptional up-regulation relative to adjacent benign tissue. Figure 4D shows the frequency with which gene isoforms catalyzing different glycolytic steps were among the four topmost up-regulated glycolytic genes in a given patient. Consistent with our findings regarding flux control in cultured cells, we find that the most overexpressed glycolytic genes in human cancer encode for glucose import (GLUT1 and GLUT5), glucose phosphorylation (HK2 and HK3), FBP production (PFKFB4), PEP production (ENO2), and lactate export (MCT1 and MCT4). Except for enolase, which catalyzes the chemically challenging dehydration of 2-phosphoglycerate to PEP, these are exactly the four steps that we identified as being flux-controlling in the cell culture studies.
Analysis of published gene and protein expression data in lymphocyte, macrophage, and endothelial cell activation, which are physiological contexts where glycolysis is up-regulated, also reveals a consistent up-regulation of at least one isozyme catalyzing glucose uptake, glucose phosphorylation, FBP production, and lactate secretion (Jeong et al., 2017; Jha et al., 2015; Ron-Harel et al., 2016; Wang et al., 2011) (Figure S4B). Thus, activation of glycolysis in mammals, either by physiological stimuli or cancer, is associated with induction of the four flux-controlling steps identified by the systematic overexpression analysis.
DISCUSSION
Glycolysis is the earliest described biochemical pathway. Our current understanding reflects the combined efforts of generations of scientists, with seminal contributions including Louis Pasteur’s characterization of the inputs and outputs in the 1850s and Gustav Embden’s outline of the reaction steps in the 1930s. Motivated in part by Otto Warburg’s discovery of increased glycolytic rate in cancer in the 1920s, there is widespread and longstanding interest in understanding how flux through glycolysis is controlled. This interest is mirrored by the many thousands of studies on the topic. Nevertheless, the resulting body of primary literature is mainly composed of papers focusing on individual enzymes, with every glycolytic enzyme proposed to be rate-limiting in some study, and phosphofructokinase emerging as the consensus main rate-limiting step in textbook descriptions of the pathway.
Systematic assessment of pathway regulation by metabolic control analysis, however, has revealed that rate-limiting steps are an oversimplification (Fell, 1992, 1998; Kacser and Burns, 1973). Indeed, as much as every pathway step must be thermodynamically forward driven and thus consume some of the available pathway free energy, every step also exerts at least some modicum of flux control (Fell, 1998; Noor et al., 2014; Park et al., 2016). Therefore, a pragmatic understanding of metabolic control requires figuring out which steps substantially control flux.
Here we assessed flux control in mammalian glycolysis using three approaches: glycolytic enzyme overexpression, optogenetic activation of Ras, and analysis of gene expression data from human tumors. The first of these approaches is particularly informative, as it directly probes the relationship between enzyme levels and flux, with our data covering each step of glycolysis. All three approaches consistently point to flux through glycolysis being controlled by glucose uptake and phosphorylation, FBP production, and lactate export, and not by lower glycolytic enzymes including pyruvate kinase.
Our measurements of flux control apply to the context in which the enzyme overexpression was carried out (i.e., iBMK and 3T3 cells). This reflects flux control coefficients depending on relative enzyme concentrations. As the level or activity of an enzyme goes down (compared to pathway flux), its flux control goes up. A relatively high basal capacity for glucose transport and lactate secretion, relative to glucose phosphorylation and FBP production, likely explains why GLUT and MCT do not exert flux control in 3T3 cells. Moreover, while we do not find evidence for substantial flux control in any of the lower glycolytic enzymes, these enzymes may exert flux control in cellular contexts where their relative activities are lower. For example, aldolase A may gain flux control in contexts where it is substantially inactivated by binding to the actin cytoskeleton (Hu et al., 2016). GAPDH, whose catalytic cysteine can be oxidized, may exert flux control in the context of oxidative stress (Liberti et al., 2017; Ralser et al., 2007, 2009; Shestov et al., 2014; Yun et al., 2015). More generally, any enzyme of glycolysis can gain flux control if sufficiently inhibited, e.g., by genetic or pharmacological manipulation. For this reason, while studies examining the effects of enzyme depletion are valuable to understand gene essentiality, they do not shed light on intrinsic flux control (Small and Kacser, 1993a). In contrast, for the specific cell line in which they are conducted, our overexpression studies yield quantitative estimates of flux control coefficients.
In considering how overexpression of a single enzyme increases glycolysis, it is important to keep in mind that increased pathway flux requires enhancement of flux through every enzymatic step of the pathway (Hackett et al., 2016). A straightforward way to achieve this is increased expression of each pathway enzyme. Overexpression of a single enzyme may, however, drive flux through upstream and downstream pathway steps by changing metabolite levels. This appears to be the case for hexokinase and phosphofructokinase. The former increases all glycolytic intermediates. The latter depletes hexose phosphates (thereby relieving product inhibition of glucose import and phosphorylation) and increases FBP and all downstream metabolites (increasing substrate to drive these reactions). It remains unclear how GLUT overexpression drives downstream glycolytic reactions (or MCT upstream ones), as neither transporter markedly alters glycolytic intermediate concentrations.
The lag between GLUT expression and flux response raises the possibility of adaptation being involved in inducing the flux increase. Consistent with this, once we include the positive flux control coefficient for hexokinase that was masked by energy stress in our initial measurements, we observe a surplus of flux control in iBMK cells (i.e. sum of flux control coefficients greater than 1), suggesting that overexpression of certain enzymes may activate other enzymes beyond simply providing substrate or consuming product. Notably, near maximal glycolytic rate in iBMK cells is achieved by simultaneous up-regulation of both FBP production and a transport step (but not by concomitant increase of both flux-controlling transport steps). The reasons for these non-linear interactions between different steps in glycolysis are not evident from the individual flux control coefficients and are an important topic for future research.
As glycolytic rate is enhanced by overexpression of any particular flux-controlling enzyme, flux control will tend to transfer to other pathway steps. This may explain why HIF induces a broad spectrum of glycolytic enzymes including also phosphoglycerate kinase and LDH, and why tumors overexpress many glycolytic enzymes including also aldolase and enolase (Graham et al., 2017; Hu et al., 2013; Nilsson et al., 2014), and why tumor types associated with particularly high glycolytic rates due to mitochondrial defects (i.e., renal and thyroid cancer) overexpress hexokinase, even though excessive hexokinase activity can be toxic (Abu-Amero et al., 2005; Davis et al., 2014; Su et al., 2016).
While our study does not address the extent of mitochondrial respiration in cancer cells, our finding that overexpression of certain glycolytic enzymes is sufficient to drive glycolysis without altering glucose entry into the TCA cycle is consistent with emerging evidence that control of glycolytic flux and TCA cycle flux are substantially independent (Faubert et al., 2017; Hui et al., 2017). This reinforces the appropriateness of defining the Warburg effect as aerobic glycolysis in cancer, a robust clinical phenotype proven by FDG-PET imaging.
In summary, we provide a thorough experimental assessment of glycolytic flux control in two mammalian cell lines. We find that flux control is partially distributed, with four of the 12 steps collectively dominant. The four flux controlling steps reside at the top (glucose import), at the two committed phosphorylation steps (hexokinase and phosphofructokinase) and at the bottom (lactate export) of glycolysis. In contrast, at least in these cells, lower glycolytic enzymes do not substantially control glycolytic flux. The identified flux controlling enzymes stand out for being transcriptionally induced by acute Ras signaling and for being strongly up-regulated in human tumors, immune cell activation, and angiogenesis, arguing for the generality of their biological importance. By providing a systematic assessment of glycolytic flux control in mammalian cells, we hope that this work helps both to contextualize research on individual glycolytic enzymes and to motivate comprehensive studies of flux control in additional pathways.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Joshua D. Rabinowitz (joshr@princeton.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All cell lines used in this study are listed in the Key Resources Table and were cultured in standard Dulbecco’s Modified Eagle Medium (DMEM; with 4.5 g/L glucose and 0.584 g/L L-glutamine, but without sodium pyruvate) supplemented with 10% fetal bovine serum (FBS) at 37 °C and 5% CO 2. For passaging, cells were washed once with 1x phosphate buffer saline (PBS) and detached by trypsin/EDTA (0.05%). For glycolytic flux measurements and metabolomics analysis, DMEM was supplemented with 10% dialyzed FBS (dFBS).
KEY RESOURCES TABLE
The table highlights the genetically modified organisms and strains, cell lines, reagents, software, and source data essential to reproduce results presented in the manuscript. Depending on the nature of the study, this may include standard laboratory materials (i.e., food chow for metabolism studies), but the Table is not meant to be comprehensive list of all materials and resources used (e.g., essential chemicals such as SDS, sucrose, or standard culture media don’t need to be listed in the Table). Items in the Table must also be reported in the Method Details section within the context of their use. The number of primers and RNA sequences that may be listed in the Table is restricted to no more than ten each. If there are more than ten primers or RNA sequences to report, please provide this information as a supplementary document and reference this file (e.g., See Table S1 for XX) in the Key Resources Table.
Please note that ALL references cited in the Key Resources Table must be included in the References list. Please report the information as follows:
REAGENT or RESOURCE: Provide full descriptive name of the item so that it can be identified and linked with its description in the manuscript (e.g., provide version number for software, host source for antibody, strain name). In the Experimental Models section, please include all models used in the paper and describe each line/strain as: model organism: name used for strain/line in paper: genotype. (i.e., Mouse: OXTRfl/fl: B6.129(SJL)-Oxtrtm1.1Wsy/J). In the Biological Samples section, please list all samples obtained from commercial sources or biological repositories. Please note that software mentioned in the Methods Details or Data and Software Availability section needs to be also included in the table. See the sample Table at the end of this document for examples of how to report reagents.
SOURCE: Report the company, manufacturer, or individual that provided the item or where the item can obtained (e.g., stock center or repository). For materials distributed by Addgene, please cite the article describing the plasmid and include “Addgene” as part of the identifier. If an item is from another lab, please include the name of the principal investigator and a citation if it has been previously published. If the material is being reported for the first time in the current paper, please indicate as “this paper.” For software, please provide the company name if it is commercially available or cite the paper in which it has been initially described.
-
IDENTIFIER: Include catalog numbers (entered in the column as “Cat#” followed by the number, e.g., Cat#3879S). Where available, please include unique entities such as RRIDs, Model Organism Database numbers, accession numbers, and PDB or CAS IDs. For antibodies, if applicable and available, please also include the lot number or clone identity. For software or data resources, please include the URL where the resource can be downloaded. Please ensure accuracy of the identifiers, as they are essential for generation of hyperlinks to external sources when available. Please see the Elsevier list of Data Repositories with automated bidirectional linking for details. When listing more than one identifier for the same item, use semicolons to separate them (e.g. Cat#3879S; RRID: AB_2255011). If an identifier is not available, please enter “N/A” in the column.
-
○
A NOTE ABOUT RRIDs: We highly recommend using RRIDs as the identifier (in particular for antibodies and organisms, but also for software tools and databases). For more details on how to obtain or generate an RRID for existing or newly generated resources, please visit the RII or search for RRIDs.
-
○
Please use the empty table that follows to organize the information in the sections defined by the subheading, skipping sections not relevant to your study. Please do not add subheadings. To add a row, place the cursor at the end of the row above where you would like to add the row, just outside the right border of the table. Then press the ENTER key to add the row. Please delete empty rows. Each entry must be on a separate row; do not list multiple items in a single table cell. Please see the sample table at the end of this document for examples of how reagents should be cited.
TABLE FOR AUTHOR TO COMPLETE
Please upload the completed table as a separate document. Please do not add subheadings to the Key Resources Table. If you wish to make an entry that does not fall into one of the subheadings below, please contact your handling editor. (NOTE: For authors publishing in Current Biology, please note that references within the KRT should be in numbered style, rather than Harvard.)
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| ALDOA: Aldolase A (D73H4) Rabbit mAb | Cell Signaling | Cat# 8060 |
| copGFP: TurboGFP Polyclonal Antibody | Thermo Fisher | Cat# PA5-22688 |
| ENO1: Enolase-1 Antibody | Cell Signaling | Cat# 3810 |
| GAPDH: GAPDH (D16H11) XP® Rabbit mAb | Cell Signaling | Cat# 5174 |
| GLUT1: Glut1 (D3J3A) Rabbit mAb | Cell Signaling | Cat# 12939 |
| GLUT3: Anti GLUT3 [EPR10508(N)] antibody-N terminal | Abcam | Cat# ab191071 |
| HK2: Hexokinase II (C64G5) Rabbit mAb | Cell Signaling | Cat# 2867 |
| LDHA: LDHA (C4B5) Rabbit mAb | Cell Signaling | Cat# 3582 |
| PFKFB2: PFKFB2 (D7G5R) Rabbit mAb | Cell Signaling | Cat# 13045 |
| PFKFB3: PFKFB3 (D7H4Q) Rabbit mAb | Cell Signaling | Cat# 13123 |
| PFKP: PFKP (D4B2) Rabbit mAb | Cell Signaling | Cat# 8164 |
| PGAM1: PGAM1 (D3J9T) Rabbit mAb | Cell Signaling | Cat# 12098 |
| PKM1/2: PKM1/2 (C103A3) Rabbit mAb | Cell Signaling | Cat# 3190 |
| PKM2: PKM2 (D78A4) XP® Rabbit mAb | Cell Signaling | Cat# 4053 |
| TIGAR: TIGAR (D3F4A) Rabbit mAb | Cell Signaling | Cat# 14751 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 2x PfuUltra II Hotstart PCR Master Mix | Agilent | Cat# 600850 |
| Agarose (UltraPure™) | Thermo Fisher | Cat# 16500500 |
| Ampicillin | Sigma | Cat# 5354 |
| Carbenicillin | Sigma | Cat# C1389 |
| D-GLUCOSE (U-13C6, 99%) | CIL | Cat# 1396 |
| Dialyzed Fetal Bovine Serum | Sigma | Cat# F0392 |
| Dimethyl sulfoxide (DMSO) | Sigma | Cat# 5879 |
| Dulbecco’s Modification of Eagle’s Medium (DMEM), Powder | Corning | Cat# 90-113-PB |
| Dulbecco’s Modified essential medium (DMEM) | Corning | Cat# 10-017 |
| Fetal Bovine Serum | Sigma | Cat# F2442 |
| FuGENE® HD Transfection Reagent | Promega | Cat# E2311 |
| Gibson Assembly® Cloning Kit | NEB | Cat# E5510S |
| Glucose (Dextrose) | Sigma | Cat# 9434 |
| HEPES | Sigma | Cat# H0887 |
| HindIII-HF® | NEB | Cat# R3104 |
| In-Fusion® HD Cloning Plus | Clontech | Cat# 638910 |
| L-Glutamine | Sigma | Cat# G6392 |
| LB Broth (Miller) | Sigma | Cat# L3522 |
| Lipofectamine™ 2000 Transfection Reagent | Thermo Fisher | Cat# 11668019 |
| Methanol, Optima™ LC/MS Grade | Fisher Chemical | Cat# A456-4 |
| NheI-HF® | NEB | Cat# R3131 |
| Oligomycin A | Sigma | Cat# 75351 |
| Opti-MEM™ I Reduced Serum Medium | Thermo Fisher | Cat# 31985070 |
| PBS without calcium, magnesium | Hyclone | Cat# SH30256.01 |
| Phenol red | Sigma | Cat# 3532 |
| Phosphatase inhibitors: PhosStop EASYpack | Roche | Cat# 4906845001 |
| Phycocyanobilin (PCB) | Frontier Scientific | Cat# P14137 |
| Polybrene | Millipore | Cat# TR-1003-G |
| Protease inhibitors: cOmplete Tablets, Mini, EASYpack | Roche | Cat# 4693124001 |
| RIPA Buffer (10X) | Cell Signaling | Cat# 9806 |
| Sodium bicarbonate | Sigma | Cat# 6014 |
| SuperSignal™ ELISA Pico Chemiluminescent Substrate | Thermo Fisher | Cat# 37070 |
| Trypan Blue Solution, 0.4% | Thermo Fisher | Cat# 15250061 |
| Trypsin-EDTA (0.05%), phenol red | Thermo Fisher | Cat# 25300054 |
| Water | Sigma | Cat# 4502 |
| Critical Commercial Assays | ||
| Colorimetric GAPDH Assay | ScienCell | Cat# 8148 |
| Hexokinase Colorimetric Assay Kit | Sigma-Aldrich | Cat# MAK091 |
| LDH Assay Kit / Lactate Dehydrogenase Assay Kit (Colorimetric) | Abcam | Cat# 102526 |
| Pierce™ BCA Protein Assay Kit | Thermo Fisher | Cat# 23225 |
| Pyruvate Kinase (PK) Assay Kit | Abcam | Cat# 83432 |
| QIAGEN HiSpeed Plasmid Maxi Kit | Qiagen | Cat# 12663 |
| QIAprep Spin Miniprep Kit | Qiagen | Cat# 27104 |
| Wizard® SV Gel and PCR Clean-Up System | Promega | Cat# A9282 |
| Experimental Models: Organisms/Strains | ||
| HEK293T LX | Clontech Laboratories | Cat# 632180 |
| Immortalized baby mouse kidney cell (iBMK D3) | Eileen White Lab, Rutgers University | N/A |
| Immortalized baby mouse kidney cell H-RasV12G (iBMK Ras) | Eileen White Lab, Rutgers University | N/A |
| Immortalized baby mouse kidney cell myr-Akt (iBMK Akt) | Eileen White Lab, Rutgers University | N/A |
| NIH3T3 cells | ATCC | Cat# CRL-1658 |
| NIH3T3 OptoSOS (NIH 3T3 + Phy-mCherry-CAAX + YFP-SOS2cat) | Jared Toettcher Lab, Princeton University | N/A |
| Oligonucleotides | ||
| Primers used for cloning (see Table S1) | This Paper | N/A |
| Recombinant DNA | ||
| ALDOA: MGC Human ALDOA Sequence-Verified cDNA (CloneId:3459009) | Dharmacon | MHS6278-202755903 |
| BPGM: pEGFP-N1 DNA plasmid expressing FLAG-BPGM | Tom Muir Lab, Princeton University | N/A |
| copGFP: PB-Cuo-MCS-IRES-GFP-EF1α-CymR-Puro Inducible cDNA Cloning and Expression Vector | System Biosciences | Cat# PBQM812A-1 |
| ENO1: MGC Human ENO1 Sequence-Verified cDNA (CloneId:3447583) | Dharmacon | Cat# MHS6278-202755590 |
| FBP1: FBP1 (NM_000507) Human cDNA Clone | Origene | Cat# SC119828 |
| GAPDH: MGC Human GAPDH Sequence-Verified cDNA (CloneId:3869809) | Dharmacon | Cat# MHS6278-202756436 |
| GLUT1: MGC Human SLC2A1 Sequence-Verified cDNA (CloneId:40085220) | Dharmacon | Cat# MHS6278-211690539 |
| GLUT3: MGC Human SLC2A3 Sequence-Verified cDNA (CloneId:4396508) | Dharmacon | Cat# MHS6278-202757852 |
| GLUT5: MGC Human SLC2A5 Sequence-Verified cDNA (CloneId:2966459) | Dharmacon | Cat# MHS6278-202826673 |
| GPI: MGC Human GPI Sequence-Verified cDNA (CloneId:2906270) | Dharmacon | Cat# MHS6278-202826268 |
| HK2: MGC Human HK2 Sequence-Verified cDNA (CloneId:5586773) | Dharmacon | Cat# MHS6278-202759113 |
| LDHA: MGC Human LDHA Sequence-Verified cDNA (CloneId:4096518) | Dharmacon | Cat# MHS6278-202840043 |
| Lenti-Envelope: pMD2.G | Addgene | Cat# 12259 |
| Lenti-Packaging: pCMVdR8.91 | Trono lab, École Polytechnique Fédérale de Lausanne, Switzerland | N/A |
| Lenti-Vector: pHR SFFVp IRES-CIBN-BFP-CAAX | Jared Toettcher lab, Princeton University | N/A |
| MCT4: MGC Human SLC16A3 Sequence-Verified cDNA (CloneId:8327735) | Dharmacon | Cat# MHS6278-211688899 |
| PDK1: PDK1 (NM_002610) Human cDNA Clone | Origene | Cat# SC321678 |
| PFKFB1: MGC Human PFKFB1 Sequence-Verified cDNA (CloneId:40000713) | Dharmacon | Cat# MHS6278-211689324 |
| PFKFB2: MGC Human PFKFB2 Sequence-Verified cDNA (CloneId:30915381) | Dharmacon | Cat# MHS6278-202856883 |
| PFKFB3: PFKFB3 (NM_004566) Human cDNA Clone | Origene | Cat# SC117283 |
| PFKFB4: MGC Human PFKFB4 Sequence-Verified cDNA (CloneId:2900872) | Dharmacon | Cat# MHS6278-202755424 |
| PFKP: MGC Human PFKP Sequence-Verified cDNA (CloneId:5180268) | Dharmacon | Cat# MHS6278-202800861 |
| PGAM1: pReceiver M11 DNA plasmid expressing FLAG-PGAM1 | Tom Muir, Princeton University | N/A |
| PGK1: MGC Human PGK1 Sequence-Verified cDNA (CloneId:3917985) | Dharmacon | Cat# MHS6278-202757198 |
| PKM1: Homo sapiens pyruvate kinase, muscle (PKM), transcript variant 2, mRNA. | Genecopoeia | Cat# Z5842 |
| PKM2: Homo sapiens pyruvate kinase, muscle (PKM), transcript variant 1, mRNA. | Genecopoeia | Cat# Z7438 |
| TIGAR: TIGAR (NM_020375) Human cDNA Clone | Origene | Cat# SC320794 |
| TPI1: Triosephosphate isomerase (TPI1) (NM_000365) Human cDNA Clone | Origene | Cat# SC111041 |
| Other | ||
| Cell culture plates and dishes | Falcon/Cell Treat | N/A |
| LB Agar plates with 100 μg/mL Ampicillin | Teknova | Cat# L1004 |
| PCV Packed Cell Volume Tube | TPP | Cat# 87005 |
| Seahorse XF24 Cell Culture Microplates | Agilent | Cat# 100777-004 |
TABLE WITH EXAMPLES FOR AUTHOR REFERENCE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-Snail | Cell Signaling Technology | Cat#3879S; RRID: AB_2255011 |
| Mouse monoclonal anti-Tubulin (clone DM1A) | Sigma-Aldrich | Cat#T9026; RRID: AB_477593 |
| Rabbit polyclonal anti-BMAL1 | This paper | N/A |
| Bacterial and Virus Strains | ||
| pAAV-hSyn-DIO-hM3D(Gq)-mCherry | Krashes et al., 2011 | Addgene AAV5; 44361-AAV5 |
| AAV5-EF1a-DIO-hChR2(H134R)-EYFP | Hope Center Viral Vectors Core | N/A |
| Cowpox virus Brighton Red | BEI Resources | NR-88 |
| Zika-SMGC-1, GENBANK: KX266255 | Isolated from patient (Wang et al., 2016) | N/A |
| Staphylococcus aureus | ATCC | ATCC 29213 |
| Streptococcus pyogenes: M1 serotype strain: strain SF370; M1 GAS | ATCC | ATCC 700294 |
| Biological Samples | ||
| Healthy adult BA9 brain tissue | University of Maryland Brain & Tissue Bank; http://medschool.umaryland.edu/btbank/ | Cat#UMB1455 |
| Human hippocampal brain blocks | New York Brain Bank | http://nybb.hs.columbia.edu/ |
| Patient-derived xenografts (PDX) | Children’s Oncology Group Cell Culture and Xenograft Repository | http://cogcell.org/ |
| Chemicals, Peptides, and Recombinant Proteins | ||
| MK-2206 AKT inhibitor | Selleck Chemicals | S1078; CAS: 1032350-13-2 |
| SB-505124 | Sigma-Aldrich | S4696; CAS: 694433-59-5 (free base) |
| Picrotoxin | Sigma-Aldrich | P1675; CAS: 124-87-8 |
| Human TGF-β | R&D | 240-B; GenPept: P01137 |
| Activated S6K1 | Millipore | Cat#14-486 |
| GST-BMAL1 | Novus | Cat#H00000406-P01 |
| Critical Commercial Assays | ||
| EasyTag EXPRESS 35S Protein Labeling Kit | Perkin-Elmer | NEG772014MC |
| CaspaseGlo 3/7 | Promega | G8090 |
| TruSeq ChIP Sample Prep Kit | Illumina | IP-202-1012 |
| Deposited Data | ||
| Raw and analyzed data | This paper | GEO: GSE63473 |
| B-RAF RBD (apo) structure | This paper | PDB: 5J17 |
| Human reference genome NCBI build 37, GRCh37 | Genome Reference Consortium | http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/human/ |
| Nanog STILT inference | This paper; Mendeley Data | http://dx.doi.org/10.17632/wx6s4mj7s8.2 |
| Affinity-based mass spectrometry performed with 57 genes | This paper; and Mendeley Data | Table S8; http://dx.doi.org/10.17632/5hvpvspw82.1 |
| Experimental Models: Cell Lines | ||
| Hamster: CHO cells | ATCC | CRL-11268 |
| D. melanogaster: Cell line S2: S2-DRSC | Laboratory of Norbert Perrimon | FlyBase: FBtc0000181 |
| Human: Passage 40 H9 ES cells | MSKCC stem cell core facility | N/A |
| Human: HUES 8 hESC line (NIH approval number NIHhESC-09-0021) | HSCI iPS Core | hES Cell Line: HUES-8 |
| Experimental Models: Organisms/Strains | ||
| C. elegans: Strain BC4011: srl-1(s2500) II; dpy-18(e364) III; unc-46(e177)rol-3(s1040) V. | Caenorhabditis Genetics Center | WB Strain: BC4011; WormBase: WBVar00241916 |
| D. melanogaster: RNAi of Sxl: y[1] sc[*] v[1]; P{TRiP.HMS00609}attP2 | Bloomington Drosophila Stock Center | BDSC:34393; FlyBase: FBtp0064874 |
| S. cerevisiae: Strain background: W303 | ATCC | ATTC: 208353 |
| Mouse: R6/2: B6CBA-Tg(HDexon1)62Gpb/3J | The Jackson Laboratory | JAX: 006494 |
| Mouse: OXTRfl/fl: B6.129(SJL)-Oxtrtm1.1Wsy/J | The Jackson Laboratory | RRID: IMSR_JAX:008471 |
| Zebrafish: Tg(Shha:GFP)t10: t10Tg | Neumann and Nuesslein-Volhard, 2000 | ZFIN: ZDB-GENO-060207-1 |
| Arabidopsis: 35S::PIF4-YFP, BZR1-CFP | Wang et al., 2012 | N/A |
| Arabidopsis: JYB1021.2: pS24(AT5G58010)::cS24:GFP(-G):NOS #1 | NASC | NASC ID: N70450 |
| Oligonucleotides | ||
| siRNA targeting sequence: PIP5K I alpha #1: ACACAGUACUCAGUUGAUA | This paper | N/A |
| Primers for XX, see Table SX | This paper | N/A |
| Primer: GFP/YFP/CFP Forward: GCACGACTTCTTCAAGTCCGCCATGCC | This paper | N/A |
| Morpholino: MO-pax2a GGTCTGCTTTGCAGTGAATATCCAT | Gene Tools | ZFIN: ZDB-MRPHLNO-061106-5 |
| ACTB (hs01060665_g1) | Life Technologies | Cat#4331182 |
| RNA sequence: hnRNPA1_ligand: UAGGGACUUAGGGUUCUCUCUAGGGACUUAGGGUUCUCUCUAGGGA | This paper | N/A |
| Recombinant DNA | ||
| pLVX-Tight-Puro (TetOn) | Clonetech | Cat#632162 |
| Plasmid: GFP-Nito | This paper | N/A |
| cDNA GH111110 | Drosophila Genomics Resource Center | DGRC:5666; FlyBase:FBcl0130415 |
| AAV2/1-hsyn-GCaMP6-WPRE | Chen et al., 2013 | N/A |
| Mouse raptor: pLKO mouse shRNA 1 raptor | Thoreen et al., 2009 | Addgene Plasmid #21339 |
| Software and Algorithms | ||
| Bowtie2 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Samtools | Li et al., 2009 | http://samtools.sourceforge.net/ |
| Weighted Maximal Information Component Analysis v0.9 | Rau et al., 2013 | https://github.com/ChristophRau/wMICA |
| ICS algorithm | This paper; Mendeley Data | http://dx.doi.org/10.17632/5hvpvspw82.1 |
| Other | ||
| Sequence data, analyses, and resources related to the ultra-deep sequencing of the AML31 tumor, relapse, and matched normal. | This paper | http://aml31.genome.wustl.edu |
| Resource website for the AML31 publication | This paper | https://github.com/chrisamiller/aml31SuppSite |
METHOD DETAILS
All reagents and resources used in this study are listed in the Key Resources Table.
Cloning of transient overexpression and lentivirus constructs
All purchased human cDNAs of the 24 glycolytic enzymes are listed above and corresponding primers are listed in Table S1. For cloning into the pCMV-6-A-puro mammalian expression vector, the vector was digested by HindIII (5′end) and NotI (3′end) restriction enzymes for 4.5 h at 37 °C. The digested vector was run on a 1% agarose gel (with ethidium bromide) and the cut band was gel purified using the Wizard® SV Gel and PCR Clean-Up System. The open reading frame (ORF) of a given gene was amplified by polymerase chain reaction (PCR) using corresponding primers and the 2x PfuUltra II Hotstart PCR Master Mix according to manufacturer’s instructions. PCR products were cleaned up with the Wizard® SV Gel and PCR Clean-Up System before Gibson Assembly. For all Gibson Assembly reactions, a molar ratio of 1:3 (vector:insert) was used and the reaction was performed at 50 °C for 1 h as per manufacturer’s instructions. Assembled constructs were transformed into NEB 5-alpha competent E. coli cells and grown on agar plates (100 μg/mL ampicilin) at 37 °C overnight. Single colonies were inoculated in Luria broth (100 μg/mL ampicilin) overnight and plasmid DNA was extracted using the QIAprep Spin Miniprep Kit. Constructs were sequence verified by Sanger sequencing (Genewiz, South Plainfield, NJ, USA) and stocks for all experiments were prepared using the QIAGEN Maxi Kit.
For lentivirus transductions, ORFs were cloned into a pHR’ SFFVp lentiviral vector, upstream of IRES CIBN-BFP-CAAX, using the In-Fusion® HD Cloning Kit according to the manufacturer’s instructions. Briefly, the vector was linearized by backbone PCR and the ORFs were amplified by PCR as described above. The linearized vector and PCR products were run on a 1% agarose gel (with ethidium bromide) and the cut bands were cleaned up with the Wizard® SV Gel and PCR Clean-Up System. Using a molar ratio of 1:3 (vector:insert), all In-Fusion reactions were performed at 50 °C for 15 min as per manufacturer’s instructions. Assembled constructs were transformed into Clontech Stellar competent E. coli cells and grown on agar plates (50 μg/mL carbenicillin) at 37 °C overnight. Single colonies were inoculated in Lu ria broth (50 μg/mL carbenicillin) overnight. Plasmid DNA was prepared by the QIAprep Spin Miniprep Kit and constructs were sequence verified by Sanger sequencing (Genewiz, South Plainfield, NJ, USA).
Transient overexpression of glycolytic genes in iBMK cells
iBMK cells were transfected using Lipofectamine 2000 as described in the manufacturer’s protocol. 24 h before transfection, 0.5 μl packed cell volume (PCV) iBMK cells were plated per well in a 6-well plate in DMEM (10% dFBS). For each well, 2.5 μg of plasmid DNA, 10 μl of transfection reagent and 200 μl of OptiMEM were used. The DNA to lipofectamine ratio was kept equal for all experiments. For coexpression experiments, the total amount of plasmid DNA was 2.5 μg and the amount of the different constructs was kept equivalent. The single expression controls were adjusted to the lowest concentration and mixed with vector plasmid DNA to keep the total amount of plasmid DNA at 2.5 μg. Correspondingly, serial dilutions of a given construct were supplemented with vector plasmid DNA to keep the 2.5 μg total plasmid DNA amount. iBMK cells were incubated with the plasmid DNA-lipofectamine complexes in 2 ml DMEM (10% dFBS) for 6 h before switching to fresh DMEM (10% dFBS). Metabolomics experiments were performed in 6 cm dishes. For transfection in 6 cm dishes, 1 μl PCV iBMK cells were plated and grown overnight before transfection with 8 μg of plasmid DNA, 20 μl of lipofectamine as well as 500 μl of OptiMEM incubated in 5 ml of DMEM (10% dFBS) for 6 h.
Number of viable cells was counted at the indicated time points. Cells were transfected in 6-well plates as described above and viable cells were counted using Trypan blue exclusion assays and a Countess™ II FL Automated Cell Counter (ThermoFisher, MA).
Western Blotting
iBMK cells were collected at the indicated time points after transfection. Cells were washed once with ice cold 1x PBS and scraped into 200 μl RIPA buffer supplemented with phosphatase and protease inhibitors. Collected iBMK cells were vortexed, incubated for 10 min on ice and centrifuged at 17,000 g at 4 °C. Supernatant was transferred to a fresh tube and protein content was determined by Pierce™ BCA Protein Assay Kit. 10 μg of protein per sample was analyzed by SDS-PAGE and western blotting. Primary and secondary antibodies (horse-radish peroxidase conjugated) were used at 1:1000 and 1:10,000 respectively. The SuperSignal™ ELISA Pico Chemiluminescent Substrate was used for protein detection.
Glycolytic flux measurements
Glucose and lactate measurements were performed in 6-well plates between 24 h and 30 h post transfection unless stated otherwise. Cells were washed twice with 2 ml of 1x PBS before 1 ml of fresh DMEM (10% dFBS) was added. After 6 h of incubation, 300 μl of supernatant was collected and analyzed by a YSI 2900D Biochemistry Analyzer (YSI, Yellow Springs, OH, USA). Measured glucose consumption and lactate secretion were normalized to the average PCV during the 6 h measurement interval as determined based on the terminal PCV measurement at 30 h and the known growth rate (0.028 h−1) of GFP expressing cells (Figure S1A):
| (Equation 3) |
Measurement of the secreted lactate to consumed glucose ratio was done in 5 mM glucose conditions. To make growth medium with 5 mM glucose, DMEM powder without glucose, glutamine, or phenol red was dissolved in deionized water and supplemented with L-glutamine (0.584 g/l), phenol red (0.015 g/l), sodium bicarbonate (3.7 g/l), glucose (0.9 g/l) and 10% dFBS. Data was presented either as absolute values or the percentage change normalized to the vector control as indicated in the corresponding figure panels.
Measurement of oxygen consumption rates
Oxygen consumption rates were measured using a Seahorse XF24 extracellular flux analyzer (Agilent, Santa Clara, CA). Briefly, 100 ul of a 0.5 μlcells ml−1 solution were plated per well in a XF24 cell culture microplate. The next day, cells were transfected with Vector, GFP, GLUT3, PFKP, GAPDH, PKM2, LDHA and MCT4 constructs as described above. 24 h post transfection, cells were equilibrated for 1 h in DMEM supplemented with 25 mM glucose and L-glutamine (0.584 g/l) in a non-CO2 incubator. Oxygen consumption rates were monitored at baseline (5 measurements) and were normalized to cell number as determined by the CyQUANT Cell Proliferation Assay Kit.
Experiments were performed with four replicates per overexpression condtion and the average of three independent experiments from different days were reported with standard errors.
Calculation of flux control coefficients
Flux control coefficients for every step in glycolysis were calculated using the relationship described by Small and Kacser accounting for large changes in enzymatic activity (Small and Kacser, 1993a):
| (Equation 1.1) |
where is the fold-change in glycolytic flux J as a function of change in enzyme activity rE. The derivation of this equation assumes an unbranched pathway with linear reaction kinetics, neither of which applies to glycolysis. The equation is, nevertheless, a valid approximation, as long as flux is measured through the branch in which the enzyme variation occurred (Small and Kacser, 1993a, 1993b). This is true in our case as we are modulating glycolytic enzymes and measuring glycolytic flux. Moreover, based on numerical simulations, the approximation is robust to enzymes that follow Michaelis-Menten as opposed to linear reaction kinetics, with typical deviations < 10% (Small and Kacser, 1993a, 1993b). Thus, we utilized this equation to approximate flux control coefficients.
Solving Equation (1.1) for results in the following equation:
| (Equation 1.2) |
With the assumption that changes in protein levels of a given enzyme E are directly proportional to changes in enzyme activities rE, and since ectopic overexpression generally results in large changes in protein levels, the term approximates 1 and Equation 1.2 can be simplified:
| (Equation 2) |
Equation 2 is shown in Figure 1A. for a given overexpressed enzyme E were calculated using Equation 2 based on the determined fold-change in lactate secretion from experiments performed in 25 mM glucose conditions. Results were presented as the mean with 95% confidence intervals from n≥6 biological replicates of at least two independent experiments. For the calculation of the sum of determined in iBMK cells, only constructs exhibiting positive flux control in both 5 mM and 25 mM glucose conditions were considered:
| (Equation 4.1) |
whereby for different gene isoforms acting on the same glycolytic step were averaged:
| (Equation 4.2) |
| (Equation 4.3) |
| (Equation 4.4) |
Oligomycin treatment
Analogous to the transient overexpression experiments, 0.5 μl PCV iBMK cells were plated per well of a 6-well plate in DMEM (10% dFBS) the day before the experiment. The next day, iBMK cells were washed twice with 1x PBS and incubated with 1 ml of fresh DMEM (10% dFBS) supplemented with 1 μM oligomycin. Supernatant was analyzed after 6 h of incubation and measured lactate was normalized to PCV. Data was presented as the percentage change in lactate secretion compared to vector control.
Metabolomic analysis using mass spectrometry
Metabolomics experiments were performed in 6 cm dishes. 24 h post transfection, iBMK cells were switched to fresh DMEM (10% dFBS) and incubated for another 2 h before extraction. Supernatant was subsequently aspirated and metabolism was directly quenched on dry ice using −20 °C methanol:water (80 :20 v/v %). Quenched cells were left on dry ice for at least 10 min before scraping and collecting them into fresh Eppendorf tubes. Samples were vortexed and centrifuged at 17,000 g at 4 °C for 10 min. Supernatant was transferred to a fresh Eppendorf tube and dried under a stream of nitrogen. Freshly extracted samples were subjected to liquid chromatography mass spectrometry (LC-MS) analysis on the same day.
For labeling experiments using U-13C-glucose, cells were washed twice with 1x PBS and switched into the glucose labeled cell culture medium 24 h post transfection. The cells were kept in the labeled medium for 6 h before quenching. To make U-13C-glucose-containing cell growth medium, DMEM powder was dissolved in deionized water and supplemented with L-glutamine (0.584g/l), phenol red (0.015g/l), sodium bicarbonate (3.7g/l), U-13C-glucose (4.5 g/l) and 10% dFBS.
For LC-MS, dried samples were reconstituted in HPLC grade water at 40 μl water per 1 μl PCV cells. At least 50 μl of sample were transferred to a fresh MS vial and 10 μl of sample were injected. Negatively charged metabolites were analyzed by reverse-phase ion-pairing chromatography coupled to an Exactive Orbitrap mass spectrometer (ThermoFisher Scientific, San Jose, CA, USA). For LC separation, a previously described method was modified (Lu et al., 2010) and an Atlantis T3 column (150mm × 2.1mm, 3 m particle size, Waters, Milford, MA) was used. Solvent A consisted of 97:3 water:methanol (v/v %) with 10mM tributylamine and 15mM acetic acid, and solvent B was 100% methanol. The LC gradient was run as following: 0 min, 0% B, 200 μl/min; 2 min, 0% B, 200 μl/min; 4 min, 20% B, 200 μl/min; 13 min, 80% B, 200 μl/min; 17 min, 100% B, 200 μl/min; 17.5 min, 100% B, 300 μl/min; 20 min, 100% B, 300 μl/min; 20.5 min, 0% B, 300μl/min; 24 min, 0% B, 300 μl/min; 25 min, 0% B, 200 μl/min. The column and autosampler temperatures were kept at 25 °C and 5 °C, respectively. The mass spectrometer was operated in negative ion mode with resolving power of 100,000 at m/z 200 and scanning range of m/z 75-1000.
Data were analyzed using the open source software MAVEN (Melamud et al., 2010) and presented as log2 fold-changes compared to vector control unless stated otherwise. Previously determined absolute metabolite concentrations from iBMK cells (Park et al., 2016) were used to convert the measured metabolite intensities into absolute concentrations (assuming any effect of the empty vector expression on absolute metabolite concentrations is small). Adenylate energy charge was calculated using the following equation:
| (Equation 5) |
Enzyme assays for HK, GAPDH, PKM and LDH activities
Cell culture for enzyme activity assays was performed in 6-well plates and iBMK cells were collected 24 h post transfection. Briefly, cells were washed once with ice-cold 1x PBS and directly scraped in 200 μl ice-cold assay buffer supplemented with phosphatase and protease inhibitors. Cells were lysed by sonication and whole cell lysates were centrifuged at 17,000 g at 4 °C for 10 min. The supernatant was transferred to a fresh Eppendorf tube and 10 μl of whole cell lysate were used for enzyme assays. For GAPDH, PKM and LDH activity assays, cell lysates were diluted 50-fold. Colorometric assay kits from Sigma (HK), ScienCell (GAPDH) and Abcam (PKM and LDH) were used to measure enzyme activities according to manufacturer’s instructions. Enzyme activities were normalized to the protein content as determined by the Pierce™ BCA Protein Assay Kit.
Overexpression of glycolytic genes in 3T3 cells by lentivirus transduction
Lentivirus production was performed in 6-well plates, with one plate (or 6 wells) used per individual construct. 30% to 40% confluent HEK293T LX cells were transfected with 1.33 μg of pCMVdR8.91 (packaging), 0.17 μg pMD2.G (envelope) and 1.5 μg of construct DNA using 9 μl of FuGENE. Supernatant was collected between 48 h and 52 h after transfection and filtered using a 0.45 m filter before transduction. Freshly-produced virus was directly used to transduce 3T3 cells on the same day.
0.3 μl PCV of 3T3 cells were plated per well of a 6-well plate in DMEM (10% dFBS) the day prior to transduction. Cell growth medium was aspirated and 700 μl of freshly-produced virus supplemented with polybrene (5 μg/ml) and HEPES (10mM) was added directly to each well. Cells were incubated for 4 h to 6 h before adding 2 ml of fresh DMEM (10% dFBS; 5 μg/ml polybrene) to the inoculum. Lactate secretion was measured between 42 h and 48 h after transduction as described above. Transduction efficiency was assessed using confocal imaging of blue fluorescent protein (BFP), which was expressed on the same lentiviral vecor downstream of the target glycolytic gene using an IRES, and GFP. Briefly, transduced 3T3 cells were harvested via trypsinization, seeded on black glass-bottom 96 well plates (Cellvis, Mountain View, CA, USA), and allowed to adhere for at least 6 h in normal growth medium. Subsequently, live-cell imaging was carried out with a Nikon Eclipse Ti Spinning Disk Confocal Microscope (Nikon Instruments, Melville, NY, USA), using a 20x objective and 405 nm (BFP) and 488 nm (GFP) laser lines.
Optogenetic stimulation of Ras signaling in 3T3 Opto-SOS cells
Optogenetics experiments were performed as previously described (Goglia et al., 2017). Briefly, clonal 3T3 cells expressing the light-activatable Opto-SOS system were plated in DMEM (10% dFBS) and grown overnight. The next day, 4 h to 6 h prior to stimulation, the cells were washed twice with 1×PBS and serum starved in fresh DMEM (without FBS) supplemented with 10 μm phycocyanobilin (PCB). For all steps following PCB addition, the cells were carefully protected from ambient light (e.g., experiments performed in low-light conditions and plates wrapped in aluminum foil) to prevent activation of the Opto-SOS system. Subsequently, the cells were either exposed to 650 nm red light (stimulated) or were left in the dark (unstimulated) until the end of the experiment. Glycolytic flux measurements were performed in 6-well plates and change in lactate secretion was assessed after 6 h of stimulation as described above. For metabolomics experiments, cells were grown in 6 cm dishes and intracellular metabolites were extracted and analyzed after 4 h and 24 h of stimulation as described earlier. Cell growth was determined by measuring change in PCV at 24 h post-stimulation.
Kinetic glycolytic flux profiling of stimulated and unstimulated cells using U-13C-glucose was performed in 6 cm dishes. Serum starved, PCB-treated cells were either pre-exposed to activating red light or left in the dark for 30 min before washing twice with 1×PBS and switching to 2 ml of U-13C-glucose-containing DMEM. Cells were subsequently collected at the indicated time points and subjected to LC-MS analysis as described above.
RNAseq analysis of 3T3 Opto-SOS cells
Changes in gene expression of 3T3 Opto-SOS cells upon Ras activation were reanalyzed from a previously published dataset (GEO accession # GSE100816) (Wilson et al., 2017). The reads for the light activated samples (0, 30, 60, and 120 min) were analyzed using the Galaxy Workflow system (Afgan et al., 2016) and custom scripts (https://bitbucket.org/princeton_genomics/ltanner_glycolosis_flux_rnaseq/) implemented in R (R Core Team, 2017). Briefly, adapters were removed and reads were quality trimmed using Trim Galore! V0.4.3.1 with default parameters (Krueger, 2017). The trimmed reads were mapped to the GRCm38/mm10 genome using Tophat 2.1.1 (Kim et al., 2013). The GTF option was used with the genes.gtf file from Illumina’s iGenomes (https://support.illumina.com/sequencing/sequencing_software/igenome.html) and the no-novel-juncs option was specified to ensure all samples used the same set of splice junctions. FeatureCounts 1.4.6.p5 (Liao et al., 2014) was used to determine the read counts for each gene specified in the iGenomes gtf file. Finally, DESeq2 1.16.1 (Love et al., 2014) was used to calculate normalization factors for each sample. Technical replicates were combined and genes with counts lower than 200 were excluded from further analysis. Differential expression was calculated at each time point compared to 0 min and the data was presented as log2 fold-ratios. Gene set enrichment analysis (GSEA) was performed using the GSEA software (Mootha et al., 2003; Subramanian et al., 2005). Briefly, only genes exhibiting a two-fold change on at least one time point were chosen and enrichment for hallmark gene sets and KEGG annotated gene sets was calculated.
Gene expression analysis of TCGA data
RNAseq data from human solid tumor tissue samples were downloaded from The Cancer Genome Atlas (TCGA) Research Network (http://cancergenome.nih.gov/) using the open-source software TCGA-assembler (Lu et al., 2010) executed in R (R Core Team, 2017). Only primary tumor samples (TCGA barcode identifier: 01) with paired adjacent normal tissue samples (TCGA barcode identifier: 11) were considered for further analysis. This resulted in a final set of 666 paired human solid tumor tissue samples from 19 different tumor types (Table S2).
A list of 40 glycolytic enzymes and transporters was manually curated based on annotation in KEGG pathways (Kanehisa and Goto, 2000; Kanehisa et al., 2016, 2017). Gene expression of primary tumor tissue samples was normalized to the corresponding adjacent healthy tissue samples and the normalized values were presented as a log2 fold ratio. Gene and samples were clustered by pearson correlation distances (centered) with average-linkage implemented in Cluster3.0 (de Hoon et al., 2004). Calculated dendrograms and heat maps were subsequently visualized using Java TreeView (Saldanha, 2004). The clustered dendrogram heat map was generated using aheatmap command in the R package NMF (Gaujoux and Seoighe, 2010).
For rank order analysis of the 40 glycolytic genes, the average fold-expression change relative to benign adjacent tissue of different glycolytic genes across all samples of a given tumor type was ranked from highest to lowest. The topmost up-regulated GLUT, PFK and MCT isoforms were highlighted until at least one gene isoform of each of these three glycolytic steps was covered.
To determine the four most up-regulated glycolytic steps in each tumor sample, gene isoforms were ranked based on their extent of up-regulation relative to adjacent benign tissue. Subsequently, the frequency with which gene isoforms were among the four topmost up-regulated glycolytic genes was tabulated. Gluconeogenic and anti-glycolytic (FBP1, PFKFB2 &TIGAR), poorly annotated (HKDC1) and pyruvate dehydrogenase (PDHA1 & PDHB) gene isoforms were excluded from this analysis (Table S3). A Fisher’s exact ttest (with Bonferroni correction for multiple hypothesis testing) was performed to probe the statistical significance of whether a given isoform is more likely to rank in the four top most up-regulated genes than expected.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analyses were done in R (R Core Team, 2017). The following packages were additionally used for general data preparation: dplyr (Wickham and Francois, 2016), plotrix (Lemon, 2006), reshape/reshape2 (Wickham, 2007), stringr (Wickham, 2017a) and tidyr (Wickham, 2017b). For all experiments, mean and 95% confidence intervals of at least n=3 biological replicates were plotted unless stated otherwise. Statistically significant differences (p<0.05) were calculated by two-tailed unpaired Student’s t test and multiple hypothesis correction (q<0.05) was performed using the qvalue package (Storey, 2015). Michaelis-Menten curves were fitted using the nls function and R2 for fits were calculated by the residual sum of squares (RSS) and total sum of squares (TSS) using the following formula:
| (Equation 6) |
95% confidence intervals of the Michaelis-Menten fit were calculated using the drc package (Ritz et al., 2015). P values for Michaelis-Menten fits were calculated by linear regression of the predicted versus measured values. Metabolomics heat maps were generated with the pheatmap package (Kolde, 2015). All other plots were generated using ggplot2 (Wickham, 2009) and all the figures were formatted in Illustrator (Adobe, San Jose, CA, USA).
Supplementary Material
Table S1: Primers used for cloning, Related to Star Methods.
Highlights.
Systematic analysis identifies four key flux-controlling steps in glycolysis
Hexokinase and phosphofructokinase promote glycolytic flux
Glucose and lactate transport (but not lower glycolysis) also control pathway flux
Activation of glycolysis in cancer is achieved by induction of the four key steps
Acknowledgments
The authors thank Wenyun Lu for help with mass spectrometry analysis, Max Homilius for help with the TCGA data analysis, and Gregory S. Ducker and the other Rabinowitz lab members for helpful comments and discussions. This work was supported by grants from the NIH (R01 CA163591 and DP1 DK113643) and from Stand Up To Cancer (Dream Team Translational Research Grant SU2C-AACR-DT2016) to J.D.R., by grants from the NIH (DP2EB024247) and from the American Cancer Society (IRG-15-168-01) to J.E.T., by an Early Postdoc Mobility fellowship from the Swiss National Science Foundation (P2SKP3-148484) to L.B.T., and by an NIH National Cancer Institute fellowship (F30CA206408) to A.G.G. Stand Up To Cancer is a program of the Entertainment Industry Foundation, administered by the American Association for Cancer Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
Conceptualization, L.B.T., J.E.T. and J.D.R.; Investigation, L.B.T., A.G.G., M.H.W. and T.S.; Formal Analysis, L.B.T., L.R.P., J.O.P. and J.D.R.; Resources, E.H.W., J.E.T. and J.D.R.; Writing – Original Draft, L.B.T, A.G.G. and J.D.R. with inputs from the other authors; Writing – Review & Editing, L.B.T, A.G.G. and J.D.R.; Funding Acquisition, J.E.T. and J.D.R.; Supervision, J.E.T. and J.D.R.
DECLARATION OF INTERESTS
The authors declare no competing interests.
References
- Abdelmalek MF, Lazo M, Horska A, Bonekamp S, Lipkin EW, Balasubramanyam A, Bantle JP, Johnson RJ, Diehl AM, Clark JM, et al. Higher dietary fructose is associated with impaired hepatic adenosine triphosphate homeostasis in obese individuals with type 2 diabetes. Hepatology. 2012;56:952–960. doi: 10.1002/hep.25741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abu-Amero KK, Alzahrani AS, Zou M, Shi Y. High frequency of somatic mitochondrial DNA mutations in human thyroid carcinomas and complex I respiratory defect in thyroid cancer cell lines. Oncogene. 2005;24:1455–1460. doi: 10.1038/sj.onc.1208292. [DOI] [PubMed] [Google Scholar]
- Van Aelst L, Hohmann S, Bulaya B, de Koning W, Sierkstra L, Neves MJ, Luyten K, Alijo R, Ramos J, Coccetti P. Molecular cloning of a gene involved in glucose sensing in the yeast Saccharomyces cerevisiae. Mol Microbiol. 1993;8:927–943. doi: 10.1111/j.1365-2958.1993.tb01638.x. [DOI] [PubMed] [Google Scholar]
- Afgan E, Baker D, van den Beek M, Blankenberg D, Bouvier D, Čech M, Chilton J, Clements D, Coraor N, Eberhard C, et al. The {Galaxy} platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res. 2016;44:W3–W10. doi: 10.1093/nar/gkw343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensaad K, Tsuruta A, Selak MA, Vidal MNC, Nakano K, Bartrons R, Gottlieb E, Vousden KH. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006;126:107–120. doi: 10.1016/j.cell.2006.05.036. [DOI] [PubMed] [Google Scholar]
- Berg JM, Jeremy M, Tymoczko JL, Stryer L, Stryer L. Biochemistry. W.H. Freeman; 2002. [Google Scholar]
- Birsoy K, Possemato R, Lorbeer FK, Bayraktar EC, Thiru P, Yucel B, Wang T, Chen WW, Clish CB, Sabatini DM. Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature. 2014;508:108–112. doi: 10.1038/nature13110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bluemlein K, Grüning NM, Feichtinger RG, Lehrach H, Kofler B, Ralser M. No evidence for a shift in pyruvate kinase PKM1 to PKM2 expression during tumorigenesis. Oncotarget. 2011;2:393–400. doi: 10.18632/oncotarget.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck MD, Sowell RT, Kaech SM, Pearce EL. Metabolic Instruction of Immunity. Cell. 2017;169:570–586. doi: 10.1016/j.cell.2017.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, Fleming MD, Schreiber SL, Cantley LC. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452:230–233. doi: 10.1038/nature06734. [DOI] [PubMed] [Google Scholar]
- Collard F, Baldin F, Gerin I, Bolsée J, Noël G, Graff J, Veiga-da-Cunha M, Stroobant V, Vertommen D, Houddane A, et al. A conserved phosphatase destroys toxic glycolytic side products in mammals and yeast. Nat Chem Biol. 2016;12:601–607. doi: 10.1038/nchembio.2104. [DOI] [PubMed] [Google Scholar]
- Dai Z, Shestov AA, Lai L, Locasale JW. A Flux Balance of Glucose Metabolism Clarifies the Requirements of the Warburg Effect. Biophys J. 2016;111:1088–1100. doi: 10.1016/j.bpj.2016.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV. Links between metabolism and cancer. Genes Dev. 2012;26:877–890. doi: 10.1101/gad.189365.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis CF, Ricketts CJ, Wang M, Yang L, Cherniack AD, Shen H, Buhay C, Kang H, Kim SC, Fahey CC, et al. The Somatic Genomic Landscape of Chromophobe Renal Cell Carcinoma. Cancer Cell. 2014;26:319–330. doi: 10.1016/j.ccr.2014.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayton TL, Jacks T, Vander Heiden MG. PKM2, cancer metabolism, and the road ahead. EMBO Rep. 2016;17:1721–1730. doi: 10.15252/embr.201643300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bock K, Georgiadou M, Schoors S, Kuchnio A, Wong BW, Cantelmo AR, Quaegebeur A, Ghesquière B, Cauwenberghs S, Eelen G, et al. Role of PFKFB3-Driven Glycolysis in Vessel Sprouting. Cell. 2013;154:651–663. doi: 10.1016/j.cell.2013.06.037. [DOI] [PubMed] [Google Scholar]
- DeBerardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv. 2016;2:e1600200–e1600200. doi: 10.1126/sciadv.1600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernandes JR, De Meirsman C, Rolland F, Winderickx J, de Winde J, Brandão RL, Thevelein JM. During the initiation of fermentation overexpression of hexokinase PII in yeast transiently causes a similar deregulation of glycolysis as deletion of Tps1. Yeast. 1998;14:255–269. doi: 10.1002/(SICI)1097-0061(199802)14:3<255::AID-YEA228>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Everts B, Amiel E, Huang SCC, Smith AM, Chang CH, Lam WY, Redmann V, Freitas TC, Blagih J, van der Windt GJW, et al. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKε supports the anabolic demands of dendritic cell activation. Nat Immunol. 2014;15:323–332. doi: 10.1038/ni.2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J, Kamphorst JJ, Mathew R, Chung MK, White E, Shlomi T, Rabinowitz JD. Glutamine-driven oxidative phosphorylation is a major ATP source in transformed mammalian cells in both normoxia and hypoxia. Mol Syst Biol. 2014;9:712–712. doi: 10.1038/msb.2013.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faubert B, Li KY, Cai L, Hensley CT, Kim J, Zacharias LG, Yang C, Do QN, Doucette S, Burguete D, et al. Lactate Metabolism in Human Lung Tumors. Cell. 2017;171:358–371.e9. doi: 10.1016/j.cell.2017.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fell DA. Metabolic control analysis: a survey of its theoretical and experimental development. Biochem J. 1992;286(Pt 2):313–330. doi: 10.1042/bj2860313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fell DA. Increasing the flux in metabolic pathways: A metabolic control analysis perspective. Biotechnol Bioeng. 1998;58:121–124. doi: 10.1002/(sici)1097-0290(19980420)58:2/3<121::aid-bit2>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- Flier JS, Mueckler MM, Usher P, Lodish HF. Elevated levels of glucose transport and transporter messenger RNA are induced by ras or src oncogenes. Science. 1987;235:1492–1495. doi: 10.1126/science.3103217. [DOI] [PubMed] [Google Scholar]
- Gaglio D, Metallo CM, Gameiro PA, Hiller K, Danna LS, Balestrieri C, Alberghina L, Stephanopoulos G, Chiaradonna F. Oncogenic K-Ras decouples glucose and glutamine metabolism to support cancer cell growth. Mol Syst Biol. 2011;7:523. doi: 10.1038/msb.2011.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garland PB, Randle PJ, Newsholme EA. Citrate as an Intermediary in the Inhibition of Phosphofructokinase in Rat Heart Muscle by Fatty Acids, Ketone Bodies, Pyruvate, Diabetes and Starvation. Nature. 1963;200:169–170. doi: 10.1038/200169a0. [DOI] [PubMed] [Google Scholar]
- Gaujoux R, Seoighe C. A flexible R package for nonnegative matrix factorization. BMC Bioinformatics. 2010;11:367. doi: 10.1186/1471-2105-11-367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerin I, Noël G, Bolsée J, Haumont O, Van Schaftingen E, Bommer GT. Identification of TP53-induced glycolysis and apoptosis regulator (TIGAR) as the phosphoglycolate-independent 2,3-bisphosphoglycerate phosphatase. Biochem J. 2014;458:439–448. doi: 10.1042/BJ20130841. [DOI] [PubMed] [Google Scholar]
- Goglia AG, Wilson MZ, DiGiorno DB, Toettcher JE. Optogenetic Control of Ras/Erk Signaling Using the Phy-PIF System. Methods Mol Biol. 2017;1636:3–20. doi: 10.1007/978-1-4939-7154-1_1. [DOI] [PubMed] [Google Scholar]
- González MI, Blázquez MA, Gancedo C, Stucka R, Feldmann H. Molecular cloning ofCIF1, a yeast gene necessary for growth on glucose. Yeast. 1992;8:183–192. doi: 10.1002/yea.320080304. [DOI] [PubMed] [Google Scholar]
- Graham NA, Minasyan A, Lomova A, Cass A, Balanis NG, Friedman M, Chan S, Zhao S, Delgado A, Go J, et al. Recurrent patterns of DNA copy number alterations in tumors reflect metabolic selection pressures. Mol Syst Biol. 2017;13:914. doi: 10.15252/msb.20167159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackett SR, Zanotelli VRT, Xu W, Goya J, Park JO, Perlman DH, Gibney PA, Botstein D, Storey JD, Rabinowitz JD. Systems-level analysis of mechanisms regulating yeast metabolic flux. Science (80−) 2016;354:aaf2786–aaf2786. doi: 10.1126/science.aaf2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay N. Reprogramming glucose metabolism in cancer: can it be exploited for cancer therapy? Nat Rev Cancer. 2016;16:635–649. doi: 10.1038/nrc.2016.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, DeBerardinis RJ. Understanding the Intersections between Metabolism and Cancer Biology. Cell. 2017;168:657–669. doi: 10.1016/j.cell.2016.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science (80−) 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinrich R, Rapoport TA. A linear steady-state treatment of enzymatic chains. General properties, control and effector strength. Eur J Biochem. 1974;42:89–95. doi: 10.1111/j.1432-1033.1974.tb03318.x. [DOI] [PubMed] [Google Scholar]
- de Hoon MJL, Imoto S, Nolan J, Miyano S. Open source clustering software. Bioinformatics. 2004;20:1453–1454. doi: 10.1093/bioinformatics/bth078. [DOI] [PubMed] [Google Scholar]
- Hu H, Juvekar A, Lyssiotis CA, Lien EC, Albeck JG, Oh D, Varma G, Hung YP, Ullas S, Lauring J, et al. Phosphoinositide 3-Kinase Regulates Glycolysis through Mobilization of Aldolase from the Actin Cytoskeleton. Cell. 2016;164:433–446. doi: 10.1016/j.cell.2015.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Locasale JW, Bielas JH, O’Sullivan J, Sheahan K, Cantley LC, Vander Heiden MG, Vitkup D. Heterogeneity of tumor-induced gene expression changes in the human metabolic network. Nat Biotechnol. 2013;31:522–529. doi: 10.1038/nbt.2530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui S, Ghergurovich JM, Morscher RJ, Jang C, Teng X, Lu W, Esparza LA, Reya T, Le Zhan Yanxiang, Guo J, et al. Glucose feeds the TCA cycle via circulating lactate. Nature. 2017;551:115–118. doi: 10.1038/nature24057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huijing F, Slater EC. The use of oligomycin as an inhibitor of oxidative phosphorylation. J Biochem. 1961;49:493–501. doi: 10.1093/oxfordjournals.jbchem.a127334. [DOI] [PubMed] [Google Scholar]
- Ishimoto T, Lanaspa MA, Le MT, Garcia GE, Diggle CP, Maclean PS, Jackman MR, Asipu A, Roncal-Jimenez CA, Kosugi T, et al. Opposing effects of fructokinase C and A isoforms on fructose-induced metabolic syndrome in mice. Proc Natl Acad Sci U S A. 2012;109:4320–4325. doi: 10.1073/pnas.1119908109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong HW, Hernández-Rodríguez B, Kim J, Kim KP, Enriquez-Gasca R, Yoon J, Adams S, Schöler HR, Vaquerizas JM, Adams RH. Transcriptional regulation of endothelial cell behavior during sprouting angiogenesis. Yeast. 2017;14:255–269. doi: 10.1038/s41467-017-00738-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha AK, Huang SCC, Sergushichev A, Lampropoulou V, Ivanova Y, Loginicheva E, Chmielewski K, Stewart KM, Ashall J, Everts B, et al. Network Integration of Parallel Metabolic and Transcriptional Data Reveals Metabolic Modules that Regulate Macrophage Polarization. Immunity. 2015;42:419–430. doi: 10.1016/j.immuni.2015.02.005. [DOI] [PubMed] [Google Scholar]
- Johnson RJ, Nakagawa T, Sanchez-Lozada LG, Shafiu M, Sundaram S, Le M, Ishimoto T, Sautin YY, Lanaspa MA. Sugar, Uric Acid, and the Etiology of Diabetes and Obesity. Diabetes. 2013;62:3307–3315. doi: 10.2337/db12-1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kacser H, Burns JA. The control of flux. Symp Soc Exp Biol. 1973;27:65–104. [PubMed] [Google Scholar]
- Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Sato Y, Kawashima M, Furumichi M, Tanabe M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016;44:D457–D462. doi: 10.1093/nar/gkv1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017;45:D353–D361. doi: 10.1093/nar/gkw1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. {TopHat}2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochanowski K, Volkmer B, Gerosa L, Haverkorn van Rijsewijk BR, Schmidt A, Heinemann M. Functioning of a metabolic flux sensor in Escherichia coli. Proc Natl Acad Sci. 2013;110:1130–1135. doi: 10.1073/pnas.1202582110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolde R. Pheatmap: Pretty Heatmaps 2015 [Google Scholar]
- Kotte O, Zaugg JB, Heinemann M. Bacterial adaptation through distributed sensing of metabolic fluxes. Mol Syst Biol. 2010;6:355. doi: 10.1038/msb.2010.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger FB. Bioinformatics - Trim Galore! 2017 [Google Scholar]
- Lardy HA, Johnson D, McMurray WC. Antibiotics as tools for metabolic studies. I. A survey of toxic antibiotics in respiratory, phosphorylative and glycolytic systems. Arch Biochem Biophys. 1958;78:587–597. doi: 10.1016/0003-9861(58)90383-7. [DOI] [PubMed] [Google Scholar]
- Lemon J. Plotrix: a package in the red light district of R. R-News. 2006;6:8–12. [Google Scholar]
- Li B, Qiu B, Lee DSM, Walton ZE, Ochocki JD, Mathew LK, Mancuso A, Gade TPF, Keith B, Nissim I, et al. Fructose-1,6-bisphosphatase opposes renal carcinoma progression. Nature. 2014;513:251–255. doi: 10.1038/nature13557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W. {featureCounts}: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- Liberti MV, Locasale JW. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem Sci. 2016;41:211–218. doi: 10.1016/j.tibs.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberti MV, Dai Z, Wardell SE, Baccile JA, Liu X, Gao X, Baldi R, Mehrmohamadi M, Johnson MO, Madhukar NS, et al. A Predictive Model for Selective Targeting of the Warburg Effect through GAPDH Inhibition with a Natural Product. Cell Metab. 2017;26:648–659.e8. doi: 10.1016/j.cmet.2017.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llorente P, Marco R, Sols A. Regulation of Liver Pyruvate Kinase and the Phosphoenolpyruvate Crossroads. Eur J Biochem. 1970;13:45–54. doi: 10.1111/j.1432-1033.1970.tb00897.x. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for {RNA}-seq data with {DESeq}2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Clasquin MF, Melamud E, Amador-Noguez D, Caudy AA, Rabinowitz JD. Metabolomic Analysis via Reversed-Phase Ion-Pairing Liquid Chromatography Coupled to a Stand Alone Orbitrap Mass Spectrometer. Anal Chem. 2010;82:3212–3221. doi: 10.1021/ac902837x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunt SY, Vander Heiden MG. Aerobic Glycolysis: Meeting the Metabolic Requirements of Cell Proliferation. Annu Rev Cell Dev Biol. 2011;27:441–464. doi: 10.1146/annurev-cellbio-092910-154237. [DOI] [PubMed] [Google Scholar]
- Marín-Hernández A, Rodríguez-Enríquez S, Vital-González PA, Flores-Rodríguez FL, Macías-Silva M, Sosa-Garrocho M, Moreno-Sánchez R. Determining and understanding the control of glycolysis in fast-growth tumor cells. FEBS J. 2006;273:1975–1988. doi: 10.1111/j.1742-4658.2006.05214.x. [DOI] [PubMed] [Google Scholar]
- Melamud E, Vastag L, Rabinowitz JD. Metabolomic Analysis and Visualization Engine for LC–MS Data. Anal Chem. 2010;82:9818–9826. doi: 10.1021/ac1021166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstråle M, Laurila E, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- Newsholme EA, Sugden PH, Williams T. Effect of citrate on the activities of 6-phosphofructokinase from nervous and muscle tissues from different animals and its relationships to the regulation of glycolysis. Biochem J. 1977;166:123–129. doi: 10.1042/bj1660123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson R, Jain M, Madhusudhan N, Sheppard NG, Strittmatter L, Kampf C, Huang J, Asplund A, Mootha VK. Metabolic enzyme expression highlights a key role for MTHFD2 and the mitochondrial folate pathway in cancer. Nat Commun. 2014;5:3128. doi: 10.1038/ncomms4128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noor E, Bar-Even A, Flamholz A, Reznik E, Liebermeister W, Milo R. Pathway Thermodynamics Highlights Kinetic Obstacles in Central Metabolism. PLoS Comput Biol. 2014;10:e1003483. doi: 10.1371/journal.pcbi.1003483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onodera Y, Nam JM, Bissell MJ. Increased sugar uptake promotes oncogenesis via EPAC/RAP1 and O-GlcNAc pathways. J Clin Invest. 2014;124:367–384. doi: 10.1172/JCI63146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JO, Rubin SA, Xu YF, Amador-Noguez D, Fan J, Shlomi T, Rabinowitz JD. Metabolite concentrations, fluxes and free energies imply efficient enzyme usage. Nat Chem Biol. 2016;12:482–489. doi: 10.1038/nchembio.2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patra KC, Wang Q, Bhaskar PT, Miller L, Wang Z, Wheaton W, Chandel N, Laakso M, Muller WJ, Allen EL, et al. Hexokinase 2 Is Required for Tumor Initiation and Maintenance and Its Systemic Deletion Is Therapeutic in Mouse Models of Cancer. Cancer Cell. 2013;24:213–228. doi: 10.1016/j.ccr.2013.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peeters K, Van Leemputte F, Fischer B, Bonini BM, Quezada H, Tsytlonok M, Haesen D, Vanthienen W, Bernardes N, Gonzalez-Blas CB, et al. Fructose-1,6-bisphosphate couples glycolytic flux to activation of Ras. Nat Commun. 2017;8:922. doi: 10.1038/s41467-017-01019-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prior IA, Lewis PD, Mattos C. A Comprehensive Survey of Ras Mutations in Cancer. Cancer Res. 2012;72:2457–2467. doi: 10.1158/0008-5472.CAN-11-2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racker E, Resnick RJ, Feldman R. Glycolysis and methylaminoisobutyrate uptake in rat-1 cells transfected with ras or myc oncogenes. Proc Natl Acad Sci U S A. 1985;82:3535–3538. doi: 10.1073/pnas.82.11.3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing 2017 [Google Scholar]
- Ralser M, Wamelink MM, Kowald A, Gerisch B, Heeren G, Struys EA, Klipp E, Jakobs C, Breitenbach M, Lehrach H, et al. Dynamic rerouting of the carbohydrate flux is key to counteracting oxidative stress. J Biol. 2007;6:10. doi: 10.1186/jbiol61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ralser M, Wamelink MMC, Latkolik S, Jansen EEW, Lehrach H, Jakobs C. Metabolic reconfiguration precedes transcriptional regulation in the antioxidant response. Nat Biotechnol. 2009;27:604–605. doi: 10.1038/nbt0709-604. [DOI] [PubMed] [Google Scholar]
- Ritz C, Baty F, Streibig JC, Gerhard D. Dose-Response Analysis Using R. PLoS One. 2015;10:e0146021. doi: 10.1371/journal.pone.0146021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron-Harel N, Santos D, Ghergurovich JM, Sage PT, Reddy A, Lovitch SB, Dephoure N, Satterstrom FK, Sheffer M, Spinelli JB, et al. Mitochondrial Biogenesis and Proteome Remodeling Promote One-Carbon Metabolism for T Cell Activation. Cell Metab. 2016;24:104–117. doi: 10.1016/j.cmet.2016.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ros S, Schulze A. Balancing glycolytic flux: the role of 6-phosphofructo-2-kinase/fructose 2,6-bisphosphatases in cancer metabolism. Cancer Metab. 2013;1:8. doi: 10.1186/2049-3002-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldanha AJ. Java Treeview–extensible visualization of microarray data. Bioinformatics. 2004;20:3246–3248. doi: 10.1093/bioinformatics/bth349. [DOI] [PubMed] [Google Scholar]
- Shestov AA, Liu X, Ser Z, Cluntun AA, Hung YP, Huang L, Kim D, Le A, Yellen G, Albeck JG, et al. Quantitative determinants of aerobic glycolysis identify flux through the enzyme GAPDH as a limiting step. Elife. 2014;3:1278. doi: 10.7554/eLife.03342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim H, Dolde C, Lewis BC, Wu CS, Dang G, Jungmann RA, Dalla-Favera R, Dang CV. c-Myc transactivation of LDH-A: implications for tumor metabolism and growth. Proc Natl Acad Sci U S A. 1997;94:6658–6663. doi: 10.1073/pnas.94.13.6658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small JR, Kacser H. Responses of metabolic systems to large changes in enzyme activities and effectors. 1. The linear treatment of unbranched chains. Eur J Biochem. 1993a;213:613–624. doi: 10.1111/j.1432-1033.1993.tb17801.x. [DOI] [PubMed] [Google Scholar]
- Small JR, Kacser H. Responses of metabolic systems to large changes in enzyme activities and effectors. 2. The linear treatment of branched pathways and metabolite concentrations. Assessment of the general non-linear case. Eur J Biochem. 1993b;213:625–640. doi: 10.1111/j.1432-1033.1993.tb17802.x. [DOI] [PubMed] [Google Scholar]
- Storey JD. Q-value estimation for false discovery rate control 2015 [Google Scholar]
- Su X, Wang W, Ruan G, Liang M, Zheng J, Chen Y, Wu H, Fahey T, Guan M, Teng L. A Comprehensive Characterization Mitochondrial Genome in Papillary Thyroid Cancer. Int J Mol Sci. 2016;17:1594. doi: 10.3390/ijms17101594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tejwani GA. Regulation of fructose-bisphosphatase activity. Adv. Enzymol. Relat. Areas Mol Biol. 1983;54:121–194. doi: 10.1002/9780470122990.ch3. [DOI] [PubMed] [Google Scholar]
- Toettcher JE, Weiner OD, Lim WA. Using optogenetics to interrogate the dynamic control of signal transmission by the Ras/Erk module. Cell. 2013;155:1422–1434. doi: 10.1016/j.cell.2013.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres NV, Mateo F, Meléndez-Hevia E, Kacser H. Kinetics of metabolic pathways. A system in vitro to study the control of flux. Biochem J. 1986;234:169–174. doi: 10.1042/bj2340169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, McCormick LL, Fitzgerald P, Chi H, Munger J, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity. 2011;35:871–882. doi: 10.1016/j.immuni.2011.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- Webb BA, Forouhar F, Szu FE, Seetharaman J, Tong L, Barber DL. Structures of human phosphofructokinase-1 and atomic basis of cancer-associated mutations. Nature. 2015;523:111–114. doi: 10.1038/nature14405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H. Reshaping data with the reshape package. J Stat Softw. 2007;21 [Google Scholar]
- Wickham H. ggplot2: Elegant Graphics for Data Analysis. New York: Springer-Verlag; 2009. [Google Scholar]
- Wickham H. Stringr: Simple, Consistent Wrappers for Common String Operations 2017a [Google Scholar]
- Wickham H. Tidyr: Easily Tidy Data with “spread()” and “gather()” Functions 2017b [Google Scholar]
- Wickham H, Francois R. Dplyr: A Grammar of Data Manipulation 2016 [Google Scholar]
- Wilson MZ, Ravindran PT, Lim WA, Toettcher JE. Tracing Information Flow from Erk to Target Gene Induction Reveals Mechanisms of Dynamic and Combinatorial Control. Mol Cell. 2017 doi: 10.1016/j.molcel.2017.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi W, Clark PM, Mason DE, Keenan MC, Hill C, Goddard WA, Peters EC, Driggers EM, Hsieh-Wilson LC. Phosphofructokinase 1 Glycosylation Regulates Cell Growth and Metabolism. Science (80−) 2012;337:975–980. doi: 10.1126/science.1222278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, Locasale JW, Son J, Zhang H, Coloff JL, et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell. 2012;149:656–670. doi: 10.1016/j.cell.2012.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu P, Wilhelm K, Dubrac A, Tung JK, Alves TC, Fang JS, Xie Y, Zhu J, Chen Z, De Smet F, et al. FGF-dependent metabolic control of vascular development. Nature. 2017;545:224–228. doi: 10.1038/nature22322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun J, Rago C, Cheong I, Pagliarini R, Angenendt P, Rajagopalan H, Schmidt K, Willson JKV, Markowitz S, Zhou S, et al. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science. 2009;325:1555–1559. doi: 10.1126/science.1174229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun J, Mullarky E, Lu C, Bosch KN, Kavalier A, Rivera K, Roper J, Chio IIC, Giannopoulou EG, Rago C, et al. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science. 2015;350:1391–1396. doi: 10.1126/science.aaa5004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CS, Hawley SA, Zong Y, Li M, Wang Z, Gray A, Ma T, Cui J, Feng JW, Zhu M, et al. Fructose-1,6-bisphosphate and aldolase mediate glucose sensing by AMPK. Nature. 2017;548:112–116. doi: 10.1038/nature23275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong WX, Rabinowitz JD, White E. Mitochondria and Cancer. Mol Cell. 2016;61:667–676. doi: 10.1016/j.molcel.2016.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zu XL, Guppy M. Cancer metabolism: facts, fantasy, and fiction. Biochem. Biophys. Res Commun. 2004;313:459–465. doi: 10.1016/j.bbrc.2003.11.136. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: Primers used for cloning, Related to Star Methods.